

Abstract
Triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy (NAC) represents a major clinical challenge; therefore, delineating tumor heterogeneity can provide novel insight into resistance mechanisms and potential therapeutic targets. Herein, we identified the transcriptional landscape associated with TNBC resistance to NAC at the single-cell level by analyzing publicly available transcriptome data from more than 5,000 single cells derived from four extinction (responders) and four persistence (non-responders) patients, revealing remarkable tumor heterogeneity. Employing iterative clustering and guide-gene selection (ICGS) and uniform manifold approximation and projection (UMAP), we classified TNBC single cells into several clusters based on their distinct gene signatures. The presence of clusters indicative of immune cell activation was a hallmark of the extinction group pre-NAC, while post NAC, the extinction tissue consisted mostly of breast, omental fat, and fibroblasts. The persistent gene signatures of pre-NAC resembled the gene signature of lung epithelial, mammary, and salivary glands and acute myeloid leukemia blast cells, which were associated with enhanced cellular movement and activation of FOXM1, NOTCH1, and MYC and suppression of tumor necrosis factor (TNF) and IFNG mechanistic networks. Multivariate survival analysis identified persistence-derived three-gene signature (KIF5BhighHLA-ClowIGHG2low) predictive of relapse-free survival (hazard ratio [HR]: 2.2 [1.6–3.2, p < 0.0001]) in a second cohort of 360 TNBC patients. Mechanistically, loss of function of several upregulated genes in the persistent group (BYSL, FDPS, ENO1, MED20, MRPL9, MRPL37, NDUFB11, PMVK, MYC, and GSTP1) inhibited MDA-MB-231 and BT-549 TNBC models’ colony-forming unit (CFU) potential and enhanced their sensitivity to paclitaxel. Our data unraveled the transcriptional portrait associated with NAC resistance, identified several key genes, and suggested their potential utilization as prognostic markers and therapeutic targets in TNBC.
Keywords: triple-negative breast cancer, TNBC, neoadjuvant chemotherapy, NAC, resistance, single cell, gene signature
Graphical Abstract
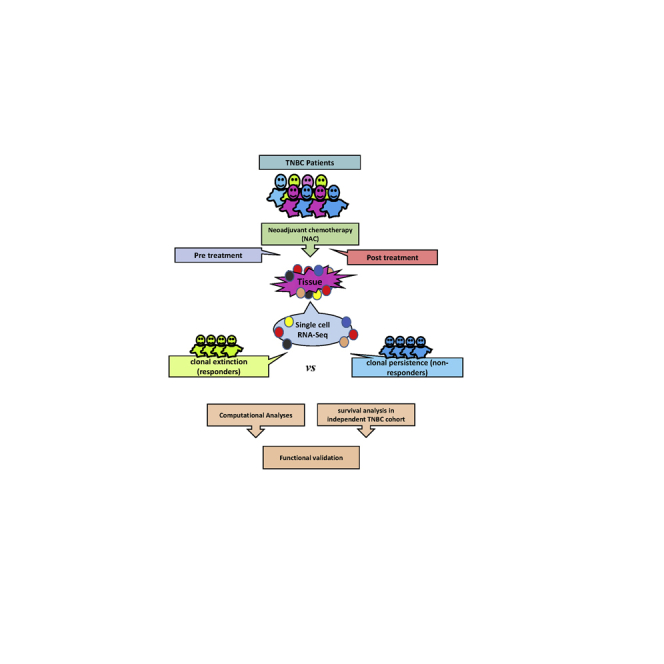
TNBC patients exhibit variable responses to neoadjuvant chemotherapy (NAC), suggesting an underlying clonal heterogeneity. Employing single-cell transcriptome analysis of TNBC responders and non-responders to NAC, we identified gene signature(s) associated with NAC resistance and survival and highlighted the presence of immune infiltration as the hallmark of NAC responsiveness.
