

Abstract
Background
Ghrelin may ameliorate cancer cachexia (CC) by preventing anorexia, muscle, and fat loss. However, the mechanisms mediating these effects are not fully understood. This study characterizes the pathways involved in muscle mass and strength loss in the Lewis lung carcinoma (LLC)‐induced cachexia model, and the effects of ghrelin in mice with or without its only known receptor: the growth hormone secretagogue receptor‐1a ((GHSR‐1a), Ghsr +/+ and Ghsr −/− ).
Methods
Five to 7‐month‐old male C57BL/6J Ghsr +/+ and Ghsr −/− mice were inoculated with 1 × 106 heat‐killed (HK) or live LLC cells (tumour implantation, TI). When tumours were palpable (7 days after TI), tumour‐bearing mice were injected with vehicle (T + V) or ghrelin twice/day for 14 days (T + G, 0.8 mg/kg), while HK‐treated mice were given vehicle (HK + V). Body weight and grip strength were evaluated before TI and at termination (21 days after TI). Hindlimb muscles were collected for analysis.
Results
Less pronounced body weight (BW) loss (87.70 ± 0.98% vs. 83.92 ± 1.23%, percentage of baseline BW in tumour‐bearing Ghsr +/+ vs. Ghsr −/− , P = 0.008), and lower upregulation of ubiquitin‐proteasome system (UPS, MuRF1/Trim63, 5.71 ± 1.53‐fold vs. 9.22 ± 1.94‐fold‐change from Ghsr +/+ HK + V in tumour‐bearing Ghsr +/+ vs. Ghsr ‐/‐ , P = 0.036) and autophagy markers (Becn1, Atg5, Atg7, tumour‐bearing Ghsr +/+ < Ghsr −/− , all P < 0.02) were found in T + V Ghsr +/+ vs. Ghsr −/− mice. Ghrelin attenuated LLC‐induced UPS marker upregulation in both genotypes, [Trim63 was decreased from 5.71 ± 1.53‐fold to 1.96 ± 0.47‐fold in Ghsr +/+ (T + V vs. T + G: P = 0.032) and 9.22 ± 1.94‐fold to 4.72 ± 1.06‐fold in Ghsr −/− (T + V vs. T + G: P = 0.008)]. Only in Ghsr +/+ mice ghrelin ameliorated LLC‐induced grip strength loss [improved from 89.24 ± 3.48% to 97.80 ± 2.31% of baseline (T + V vs. T + G: P = 0.042)], mitophagy markers [Bnip3 was decreased from 2.28 ± 0.56 to 1.38 ± 0.14‐fold (T + V vs. T + G: P ≤ 0.05)], and impaired mitochondrial respiration [State 3u improved from 698.23 ± 73.96 to 934.37 ± 95.21 pmol/min (T + V vs. T + G: P ≤ 0.05)], whereas these markers were not improved by ghrelin Ghsr −/− . Compared with Ghsr +/+ , Ghsr −/− tumour‐bearing mice also showed decreased response to ghrelin in BW [T + G‐treated Ghsr +/+ vs. Ghsr −/− : 91.75 ± 1.05% vs. 86.18 ± 1.13% of baseline BW, P < 0.001)], gastrocnemius (T + G‐treated Ghsr +/+ vs. Ghsr −/− : 96.9 ± 2.08% vs. 88.15 ± 1.78% of Ghsr +/+ HK + V, P < 0.001) and quadriceps muscle mass (T + G‐treated Ghsr +/+ vs. Ghsr −/− : 96.12 ± 2.31% vs. 88.36 ± 1.94% of Ghsr +/+ HK + V, P = 0.01), and gastrocnemius type IIA (T + G‐treated Ghsr +/+ vs. Ghsr −/− : 1250.49 ± 31.72 vs. 1017.62 ± 70.99 μm2, P = 0.027) and IIB fibre cross‐sectional area (T + G‐treated Ghsr +/+ vs. Ghsr −/− : 2496.48 ± 116.88 vs. 2183.04 ± 103.43 μm2, P = 0.024).
Conclusions
Growth hormone secretagogue receptor‐1a mediates ghrelin's effects on attenuating LLC‐induced weakness but not muscle mass loss by modulating the autophagy‐lysosome pathway, mitophagy, and mitochondrial respiration.
Keywords: Cachexia, Wasting, Autophagy, Mitochondria
Introduction
Cancer cachexia (CC) is a multifactorial syndrome characterized by muscle loss that leads to functional impairment. 1 , 2 , 3 It is associated with devastating complications, such as intolerance to cancer treatments, poor quality of life, and increased morbidity and mortality. 4 , 5 In spite of its relevance, the mechanisms leading to cancer‐induced muscle wasting are incompletely understood, and this is a barrier to developing effective treatments for this condition.
Ghrelin, a circulating hormone secreted mainly by the stomach, is known to stimulate appetite and growth hormone secretion by binding to the growth hormone secretagogue receptor (GHSR)‐1a, its only known receptor to date. 6 In animal models of muscle wasting induced by tumour, chemotherapy, fasting, or denervation, ghrelin prevents the loss of muscle mass and function 7 , 8 , 9 and is currently considered a promising treatment for CC. 10 , 11 Interestingly, emerging data suggest that some of the ghrelin's effects are independent of GHSR‐1a. 12 , 13 GHRS‐1a is not expressed in skeletal muscle, 14 , 15 cisplatin‐induced C2C12 myotube atrophy is prevented by ghrelin treatment in the absence of GHSR‐1a, 7 and ghrelin prevented fasting‐induced muscle atrophy and improved protein synthesis in GHSR‐1a‐deficient mice. 8 However, this has not been well‐characterized in tumour‐induced cachexia. There is a pressing need to understand how GHSR‐1a mediates the effects of ghrelin on attenuating muscle wasting in CC, and to characterize these GHSR‐1a‐dependent and GHSR‐1a‐independent pathways given that ghrelin and agonists of the GHSR‐1a are in clinical development for this indication. 10 , 16 , 17
The relationship between muscle mass and function changes in CC is not well‐understood. The loss of muscle strength and function is accompanied by the loss of muscle mass, but muscle mass and function do not respond in parallel to interventions in clinical studies. 17 Muscle mass loss is often seen as a result of the imbalance between protein synthesis and degradation. The increased protein degradation in CC is regulated by several major catabolic pathways, including the ubiquitin‐proteasome system (UPS) and the autophagy pathway. 3 , 18 , 19 UPS is known as a major player in myofibrillar protein degradation that leads to muscle mass loss, and ghrelin has been shown to suppress UPS activation in CC. 7 , 8 Autophagy homeostasis is essential for maintaining muscle quantity and quality. Under basal conditions, autophagy removes damaged organelles, protein aggregates, and pathogens to maintain muscle quality. 20 During illness or in conditions of negative energy balance, such as caloric restriction, 21 , 22 chemotherapy administration, 23 and denervation, 24 , 25 excessive activation of autophagy triggers selective removal of myofibrillar and structural proteins, 19 , 26 and mitochondria (mitophagy). 27 , 28 Excessive degradation of these proteins and mitophagy contributes to muscle atrophy and mitochondrial dysfunction in skeletal muscle. 28 , 29 Autophagy is thought to play a role in CC as increased autophagy markers have been found in animal models 30 , 31 and CC patients, 32 but its association with muscle function and the potential role of ghrelin in altering autophagy is not known. In addition, mitochondrial dysfunction also contributes to diminished muscle function. However, only a few studies have shown decreased muscle mitochondrial respiration in rodent models of CC, 33 , 34 and the mechanisms are not fully understood. The effect of ghrelin and GHSR agonists on restoring mitochondria function has only been reported in a few preclinical studies where ghrelin combined with higher food intake improved mitochondrial oxidative capacity in rats with chronic kidney disease 35 or chronic heart failure. 36 Also, GHSR agonists improved cisplatin‐induced autophagy overactivation and mitochondrial dysfunction in rats by modulating mitochondrial biogenesis, mitochondrial dynamics, and reactive oxygen species (ROS) production. 37 Whether ghrelin plays a role in modifying mitochondrial function during cancer cachexia is not known.
The objectives of this study were (i) to characterize the role of GHSR‐1a on modulating tumour‐induced activation of catabolic pathways in skeletal muscle, including UPS and autophagy, as well as mitochondrial dysfunction, and (ii) to determine the extent to which GHSR‐1a mediates the effects of exogenous ghrelin administration on preventing muscle mass and function loss in CC and the mechanisms mediating these effects. We show that GHSR‐1a mediates ghrelin's effects on attenuating LLC‐induced loss of muscle strength but not muscle mass by modulating the autophagy‐lysosome pathway, mitophagy, and mitochondrial respiration.
Methods
Animals
Adult (5‐ to 7‐month‐old) male growth hormone (GH) secretagogue receptor (GHSR)‐1a wild type (Ghsr +/+ ) and knock out (Ghsr −/− ) mice on a C57BL/6J background were used for the current study as previously described. 15 Ghsr −/− mice were originally generated on a 129Sv and C57BL/6J background and subsequently bred into C57BL/6J mice for at least 10 generations. To determine whether both N10 Ghsr +/+ and Ghsr −/− mice were congenic, the mice were analysed for 110 microsatellite markers (Charles River Laboratory, Wilmington, MA). Besides the gene deletion, all other markers were 100% identical to those characteristics of C57BL/6J mice, indicating that the N10 null mice are congenic. These procedures were performed in Dr. Roy G. Smith's Laboratory and described previously. 38 The Ghsr −/− and Ghsr +/+ mice used in our experiments were generated from these established homozygous lines individually (approximately five generations), and the genotype of these mice was confirmed. Mice were individually housed and maintained on a 12/12 light/dark cycle (lights on at 6 a.m.). A week prior to the experiments, mice were acclimated to their cages and human handling. All experiments were conducted with the approval of the Institutional Animal Care and Use Committee at VA Puget Sound Health Care System and in compliance with the NIH Guidelines for Use and Care of Laboratory Animals.
