

Abstract
SURF1 (surfeit locus protein 1)-related Leigh syndrome is an early-onset neurodegenerative disorder, characterized by reduction in complex IV activity, resulting in disrupted mitochondrial function. Currently, there are no treatment options available. To test our hypothesis that adeno-associated viral vector serotype 9 (AAV9)/human SURF1 (hSURF1) gene replacement therapy can provide a potentially meaningful and long-term therapeutic benefit, we conducted preclinical efficacy studies using SURF1 knockout mice and safety evaluations with wild-type (WT) mice. Our data indicate that with a single intrathecal (i.t.) administration, our treatment partially and significantly rescued complex IV activity in all tissues tested, including liver, brain, and muscle. Accordingly, complex IV content (examined via MT-CO1 protein expression level) also increased with our treatment. In a separate group of mice, AAV9/hSURF1 mitigated the blood lactic acidosis induced by exhaustive exercise at 9 months post-dosing. A toxicity study in WT mice showed no adverse effects in either the in-life portion or after microscopic examination of major tissues up to a year following the same treatment regimen. Taken together, our data suggest a single dose, i.t. administration of AAV9/hSURF1 is safe and effective in improving biochemical abnormalities induced by SURF1 deficiency with potential applicability for SURF1-related Leigh syndrome patients.
Keywords: SURF1, Leigh syndrome, gene therapy, adeno-associated virus, AAV
Graphical abstract
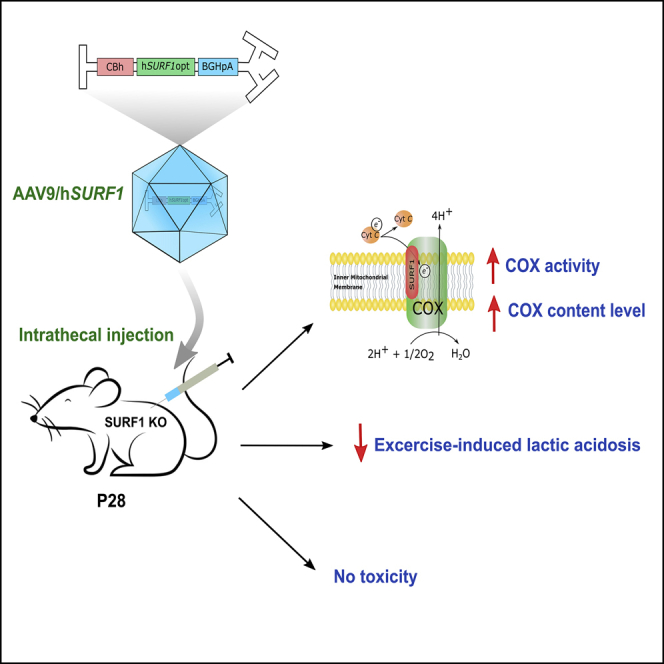
This study evaluated the safety and efficacy of SURF1 gene transfer using AAV9 vectors to restore biochemical abnormalities in a SURF1 Leigh syndrome mouse model. Our data suggested that gene replacement therapy through intrathecal administration of AAV9/hSURF1 is worthy of further development as a potential treatment for SURF1-related Leigh syndrome patients.
Introduction
Leigh syndrome (LS) (OMIM: 256000), or subacute necrotizing encephalomyopathy, is a recessively early onset mitochondrial disorder. Patients show asymmetric necrotic lesions in basal ganglia, diencephalon, brain stem, and spinal cord, and patients generally develop motor and neurological disorders during early infancy.1,2 One of the most common causes of LS is from loss-of-function mutations in the nuclear genome coding the SURF1 (surfeit locus protein 1) gene.3,4 Mutations of SURF1 are typically autosomal recessive, in which a patient inherits one defective copy from each healthy carrier parent.4 SURF1 protein localizes in the inner mitochondrial membrane and is predicted to form two transmembrane helices with a connecting loop facing the intermembrane space.5 Evidence suggested that SURF1 may contribute to assembly or stabilization of complex IV (cIV)/cytochrome c oxidase (COX), the fourth complex of the mitochondrial oxidative phosphorylation (OXPHOS) system.6,7 In most cases, patients with SURF1-related LS show 10%–20% remaining COX activity as examined with muscle or skin fibroblasts.1,8,9 Additionally, 81% of patients show elevated blood lactate levels.9
So far, there is no approved treatment for this devastating disease besides palliative care. Therapies for most patients with mitochondrial disorders in general are limited to preventing or treating complications. During the last two decades, viral vector-based gene therapy has been extensively explored as a potential treatment for genetic diseases.10 In particular, the recombinant adeno-associated viral vector (AAV) has been shown in numerous studies to be a safe and efficacious vector for delivering a transgene to various tissues with tissue specificity from different serotypes.11 The AAV serotype 9 (AAV9) has become a favored choice for CNS gene transfer, due to its ability to mediate widespread transgene expression across the entire CNS tissues of both rodents and non-human primates (NHPs).12, 13, 14, 15 The AAV9 vector has been used in clinical trials for multiple neurological diseases including spinal muscular atrophy (SMA), which was approved in 2017 by the US Food and Drug Administration (FDA), GM1 gangliosidoses (ClinicalTrials.gov: NCT03952637), CLN7 Batten disease (ClinicalTrials.gov: NCT04737460), and several others.11 Our group developed the first-in-human intrathecal (i.t.)-administered AAV clinical trial for giant axonal neuropathy starting in 2015 (ClinicalTrials.gov: NCT02362438).16,17
In this study, we developed a gene transfer vector, in which AAV9 is used to deliver a codon-optimized human SURF1 (hSURF1opt) gene (AAV9/hSURF1) as a potential gene therapy for SURF1-related LS patients. We used the SURF1 knockout (KO) mice as the disease model to evaluate the efficacy of the vector. The mouse model has a frameshift mutation in exon 7 of SURF1, which causes a total loss of SURF1 protein expression.18 Previous studies demonstrated several manifestations of dysfunctional mitochondria such as about 50% reduced complex IV/ COX activity, reduced MT-CO1 (mitochondria-encoded COX subunit 1) protein expression in multiple organs compared to age-matched wild-type (WT) mice, and exercise-induced blood lactic acidosis.18, 19, 20 This experimental therapy is designed to broadly express the hSURF1opt gene in the CNS and thereby rescue the reduced COX activity and MT-CO1 protein level in disease-related tissues, as well as mitigate blood lactic acidosis induced by exhaustive exercise. To explore potential risks associated with the treatment, we also conducted a non-good laboratory practice (GLP) toxicology study in WT C57BL/6J mice utilizing the same dose and administration routes as we did in the efficacy study. Our data suggest that a single dose of i.t. delivered AAV9/hSURF1 is safe, effective, and sufficient to mitigate SURF1 deficiency-related dysfunctions in the SURF1 KO mouse model. These findings strongly support that gene replacement therapy through i.t. administration of AAV9/hSURF1 has the potential for further development as a treatment for SURF1-related LS patients.