Introduction
Breast cancer (BC) is one of the most common cancer types among women with an estimated incidence of 1.5 million new cases per year globally, hence it remains a common cause of death worldwide.1 The current molecular classification of BC is based on expression of sex hormone receptors and human epidermal growth factor receptor 2 (HER2), which have remarkable implications in diagnosis and treatment choice.2 Recent approaches of molecular classification of BC based on gene signatures are gaining momentum.3,4 Triple-negative breast cancer (TNBC) represents 15%–20% of invasive BCs and is characterized by the lack of expression of estrogen and progesterone receptors and lack of amplification of HER2. Those patients do not benefit from endocrine or anti-HER2 therapies; therefore, chemotherapy and surgery are the main treatment modality for TNBC patients.5 Neoadjuvant chemotherapy (NAC) is mainly administered to facilitate breast-conserving surgery and to eliminate clinically silent micro-metastases. TNBC patients with stages I–III receive NAC; however, only approximately 50% achieve pathological complete response (pCR) with favorable recurrence-free survival,6,7 whereas remaining patients with residual disease have high rates of metastatic recurrence and significantly worse survival. Therefore, defining the molecular signature that can predict the response of TNBC patients to chemotherapy can aid in personalizing their medical care choices.8
A large number of studies revealed TNBC tumors to harbor various somatic mutations resulting in higher degree of tumor heterogeneity.9 Several mechanisms have been implicated in BC heterogeneity, including differentiation state of the cell of origin, cell plasticity, genetic evolution, and the tumor microenvironment niche.10, 11, 12, 13, 14, 15 A recent study by Kim et al.16 described two categories of TNBC patients in response to NAC. The first group was referred to as “clonal extinction,” which exhibited disappearance of tumor cells post-treatment based on mutation analysis pre- and post-NAC treatment. The second group was referred to as “clonal persistence,” in which certain tumor clones based on DNA mutation analysis existed pre-NAC and were subsequently enriched in response to NAC treatment, suggesting lack of response of those patients to the treatment. In the current study, we employed various computational pipelines (iterative clustering and guide-gene selection [ICGS], uniform manifold approximation and projection [UMAP] dimensionality reduction, and ingenuity pathway analysis [IPA]) to delineate TNBC heterogeneity and identified their transcriptional landscape in response to NAC at the single-cell level. Data from the single-cell analysis was further validated in a large cohort of TNBC patients (360 patients), leading to the identification of a three-gene signature predictive of TNBC relapse-free survival (RFS). Mechanistically, we implicated several persistence-derived genes in paclitaxel (PTX) resistance employing the MDA-MB-231 and BT-549 TNBC models.
Results
Single-cell RNA sequencing and ICGS reveal TNBC intratumor heterogeneity pre- and post-NAC treatment
To investigate TNBC heterogeneity at the single-cell level, we analyzed single-cell transcriptome data from patients classified as extinction (responders, n = 4) and persistence (non-responders, n = 4) pre- and post-NAC treatment. We employed the ICGS2 algorithm, which classifies interconnected gene elements through pairwise correlations of variable genes, followed by various series of HOPACH heatmap clustering of genes and cells, and finally determination of representative marker genes associated with each phenotype (persistence versus extinction). Unsupervised transcriptome analysis of 719 extinction and 525 persistence single cells pre-NAC and 894 extinction and 687 persistence single cells post-NAC identified 13 clusters, each with distinct molecular signature (Figure 1A; Table S1). MarkerFinder subsampled ordered in heatmap is highlighting differentially expressed genes in each cell population within the persistence and extinction group pre- and post-NAC, where expression level of each gene (log2) is depicted according to the color scale. Statistically enriched Gene Ontology (GO) terms associated with each gene cluster relates common proclaimed cell types from GO-Elite analysis. The lower legend indicates the origin of each cell (red: persistence pre-NAC; blue: extinction pre-NAC; gray: extinction post-NAC; and maroon: persistence post-NAC; Figure 1A). In our data, C1 and C12 clusters were predominantly present in the pre- and post-NAC persistence group and were highly enriched in genes resembling acute myeloid leukemia blast cells and breast (C1) and bone marrow lymphoid natural killer (NK) cells (C12) (Figure 1A). In contrast, the presence of activated B cells and monocyte progenitors (C5) was a hallmark of the extinction group pre-NAC. Remarkably, we observed three clusters (C31, C22, and C14) that were enriched in the extinction, and to a lesser extent in the persistence, post-NAC. Those clusters denote CD56+ NK cells, myeloid leukemia pre-leukemic hematopoietic stem cells (C31), lung macrophage and fibroblast (C22), and macrophages and stimulated macrophages (C14). To further select features and delineate cell populations, we subjected the data from the same cell populations to UMAP dimension reduction analysis, which revealed a total of 13 clusters (Figure 1B). Remarkably, density-based clustering of C12 and C1 showed the clear distinctive pattern among other clusters without overlapping.