Tumour implantation and ghrelin administration
The procedures of tumour implantation (TI) and ghrelin intervention were described previously. 7 , 15 Briefly, Lewis lung carcinoma (LLC) cells (1 × 106 cells American Type Culture Collection, Manassas, VA) were implanted subcutaneously into the right flank of mice in tumour‐bearing groups, while the mice in control groups were injected with equal volume and number of heat‐killed LLC cells (HK) in the same area. When the tumour was palpable (~1 cm in diameter, about 7 days after TI), the tumour‐bearing mice were treated with either acylated ghrelin (Anaspect, Fremont, CA) at a dose of 0.8 mg/kg (tumour + ghrelin, T + G) or saline solution (tumour + vehicle, T + V), s.q., twice daily, at 8 a.m. and 5 p.m., for approximately 14 days; while mice in HK groups received saline solution (15 mcl, HK + V), s.q., twice/day for 2 weeks until the endpoint (21 days after TI). Our dose selection was based on our group's experience and that of other groups. 7 , 15 , 39 In brief, the dose selected for this study has been shown to be well‐tolerated and to induce a 5–10% weight gain and a ~10% increase in food intake. 39 These changes are clinically relevant because they are similar to those seen in recent human trials where ghrelin mimetics are being used. 10 Mice were euthanized by CO2 on Day 21 after TI, approximately 14 days after the tumour was noted. Hindlimb muscles were collected after euthanasia. The right gastrocnemius/plantaris (GAS/PL) muscles were preserved in Optimal Cutting Temperature (OCT) for histology. The left side of GAS/PL muscle was saved for biochemical analysis. For the subsequent autophagy flux experiment, a separate set of mice from each group also received vehicle or colchicine treatment (intraperitoneal injection, 0.4 μg/g/day) for 2 days before euthanasia (Day 19 and 20 after TI). 40 For the mitochondrial respiration tests, plantaris (PL) muscles were taken while mice were under anaesthesia with isoflurane. These animals were terminated by cervical dislocation after tissue collection.
Body weight and lean body mass
Body weight (BW) was assessed before TI and at the endpoint (about 3 weeks after TI and before termination). The changes in BW are expressed as carcass weight at the endpoint (weight at endpoint minus tumour mass) normalized to baseline BW in percentage. Baseline LBM was measured by nuclear magnetic resonance (NMR, Bruker optics, The Woodlands, TX) before TI. Muscle mass was determined by the average muscle wet weight of bilateral muscles and normalized to baseline LBM.
Grip strength
Grip strength was measured before TI and right before euthanasia by a grip strength meter with a digital force gauge (Columbus Instruments, Columbus, OH). Forelimb grip strength was accessed by allowing the mouse to grasp a pull bar connected to a force gauge by only using its forelimbs. The maximum grip strength was recorded in the force gauge in kilograms. The test was performed three times at a 1 min interval. Maximum grip strength was recorded, and the final result is expressed as grip strength at endpoint normalized to baseline grip strength in percentage.
Immunohistochemistry
The cross‐sectional area (CSA) of individual fibres from the GAS and PL muscle was determined as previously described. 41 , 42 , 43 Briefly, the OCT‐mounted GAS/PL muscle was sliced at 10 μm using a Cryostat (Leica CM3050S, Nußloch, Germany) at −25°C. All muscles were transected at the mid‐belly area, the largest cross‐section of the whole muscle. The muscle sections were dehydrated for 30 min, blocked with 10% goat serum in PBS for 1 h. Primary antibodies were applied for 2 h at room temperature. Primary antibodies and dilutions were used as follows: BA‐F8 (1:50), which detects myosin heavy chain (MHC)‐I fibres; SC‐71 (1:600), which detects MHC‐IIA fibres; and BF‐F3 (1:100), which detects type MHC‐IIB fibres (Developmental Studies Hybridoma Bank, Iowa City, IA). After three washes in PBS, sections were incubated in the corresponding secondary antibodies (Thermo Fisher Scientific, Waltham, MA) for 1 h: Alexa 350 IgG2b (1:500) for MHC‐I (blue); Alexa 488 IgG1 (1:500) for MHC‐IIA (green); and Alexa 555 IgM (1:500) for MHC‐IIB (red). After another three washes in PBS, the sections were mounted with Prolong Gold AntiFade reagent (Thermo Fisher Scientific).
We obtained a 10× stitched whole cross‐sectional image of the GAS and plantaris (PL) muscle complex (6 × 6 sections with 165.75 μm overlap) by using Nikon Ni‐E microscope and NIS‐Elements software (Nikon, Tokyo, Japan). PL is located between the deep areas of medial and lateral GAS muscle flaps and can be identified by its epimysium and a blood vessel between PL and medial GAS flap. To analyse the CSA of GAS, 200 type IIA and 300 type IIB fibres were randomly selected from the deep areas from both medial and lateral GAS, where both IIA and IIB fibres are present (three to four sections from deep GAS muscle areas parallel to PL, from both sides), and traced by a blinded trained researcher using ImageJ analysis software (National Institutes of Health, http://rsb.info.nih.gov/ij/). Each section is right next to the other and joined by a 165.75 μm overlap stitch. The overlapping area is automatically merged by the NIS‐Elements software to prevent double‐analysing. To analyse CSA of PL, approximately 100 type IIA, 200 type IIB, and 60–80 type IIX or IIA/X fibres from the whole PL area were analysed using the same methods. The percentage of each fibre type in PL muscle was analysed by using the Cell Counter plugin from the ImageJ analysis software.
Succinate dehydrogenase staining
The succinate dehydrogenase (SDH) staining protocol is adapted from Dr. Daniel P. Kelly's lab. 44 Briefly, the GAS/PL sections were incubated with a mixed solution with 10 ml 0.2 M Phosphate Buffer, pH 7.6 (0.2 M NaH2PO4, 0.2 M Na2HPO4), 270 mg sodium succinate, and 10 mg nitro blue tetrazolium (NBT), for 30 min at 37°C. After three washes with dH2O, unbound NBT was washed off by three exchanges of the acetone solutions in increasing (30%, 60%, and 90% acetone) followed by several rinses of dH2O and mounting with an aqueous mounting medium. SDH+ (dark purple) and SDH− fibres in PL muscles were counted by two trained and blinded researchers using the Cell Counter plugin from the ImageJ analysis software.
Real‐time reverse transcription‐quantitative polymerase chain reaction
Half of the GAS/PL muscle from the left side of the animal was saved in RNAlater® (Qiagen, Valencia, CA) after harvesting. RNA was isolated by using Qiagen RNeasy mini kit (Qiagen). Transcription levels of the isolated RNA were identified by BioTek Cytation 5. Total RNA was reverse transcribed to cDNA by QuantiTect Reverse Transcription Kit (Qiagen). RT‐PCR was detected by an ABI 7500 instrument (Applied Biosystems, Foster City, CA) by using predesigned Taqman Expression Assays (Thermo Fisher Scientific, Waltham, MA). The quantification of genes of interest was normalized to a reference gene glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and expressed as a relative fold‐change of the Ghsr +/+ HK + V group by a standard 2‐ΔCT method. The following Taqman primers from Thermo Fisher Scientific (4331182) were used in this study: Gapdh (Mm99999915_g1), Ghsr (Mm00616415_m1), Fbxo32/Atrogin1 (Mm00499523_m1), Trim63/MuRF1 (Mm01185221_m1), Becn1 (Mm01265461_m1), Atg5 (Mm01187303_m1), Atg7 (Mm00512209_m1), Bnip3 (Mm01275600_g1), and Ppargc1a (Mm01208835_m1).
Western blotting
Western blotting was performed to identify the protein content of LC3B, p62, phospho‐AKT (Ser473), total AKT, oxidative phosphorylation (OXPHOS), and peroxisome proliferator‐activated receptor‐gamma coactivator (PGC‐1α) in GAS/PL muscles. A portion of GAS/PL muscle (~60 mg) was homogenized in RIPA buffer (Thermo Fisher Scientific) with a cocktail of protease and phosphatase inhibitors (Thermo Fisher Scientific). The homogenate was centrifuged at 10 000 g for 15 min at 4°C, and the supernatant was collected as the protein extraction. The protein concentration was further quantified by a bicinchoninic acid (BCA) assay (Thermo Fisher Scientific) using bovine serum albumin (BSA) as a standard. Prior to electrophoresis, protein extractions were diluted with 5× Lane Marker Reducing Sample Buffer (Thermo Fisher Scientific) and heated at 95°C for 4 min; 50 μg protein was loaded onto 4–15% Criterion™ TGX™ Precast Midi Protein Gels (BIO‐RAD, Hercules, CA) and separated by using a BIO‐RAD Criterion™ Cell electrophoresis system (165‐6001). The proteins were then transferred to nitrocellulose membranes by a BIO‐RAD Criterion™ Blotter (170‐4070) at 100 V, 4°C, for 30 min. After blocking in 5% non‐fat dry milk in TBS/T at room temperature for 1 h, membranes were incubated with primary antibodies overnight at 4°C. The following primary antibodies and dilutions were used for the experiments: anti‐LC3B antibody (1:500. NB100‐2220, Novus Biologicals, CO); anti‐SQSTM1/p62 antibody (1:1000. Ab56416, Abcam, Cambridge, MA); GAPDH (D16H11) XP® Rabbit mAb (horseradish peroxidase (HRP)‐Conjugate, 1:2000. 8884, Cell Signalling, Beverly, MA); Phospho‐Akt (Ser473) (D9E) XP® Rabbit mAb (1:1000. 4060, Cell Signalling); Akt (pan) (C67E7) Rabbit mAb (1:1000. 4691, Cell Signalling); Anti‐PGC1 alpha antibody (1:1000, ab54481, Abcam); Total OXPHOS Rodent WB Antibody Cocktail (1:1000, ab110413, Abcam); Anti‐beta Actin antibody (1: 2000, ab8227, Abcam). Following incubation, the membranes were washed and then probed with the corresponding HRP‐conjugated secondary antibodies (except GAPDH). The membranes were developed using SuperSignal™ West Dura Extended Duration Substrate (Thermo Fisher Scientific) and imaged by ImageQuant LAS 4000 (GE Health Care, Chicago, IL). The quantification of densitometry was analysed by ImageJ and expressed as a ratio over GAPDH.