Results
Design and expression of a gene transfer vector expressing SURF1
The gene transfer vector was designed as a self-complementary AAV9 (scAAV9) vector carrying hSURF1opt transgene cDNA, the expression of which is controlled by the hybrid chicken β-actin (CBA) (CBh) promoter and the bovine growth hormone (BGH) polyadenylation (poly(A)) (BGHpA) tail (Figure 1A). The modified scAAV9 was chosen here, as it was suggested to transduce 10-fold more cells than did single-stranded AAV9 (ssAAV9).14 The transgene insert is bounded by the AAV2 inverted terminal repeats (ITRs), in which the 5′ ITR is mutated to delete the D element, including the terminal resolution site, to prevent the initiation of replication from the 5′ end.21 Thus, the packing capacity of scAAV9 is limited to only about 2.2 kb of foreign DNA. In this plasmid, the size of the hSURF1opt gene and CBh promoter are 921 and 800 bp, respectively, which are small enough to fit into a self-complementary genome configuration with the BGHpA. The CBh promoter was chosen, as it induced ubiquitous, sustained, and relatively strong hSURF1opt expression.22 Even though SURF1-related LS is a neurodegenerative disease, with the brain and spinal cord being the most damaged tissue, SURF1 is ubiquitously expressed in all organs. Thus, use of the CBh promoter facilitates universal expression of functional SURF1 protein. CBh-hSURF1opt plasmid was tested by transfecting it into HEK293 cells, and Figures 1B and 1C show that the plasmid induced both mRNA and protein expression in HEK293 cells.
Figure 1.

AAV9/hSURF1 vector design for expressing human SURF1
(A) Schematic diagram of the AAV9/hSURF1 gene transfer cassette using a CBh promoter, the full-length, codon-optimized human SURF1 cDNA, and the BGH poly(A) tail. (B) RT-PCR of SURF1 mRNA from HEK293 cells transfected with CBh-hSURF1opt plasmid or CBh-GFP control plasmid for 48 h. (C) Western blot of SURF1 expression from HEK293 cells transfected with CBh-hSURF1opt plasmid or CBh-GFP control plasmid for 48 h.
Intrathecal delivery of AAV9/hSURF1 improved COX activity deficiency
As SURF1 plays a critical role in COX holoenzyme assembly, COX activity deficiency becomes one of the hallmarks of SURF1-related LS patients.6,9 In clinical studies, COX activity is generally measured in muscle or skin biopsies of patients, which show 20%–30% remaining COX activity.9 Previous studies suggest that COX activity was reduced in multiple tissues of SURF1 KO mice compared with WT mice.18,19,23 Thus, we intended to confirm these findings and examine whether AAV9/hSURF1 can rescue COX activity deficiency in brain (cerebrum and cerebellum), liver, and muscle tissues. SURF1 KO mice were treated with AAV9/hSURF1 at a highest dose (8 × 1011 vector genomes [vg]/mouse, KO+High), 4-fold lower dose (2 × 1011 vg/mouse, KO+Low), and vehicle (KO+Vehicle) through i.t. administration. A fourth group of mice were treated with a combination of high dose i.t. and the same dose intravenously (i.v.) (8 × 1011 vg + 8 × 1011 vg)/mouse, KO+High+i.v.), as other studies suggested that combining two different administration routes may enhance the treatment effects.24 All mice were treated at 4 weeks of age and tissues were collected 4 weeks later. As shown in Figure 2, COX activity of SURF1 KO mice was reduced approximately 50% compared with that of WT mice in all tissues except muscle, which was reduced by 35% (p < 0.001). These results are consistent with previous findings.18,25 In the cerebrum (Figure 2A), the low-dose treatment increased by 4% of the KO+Vehicle level (p = 0.3896 compared to the KO+Vehicle group), and the high-dose treatment and combination of high dose i.t. and i.v. treatment increased by 28% and 31%, respectively, of the level of the KO+Vehicle group (p = 0.0362 and 0.0127, respectively). In the cerebellum (Figure 2B), both the low-dose and high-dose treatments increased COX activity by 10% and 12% (p = 0.1528 and 0.1594, respectively, compared with the KO+Vehicle group), while treating the mice i.v. along with high-dose i.t. increased COX activity by 20% (p = 0.0453 compared with the KO+Vehicle group). In the liver, as shown in Figure 2C, COX activity was increased by 43% with low-dose treatment (p = 0.0007 compared with the KO+Vehicle group). Both the high dose and the combination of i.t. and i.v. treatment increased the activity by about 65% (p = 0.0022 and p = 0.0001, respectively, compared with the KO+Vehicle group). As many SURF1-related LS patients also show muscular dystrophy, we also tested the change of COX activity in skeletal muscle. As shown in Figure 2D, COX activity of KO mice showed a 35% reduction compared with WT mice (p < 0.0001), and all of our treatments showed significant improvement in COX activity compared with vehicle-treated KO mice (p = 0.0497 for KO+Low, p = 0.001 for KO+High, and p = 0.0089 for KO+High+i.v.). Thus, our data showed that both high-dose treatment through i.t. and a combination of i.t. and i.v. treatment are effective in improving COX activity deficiency. However, the additional i.v. dose did not show further improvement in any tissue tested over that achieved with a single i.t. dose alone. Taken together, these data suggest that treating SURF1 KO mice with a single high dose of AAV9/hSURF1 through i.t. administration is effective in increasing COX activity in target tissues, which is the underlying driver of the disease pathology.
Figure 2.