Figure 1.

Heterogeneity of TNBC single cells revealed through ICGS2 and UMAP dimensionality reduction analysis of TNBC-derived single cells pre- and post-NAC
(A) Unsupervised single-cell population identification using ICGS2 algorithm conducted on 719 extinction (responders) and 525 persistence (non-responders) single cells pre-NAC and 894 extinction (responders) and 687 persistence (non-responders) single cells post-NAC. Data are presented as heatmap with the enriched cell population indicated on the left legend and the corresponding single-cell cluster on top. Color scale displays differential gene expression (log2). Lower legend indicated cell origin. (B) UMAP dimensionality reduction analysis revealing 13 cell clusters pre- and post-NAC. EXT_POST, extinction post-NAC; EXT_PRE, extinction pre-NAC; PER_PRE, persistence pre-NCA; PER_POST, persistence post-NAC.
Comparative analysis of persistence versus extinction single-cell transcriptome pre-NAC revealed remarkable differences in gene expression and enriched GO terms
To devise a gene signature associated with resistance to NAC, we compared the transcriptome of persistence and extinction TNBC-derived single cells pre-NAC. Hierarchical clustering of persistence (n = 534) and extinction (n = 781) single cells shows remarkable differences between the two groups at the transcriptome level (Figure 2A; Table S2). Enrichment in GO categories associated with response to interferon gamma, gene expression, and regulation of cell death was prominent in the persistence group. In contrast, GO terms associated with regulation of cell division, response to protein stimulus, and serotonin metabolic processes were underrepresented in the persistence group. Immune response was most prominent in genes enriched in the extinction group. Differentially expressed genes between the persistence and extinction group were also reflected employing a volcano plot (Figure 2B).
Figure 2.

Comparative analysis of the transcriptional landscape in persistence and extinction TNBC-derived single cells pre-NAC
(A) Hierarchical clustering of TNBC-derived single cells from persistence (n = 534) and extinction (n = 781) group pre-NAC. Each column represents one cell, and each row represents a gene. Expression level of each gene (log2) in a single cell is depicted according to the color scale. (B) Volcano plot illustrating the upregulated (red) and downregulated (blue) genes in the persistence versus extinction group pre-NAC. Validation of top 10 upregulated (C) and top 10 downregulated (D) genes in a second cohort consisting of 782 extinction and 535 persistence TNBC-derived single cells.
To validate the data obtained from the discovery cohort, we validated the expression of the top 10 upregulated (CALML5, AZGP1, S100P, IRX1, IRX2, NES, AC013457.1, ACTA2, PIK3R1, and LRP11) and top 10 downregulated (FTL, B2M, HLA-DRA, HGB1, LCP1, CXCL9, CD74, IGHG1, CD44, and UBD) genes using a second cohort consisting of 782 extinction and 535 persistence pre-NAC TNBC-derived single cells (Figures 2C and 2D), respectively.