Mitochondria isolation and mitochondrial respiration measurements
The mitochondrial respiration tests were done in a separate cohort of mice as it requires plantaris muscles from both sides, and experiments for identifying molecular markers and CSA require a complete set of GAS/PL muscles. Therefore, CSA and molecular markers are from a set of mice separate from mice used for mitochondrial respiration. However, other data including BW, grip strength, muscle wet weight are combined data from both sets of mice as there was no difference in these outcomes between the two cohorts. The mitochondria isolation method is adapted from an established protocol published previously. 45 Plantaris muscles were harvested while the mouse was under deep anaesthesia by isoflurane and immediately saved in mitochondria isolation buffer (MIB) (210 mM sucrose, 2 mM EGTA, 40 mM NaCl, 30 mM HEPES, pH 7.4) on ice. The muscle was then homogenized in MIB by Kimble homogenizer and centrifuged at 900 g and 4°C for 10 min. The supernatant was collected and centrifuged again at 10 000 g and 4°C for 10 min. The mitochondrial pellet was collected and resuspended in MIB, and protein concentration was identified by Pierce Rapid Gold BCA Protein Assay (Thermo Fisher Scientific). After another centrifuge at 10 000 g and 4°C for 10 min, isolated mitochondria were resuspended in mitochondrial assay solution (MAS), 70 mM sucrose, 220 mM d‐mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, 0.2% fatty‐acid free BSA, pH 7.4; Sigma‐Aldrich, Carlsbad, CA, USA) with substrates (5 mM malate and 5 mM pyruvate).
Mitochondrial respiration was detected by Agilent Seahorse XFe24 Extracellular Flux Assay Kit (Agilent Technologies, Santa Clara, CA) by using the methods adapted from Rodger's protocol. 46 , 47 The day prior to the Seahorse experiment, XFe24 sensor cartridges were hydrated and incubated in a non‐CO2 incubator overnight following the manufacturer's instructions. On the day of the experiment, the isolated mitochondria in MAS with substrates were plated in Agilent Seahorse XF24 Cell Culture Microplates (7.5 μg/well in triplicates). After centrifugation at 2000 g and 4°C for 20 min, an additional MAS with substrates were added to each well and made a final volume of 500 μL in each well; 50 μL of adenosine diphosphate (ADP), 55 μL oligomycin, 60 μL carbonyl cyanide‐p‐trifluoromethoxy‐phenylhydrazone (FCCP), and 65 μL of antimycin A were loaded into the cartridge plate and injected into the cell plate in sequence with final concentrations as follows: ADP 4 mM, oligomycin 2 μM, FCCP 4 μM, and antimycin A 2 μM. Oxygen consumption rate ((OCR), pmol/min) was measured using an XFe24 Seahorse Instrument (Agilent Technologies) in real‐time. Two measures were made in each mitochondrial respiration state: basal (state 2 respiration), phosphorylating respiration in the presence of ADP (state 3 respiration), resting respiration in the presence of oligomycin (state 4o respiration), maximal uncoupling respiration in the presence of FCCP (state 3u respiration) and electron transport chain‐unrelated respiration in the presence of complex III inhibitor antimycin A.
Plasma insulin‐like growth factor‐1 levels
Blood samples were collected after euthanasia and then processed into plasma. Insulin‐like growth factor‐1 (IGF‐1) levels in the plasma were measured by Mouse Magnetic Luminex Assay at the Hormone Assay and Analytical Services Core at Vanderbilt University Medical Center.
Statistics
Two‐way analysis of variance (ANOVA) was performed to identify differences between genotypes (Ghsr +/+ vs. Ghsr −/− ) across treatments (HK + V, T + V, and T + G) followed by Fisher's least significant difference (LSD) post hoc test (P < 0.05). Spearman correlations were used to determine associations between grip strength and p62 or Bnip3 expression in all the animals. Values are presented in mean ± SE. All statistical testing was performed using IBM SPSS version 18 software.
Results
Ghsr is not expressed in skeletal muscles from either Ghsr +/+ or Ghsr −/− mice
In agreement with previous reports, 14 , 15 Ghsr mRNA was expressed in brain tissue but not in skeletal muscle in Ghsr +/+ mice. Also, no Ghsr mRNA was detected in brain or muscle tissue in Ghsr −/− mice (Supporting Information, Figure S1).
Ghrelin requires the presence of growth hormone secretagogue receptor‐1a to restore muscle strength but not muscle mass in Lewis lung carcinoma‐induced cachexia
Lewis lung carcinoma‐tumour implantation induced significant decreases in body weight (BW), grip strength, and muscle mass in both genotypes (Figure 1A,B). Ghsr −/− mice showed a more pronounced loss in BW compared with Ghsr +/+ mice, suggesting a role of GHSR‐1a in protecting against BW loss induced by LLC. Administration of ghrelin attenuated LLC‐induced loss in BW and prevented the decrease in grip strength in Ghsr +/+ animals, whereas no significant effect of ghrelin was found in these measures in the Ghsr −/− tumour‐bearing mice (Figure 1A). Ghrelin also mitigated LLC‐induced muscle mass loss in soleus (trend), gastrocnemius (GAS), plantaris (PL), tibialis anterior (TA), and quadriceps (Quad) in Ghsr +/+ mice. These effects of ghrelin were attenuated and only partially seen in Ghsr −/− , where ghrelin only partially ameliorated tumour‐induced muscle wasting in Quad (Figure 1B).
Figure 1.
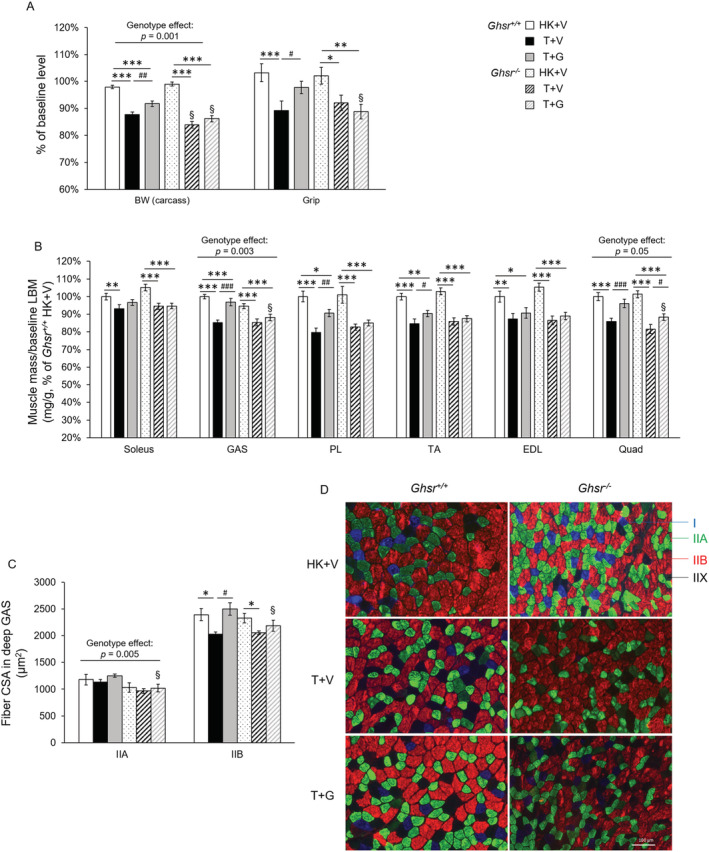
Body weight (BW), grip strength, and muscle mass in Ghsr +/+ and Ghsr −/− mice. HK + V: Heat‐killed + vehicle; T + V: Tumour + vehicle; T + G: Tumour + ghrelin. (A) BW (carcass weight without tumour), and grip strength were measured at endpoint (~3 weeks after tumour implantation) and are shown as % of their baseline levels (before tumour implantation, N = 28–34/group). (B) Muscle mass of hindlimb muscles. The average wet weight of bilateral muscles was measured at the endpoint and expressed as the ratio of muscle wet weight to baseline lean body mass (LBM, mg/g) and normalized to the Ghsr +/+ HK + V group. (N = 13–15/group for GAS and PL muscle mass; N = 27–35/group for other muscles). (C, D) cross‐sectional area (CSA) of myosin heavy chain (MHC) type IIA and IIB fibres in the deep area of GAS muscles at endpoint (μm2). 200–300 individual fibres/fibre type were analysed per animal for CSA (N = 4). (D) Representative images of immunohistochemistry staining MHC I (blue), MHC IIA (green), MHC IIB (red), and MHC IIX (blank, not being stained) fibres in the deep area of GAS muscles. Data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. *, **, and *** denote significant differences comparing to HK + V within the same genotype; #, ##, and ## denote significant differences compared with T + V within the same genotype. *P < 0.05, **P < 0.01, and ***P < 0.001. # P < 0.05, ## P < 0.01, and ### P < 0.001. The main effects of genotype (P < 0.05) are shown in P‐values above the corresponding figures. §: Genotype difference between Ghsr −/− and Ghsr +/+ within the same treatment group (P < 0.05).
We analysed muscle fibre cross‐sectional area (CSA) in the deep area of GAS muscles. The decrease in fibre CSA induced by LLC implantation was greater in type IIB (fast‐twitching, glycolytic) than in type IIA (slow‐twitching, oxidative) muscle fibres (Figure 1C,D). LLC‐induced atrophy in type IIB fibres, the predominant fibre type in GAS muscle, was fully prevented in Ghsr +/+ but only partially attenuated in Ghsr −/− mice (Figure 1D), suggesting a combination of GHSR‐1a dependent and independent effects of ghrelin on preserving muscle mass loss. Interestingly, a genotype difference was observed as a main effect in type IIA fibres that were smaller in the absence of GHSR‐1a (Figure 1C).