AAV9/hSURF1 improved COX activity in SURF1 KO mice at 4 weeks post-injection
(A–D) COX activity of cerebrum (n = 10–18 per group) (A), cerebellum (n = 10–18 per group) (B), liver (n = 5–8 per group) (C), and muscle (n = 10–14 per group) (D) of WT and SURF1 KO mice with assigned treatments. All data were normalized to the average of WT mice. Each data point represents measurement from an individual animal, with bars representing the mean ± SEM (standard error of the mean). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 compared with WT mice using Dunn’s multiple comparison test following a Kruskal-Wallis test for non-normal data, or Tukey’s multiple comparison method following an ordinary one-way ANOVA for normal data. A Shapiro-Wilk’s test was used for the normality of the data distribution, and a Brown-Forsythe test was used for homogeneity of variance. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with KO+Vehicle mice using a one-tailed Student’s t test. A dashed line indicates the level of KO+Vehicle, and the percentage of improvement compared with the KO+Vehicle group from each treatment is provided at the bottom of the respective column.
SURF1 mRNA expression and COX content level in the CNS of mice
To support our findings on COX activity, we evaluated hSURF1opt mRNA expression and MT-CO1 protein expression level in the brain and spinal cord using the tissues from the same mice. A customized probe was used in RNAscope to specifically detect mRNA of the codon-optimized hSURF1 in AAV9/hSURF1-treated mice. Figure 3A shows representative images of hSURF1opt mRNA and MT-CO1 staining. As the probe was specific to hSURF1opt, no signal could be detected in WT or vehicle-treated KO mice. Human SURF1opt mRNA was successfully expressed in all disease-relevant brain areas, the cervical spinal cord (furthest from the injection site), and the lumbar spinal cord (closest to the injection site) of AAV9/hSURF1-treated animals (Figure 3B). Similar to the effects on COX activity, the combinational treatment of i.t. and i.v. administration did not significantly improve the mRNA expression level over i.t. administration alone. Previous studies have shown that the expression level of MT-CO1 protein correlates with COX activity,26 and in SURF1 KO mice, the expression of MT-CO1 is reduced.20 As shown in Figure 3C, the expression of MT-CO1 is lowest in vehicle-treated KO mice and highest in WT mice, while the levels of AAV9/hSURF1-treated animals were between those two. This trend of improvement in MT-CO1 expression is significantly correlated with the trend of COX activity level in both cerebrum (Figure 3D) and cerebellum (Figure 3E). Graphs with individual values from all the analyses are shown in Figure S1 (hSURF1opt mRNA) and Figure S2 (MT-CO1 expression).
Figure 3.

hSURF1opt mRNA and MT-CO1 protein expression in CNS tissues
(A) Representative images of hSURF1opt mRNA staining in brain areas (striatum, hypothalamus, thalamus, midbrain, cerebellum, and hindbrain) and spinal cord sections (cervical cord and lumbar cord). The red dots are positive mRNA staining. The same brain slice was stained with MT-CO1 protein using IHC staining, which shows the brown color. Under the same treatment group, all images are from a single animal and are representative of all animals. (B) hSURF1opt mRNA is expressed in the CNS tissues of SURF1 KO mice. The axis of the Arachne plot represents the percent area staining positive for hSURF1opt. The abundance of hSURF1opt mRNA in WT and KO+Vehicle groups are close to 0 percent. THM, thalamus, hypothalamus, and midbrain; PM, pons and medulla; CC, cervical cord; LC, lumbar cord. See also Figure S1. (C) Abundance of MT-CO1 protein expression. The axis of the Arachne plot represents the percent area staining positive for MT-CO1 protein. See also Figure S2. (D and E) The correlation between percent area staining positive for MT-CO1 protein expression with COX activity of affected regions in cerebrum and cerebellum.
Intrathecal delivery of AAV9/hSURF1 ablated exhaustive exercise-induced lactic acidosis in SURF1 KO mice
LS patients with SURF1 deficiency often show elevated blood lactate. Previous studies reported that SURF1 KO mice also exhibited elevated blood lactate levels, especially after exhaustive exercise.19 Thus, to evaluate whether gene therapy can correct the blood lactate level, we treated another group of mice with the same high (8 × 1011 vg/mouse) and low (2 × 1011 vg/mouse) dose of AAV9/hSURF1 through i.t. administration and examined blood lactate levels both at rest and after exhaustive exercise. The study scheme is shown in Figure 4A. The mice were tested at two ages, 10 weeks (6 weeks after treatment) and 10 months (9 months after treatment). Blood lactate was examined both before and after running on a treadmill until exhaustion, and the change of blood lactate level was recorded as ΔLactate. There were no differences in their running time (Figure 4B) or resting lactate level among groups at both ages (Figure 4C). However, as shown in Figure 4D, ΔLactate of SURF1 KO mice was significantly higher than that of WT animals when tested at 10 months of age (p < 0.001). Both low-dose and high-dose treatments significantly reduced ΔLactate of KO mice compared with vehicle-treated KO mice (p < 0.01 and p < 0.001, respectively). Furthermore, we collected their tissues at 18 months of age and examined COX activity of cerebrum and liver. We obtained similar trends of improvement as found in previous analysis from the 8-week-old mice (Figures 4E and 4F). Taken together, our data suggest that both low and high doses of AAV9/hSURF1 treatment administered i.t. mitigated abnormal lactic acidosis during exhaustive exercise in SURF1 KO mice without affecting their gross physical function.
Figure 4.

AAV9/hSURF1 normalized exhaustive exercise-induced lactic acidosis in SURF1 KO mice
(A) Scheme of study design. (B) Running time on treadmill until exhaustion. (C) Resting blood lactate level before treadmill running. (D) Change of lactate after running on treadmill until exhaustion. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 compared with KO+Vehicle mice using a Tukey’s multiple comparison test following two-way ANOVA. (E and F) COX activity of cerebrum (E) (n = 8–10 per group) and liver (F) (n = 9–10 per group) of mice at 18 months of age. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 compared with WT mice using Dunn’s multiple comparison method following a Kruskal-Wallis test, since KO+High mice did not pass the normality test using the Shapiro-Wilk’s test. #p < 0.05, ###p < 0.001 compared with KO+Vehicle mice using a one-tailed Student’s t test. A dashed line in (E) and (F) indicates the level of KO+Vehicle, and the percentage of improvement compared with the KO+Vehicle group from each treatment is provided at the bottom of the respective column. Each data point represents measurement from an individual animal, with bars representing the mean ± SEM.