IPA revealed remarkable differences in enriched functional categories and pathways in persistence versus extinction group
To gain better insight into the biological processes enriched in the persistence versus extinction groups, we subjected upregulated genes in the persistence or extinction group to IPA, which revealed activation of several canonical pathways in each group, respectively (Tables S3 and S6). The top 20 enriched canonical pathways in the persistence group are presented in Figure 3A, which highlight pathways involved in organelle development and cellular function, like proliferation, migration, and invasion, including oxidative phosphorylation, Integrin and Integrin-linked kinase (ILK) signaling, GP6, EIF2, Rho family GTPases, vascular endothelial growth factor (VEGF), glycolysis, gluconeogenesis, TCA cycle, and cholesterol biosynthesis to be predominant in the persistence group (Figure 3A; Table S3). In contrast, canonical pathways underrepresented in the persistence group (enriched in the extinction group) were mostly involved in dendritic cell maturation, T cell signaling pathway, immune response, PKC9 signaling in T lymphocyte, inducible co-stimulator (ICOS)-ICOS (CD278) signaling costimulatory molecule expressed on T helper cells (Th1 and Th2 cells), calcium-induced T lymphocyte apoptosis, Th1 pathway, phospholipase C signaling, oxidative phosphorylation, interferon signaling, and BAG2 signaling pathway (Figure 3B; Table S4). Further IPA-downstream effects analysis (DEA) revealed a number of affected downstream biological functions that are likely to be affected by transcriptome data. The hierarchical heatmap portrays the affected downstream functional groups based on differentially expressed mRNAs where the major boxes with size and color coding depict a category of diseases and functions in the persistence versus extinction group.17 Top enriched functional categories in the persistence group were cellular movement and cell growth and proliferation (Figure 3C; Table S7). To explore further into the downstream functional effects of upstream regulators in each patient group, we employed the regulator effector analysis pipeline in IPA on upregulated genes in persistence versus extinction TNBC (Table S6). The network identified 7 upstream regulators (5 up: GNA12, phosphatidylinositol 3-kinase [PI3K] family, Insulin-like growth factor binding protein 2 [IGFBP2], STAT3, and EGFR; and 2 down: COL18A1 and LONP1) and 35 genes in the intermediate of hierarchy, which collectively drive oncogenic phenotype, such as cell cycle, proliferation migration, and invasion (Figure 3D). Our data identified several activated (FOXM1, NOTCH1, and MYC) and suppressed (tumor necrosis factor [TNF] and IFNG) signaling cascades as potential drivers of the predicted oncogenic phenotype in the persistence group (Figure S1).
Figure 3.

Ingenuity pathway analysis (IPA) of differentially expressed genes in persistence versus extinction group
Enriched canonical pathways based on upregulated (A) and downregulated (B) genes in persistence versus extinction TNBC groups. (C) Tree map (hierarchical heatmap) depicting affected functional categories based on upregulated genes where the major boxes represent a category of diseases and function. Illustration of cellular movement and cell growth and proliferation are shown in the lower panels. (D) Regulator effects network analysis based on IPA highlighting a role for activated (GNA12, PI3K family, IGFBP2, STAT3, and EGFR) and suppressed (COL18A1 and LONP1) upstream regulators and their roles in divining tumorigenic function.
Differences in immune-infiltrating cells are the hallmark of the extinction group
IPA analysis of enriched gene sets in the extinction group was of mainly those involved in immune cell trafficking and cell-to-cell signaling interaction (Figure 4A; Table S7). We subsequently looked for the expression of selected immune-related genes based on differential gene expression analysis in a total of 5,524 single cells derived from the extinction and persistence groups pre- and post-NAC. Our data revealed higher expression of CD19, CD8A, CD4, CD52, CD2, CD53, CD59, CD47, CD74, and CXCL9 in the extinction compared with the persistence group pre-NAC (Figure 4B). Interestingly, the number of immune-infiltrating cells declined in the post-NAC treatment in the extinction group; however, the opposite was observed in the persistence group, where the number of infiltrating immune cells increased post-NAC (Figure 4B). Of particular interest, we observed a substantial increase in the number of CD74+ cells post-NAC treatment in the persistence group (Figure 4B). Additionally, we also observed the emergence of a subset of CD47bright in the persistence group post-NAC, which otherwise was not present pre-NAC. To determine whether there are differences in the functionality of immune-infiltrating cells in the extinction versus persistence, we subjected the transcriptome from CD45+EPCAM− cells to IPA analysis, which again revealed immune cells from the extinction group to exhibit enhanced functionality compared with immune cells from the persistence group, suggesting functional impairment of immune-infiltrating cells in the persistence group (Figures 4C and 4D).
Figure 4.