Proteolytic and autophagy but not protein synthesis markers are modulated by ghrelin in Lewis lung carcinoma‐induced cachexia
Tumour implantation induced an increase in atrogenes in all groups. However, Ghsr −/− mice showed a more pronounced increase in Trim63/MuRF1 in response to tumour implantation than Ghsr +/+ (Figure 2A). A similar pattern was observed in Fbxo32/atrogin, although the genotype difference did not reach significance. Exogenous ghrelin administration partially prevented these tumour‐induced atrogene increases. These results suggest that GHSR‐1a partially prevents the LLC‐induced activation of proteolysis and that ghrelin prevents this in a GHSR‐1a‐independent manner. In Ghsr +/+ animals, there were no significant changes in autophagy markers including Becn1, Atg5 and Atg7 at the transcript level, as well as in protein 1A/1B‐light chain 3 (LC3)‐II protein levels upon tumour implantation or ghrelin treatment (Figure 2B–D). Becn1 is responsible for the initiation of autophagy. Atg7 is the E1‐like enzyme that mediates the Atg5‐Atg12 complex formation, and is essential for the conjugation of LC3I to phosphatidylethanolamine to form the LC3‐II. 48 When autophagy flux was assessed by blocking the delivery of autophagosomes to lysosomes with colchicine, LLC tumour implantation induced a significant increase in LC3II levels that was partially prevented by ghrelin treatment (Figure 2D). In Ghsr −/− mice, LLC implantation caused an increase in Becn1, Atg5, and Atg7, and these markers were also higher than in the Ghsr +/+ mice. Ghrelin administration prevented the increase in autophagy initiation (Becn1) but did not impact autophagosome formation, LC3I conjugation or activation into LC3II. The net result was that LC3II was significantly increased in Ghsr −/− mice by LLC implantation and ghrelin did not prevent this increase. Interestingly, when autophagy flux was assessed with the administration of colchicine in Ghsr −/− tumour‐bearing mice, LC3II did not increase as much as in Ghsr +/+ mice, suggesting that GHSR‐1a plays a role in inducing autophagosome degradation (Figure 2D).
Figure 2.
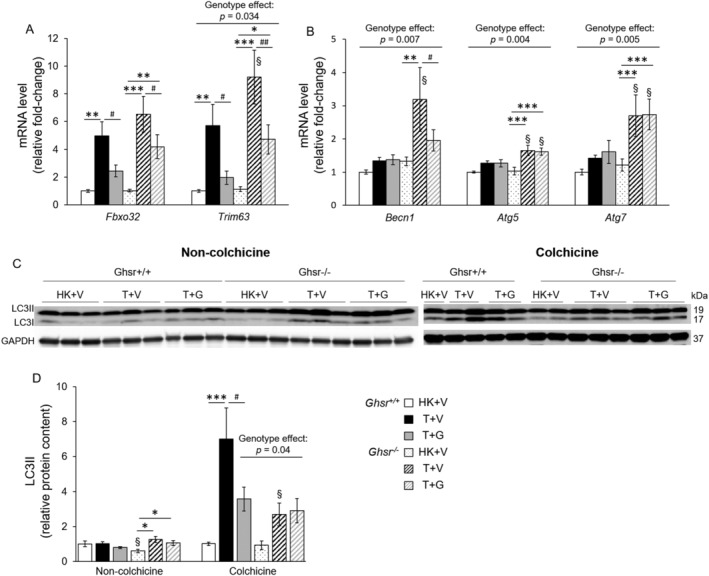
Proteolytic and autophagy markers in skeletal muscles in Ghsr +/+ and Ghsr −/− . HK + V: Heat‐killed + vehicle; T + V: Tumour + vehicle; T + G: Tumour + ghrelin. Gene expression of (A) proteolytic markers Fbxo32 and Trim63, (N = 12–14/group) and (B) autophagy markers Becn1, Atg5, and Atg7 (N = 9–12/group) in GAS/PL muscles. GAPDH was used as a reference gene, and data is expressed as a relative fold‐change of the Ghsr +/+ HK + V group. (C‐D) protein levels of LC3II in GAS/PL muscles from mice without (N = 13–15) and with treatment (N = 6–7) with the lysosome inhibitor colchicine for 2 days before euthanasia. (C) Representative Western blots of LC3II, LC3I, and GAPDH. (D) the relative protein content of LC3II. Western blots were quantified by densitometry and normalized to GAPDH. Results are presented as fold‐change of Ghsr +/+ HK + V. data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. *, **, and *** denote significant differences comparing to HK + V within the same genotype; # denotes significant differences compared with T + V within the same genotype. * P < 0.05, ** P < 0.01, and *** P < 0.001. # P < 0.05. The main effects of genotype (P < 0.05) are shown in P‐values above the corresponding figures. §: Genotype difference between Ghsr −/− and Ghsr +/+ within the same treatment group (P < 0.05).
We also detected markers of protein synthesis in response to LLC tumour implantation and ghrelin. Tumour implantation decreased phospho‐Protein kinase B (p‐AKT, Ser473)/total AKT ratio in Ghsr +/+ mice and this decrease was not prevented by ghrelin (Supporting Information, Figure S2A,B). Ghsr −/− mice without tumour (HK + V) showed lower levels of p‐AKT/total AKT, and there was no effect from either tumour or ghrelin in these mice. Insulin‐like growth factor 1 (IGF)‐1 plasma levels were lower in Ghsr −/− , and no effect was found in response to tumour implantation or ghrelin treatment in either genotype (Supporting Information, Figure S2C).
Lewis lung carcinoma induced mitophagy is prevented by ghrelin via growth hormone secretagogue receptor‐1a
p62/Sequestosome‐1 (SQSTM1) and BCL2 interacting protein 3 (Bnip3) are both involved in removing damaged mitochondria via selective autophagic degradation (mitophagy) by recruiting and binding autophagosome proteins through their cargo‐binding domains and LC3‐interacting domains. 49 The protein level of p62 (Figure 3‐A,B) and transcript level of Bnip3 ( Figure 3D) were increased by LLC implantation in both genotypes, and ghrelin attenuated these changes only in Ghsr +/+ mice. Importantly, grip strength at the time of sacrificing was inversely correlated with p62 (Figure 3C), and Bnip3 levels (Figure 3E), suggesting that elevated mitophagy is associated with decreased grip strength.
Figure 3.
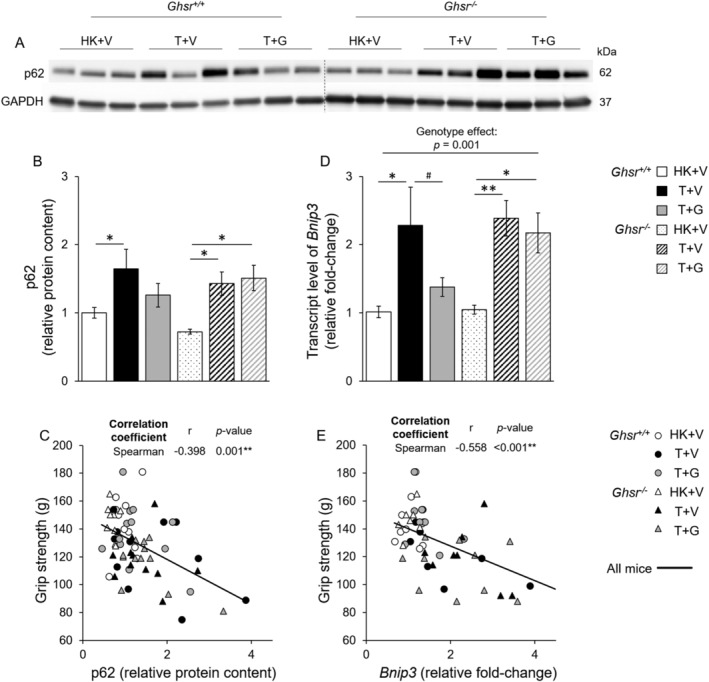
Mitophagy markers p62 and Bnip3 in skeletal muscles in Ghsr +/+ and Ghsr −/− mice. HK + V: Heat‐killed + vehicle; T + V: Tumour + vehicle; T + G: Tumour + ghrelin. (A, B) protein levels of p62 in GAS/PL muscles in mice from each group (N = 9–12/group). (A) Representative Western blots of p62 and GAPDH. A dashed line indicates images acquired from different blots. (B) P62 relative protein content. Western blots were quantified by densitometry and normalized to GAPDH. Results are presented as fold‐change of Ghsr +/+ HK + V. (C) Correlation between grip strength and p62 relative protein content (r = −0.398, P = 0.001). (D) Gene expression of Bnip3 in GAS/PL muscles in mice from each group (N = 7–10/group). GAPDH was used as a reference gene, and data are expressed as a relative fold‐change of the Ghsr +/+ HK + V group. (E) Correlation between grip strength and Bnip3 expression (r = −0.558, P = 0.001). (B and D) data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. * and ** denote significant differences comparing to HK + V within the same genotype; # denotes significant differences compared with T + V within the same genotype. * P < 0.05, ** P < 0.01. # P < 0.05. The main effects of genotype (P < 0.05) are shown in P‐values above the corresponding figures. §: Genotype difference between Ghsr −/− and Ghsr +/+ within the same treatment group (P < 0.05). (C and E) correlations were assessed with the spearman correlation coefficient test. ** P < 0.01.
Ghrelin prevented the Lewis lung carcinoma‐induced decrease in mitochondrial respiration and growth hormone secretagogue receptor‐1a has a protective role in preserving mitochondrial respiration
In Ghsr +/+ mice, LLC implantation reduced State 3 (ADP‐dependent respiration), and State 3u (maximal uncoupled respiration) mitochondrial respiration. These changes were partially ameliorated by ghrelin although it only reached significance for State 3u (Figure 4A). No effect of tumour or ghrelin was found in Ghsr −/− mice, and GHSR‐1a deletion was associated with lower mitochondrial respiration at all respiration states except for State 3u compared with Ghsr +/+ mice.
Figure 4.