Intrathecal delivery of AAV9/hSURF1 is safe in WT mice
When considering the translation of this gene therapy strategy to patients, it is important to assess the long-term safety of the AAV9/hSURF1 vector. WT C57BL/6J mice were treated with the same regimen as in the efficacy study and they were monitored for gross changes, including body weight, survival, and general body conditions, for a year. The gene therapy treatment did not confer significant differences in body weight growth in either sex (Figures 5A and 5B). Body weight from each mouse at each time point is shown in Table S1. Serum from 4 weeks and 12 months post-dosing were analyzed for blood biochemistry, including total bilirubin (TBIL), blood urea nitrogen (BUN), albumin (ALB), creatine kinase (CK), and aspartate aminotransferase (AST). Levels of these biochemicals indicate the function of kidney and liver. None of above showed significant changes induced by the treatment (Figures 5C–5G). No outward signs of body condition change were observed during the duration of the study. 12 months post-dosing, tissues from all 30 mice were collected and evaluated for histopathology abnormalities. We summarize all of the findings in Table S2, and none of the pathological signs was attributed to AAV9/hSURF1, but rather they were typical findings in aged mice. Thus, our study showed that doses up to 8 × 1011 vg/mouse is safe for WT C57BL/6J mice for at least 12 months.
Figure 5.

AAV9/hSURF1 is safe in WT C57BL/6J mice
(A and B) Body weight of male (A) and female (B) mice up to a year after dosing. (C–G) Blood chemistry tests 4 weeks and 12 months after dosing. Data are shown as mean ± SEM.
Discussion
In this proof-of-concept study, we have designed and demonstrated AAV9/hSURF1 gene therapy as a translationally relevant option as a potential treatment for SURF1-related LS. On the basis of results obtained in the present mouse study, a one-time i.t. injection of AAV9/hSURF1 vectors, driven by the strong ubiquitous CBh promoter, has resulted in codon-optimized human SURF1 mRNA expression in multiple relevant brain and spinal cord regions. The treatment partially corrected COX activity deficiency, as well as abnormal lactate acidosis induced by exhaustive exercise in the SURF1 KO mice. These SURF1 KO mice and WT mice receiving the treatment did not show any signs of clinical or histological toxicity, supporting the general safety of AAV9/hSURF1. As a proof of concept, our results support the potential of this gene therapy strategy as a treatment for patients with SURF1-related LS.
A major limitation of this study is that we were not able to identify physiological phenotypes in the mouse model we used. In the past, there have been two SURF1 KO mouse models generated.7,18 The first one is a constitutive KO model with exons 5–7 replaced by a neomycin (NEO) resistance cassette. Unfortunately, 90% of mice generated from this mouse line were embryonic lethal,7 but further studies suggested this might have been due the effects of the neomycin resistance gene cassette on the expression of neighboring genes.18 To resolve the issue, another mouse model was generated with a frameshift mutation in exon 7 of the SURF1 gene and the neomycin cassette was removed,18 and this is the mouse model we used in our study. Surprisingly, these mice showed prolonged lifespan (longer than WT) and several other phenotypes indicating they do not fully recapitulate signs of human patients. We summarize the findings from published studies on this mouse model in Table 1. Based on these previous characterizations, we chose to test endurance capacity on a treadmill as the major phenotypic outcome measure. However, even with a large group of mice, we were not able to detect significant differences between WT and SURF1 KO mice regarding their running time or distance until exhaustion at either 10 weeks or 10 months of age. We have also tested the same mice on an accelerating rotarod, but consistent with some previous reports there was no significant difference between WT and SURF1 KO mice (Figure S3). Thus, due to the animal model limitations, we were constrained to using biochemical readouts as the major indicators of therapeutic efficacy.
Table 1.
Phenotypes of SURF1 KO mice identified in previous studies
| Study no. | Mouse age | Sex | Behavioral/physiological test | Function tested | Change in SURF1 KO mice | References |
|---|---|---|---|---|---|---|
| 1 | 3 months | F+M | rotarod test | motor skill | ↓ | 18 |
| 2 | 6–7 months | M | Y-maze | working memory | ↑ | 27 |
| object recognition | long-term memory | ↑ | ||||
| elevated maze | basal anxiety | ↔ | ||||
| 3 | 5–6 months | M | open field activity | basal activity | ↔ | 19 |
| treadmill test | endurance capacity | ↓↓ | ||||
| grip strength | muscle strength | ↓ | ||||
| 4 | 28 months | F | grip strength | muscle strength | ↔ | 23 |
| rotarod test | motor skill | ↔ | ||||
| sleep/wake pattern | basal activity | ↔ | ||||
| open field activity | basal activity | ↔ | ||||
| 5 | 3 months | F+M | treadmill test | endurance capacity | ↔ | 20 |
F, female; M, male. ↓, decrease; ↑, increase; ↔, no differences; the number of arrows indicate the degree of changes.
SURF1 plays a critical role in assembling the COX holoenzyme. Thus, it is mechanistically sound to predict that the loss of SURF1 will affect COX activity. SURF1 patients generally have 20%–30% COX activity remaining.9 Our study and those of others show that the SURF1 KO mice have about 40%–50% COX activity remaining.18 One might speculate that the difference of 20%–30% residual COX activity in humans compared to 40%–50% COX activity in the mice contributes to the profound difference in disease symptoms. The treatment exerted an approximately 20%–30% improvement in COX activity of brain and muscle over KO levels, with a higher degree of improvement in liver. We further confirmed the findings in the brain by using histological staining to examine the expression level of MT-CO1, which is commonly used as a marker for COX content level. The expression of MT-CO1 protein also showed similar trends of improvement in all disease-related brain areas, reinforcing and verifying the observed increases in COX activity. We also examined COX activity of the second cohort of mice at 18 months of age (17 months post-dosing), which showed a similar trend as the data we acquired from 8-week-old mice (4 weeks post-injection). These data suggest that improvements in COX activity from the treatment have been maintained for at least 17 months. As SURF1 is a membrane-bound protein, it is expected to function in a cell-autonomous manner, meaning that the treatment can only exert cell-autonomous effects. Thus, this degree of improvement corresponds to our expectation of the number of cells transduced by AAV9 vectors (approximately 5%–20% across the brain).12 While we did not test this directly, considering the expected percentage of cells transduced with the observed increase in COX activity, we speculate that the expressed SURF1 transgene resulted in near-complete rescue of COX activity in any transduced cell. Due to limitations of the available animal models (i.e., a lack of behavioral phenotypes), we could not directly test whether AAV9/hSURF1 treatment leads to behavioral improvements. Our future work involves using genetic or chemical approaches to further reduce COX activity to determine whether more severe phenotypes can be generated in the mice, which we would predict could be rescued by AAV9/hSURF1 gene therapy.