Immune infiltration is the hallmark of the extinction group
(A) Tree map (hierarchical heatmap) depicting affected functional categories based on downregulated genes in the persistence group (upregulated in the extinction group) where the major boxes represent a category of diseases and function. Illustration of immune cell trafficking and cell-to-cell signaling and interaction are shown in the lower panels. (B) The expression of CD19, CD8A, CD4, CD52, CD2, CD53, CD59, CD47, CD74, and CXCL9 in the extinction (EXT) compared with the persistence (PER) group pre- and post-NAC. ∗p < 0.01, ∗∗p < 0.001, ∗∗∗p < 0.0001. Tree map (hierarchical heatmap) depicting activation of immune cell trafficking (C) and hematological system development and function (D).
Clinical relevance of persistence versus extinction-derived gene signature on TNBC patients’ survival
Comparative analysis of persistence versus extinction of TNBC subjects identified 788 upregulated and 244 downregulated genes in the persistence cohort (Figure 4; Table S2). Genes enriched in the persistence or extinction group were subsequently subjected to survival analysis on a second cohort of 360 TNBC patients. Raw RNA sequencing (RNA-seq) data from this cohort were retrieved from the Sequence Read Archive (SRA): SRP157974 dataset and were subsequently aligned to the human genome (hg38), and gene expression data (log2) for the persistence- and extinction-derived gene signatures were subjected to univariate survival analysis. Data presented in Figure 5 revealed several genes upregulated in the persistence group (CD55, KIF5B, ZFAS1, CAMTA1, LRPPRC, RSL24D1, NEBL, COXC6, PTK2, TCEA1, GDI2, NUDT5, and CD46) to correlate with worse RFS in TNBC patients (Figure 5A). In contrast, the expression of several genes upregulated in the extinction group (IGHG2, IGHM, IGLC1, HLA-C, IGHG4, IGHGP, ITGB2, HLA-DPB1, HLA-A, and SLAMF7) showed better prognosis (Figure 5B). IPA of enriched gene sets in the extinction group was of mainly those involved in immune cell trafficking and function (Figure 4A; Table S7), correlated with better RFS in the same TNBC cohort (Figure 5B). We subsequently employed multivariate analysis using stepwise Cox regression model and identified three-gene signature (KIF5BhighHLA-ClowIGHG2low) as best predictor of RFS (Figure 5C). The three-gene signature was superior to other variables, such as tumor size (continuous), age (old versus young), intrinsic subtype (basal versus others), and treatment (PTX and radiotherapy), in predicting RFS (p < 0.0001, hazard ratio [HR] = 2.2 [1.6–3.2]) (Table 1).
Figure 5.

Univariate and multivariate recurrence-free survival analysis in 360 TNBC patients
A cohort of 360 patients were divided into high and low based on median gene expression derived from the persistence (A) or extinction (B) groups using Kaplan-Meier survival analysis. Hazard ratio and log rank p value are indicated on each plot. (C) The prognostic value of three-gene signature (KIF5BhighHLA-ClowIGHG2low) is indicated.
Table 1.