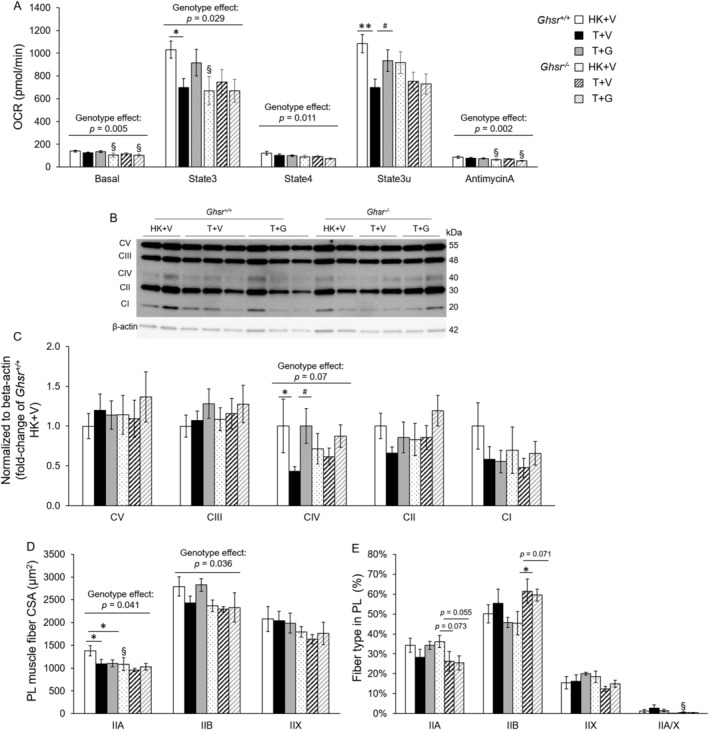
Oxygen consumption rate, OXPHOS complexes and fibre CSA and typing in plantaris muscles in Ghsr +/+ and Ghsr −/− mice. (A) Oxygen consumption rate (OCR, pmol/min) in mitochondria isolated from plantaris muscles in Ghsr +/+ and Ghsr −/− mice. HK + V: Heat‐killed + vehicle; T + V: Tumour + vehicle; T + G: Tumour + ghrelin. OCR was measured in five mitochondrial respiration states (N = 13–16/group). (B, C) protein levels of OXPHOS in isolated mitochondria in PL muscles from each group (N = 9–12/group). (B) Representative Western blots of OXPHOS complexes and β‐actin. (B) OXPHOS complexes relative protein content. Western blots were quantified by densitometry and normalized to β‐actin. Results are presented as fold‐change of Ghsr +/+ HK + V (N = 9/group). (D) Cross‐sectional area (CSA) of myosin heavy chain (MHC) type IIA, IIB, IIX, and IIA/X fibres in the PL muscles at endpoint (μm2). One hundred type IIA, 200 type IIB, and 60–80 type IIX or IIA/X individual fibres were analysed per animal for CSA (N = 4/group). (E) Percentage of each fibre type in PL muscles. Type IIA, IIB, IIX, and IIA/X fibres were counted and normalized to total fibre number in PL muscles (expressed as %, N = 4). Data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. * and ** denote significant differences comparing to HK + V within the same genotype; # denotes significant differences compared with T + V within the same genotype. * P < 0.05, ** P < 0.01. # P < 0.05. The main effects of genotype (P < 0.05) are shown in P‐values above the corresponding figures. §: Genotype difference between Ghsr −/− and Ghsr +/+ within the same treatment group (P < 0.05).
We did not observe any differences in the percentage of Succinate Dehydrogenase (SDH, complex II of the mitochondrial electron transport chain) positive fibres in PL across groups (Supporting Information, Figure S3A). Also, there were no differences in protein levels of the mitochondrial biogenesis marker peroxisome proliferator‐activated receptor‐gamma coactivator (PGC‐1α) in GAS/PL muscle of Ghsr +/+ mice. In Ghsr −/− mice, PGC‐1α protein level was lower in HK + V compared with Ghsr +/+ mice but increased with tumour implantation (Supporting Information, Figure S3B,C). This was not affected by ghrelin administration. Similar trends were seen at the transcript level (Ppargc1a) but the differences did not reach significance (Supporting Information, Figure S3D). Lastly, we detected the protein levels of OXPHOS complexes from the isolated mitochondria of PL muscles. Complex IV (Cytochrome c oxidase), a catalytic complex in the electron transport chain, was decreased in Ghsr +/+ after tumour implantation and this change was prevented by ghrelin treatment. The absence of GHSR‐1a tended to lower (genotype effect: P = 0.07) the levels of Complex IV compared with Ghsr +/+, although no tumour or ghrelin effect was found. We did not detect any tumour or ghrelin effect on the protein levels of Complex I, II, III, and V (Figure 4B,C).
Growth hormone secretagogue receptor‐1a modulates the effect of Lewis lung carcinoma‐implantation on plantaris fibre typing and cross‐sectional area
Type IIA and IIB fibre CSA in PL muscles were smaller in Ghsr −/− mice compared with wildtype and type IIA fibre CSA was also decreased in tumour‐bearing Ghsr +/+ mice (Figure 4D). We also identified fibre type % in PL muscles. In Ghsr −/− mice, the percentage of IIA fibres was decreased (trend) and the percentage of type IIB reciprocally increased with tumour implantation. Also, Ghsr −/− tumour‐bearing mice showed less hybrid type IIA/X. These results suggest a slow‐to‐fast fibre type switch with tumour implantation in the absence of GHSR‐1a that was not prevented by ghrelin (Figure 4E).
Discussion
To this date, there are no Food and Drug Administration (FDA)‐approved drugs for CC, and several clinical trials targeting this condition have failed to meet their primary endpoints due to lack of an effect on muscle function in spite of inducing significant gains in lean mass. 10 , 17 Therefore, there is a pressing need to improve our understanding of the mechanisms mediating muscle function loss in CC. Ghrelin and agonists of its only known receptor, GHSR‐1a, show potential to ameliorate CC at least in part by preventing decreases in body weight and muscle mass, but the specific mechanisms mediating these effects have not been fully characterized. In this study, we show that ghrelin ameliorates LLC‐induced muscle mass loss and UPS activation independently of GHSR‐1a; whereas, ghrelin prevents LLC‐induced decreases in grip strength, mitophagy, and mitochondrial dysfunction via GHSR‐1a. In addition, we found that GHSR‐1a plays a role in regulating skeletal muscle fibre CSA, autophagy, and mitochondrial respiration.
The loss of muscle strength and function in CC is accompanied by loss of muscle mass. 50 However, the relationship between these parameters is likely to be non‐linear, where the loss of function may occur later in some cases but earlier in others and accelerate faster than the loss of muscle mass. 51 , 52 Also, the post‐intervention recovery of muscle mass may happen sooner than the recovery of muscle function. 51 This could potentially explain why several phase III clinical trials using GHSR‐1a agonists or other anabolic agents such as selective androgen receptor modulators failed to improve muscle function in spite of increasing muscle mass, 17 , 53 , 54 In the current study, we show that, in the absence of GHSR‐1a, ghrelin partly attenuates muscle atrophy but not the decrease in grip strength induced by LLC tumour implantation. Whereas, in the Ghsr +/+ mice, the magnitude of muscle atrophy is less than in the Ghsr −/− mice, and ghrelin prevents both tumour‐induced muscle mass and function loss. Taken together, this data is consistent with the clinical evidence that muscle mass and function are independently modulated in CC. It also suggests that both GHSR‐1a‐dependent and ‐independent pathways contribute to the effects of ghrelin, and that the presence of GHSR‐1a is required for ghrelin to improve muscle strength, with the caveat that muscle strength was only determined by grip test in these experiments. Future studies should incorporate other measures of muscle function such as assessments of individual muscle contractility and specific force.
The imbalance between protein synthesis and degradation leads to muscle atrophy, and several groups have previously suggested a potential role for protein synthesis in CC. 3 , 7 , 8 , 55 In Ghsr +/+ mice, we found a decreased p‐AKT/total AKT ratio in response to LLC tumour but no effect from ghrelin. Interestingly, we did not see a tumour or ghrelin effect in Ghsr −/− mice, but these mice had a lower ratio of p‐AKT/AKT at baseline. This genotype difference could be related to IGF‐1 levels, which is upstream of AKT and were lower in the absence of GHSR‐1a in agreement with previous reports. 56 The lack of effect of ghrelin on protein synthesis pathways has also been reported previously. Porporato et al. reported that ghrelin counteracted protein degradation rather than promoting protein synthesis as determined by S6 phosphorylation level in C2C12 cells, co‐cultured with dexamethasone. 8 Also, other studies showed that the decreased protein synthesis induced by C26 or LLC tumours cannot be restored by treatments that attenuate muscle atrophy. 57 , 58 Taken together, our data suggest that protein synthesis pathways may not be affected by ghrelin in the LLC‐induced cachexia model, although GHSR‐1a is likely to play a role in regulating their basal levels.
UPS and autophagy are overactivated in CC, 1 , 59 , 60 and our results demonstrate that they are regulated in a GHSR‐1a‐specific manner. We show that the effects of ghrelin on altering tumour‐induced upregulation of UPS do not require the presence of GHSR‐1a. This result is consistent with other non‐cancer in‐vitro and preclinical studies. 7 , 8 Previously, we showed that ghrelin prevented LLC‐induced increases in the expression of Atrogin‐1 and MuRF1, 7 two muscle‐specific E3 ubiquitin ligases in the UPS pathway known as ‘atrogenes’, which are key markers of protein degradation in skeletal muscle. 61 , 62 Here, we show that these effects are mediated through a GHSR‐1a‐independent mechanism. Our study also shows that GHSR‐1a plays a protective role in mitigating muscle atrophy and suppressing UPS activation induced by LLC tumour even in the absence of exogenous ghrelin administration given that Ghsr −/− mice exhibited more pronounced muscle loss, and higher levels of ‘atrogenes’ in skeletal muscle.