Besides COX activity, we also examined blood lactate levels in our mice. As previous studies have shown that blood lactate was lower in SURF1 KO mice than in WT mice, we expected to see a similar difference in our mice. Thus, we measured their blood lactate level once every month for a year starting at 4 weeks of age, but there were no significant differences between WT and SURF1 KO mice at any time point (data not shown). However, we observed lactic acidosis in SURF1 KO mice induced by exhaustive exercise. After running on a treadmill until exhaustion, blood lactate of SURF1 KO mice was elevated to a higher degree compared with that of WT mice, which was shown as the differences in the change of lactate levels. This difference was more significant when the mice were 10 months old (9 months post-dosing). The SURF1 KO mice that have been treated with either the low or high dose of AAV9/hSURF1 showed a change of lactate level close to that of WT, indicating that the treatment corrected this lactic acidosis. However, to identify which tissues are responsible for the rescue of lactic acidosis would require further studies.
Several features of this study shed light on the general context of a translatable AAV9-based gene therapy treatment for a mitochondrial disease with neurological manifestations. (1) In this study, we tested the effectiveness of administering AAV9 vectors through an i.t. route to mediate widespread CNS transduction. To our knowledge, this is the first time that this administration route has been tested for a mitochondrial disease. Many mitochondrial diseases have neurological manifestations.28 Thus, for a gene therapy approach, it is important to choose a vector and an administration route that can specifically address those issues as a priority. Previous studies in both mice and NHPs have supported that an AAV9 vector carrying the reporter gene, green fluorescent protein (GFP), widely distributed the transgene broadly across the CNS following a single i.t. injection.12, 13, 14, 15 Additionally, i.t. administration reduces the biodistribution of the vector to peripheral organs compared to an i.v. route, but importantly some biodistribution to peripheral organs still occurs, considering the multisystem involvement in mitochondrial diseases. (2) It is noteworthy that the treatment was administered to the mice at 4 weeks of age, an age approximately corresponding to a human teenager.29 The onset of SURF1-related LS shows a broad range, but the most severe cases show an onset of around 2 years of age. Since LS is a degenerative disease, we would predict earlier intervention to correlate with a greater therapeutic benefit. Thus, patients might show a better outcome when they are diagnosed and treated early, even before the onset of symptoms. (3) The vector genome was a self-complementary design, which improved the transduction efficiency compared to a more traditional ssAAV genome design. (4) This study evaluated an AAV9 vector solution stock produced at a concentration of 1.6 × 1014 vg/mL, which is near the estimated concentration limit of AAV9 without risking AAV vector aggregation/precipitation. This concentrated vector preparation was injected undiluted (high dose, 8 × 1011 vg/mouse) and at a 1:4 dilution (low dose, 2 × 1011 vg/mouse). Since the volume of i.t. administration is limited, escalating to a higher dose is unlikely to be feasible. Further improvements to enhance efficacy might be facilitated by optimization of the intra-CSF AAV9 administration or utilizing a more efficient AAV capsid technology if it becomes available.
Taken together, our study demonstrated that the gene therapy strategy using AAV9/hSURF1 partially but significantly corrected biochemical defects in a SURF1-related LS mouse model. There are no effective treatments for SURF1-related LS patients beyond supportive care, and we propose that this approach has the potential to improve or ameliorate disease symptoms for those patients. To achieve optimal therapeutic effects, we speculate that early treatment, high (approaching maximally feasible) doses of AAV9, and the use of a self-complementary genome design will be essential. Our studies supported these notions through examining biochemical outcome measures. Due to the limitations of the animal model, further studies may provide additional insights on the correlation between these biochemical improvements and patients’ quality of life.
Materials and methods
Animals
The SURF1 KO mice were initially generated from Dr. Massimo Zeviani’s Lab, and we acquired them as a gift from Dr. Holly Van Remmen’s Lab at the Oklahoma Medical Research Foundation. The genotypes were determined by PCR analysis using a mouse ear punch as previously described.18 The WT C57BL/6J mice were supplied from the University of Texas Southwestern Medical Center (UTSW) Animal Breeding Core. The animal studies at UTSW were conducted according to a protocol approved by the Institutional Animal Care and Use Committee (IACUC). All mice were weaned between postnatal day (PND)21 and PND28 and were provided food and water ad libitum.
Plasmids
The complete name for hSURF1opt is self-complementary CBh codon-optimized human SURF1-BGHpA. It is composed of a CBh promoter, a codon-optimized human SURF1 cDNA sequence (ATUM, Newark, CA, USA), and a BGH poly(A) tail. This construct with a GFP reporter in place of hSURF1opt has been described in multiple publications from our group across different animal models.12,14,16,22,30 The 5′ ITR was mutated to form the self-complementary structure as previously described.31
Virus production
Vectors were produced at the University of North Carolina-Chapel Hill Vector Core as described.32 Purified vectors were dialyzed in PBS (350 mM final NaCl concentration) containing 5% d-sorbitol and stored at −80°C until use. Thawed aliquots were subsequently stored at 4°C. A filter-sterilized solution of PBS (350 mM final NaCl concentration) containing 5% d-sorbitol was used as vehicle and virus dilution buffer. The viral vector was titered by qPCR and confirmed by PAGE and silver stain.33 The vectors in this study were packaged in scAAV9. The certificate of analysis from the UNC Vector Core is provided in Supplemental information.
Animal injections
AAV9 vectors were diluted in vehicle solution. For intrathecal injection, 5 μL was injected into each animal through lumbar puncture as previously described.34 For i.v. injection, 20 μL was injected to each animal through the tail vein. The i.v. injection was performed 30–60 min before the following i.t. injection.