Multivariate analyses for the prognostic value of the 3-gene signature in TNBC
| B | SE | Wald | Sig. | Exp(B) | 95.0% CI for Exp(B) |
95.0% CI for Exp(B) |
|
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| Three- gene Score | .820 | .178 | 21.256 | .000 | 2.271 | 1.602 | 3.218 |
| intrinsic subtype | .206 | .355 | .338 | .561 | 1.229 | .613 | 2.466 |
| Size | -.244 | .153 | 2.547 | .110 | .784 | .581 | 1.057 |
| Age | -.195 | .303 | .415 | .520 | .823 | .455 | 1.489 |
| Paclitaxel | .355 | .377 | .888 | .346 | 1.427 | .682 | 2.986 |
| Radiotherapy | .813 | .298 | 7.412 | .006 | 2.254 | 1.256 | 4.045 |
Targeted depletion of selected persistence-derived genes reduces TNBC colony formation and enhances PTX sensitivity
To provide additional mechanistic insight into driver genes and their role in PTX resistance, we chose 10 genes for further investigation based on our differential and pathway analysis, as well as integration with the Achilles dependency data.18 The expression of 10 upregulated genes from the discovery cohort (BYSL, ENO1, FDPS, GSTP1, MED20, MRPL9, MRPL37, MYC, NDUFB11, and PMVK) was subsequently validated in a second cohort of 782 extinction and 535 persistence TNBC-derived single cells, which was consistent with the discovery cohort (Figure 6A). The expression of the 10 genes was explored in a panel of TNBC cell lines from the CCLE database, which confirmed their expression, suggesting their suitability as cell models to study the function of those genes (Figure 6B). The expression of the 10 genes was depleted in two TNBC models (MDA-MB-231 and BT-549) employing small interfering RNA (siRNA)-mediated gene silencing as single agent or in combination with PTX (20 nM), followed by colony-forming unit (CFU) assay as a measure of the effects of gene targeting on CFU potential of the two TNBC models. Targeted depletion of BYSL and MYC caused substantial inhibition of CFU potential of the BT-549 and MDA-MB-231 models qualitatively and quantitatively, which was further enhanced in the presence of PTX (Figures 6C–6F). Knockdown of FDPS, ENO1, and PMVK caused significant inhibition of CFU potential of MDA-MB-231 and BT-549 models, which was further enhanced in the presence of PTX (Figures 6C–6F). Interestingly, depleting MRPL37, NDUFB11, and GSTP1 has minimal effects on CFU potential as single agents; however, the effects were very remarkable when combined with PTX, suggesting possible synergistic effects. Taken together, our data demonstrated a remarkable role for several persistence-derived genes in mediating PTX resistance in TNBC.
Figure 6.

Targeted depletion of selected persistence-derived genes inhibits TNBC CFU potential in vitro
(A) Expression of BYSL, FDPS, ENO1, MED20, MRPL9, MRPL9, NDUFB11, PMVK, MYC, and GSTP1 in a second cohort of single cells derived from the persistence (782 cells) and the extinction (535 cells) TNBC. (B) Expression of the selected 10 genes in a panel of TNBC cell lines from the CCLE database. Clonogenic potential of MDA-MB-231 (C and D) or BT-549 (E and F) after transfection with the indicated siRNA as single agent or in combination with paclitaxel (PTX; 20 nM). Data are representative of two experiments conducted in duplicate.
Discussion
Emerging data suggest added benefit for NAC in a fraction of TNBC patients;19 hence patients who had residual disease after NAC treatment usually have significantly shorter overall and post-recurrence survival compared with patients with pCR.7,20 Therefore, delineating the response of individual tumor-derived cells to NAC might help identify the molecular signature capable of predicting the response of TNBC patients to NAC treatment, hence offering them a more beneficial treatment option. In the current study, we characterized the transcriptional landscape from more than 5,000 single cells derived from eight TNBC patients in response to NAC treatment. Our data revealed significant changes in the transcriptome of individual cells from patients who exhibited clonal persistence, compared with patients who exhibited clonal extinction. Our data revealed large similarity in the gene expression profile in the persistence group pre- and post-NAC treatment, suggesting those tumors did not respond well to the treatment, compared with the extinction group who exhibited better response to NAC treatment as evident by distinct transcriptome portrait pre- and post-NAC. Surprisingly, clustering analysis performed on individual cells from different TNBC patients revealed close clustering of each patients’ individual cells pre- and post-NAC, suggesting the existence of patient-specific tumor signature.