Autophagy homeostasis is essential for maintaining muscle mass and quality by removing damaged organelles, protein aggregates, and pathogens under basal conditions. Under catabolic conditions, including CC, excessive activation of autophagy also induces selective removal of myofibrillar and structural proteins, 19 , 26 as well as mitophagy. 27 , 28 In brief, the process of autophagy involves autophagosome engulfing targeted organelles and proteins, and subsequently fusing with lysosomes so the engulfed organelles and proteins can be degraded by acidic lysosomal hydrolases. 63 , 64 The initiation of autophagy requires activation of Beclin‐1, which is responsible for the formation of the phagophore so it can engulf the targeted organelles and proteins in the cytoplasm to form an autophagosome. The formation of autophagosomes also requires Atg12‐Atg5 complex and Atg7‐mediated conjugation of LC3‐I to the membrane lipid phosphatidylethanolamine (PE) to form LC3‐II, an important marker for autophagy activation. 65 Excessive activation of autophagy is responsible for further protein degradation in skeletal muscle and mitophagy in CC. 34 A protective role of ghrelin in attenuating increased autophagy has been reported in C26 co‐cultured C2C12 myotubes. 66 GHSR agonists have also been found to play a role in decreasing increased autophagy in a chemotherapy‐induced cachexia rat model, 37 although another study using the same animal model did not show any chemotherapy‐ or GHSR agonist‐effect on altering autophagy. 67 To our knowledge, the role of ghrelin or GHSR‐1a agonists on modulating tumour‐induced skeletal muscle autophagy is unknown. In our study, we only found increases in autophagy markers upon tumour implantation in Ghsr −/− mice, including Becn‐1, Atg5, Atg7 at the transcript level, and LC3II at the protein level. When autophagy flux was assessed by blocking the delivery of autophagosomes to lysosomes with colchicine, LLC tumour implantation induced a significant increase in autophagy markers that was partially prevented by ghrelin in wild‐type animals. Our data suggest that the presence of GHSR‐1a promotes autophagosome degradation preventing the increase in autophagy markers induced by tumour implantation and that ghrelin administration modulates this process via this receptor. More studies will be required to confirm the relative contribution of autophagy in mediating the effects of ghrelin in LLC‐induced cachexia.
Mitophagy is the process of removing mitochondria via selective autophagy by cargo‐binding proteins. p62, a ubiquitin‐binding protein, is responsible for mitochondrial ubiquitination, 68 whereas Bnip3 plays a central role in this process by triggering mitochondrial depolarization and mitophagy. 69 , 70 Although mitophagy is considered to be essential for cleavage of damaged mitochondria to maintain muscle quality under basal conditions, 49 it has been demonstrated that accelerated mitophagy potentially contributes to mitochondrial dysfunction in skeletal muscle during CC. 28 , 34 Increased mitophagy markers have been found in preclinical CC models, including Apc Min/+, 33 C26, 34 and LLC 33 and in cancer patients. 71 Recently, Penna and coworkers found increased mitophagy and impaired mitochondrial function in skeletal muscles in C26 tumour‐bearing mice 34 ; and these changes were exacerbated and/or maintained after the promotion of autophagy by overexpression TP53INP2, suggesting that overactivation of mitophagy impairs mitochondrial function in CC. In our model, we found increased Bnip3 and p62 levels in both genotypes that were ameliorated by ghrelin administration in a GHSR‐1a‐dependent manner. Moreover, the correlation between mitophagy markers and grip strength suggests that this process mediates at least in part, the effects of ghrelin on preventing tumour‐induced grip strength loss. This also aligns with recent data showing that mitochondrial dysfunction is associated with poor recovery of muscle function but not muscle mass in non‐cancer models of muscle atrophy, 72 and results from an LLC‐induced cachexia model showing that mitophagy occurs earlier than changes in muscle mass. 33
Mitochondrial function is crucial for maintaining muscle function by providing energy (adenosine triphosphate (ATP)) for contractility 73 and by regulating calcium homeostasis in skeletal muscle. 74 , 75 In rodent models of cachexia, decreased oxidative phosphorylation (OXPHOS) capacity, 33 , 76 disrupted mitochondrial dynamics, 33 , 37 , 76 and diminished mitochondrial respiration has been observed in muscles with decreased mass and/or strength. Impairment in state 3 mitochondrial respiration has been reported in LLC‐, 33 C26‐, 34 and LP07‐induced cachexia, 77 as well as in LLC co‐cultured C2C12 myotubes. 78 However, the role of ghrelin in modulating mitochondrial function is not well‐characterized. Although a positive effect has been recently reported in sarcopenic mice lacking ghrelin, 79 there is no evidence of ghrelin's effects on mitochondrial dysfunction in cancer cachexia. We found decreased complex‐I‐driven maximal state3 respiration under coupled (ADP‐stimulated) and uncoupled (FCCP‐stimulated) conditions in wild‐type, LLC‐implanted mice. Ghrelin administration partially prevented these changes. Interestingly, all Ghsr −/− mice including controls had lower mitochondrial respiration compared with wild‐type mice and this was not affected by tumour implantation or ghrelin administration. In the context of decreased type IIA (oxidative) fibre size in Ghsr −/− mice, this suggests a role of GHSR‐1a in regulating mitochondrial respiration. Our data also is consistent with the hypothesis that the increase in muscle strength with ghrelin administration in LLC‐implanted mice is due, at least in part, to increased mitochondrial respiration mediated via GHSR‐1a.
In the current study, we did not see any difference in SDH content across groups in PL muscles 3 weeks after tumour implantation. In a similar LLC‐induced cachexia mouse model, a decrease in SDH + cells in TA muscle was found 4 weeks after tumour implantation but was not observed when assessed at weeks 1 or 3, suggesting that this is a late effect in the course of CC. 33 We also did not detect a difference in PGC‐1α protein or mRNA levels from ghrelin administration or tumour in Ghsr +/+ mice. Similarly, in Brown's study, PGC‐1α was not altered while mitochondrial respiration was diminished in response to LLC tumour 3 weeks after tumour implantation. 33 We also detected genotype differences in this marker. The lower PGC‐1α protein level in non‐tumour‐bearing Ghsr −/− mice is likely associated with its smaller IIA fibre size, and may potentially explain its lower mitochondrial respiration at several states (basal, state4, and AntimycinA) when compared with Ghsr +/+ . However, we also saw an increase of PGC‐1α in animals without GHSR‐1a in response to tumour implantation and this may be due to the more profound negative energy balance in these animals. We found that the decreased protein level of Complex IV (Cytochrome c oxidase) was prevented by ghrelin treatment; whereas lacking GHSR‐1a tended to lower Complex IV levels, although no tumour or ghrelin effect was found in Ghsr −/− . Other studies have also shown decreased Complex IV levels in different CC models, such as Apcmin/+ mice, 55 , 76 C26 mice, 80 and Walker 256 tumour‐bearing rats. 81 We did not detect any tumour or ghrelin effect on the protein levels of Complex I, II, III, and V. Although it is not clear if all the OXPHOS components respond to cachexia in the aforementioned studies, our study showed complex IV is more susceptible to LLC‐induced cachexia than other OXPHOS complexes, which is likely due to the involvement of complex IV in both complex I and II‐driven respiration and the higher abundance of complex IV than other complexes. 82 Also, this complex IV‐dominated change has been shown in human mitochondrial myopathy 83 and training‐related mitochondrial modifications in mice. 84 We showed that Ghsr −/− mice have overall smaller type IIA and IIB fibres and there may be a slow‐to‐fast fibre type transformation (IIA to IIB) in PL in response to tumour‐implantation. These fibre type differences between groups could account at least in part for the differences in mitochondrial respiration given that type IIA fibres are known to have greater mitochondrial content than type IIB fibres. 85 Taken together, we show evidence that GHSR‐1a may modulate tumour‐induced mitochondrial respiration by changing the expression of Complex IV and the slow‐twitch fibre type content; whereas ghrelin's effects on mitochondrial respiration are not mediated by PGC‐1α and are not associated with SDH protein content, at least in the current LLC tumour model. Future studies will be required to elucidate the function of the electron transport chain in more depth. 47
Tumour weights, food intake, and physical activity levels may also contribute to the progression of CC and potentially influence the effects of ghrelin. These data in this cohort of mice have been recently published. 15 Briefly, the tumour size was not affected by either genotype or ghrelin treatment (Ghsr +/+: T + V: 3.7 ± 0.5 g; T + G: 3.5 ± 0.5 g; Ghsr −/−: T + V: 4.4 ± 0.6 g; T + G: 3.0 ± 0.3 g. Two‐way ANOVA: Genotype effect: P = 0.872; Treatment effect: P = 0.115). Ghrelin attenuated the tumour‐induced decrease in food intake only when GHSR‐1a was present, confirming a GHSR‐1a‐dependent ghrelin effect on food intake. Lastly, we found that tumour implantation decreased spontaneous locomotor activity in both genotypes, and that ghrelin administration did not prevent these changes. 15 These data suggest that the effects of ghrelin on muscle cannot be explained by differences in locomotor activity levels or tumour size. An effect of food intake cannot be entirely ruled out in Ghsr +/+ . More studies will be needed to address this issue.
In our study, Ghsr −/− mice showed a more profound cachectic phenotype in response to LLC tumour implantation, including lower body weight, smaller muscle mass, and higher levels of proteolysis and autophagy when compared with wild‐type tumour‐bearing mice receiving vehicle. These results suggest a protective role of GHSR‐1a, and, potentially, of endogenous ghrelin as the ligand for this receptor, in maintaining muscle mass and function in LLC‐induced cachexia. The results also align with the therapeutic effects of exogenous ghrelin administration we report here that are partly GHSR‐1a dependent.
There are several limitations in our study. The preclinical experiments reported here will have to be validated in clinical trials to determine their clinical relevance. Our results imply the existence of an alternative receptor for ghrelin which has not been characterized so far; ongoing experiments are underway to identify this receptor. Given the presence of esterases in blood, 19 it is expected that both acyl‐ and desacyl‐ghrelin levels will increase upon acyl‐ghrelin administration. Therefore, it is possible that desacyl‐ghrelin is at least partially responsible for the GHSR‐1a independent effects of ghrelin administration. 20 As we did not process blood samples with acidification and the addition of protease inhibitors, measurements of acyl‐ghrelin were not possible. As mentioned before, we only assessed muscle strength by grip strength and more studies are needed to confirm these effects on other measures of muscle function such as muscle contractility and specific force. Also, these experiments included only male, young adult mice, and the potential contribution of sex or age as a biological variable of interest will require more studies. Lastly, we only investigated GAS and PL muscles in these mice. More studies will be required to confirm our results in other muscles.