Cell culture and plasmid expression studies
HEK293 cells were cultured in a 100-mm cell culture dish for at least five passages before transfection. The culture medium contains DMEM (without l-glutamine), 10% fetal bovine serum (FBS), 1% GlutaMAX, 1% non-essential amino acids, and 1% penicillin/streptomycin (Pen/Strep). The cells were grown in a humidified 5% CO2 incubator at 37°C.
One day before transfection, HEK293 cells were plated into a six-well plate at a density of 0.6 × 106 cells/well. The next day, cells were transfected with 1, 1.5, or 2 μg of CBh-hSURF1opt plasmid, and 1.5 μg of CBh-EGFP plasmid as a control using PEIpro (Polyplus-transfection, New York, NY, USA). Cells were collected after 48 h. Protein was extracted using radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Waltham, MA, USA) with 1× cOmplete protease inhibitor cocktail (Sigma, St. Louis, MO, USA). Protein concentration was determined using a bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Waltham, MA, USA). 30 μg of protein was loaded onto 10% SDS-PAGE gels (Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose membranes. Blots were probed with SURF1 antibody (1:500; GeneTex, Irvine, CA, USA) and β-actin antibody (1:10,000; Abcam, Cambridge, MA, USA) for 2 h at room temperature and then with goat anti-mouse (1:5,000; Invitrogen, Waltham, MA, USA; DyLight 800) or goat anti-mouse (1:5,000; Invitrogen, Waltham, MA, USA; DyLight 680). Immunofluorescence was imaged using a Bio-Rad imager.
Total mRNA was extracted using a PureLink RNA extraction kit (Thermo Fisher Scientific, Waltham, MA, USA), and 500 ng of total mRNA was converted to cDNA using SuperScript IV VILO master mix with ezDNase enzyme (Invitrogen, Waltham, MA, USA). Oligonucleotide primers (Sigma, St. Louis, MO, USA) for PCR were as follows: FqPC_hsaSurf1, 5′-GTGGATCTGATCGGCATGGT-3′; RqPC_hsaSurf1, 5′-CGTCGATGAAAATCGGCTCG-3′. The PCR products were examined on 2% agarose gel with a 1-kb DNA ladder (Invitrogen, Waltham, MA, USA).
COX activity assay
Mice were perfused with cold 1× PBS at 4 weeks post-treatment. Tissues were extracted quickly and immediately frozen on dry ice. All tissues were stored at −80°C until use. We used a complex IV/COX rodent enzyme activity microplate assay kit (Abcam, Cambridge, MA, USA) to examine the COX activity of our mice model, according to the manufacturer’s instructions. Mitochondria were extracted as previously described.35,36 After mitochondria extraction, COX is immunocaptured within the wells and activity is determined colorimetrically through the oxidation of reduced cytochrome c as an absorbance decrease at 550 nm. The absorbance of each sample was measured once every minute for 120 min, and the initial rate of decrease was calculated as COX activity. For COX activity of 8-week-old mice, assays were performed in multiple batches, in which at least one WT and one KO+Vehicle mouse were included. Each data point was then normalized to the average of all WT COX activity as relative COX activity. For COX activity of 18-month-old mice, assays were performed in three batches for each tissue. We noticed that there were significant differences among the raw values of WT COX activity of the three batches. Thus, each data point was normalized to the average of WT COX activity within each batch as relative COX activity.
Exhaustive exercise on treadmill and blood lactate test
Blood lactate following exhaustive exercise was measured by the UTSW Metabolic Phenotyping Core. All mice were familiarized to the treadmills for 2 days prior to the exercise session. On day 1, they experienced a 5-min rest on the treadmill followed by running for 5 min at the speed of 8 m/min and then for 5 min at the speed of 10 m/min. On day 2, they experienced a 5-min rest on the treadmill followed by running for 5 min at the speed of 10 m/min and then for 5 min at the speed of 12 m/min.
On day 3 (5 weeks post-dosing), mice were placed on the treadmill for 5 min at rest, followed by running with a starting speed of 10 m/min for 40 min, next by running at speeds that were increased at the rate of 1 m/min every 10 min until the speed reached 13 m/min, and finally by running at speeds that were increased at the rate of 1 m/min every 5 min until exhaustion. The exhaustion time was noted as the time at which the mice remained on the electric shock grid for 5 continuous seconds, without attempting to resume running. Blood lactate was taken before putting the mice onto the treadmill and immediately when the mice came off the treadmill. A drop of blood was collected from the tail vein, and blood lactate concentration was measured using a Lactate Plus lactate meter (Nova Biomedical, Waltham, MA, USA).
SURF1 RNAscope and MT-CO1 immunohistochemistry (IHC)
SURF1 mRNA and MT-CO1 protein levels were visualized in the brain and spinal cord for a set of treated animals. The following protocol was carried out by an individual blinded to the treatments and genotypes of all animals. Tissues ready for staining were processed and embedded in paraffin and cut into 5-μm sections on slides. Both staining (RNAscope and MT-CO1 IHC) were performed on the same slide. An RNAscope 2.5 HD assay kit (Advanced Cell Diagnostics, Newark, CA, USA) was used. Slides were deparaffinized by xylene and then xylene was removed with 200 proof ethanol. Then slides were incubated with hydrogen peroxide for 10 min at room temperature and washed with distilled water. Antigen retrieval was performed by boiling the slides in 1× target retrieval solution for 10 min, washing with distilled water, dehydrating with 200 proof ethanol, and then air-drying. Protease Plus was added to each section, incubated at 40°C for 30 min, and washed with distilled water. Then slides were incubated with an hSURF1opt RNAscope probe in a HybEZ oven at 40°C for 2 h and washed with 1× wash buffer. Then slides were incubated with AMP1–6 for 30 or 15 min following the RNAscope 2.5 HD detection kit protocol. Finally, slides were incubated with RED solution for 10 min to detect the RNAscope signals.