Employing UMAP and ICGS2 algorithms, we identified 13 cell clusters based on similarity in their transcriptome profile. We observed enrichment of activated B and dendritic cells as the main difference between the extinction and persistence groups. Therefore, it is obvious that the extinction group is characterized by substantial immune infiltration compared with the persistence group. Several immune-related genes were enriched in the extinction group, including MHC class I and II, CCL19, CXCL9, CD19, CD52, CD53, CD59, CD47, CXCL10, C1QB, CD74, and B2M. Our data are in agreement with published literature correlating the percentage of intratumoral lymphocytes infiltration to be an independent prognostic factor for pathologic complete response to NAC in BC patients.21 Interestingly, the same study identified CXCL9 among their predictive immune signature, which was also identified in our current study. García-Teijido et al.22 correlated immune infiltration with better response of TNBC patients to NAC. Notably, Ladoire et al.23 reported immune response post-NAC to predict BC survival; however, our data suggest that the prediction power of immune infiltration is pre-existing before NAC treatment. Interestingly, our study revealed a novel observation where the number of infiltrating immune cells pre-NAC was significantly higher in the clonal extinction compared with the clonal persistence group. However, although the number of infiltrating immune cells declined dramatically in the clonal extinction group, the number of immune-infiltrating cells increased in the clonal persistence group post-NAC treatment. It is plausible that the number of immune cells declined in the clonal extinction group as a sign of tumor regression; however, the increase in immune infiltration post-NAC in the clonal persistence group might reflect either tumor progression or triggered immune reaction in response to cell death inflicted by NAC treatment. CD52 is a 12-amino acid glycoprotein, which is widely expressed on the cell surface of different immune cells, including mature lymphocytes, NK cells, and antigen-presenting cells.24 Anti-CD52 antibodies have been used to treat patients with T cell leukemia and during organ transplantation.25,26 Elevated expression of CD52 in our data might suggest a favorable activation state of tumor-infiltrating immune cells. Nonetheless, our data revealed almost complete absence of B cells (CD19+) from the clonal persistence group. It is not clear why patients with increased immune infiltration benefit from NAC treatment. It is plausible that immune infiltration and cytokine release might enhance NAC efficacy, or alternatively, tumors with heavy immune infiltration might represent a different TNBC molecular subtype that responds differently to NAC treatment, which warrants further investigation. Our data also revealed the emergence of a CD47bright cell population in the post-NAC persistence group. Earlier studies have shown CD47 expression to be regulated by HIF-1 and to promote evasion of phagocytosis and maintenance of cancer stem cell phenotype in BC.27 Additionally, high expression of CD47 was recently found to be associated with epithelial-mesenchymal transition and poor prognosis in TNBC, thus corroborating our data.28
The clinical relevance of immune-related genes was further validated in a large cohort of TNBC patients (n=360). Univariate analysis identified several genes that can predict RFS in TNBC patients (CAMTA1, CD46, CD55, COX6C, GDI2, KIF5B, LRPPRC, NEBL, NUDT5, PTK2, RSL24D1, TCEA1, and ZFAS1) as poor prognostic and (HLA-A, HLA-C, HLA-DPB1, IGHG2, IGHG4, IGHGP, IGHM, IGLC1, ITGB2, and SLAMF7) as favorable prognostic markers. Previous study identified PTK2/FAK as a driver of radio-resistance in HPV-negative head and neck cancer, thus corroborating the findings from our study in the context of NAC resistance.29 Several of the favorable prognostic gene markers (enriched in the extinction group) belonged to immune regulation. Employing Cox regression survival model, we identified a three-gene signature (KIF5BhighHLA-ClowIGHG2low) predicative of TNBC RFS. Kinesin-1 heavy chain is a protein that is encoded by the KIF5B (Kinesin Family Member 5B) gene. A study by Wang et al.30 reveled binding of phospholipase D2 (PLD2) to KIF5B2 to regulate kinesin-1 motor function and BC metastasis. In lung cancer, a fusion protein of KIF5B-RET was shown to promote tumorigenesis of non-small cell lung cancers through activation of STAT3 signaling.31