In summary, we show for the first time that GHSR‐1a mediates ghrelin's effects on attenuating cancer‐induced muscle weakness but not muscle mass loss and that it does so by modulating the autophagy‐lysosome pathway, mitophagy, and mitochondrial respiration. These results fill a critical gap in our knowledge and may inform the design of future clinical trials targeting the ghrelin pathway as a therapeutic for cancer cachexia.
Funding
This work was funded by the U.S. Department of Veterans Affairs (BX002807 to J.M.G.). J.M.G. also receives research support from the Congressionally Directed Medical Research Programs (PC170059), and from the NIH (R01CA239208, R01AG061558). We thank the University of Washington Diabetes and Endocrinology Research Center (P30 DK017047) and Nutrition Obesity Research Center (P30 DK035816), and Vanderbilt Mouse Metabolic Phenotyping Center (supported in part by U24 DK59637), for their help.
Conflict of interest
Jose M. Garcia received research support from Aeterna Zentaris Inc., Helsinn Therapeutics, Inc. and Extend Biosciences. Haiming Liu, Pu Zang, Ian (In‐gi) Lee, Barbara Anderson, Anthony Christiani, Lena Strait‐Bodey, Beatrice A. Breckheimer, Mackenzie Storie, Alison Tewnion, Kora Krumm, Theresa Li, and Brynn Irwin declare that they have no conflict of interest.
Ethical guidelines statement
All authors certify that they comply with the Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 86 All animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. The manuscript does not contain clinical studies or patient data.
Supporting information
Figure S1. Ghsr mRNA expression in brain and skeletal muscles (GAS/PL) in Ghsr +/+ and Ghsr −/− animals. GAPDH was used as a reference gene, and data are expressed as ΔCt normalized by GAPDH expression in the same sample. Data are shown as mean ± SE (N = 4/group).
Figure S2. AKT in muscle and plasma IGF‐1 levels from Ghsr +/+ and Ghsr −/− mice. HK + V: heat‐killed + vehicle; T + V: tumour + vehicle; T + G: tumour + ghrelin. (A‐B) Protein levels of p‐Akt (Ser473) and total Akt in GAS/PL muscles in mice from each group (N = 4). (A) Representative Western blots of p‐Akt (Ser473) and total Akt. (B) Ratio of p‐Akt (Ser473)/total Akt. Western blots were quantified by densitometry. (C) IGF‐1 expression in plasma (ng/ml). Data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. * denote significant differences comparing to HK + V within the same genotype (p < 0.05). The main effects of genotype (p < 0.05) are shown in p‐values above the corresponding figures.
Figure S3. Oxidative capacity markers in skeletal muscles in Ghsr +/+ and Ghsr −/− . HK + V: heat‐killed + vehicle; T + V: tumour + vehicle; T + G: tumour + ghrelin. (A) Percentage of SDH + fibres in PL muscles (N = 4/group). (B) Representative Western blots of PGC‐1α and GAPDH. (C) Relative protein content of PGC‐1α. Western blots were quantified by densitometry and normalized to GAPDH. Results are presented as fold‐change of Ghsr +/+ HK + V (N = 7–10/group). (D) Relative gene expression of PGC‐1α (Ppargc1a) in GAS/PL muscles. GAPDH was used as a reference gene, and data is expressed as a relative fold‐change of the Ghsr +/+ HK + V group. Data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. * denotes significant differences comparing to HK + V within the same genotype (p < 0.05). §: Genotype difference between Ghsr −/− and Ghsr +/+ within the same treatment group (p < 0.05). N.S.: No significant difference was detected across the groups.
Data S1. Supporting Information
Liu H., Zang P., Lee I. ( I.‐g.), Anderson B., Christiani A., Strait‐Bodey L., Breckheimer B. A., Storie M., Tewnion A., Krumm K., Li T., Irwin B., and Garcia J. M. (2021) Growth hormone secretagogue receptor‐1a mediates ghrelin's effects on attenuating tumour‐induced loss of muscle strength but not muscle mass, Journal of Cachexia, Sarcopenia and Muscle, 12, 1280–1295, 10.1002/jcsm.12743
References
- 1. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 2. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 3. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 4. von Haehling S, Garfias Macedo T, Valentova M, Anker MS, Ebner N, Bekfani T, et al. Muscle wasting as an independent predictor of survival in patients with chronic heart failure. J Cachexia Sarcopenia Muscle 2020;11:1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Drescher C, Konishi M, Ebner N, Springer J. Loss of muscle mass: current developments in cachexia and sarcopenia focused on biomarkers and treatment. J Cachexia Sarcopenia Muscle 2015;6:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Muller TD, Tschop MH. Ghrelin ‐ a key pleiotropic hormone‐regulating systemic energy metabolism. Endocr Dev 2013;25:91–100. [DOI] [PubMed] [Google Scholar]
- 7. Chen JA, Splenser A, Guillory B, Luo J, Mendiratta M, Belinova B, et al. Ghrelin prevents tumour‐ and cisplatin‐induced muscle wasting: characterization of multiple mechanisms involved. J Cachexia Sarcopenia Muscle 2015;6:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 2013;123:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsubouchi H, Yanagi S, Miura A, Matsumoto N, Kangawa K, Nakazato M. Ghrelin relieves cancer cachexia associated with the development of lung adenocarcinoma in mice. Eur J Pharmacol 2014;743:1–10. [DOI] [PubMed] [Google Scholar]
- 10. Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, et al. Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo‐controlled, double‐blind trials. Lancet Oncol 2015;16:108–116. [DOI] [PubMed] [Google Scholar]
- 11. Zhang H, Garcia JM. Anamorelin hydrochloride for the treatment of cancer‐anorexia‐cachexia in NSCLC. Expert Opin Pharmacother 2015;16:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth‐hormone‐releasing acylated peptide from stomach. Nature 1999;402:656–660. [DOI] [PubMed] [Google Scholar]
- 13. Smith RG, Van der Ploeg LH, Howard AD, Feighner SD, Cheng K, Hickey GJ, et al. Peptidomimetic regulation of growth hormone secretion. Endocr Rev 1997;18:621–645. [DOI] [PubMed] [Google Scholar]
- 14. Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology 2007;148:1323–1329. [DOI] [PubMed] [Google Scholar]
- 15. Liu H, Luo J, Guillory B, Chen JA, Zang P, Yoeli JK, et al. Ghrelin ameliorates tumor‐induced adipose tissue atrophy and inflammation via Ghrelin receptor‐dependent and ‐independent pathways. Oncotarget 2020;11:3286–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia JM. What is next after anamorelin? Curr Opin Support Palliat Care 2017;11:266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, et al. Anamorelin in patients with non‐small‐cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double‐blind, phase 3 trials. Lancet Oncol 2016;17:519–531. [DOI] [PubMed] [Google Scholar]
- 18. Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016;5:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sandri M. Protein breakdown in cancer cachexia. Semin Cell Dev Biol 2016;54:11–19. [DOI] [PubMed] [Google Scholar]
- 20. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life‐long exercise. Exp Gerontol 2010;45:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010;16:1313–1320. [DOI] [PubMed] [Google Scholar]
- 23. Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin‐induced oxidative stress and proteolysis in skeletal muscle. J Appl Physiol (1985) 2011;110:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O'Leary MF, Vainshtein A, Carter HN, Zhang Y, Hood DA. Denervation‐induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis‐deficient animals. Am J Physiol Cell Physiol 2012;303:C447–C454. [DOI] [PubMed] [Google Scholar]
- 25. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007;6:472–483. [DOI] [PubMed] [Google Scholar]
- 26. Grumati P, Bonaldo P. Autophagy in skeletal muscle homeostasis and in muscular dystrophies. Cell 2012;1:325–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of protein degradation in striated muscles. J Mol Cell Cardiol 2013;55:64–72. [DOI] [PubMed] [Google Scholar]
- 28. VanderVeen BN, Fix DK, Carson JA. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid Med Cell Longev 2017;2017:3292087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carson JA, Hardee JP, VanderVeen BN. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer‐induced muscle wasting. Semin Cell Dev Biol 2016;54:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 2013;182:1367–1378. [DOI] [PubMed] [Google Scholar]
- 31. Chacon‐Cabrera A, Fermoselle C, Urtreger AJ, Mateu‐Jimenez M, Diament MJ, de Kier Joffe ED, et al. Pharmacological strategies in lung cancer‐induced cachexia: effects on muscle proteolysis, autophagy, structure, and weakness. J Cell Physiol 2014;229:1660–1672. [DOI] [PubMed] [Google Scholar]
- 32. Aversa Z, Pin F, Lucia S, Penna F, Verzaro R, Fazi M, et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci Rep 2016;6:30340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown JL, Rosa‐Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 2017;8:926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Penna F, Ballaro R, Martinez‐Cristobal P, Sala D, Sebastian D, Busquets S, et al. Autophagy exacerbates muscle wasting in cancer cachexia and impairs mitochondrial function. J Mol Biol 2019;431:2674–2686. [DOI] [PubMed] [Google Scholar]
- 35. Barazzoni R, Zhu X, Deboer M, Datta R, Culler MD, Zanetti M, et al. Combined effects of ghrelin and higher food intake enhance skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rats with chronic kidney disease. Kidney Int 2010;77:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barazzoni R, Gortan Cappellari G, Palus S, Vinci P, Ruozi G, Zanetti M, et al. Acylated ghrelin treatment normalizes skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rat chronic heart failure. J Cachexia Sarcopenia Muscle 2017;8:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sirago G, Conte E, Fracasso F, Cormio A, Fehrentz JA, Martinez J, et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin‐induced cachexia. Sci Rep 2017;7:13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun Y, Butte NF, Garcia JM, Smith RG. Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology 2008;149:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tschop M, Flora DB, Mayer JP, Heiman ML. Hypophysectomy prevents ghrelin‐induced adiposity and increases gastric ghrelin secretion in rats. Obes Res 2002;10:991–999. [DOI] [PubMed] [Google Scholar]