After finishing the RNAscope processing, slides were washed with 1× PBS twice for 5 min each, then blocked with 5% goat normal serum (GNS) for 1 h at room temperature. Then slides were incubated with anti-MT-CO1, mouse (Abcam, Cambridge, MA, USA, ab14705), diluted 1:400 in GNS at 4°C overnight. Then slides were washed with tap water and 1× PBS and incubated with biotinylated goat anti-mouse immunoglobulin G (IgG) secondary antibody (Vector Labs, BA-9200) at 1:400 for 1 h at room temperature. Slides were washed and incubated with ABC (Vector Labs, PK-6100) for 30 min at room temperature and washed with tap water and 1× PBS. Diaminobenzidine (DAB) (D4293, Sigma, St. Louis, MO, USA) was used to develop the reaction product. Finally, slides were counterstained with modified Mayer’s hematoxylin and covered with coverslips. Slides were imaged with an Aperio ImageScope.
Image analysis
Histology images were analyzed using custom analysis settings in the HALO image analysis platform (HALO2.2, Indica Labs, Albuquerque, NM, USA). A region of interest (ROI) was hand drawn on each image to allow for analysis by tissue region. Within the brain, four major regions were drawn: (1) thalamus, hypothalamus, and midbrain (THM); (2) pons and medulla (PM); (3) striatum; (4) cerebellum. For spinal cord samples, we analyzed lumbar cord (injection site) and cervical cord (farthest from injection site). A threshold for each stain was set using positive and negative control images, and the same analysis settings were applied for every image of the same stain. The percent area strongly staining for each marker of interest was recorded for each tissue/ ROI.
Blood biochemistry tests for WT mice
Blood was collected from submandibular veins of each mouse 4 weeks after dosing into 1.5-mL tubes. Blood was allowed to clot at room temperature for 2 h. The clotted material was removed by centrifugation at 5,000 rpm for 20 min at 4°C. The supernatant (serum) was transferred to a new tube and frozen immediately at −80°C until the test. The tests were conducted by the UTSW Metabolic Phenotyping Core.
Statistical analysis
Underlying assumptions checking for the continuous variables was performed prior to analysis. Specifically, we used the Shapiro-Wilk’s test for the normality of the data distribution and the Brown-Forsythe test for homogeneity of variance. Univariable comparisons were conducted using a Student’s t test or one-way ANOVA for normally distributed data with equal variance, and Wilcoxon or Kruskal-Wallis test, as a non-parametric counterpart, for non-normal data. Statistical significance was assumed at the 0.05 level, and multiple comparisons were adjusted using Tukey’s multiple comparison test for the Kruskal-Wallis test, or a Dunn’s multiple comparison test for the one-way ANOVA. For COX activity data for mice of both 8 weeks and 18 months of age, each group of AAV9-treated KO mice were compared with vehicle-treated KO mice using a one-tailed t test, as we would not expect COX activity to be reduced following treatment. A two-tailed t test was performed to evaluate associations between MT-CO1 protein expression and COX activity. Blood lactate data were analyzed with a two-way ANOVA with adjustment of multiple comparisons using Tukey’s multiple comparison test. GraphPad Prism (GraphPad, San Diego, CA, USA) was used for all statistical analyses and generating the graphs.
Acknowledgment
This work was supported by funding from the Cure SURF1 Foundation (now the Cure Mito Foundation) and Taysha Gene Therapies to S.J.G. We thank Dr. Massimo Zeviani and Dr. Holly Van Remmen for providing the initial SURF1 KO mice breeders, and the UNC Vector Core for producing the vectors used in these studies. We thank the UT Southwestern Metabolic Phenotyping Core for performing endurance capacity tests, measuring blood lactate, and conducting blood chemistry tests. We also thank Debby Szczepanski from the UTSW Animal Resource Center for performing intravenous injections for us. We acknowledge Dr. Mary Wright-Carter and her team at the UTSW Diagnostic Laboratory for conducting the toxicology evaluation on tissues from the safety study and providing the histopathological safety report.
Author contributions
Q.L. and S.J.G. contributed to the study concept and design. Q.L., M.R., and Y.H. contributed to data acquisition. Q.L. and M.L. contributed to data analysis. Q.L., M.L., and S.J.G. contributed to drafting the text and figures.
Declaration of interests
Q.L. and S.J.G. are inventors of the vectors used in this study, and the vectors are licensed to Taysha Gene Therapies. Q.L. and S.J.G. may receive inventor income from Taysha Gene Therapies. S.J.G. has received inventor income from Asklepios Biopharma, but that intellectual property was not used in this study. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.09.001.
Supplemental information
References
- 1.Rossi A., Biancheri R., Bruno C., Di Rocco M., Calvi A., Pessagno A., Tortori-Donati P. Leigh syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR Am. J. Neuroradiol. 2003;24:1188–1191. [PMC free article] [PubMed] [Google Scholar]
- 2.Lake N.J., Compton A.G., Rahman S., Thorburn D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016;79:190–203. doi: 10.1002/ana.24551. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 4.Tiranti V., Hoertnagel K., Carrozzo R., Galimberti C., Munaro M., Granatiero M., Zelante L., Gasparini P., Marzella R., Rocchi M. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bundschuh F.A., Hannappel A., Anderka O., Ludwig B. Surf1, associated with Leigh syndrome in humans, is a heme-binding protein in bacterial oxidase biogenesis. J. Biol. Chem. 2009;284:25735–25741. doi: 10.1074/jbc.M109.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hannappel A., Bundschuh F.A., Ludwig B. Role of Surf1 in heme recruitment for bacterial COX biogenesis. Biochim. Biophys. Acta. 2012;1817:928–937. doi: 10.1016/j.bbabio.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Agostino A., Invernizzi F., Tiveron C., Fagiolari G., Prelle A., Lamantea E., Giavazzi A., Battaglia G., Tatangelo L., Tiranti V., Zeviani M. Constitutive knockout of Surf1 is associated with high embryonic lethality, mitochondrial disease and cytochrome c oxidase deficiency in mice. Hum. Mol. Genet. 2003;12:399–413. doi: 10.1093/hmg/ddg038. [DOI] [PubMed] [Google Scholar]