- 40. Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of "autophagic flux" in mature skeletal muscle. Autophagy 2010;6:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baumann CW, Liu HM, Thompson LV. Denervation‐Induced Activation of the Ubiquitin‐Proteasome System Reduces Skeletal Muscle Quantity Not Quality. PLoS ONE 2016;11:e0160839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu HM, Ferrington DA, Baumann CW, Thompson LV. Denervation‐Induced Activation of the Standard Proteasome and Immunoproteasome. PLoS ONE 2016;11:e0166831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sawano S, Komiya Y, Ichitsubo R, Ohkawa Y, Nakamura M, Tatsumi R, et al. A One‐Step Immunostaining Method to Visualize Rodent Muscle Fiber Type within a Single Specimen. PLoS ONE 2016;11:e0166080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wende AR, Schaeffer PJ, Parker GJ, Zechner C, Han D‐H, Chen MM, et al. A Role for the Transcriptional Coactivator PGC‐1α in Muscle Refueling. J Biol Chem 2007;282:36642–36651. [DOI] [PubMed] [Google Scholar]
- 45. Chavez JD, Tang X, Campbell MD, Reyes G, Kramer PA, Stuppard R, et al. Mitochondrial protein interaction landscape of SS‐31. Proc Natl Acad Sci U S A 2020;117:15363–15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 2011;6:e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boutagy NE, Rogers GW, Pyne ES, Ali MM, Hulver MW, Frisard MI. Using Isolated Mitochondria from Minimal Quantities of Mouse Skeletal Muscle for High throughput Microplate Respiratory Measurements. J Vis Exp 2015;e53216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tanida I, Ueno T, Kominami E. LC3 and Autophagy. In Deretic V, ed. Totowa, NJ: Humana Press; 2008. p 77–88. [DOI] [PubMed] [Google Scholar]
- 49. Romanello V, Sandri M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front Physiol 2015;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anderson LJ, Chong N, Migula D, Sauer A, Garrison M, Wu P, et al. Muscle mass, not radiodensity, predicts physical function in cancer patients with or without cachexia. Oncotarget 2020;11:1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramage MI, Skipworth RJE. The relationship between muscle mass and function in cancer cachexia: smoke and mirrors? Curr Opin Support Palliat Care 2018;12:439–444. [DOI] [PubMed] [Google Scholar]
- 52. Anderson LJ, Yin C, Burciaga R, Lee J, Crabtree S, Migula D, et al. Assessing Cachexia Acutely after Autologous Stem Cell Transplant. Cancers (Basel) 2019;11:1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crawford J. Clinical results in cachexia therapeutics. Curr Opin Clin Nutr Metab Care 2016;19:199–204. [DOI] [PubMed] [Google Scholar]
- 54. Crawford J, Prado CM, Johnston MA, Gralla RJ, Taylor RP, Hancock ML, et al. Study Design and Rationale for the Phase 3 Clinical Development Program of Enobosarm, a Selective Androgen Receptor Modulator, for the Prevention and Treatment of Muscle Wasting in Cancer Patients (POWER Trials). Curr Oncol Rep 2016;18:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hardee JP, Counts BR, Gao S, VanderVeen BN, Fix DK, Koh HJ, et al. Inflammatory signalling regulates eccentric contraction‐induced protein synthesis in cachectic skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun YX, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci USA 2004;101:4679–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nissinen TA, Hentilä J, Penna F, Lampinen A, Lautaoja JH, Fachada V, et al. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J Cachexia Sarcopenia Muscle 2018;9:514–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Toledo M, Busquets S, Penna F, Zhou X, Marmonti E, Betancourt A, et al. Complete reversal of muscle wasting in experimental cancer cachexia: Additive effects of activin type II receptor inhibition and beta‐2 agonist. Int J Cancer 2016;138:2021–2029. [DOI] [PubMed] [Google Scholar]
- 59. Johns N, Stephens NA, Fearon KC. Muscle wasting in cancer. Int J Biochem Cell Biol 2013;45:2215–2229. [DOI] [PubMed] [Google Scholar]
- 60. Penna F, Ballaro R, Beltra M, De Lucia S, Garcia Castillo L, Costelli P. The Skeletal Muscle as an Active Player Against Cancer Cachexia. Front Physiol 2019;10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin‐1, a muscle‐specific F‐box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A 2001;98:14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 63. Sakuma K, Aoi W, Yamaguchi A. Molecular mechanism of sarcopenia and cachexia: recent research advances. Pflugers Arch 2017;469:573–591. [DOI] [PubMed] [Google Scholar]
- 64. Ezquerro S, Fruhbeck G, Rodriguez A. Ghrelin and autophagy. Curr Opin Clin Nutr Metab Care 2017;20:402–408. [DOI] [PubMed] [Google Scholar]
- 65. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2013;20:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zeng X, Chen S, Yang Y, Ke Z. Acylated and unacylated ghrelin inhibit atrophy in myotubes co‐cultured with colon carcinoma cells. Oncotarget 2017;8:72872–72885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Conte E, Camerino GM, Mele A, de Bellis M, Pierno S, Rana F, et al. Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin‐induced cachexia. J Cachexia Sarcopenia Muscle 2017;8:386–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamada T, Dawson TM, Yanagawa T, Iijima M, Sesaki H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019;15:2012–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010;11:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009;16:939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. de Castro GS, Simoes E, Lima J, Ortiz‐Silva M, Festuccia WT, Tokeshi F, et al. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers (Basel) 2019;11:1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Trevino MB, Zhang X, Standley RA, Wang M, Han X, Reis FCG, et al. Loss of mitochondrial energetics is associated with poor recovery of muscle function but not mass following disuse atrophy. Am J Physiol Endocrinol Metab 2019;317:E899–E910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rossi AE, Boncompagni S, Dirksen RT. Sarcoplasmic reticulum‐mitochondrial symbiosis: bidirectional signaling in skeletal muscle. Exerc Sport Sci Rev 2009;37:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fontes‐Oliveira CC, Busquets S, Toledo M, Penna F, Paz Aylwin M, Sirisi S, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 1830;2013:2770–2778. [DOI] [PubMed] [Google Scholar]
- 75. Dirksen RT. Sarcoplasmic reticulum‐mitochondrial through‐space coupling in skeletal muscle. Appl Physiol Nutr Metab 2009;34:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL‐6‐dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2011;300:R201–R211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fermoselle C, García‐Arumí E, Puig‐Vilanova E, Andreu AL, Urtreger AJ, de Kier Joffé EDB, et al. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp Physiol 2013;98:1349–1365. [DOI] [PubMed] [Google Scholar]
- 78. McLean JB, Moylan JS, Andrade FH. Mitochondria dysfunction in lung cancer‐induced muscle wasting in C2C12 myotubes. Front Physiol 2014;5:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu CS, Wei Q, Wang H, Kim DM, Balderas M, Wu G, et al. Protective Effects of Ghrelin on Fasting‐Induced Muscle Atrophy in Aging Mice. J Gerontol A Biol Sci Med Sci 2020;75:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front Physiol 2016;7:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. das Neves W, Alves CR, de Almeida NR, Guimarães FLR, Ramires PR, Brum PC, et al. Loss of strength capacity is associated with mortality, but resistance exercise training promotes only modest effects during cachexia progression. Life Sci 2016;163:11–22. [DOI] [PubMed] [Google Scholar]
- 82. Schägger H, Pfeiffer K. The Ratio of Oxidative Phosphorylation Complexes I–V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J Biol Chem 2001;276:37861–37867. [DOI] [PubMed] [Google Scholar]
- 83. Bleistein J, Zierz S. Partial deficiency of complexes I and IV of the mitochondrial respiratory chain in skeletal muscle of two patients with mitochondrial myopathy. J Neurol 1989;236:218–222. [DOI] [PubMed] [Google Scholar]
- 84. Shirai T, Aoki Y, Takeda K, Takemasa T. The order of concurrent training affects mTOR signaling but not mitochondrial biogenesis in mouse skeletal muscle. Physiol Rep 2020;8:e14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gouspillou G, Sgarioto N, Norris B, Barbat‐Artigas S, Aubertin‐Leheudre M, Morais JA, et al. The relationship between muscle fiber type‐specific PGC‐1alpha content and mitochondrial content varies between rodent models and humans. PLoS ONE 2014;9:e103044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ghsr mRNA expression in brain and skeletal muscles (GAS/PL) in Ghsr +/+ and Ghsr −/− animals. GAPDH was used as a reference gene, and data are expressed as ΔCt normalized by GAPDH expression in the same sample. Data are shown as mean ± SE (N = 4/group).
Figure S2. AKT in muscle and plasma IGF‐1 levels from Ghsr +/+ and Ghsr −/− mice. HK + V: heat‐killed + vehicle; T + V: tumour + vehicle; T + G: tumour + ghrelin. (A‐B) Protein levels of p‐Akt (Ser473) and total Akt in GAS/PL muscles in mice from each group (N = 4). (A) Representative Western blots of p‐Akt (Ser473) and total Akt. (B) Ratio of p‐Akt (Ser473)/total Akt. Western blots were quantified by densitometry. (C) IGF‐1 expression in plasma (ng/ml). Data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. * denote significant differences comparing to HK + V within the same genotype (p < 0.05). The main effects of genotype (p < 0.05) are shown in p‐values above the corresponding figures.
Figure S3. Oxidative capacity markers in skeletal muscles in Ghsr +/+ and Ghsr −/− . HK + V: heat‐killed + vehicle; T + V: tumour + vehicle; T + G: tumour + ghrelin. (A) Percentage of SDH + fibres in PL muscles (N = 4/group). (B) Representative Western blots of PGC‐1α and GAPDH. (C) Relative protein content of PGC‐1α. Western blots were quantified by densitometry and normalized to GAPDH. Results are presented as fold‐change of Ghsr +/+ HK + V (N = 7–10/group). (D) Relative gene expression of PGC‐1α (Ppargc1a) in GAS/PL muscles. GAPDH was used as a reference gene, and data is expressed as a relative fold‐change of the Ghsr +/+ HK + V group. Data are shown as mean ± SE. Two‐way ANOVA was performed to detect genotype and treatment differences. * denotes significant differences comparing to HK + V within the same genotype (p < 0.05). §: Genotype difference between Ghsr −/− and Ghsr +/+ within the same treatment group (p < 0.05). N.S.: No significant difference was detected across the groups.
Data S1. Supporting Information