- 8.Poyau A., Buchet K., Bouzidi M.F., Zabot M.T., Echenne B., Yao J., Shoubridge E.A., Godinot C. Missense mutations in SURF1 associated with deficient cytochrome c oxidase assembly in Leigh syndrome patients. Hum. Genet. 2000;106:194–205. doi: 10.1007/s004390051028. [DOI] [PubMed] [Google Scholar]
- 9.Wedatilake Y., Brown R.M., McFarland R., Yaplito-Lee J., Morris A.A.M., Champion M., Jardine P.E., Clarke A., Thorburn D.R., Taylor R.W. SURF1 deficiency: A multi-centre natural history study. Orphanet J. Rare Dis. 2013;8:96. doi: 10.1186/1750-1172-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S., Brown A.M., Jenkins C., Campbell K. Viral vector systems for gene therapy: A comprehensive literature review of progress and biosafety challenges. Appl. Biosaf. 2020;25:7–18. doi: 10.1177/1535676019899502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deverman B.E., Ravina B.M., Bankiewicz K.S., Paul S.M., Sah D.W.Y. Gene therapy for neurological disorders: Progress and prospects. Nat. Rev. Drug Discov. 2018;17:641–659. doi: 10.1038/nrd.2018.110. [DOI] [PubMed] [Google Scholar]
- 12.Bailey R.M., Rozenberg A., Gray S.J. Comparison of high-dose intracisterna magna and lumbar puncture intrathecal delivery of AAV9 in mice to treat neuropathies. Brain Res. 2020;1739:146832. doi: 10.1016/j.brainres.2020.146832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.-M.M., Fyfe J., Moullier P., Colle M.-A.A., Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray S.J., Matagne V., Bachaboina L., Yadav S., Ojeda S.R., Samulski R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder B.R., Gray S.J., Quach E.T., Huang J.W., Leung C.H., Samulski R.J., Boulis N.M., Federici T. Comparison of adeno-associated viral vector serotypes for spinal cord and motor neuron gene delivery. Hum. Gene Ther. 2011;22:1129–1135. doi: 10.1089/hum.2011.008. [DOI] [PubMed] [Google Scholar]
- 16.Gray S.J., Nagabhushan Kalburgi S., McCown T.J., Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey R.M., Armao D., Nagabhushan Kalburgi S., Gray S.J. Development of intrathecal AAV9 gene therapy for giant axonal neuropathy. Mol. Ther. Methods Clin. Dev. 2018;9:160–171. doi: 10.1016/j.omtm.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dell’agnello C., Leo S., Agostino A., Szabadkai G., Tiveron C., Zulian A., Prelle A., Roubertoux P., Rizzuto R., Zeviani M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 19.Pulliam D.A., Deepa S.S., Liu Y., Hill S., Lin A.-L., Bhattacharya A., Shi Y., Sloane L., Viscomi C., Zeviani M., Van Remmen H. Complex IV-deficient Surf1−/− mice initiate mitochondrial stress responses. Biochem. J. 2014;462:359–371. doi: 10.1042/BJ20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viscomi C., Bottani E., Civiletto G., Cerutti R., Moggio M., Fagiolari G., Schon E.A., Lamperti C., Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarty D.M., Fu H., Monahan P.E., Toulson C.E., Naik P., Samulski R.J. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 22.Gray S.J., Foti S.B., Schwartz J.W., Bachaboina L., Taylor-Blake B., Coleman J., Ehlers M.D., Zylka M.J., McCown T.J., Samulski R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deepa S.S., Pharaoh G., Kinter M., Diaz V., Fok W.C., Riddle K., Pulliam D., Hill S., Fischer K.E., Soto V. Lifelong reduction in complex IV induces tissue-specific metabolic effects but does not reduce lifespan or healthspan in mice. Aging Cell. 2018;17:e12769. doi: 10.1111/acel.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meo I.D., Marchet S., Lamperti C., Zeviani M., Viscomi C. AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther. 2017;24:661–667. doi: 10.1038/gt.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovářová N., Pecina P., Nůsková H., Vrbacký M., Zeviani M., Mráček T., Viscomi C., Houštěk J. Tissue- and species-specific differences in cytochrome c oxidase assembly induced by SURF1 defects. Biochim. Biophys. 2016;1862:705–715. doi: 10.1016/j.bbadis.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hallmann K., Kudin A.P., Zsurka G., Kornblum C., Reimann J., Stüve B., Waltz S., Hattingen E., Thiele H., Nürnberg P. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain. 2016;139:338–345. doi: 10.1093/brain/awv357. [DOI] [PubMed] [Google Scholar]
- 27.Lin A.-L., Pulliam D.A., Deepa S.S., Halloran J.J., Hussong S.A., Burbank R.R., Bresnen A., Liu Y., Podlutskaya N., Soundararajan A. Decreased in vitro mitochondrial function is associated with enhanced brain metabolism, blood flow, and memory in Surf1-deficient mice. J. Cereb. Blood Flow Metab. 2013;33:1605–1611. doi: 10.1038/jcbfm.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russell O.M., Gorman G.S., Lightowlers R.N., Turnbull D.M. Mitochondrial diseases: Hope for the future. Cell. 2020;181:168–188. doi: 10.1016/j.cell.2020.02.051. [DOI] [PubMed] [Google Scholar]
- 29.Fox J.G., Barthold S., Davisson M., Newcomer C.E., Quimby F.W., Smith A. Elsevier; 2006. The Mouse in Biomedical Research: Normative Biology, Husbandry, and Models. [Google Scholar]
- 30.Federici T., Taub J.S., Baum G.R., Gray S.J., Grieger J.C., Matthews K.A., Handy C.R., Passini M.A., Samulski R.J., Boulis N.M. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2012;19:852–859. doi: 10.1038/gt.2011.130. [DOI] [PubMed] [Google Scholar]
- 31.McCarty D.M., Monahan P.E., Samulski R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 32.Clément N., Grieger J.C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 2016;3:16002. doi: 10.1038/mtm.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray S.J., Choi V.W., Asokan A., Haberman R.A., McCown T.J., Samulski R.J. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr. Protoc. Neurosci. 2011;57:4.17.11–14.17.30. doi: 10.1002/0471142301.ns0417s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray S.J., Choi V.W., Asokan A., Haberman R.A., McCown T.J., Samulski R.J. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr. Protoc. Neurosci. 2011;57:4.17.11–14.17. 30. doi: 10.1002/0471142301.ns0417s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frezza C., Cipolat S., Scorrano L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 36.Sims N.R., Anderson M.F. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat. Protoc. 2008;3:1228–1239. doi: 10.1038/nprot.2008.105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.