Our IPA upstream regulators analysis revealed neurotrophins and insulin receptor signaling pathways (GNA12, PI3K family, IGFBP2, STAT3, and EGFR) are predicted to activate tumorigenicity-associated cellular development and functions, including cell cycle, proliferation, migration, and invasion in the persistence group. IGFBP2 is a pleiotropic oncogenic protein that regulates cancer development leading to abnormal activation of EGFR that subsequently triggers STAT3 signaling.32 IPA analysis on the upregulated genes revealed enrichment in cellular movement, while analysis of the downregulated genes revealed immune cell trafficking as the main affected functional category in the persistence group concurrent with activation of FOXM1, NOTCH1, and MYC and suppression of TNF and IFNG mechanistic networks (MNs). Canonical pathway analysis and bioinformatics revealed upregulated genes to be associated with enrichment of tumorigenic development, including VEGF, glycolysis, gluconeogenesis, TCA cycle, and cholesterol biosynthesis, whereas top downregulated pathways indicate lack of immune response, such as dendritic cell maturation, T cell signaling pathway, immune response, PKC9 signaling in T lymphocyte, ICOS-ICOS (CD278) signaling costimulatory molecule expressed on T helper cells (Th1 and Th2 cells), calcium-induced T lymphocyte apoptosis, Th1 pathway, interferon signaling, and BAG2 signaling pathway in persistence tumors. Inactive immune pathways in TNBC that exhibited resistance to NAC as inferred from tyrosine kinase activity, including ZAP70, LCK, SYK, and JAK2, had significantly lower phosphorylation in the TNBC persistence group.33 The ICOS (CD28) molecule expressed on activated T cells plays a serious role in controlling the immune response in BC, and overexpression of ICOSL protein is associated with poor prognoses.34
We subsequently employed and integrated an approach to identify potential mediators of NAC resistance through integration of our transcriptome data analysis and dependency data from the Achilles project, which led to the identification of 10 genes as potential drivers of the TNBC persistence phenotype (BYSL, FDPS, ENO1, MED20, MRPL9, MRPL37, NDUFB11, PMVK, MYC, and GSTP1). Targeted depletion of BYSL and MYC led to substantial inhibition of TNBC CFU potential as a single agent, while an additive or more than additive combination effects were observed for the other genes, suggesting a possible sensitizing effect. Recent data by Zhang et al.35 reported c-Myc to maintain the self-renewal and chemoresistance properties of colon cancer stem cells. Taken together, our data unraveled the transcriptional portrait associated with NAC resistance and identified several driver genes and suggest their potential utilization as prognostic markers and therapeutic targets.
Materials and methods
Dataset and bioinformatics
Single-cell raw RNA-seq data were retrieved from the SRA database under accession number SRA: SRP114962. Data were retrieved using the SRA toolkit version 2.9.2 as previously described.36 FASTQ files were subsequently pseudoaligned to the ENSEMBL hg38 assembly, and reads were counted using KALLISTO 0.42.1. Expression data (TPMs [transcripts per million] mapped reads) were subsequently subjected to differential gene analysis using 2.0-fold change and p value cut-off <0.05, ICGS2, hierarchical clustering, and UMAP dimensionality reduction as described before.17,37,38 Early cell-type predictions were created from the software GO-Elite in AltAnalyze using its previously defined cell and tissue marker gene database. This database encompasses markers for several cell types and bulk tissue samples. The MarkerFinder algorithm was employed to define specific markers associated with each phenotype based on the gene set enrichment output from GO-Elite.39 Volcano plot (scatterplot) was used to illustrate differentially expressed genes in GraphPad Prism 8.0 software (GraphPad, San Diego, CA, USA).
Gene set enrichment and modeling of gene interactions networks
Upregulated genes in the persistence or extinction groups were imported into the IPA software (Ingenuity Systems; https://digitalinsights.qiagen.com/) and were subjected to functional annotations and regulatory network analysis using upstream regulator analysis (URA) to analyze molecules upstream, which are connected to genes in the dataset via a set of either direct or indirect relationship expression changes. MNs, hypothesis networks constructs on regulators, determine with URA by connecting likely part of causal mechanism. Causal network analysis (CNA) is simplification of URA that connects target gene molecules through more than one intermediate regulator and constructs mechanistic hypotheses. DEA identifies the biological processes (disease) and functions, which are casually affected by deregulation of genes in the dataset and predict the biological process outline whether upregulated or downregulated. IPA uses precise algorithms to predict functional regulatory networks from gene expression data and provides a significance score for each network according to the fit of the network to the set of focus genes in the database. The p value is the negative log of p and represents the possibility that focus genes in the network are found together by chance.17,40
Read mapping, alignment, and survival analysis in TNBC cohort
Raw transcriptome data for 360 TNBC patients were retrieved from the SRA database (accession number SRA: SRP157974) using the SRA toolkit version 2.9.2 as described above. Paired-end RNA-seq FASTQ files were subsequently pseudoaligned to the human genome hg38, and reads were counted using KALLISTO 0.42.1 and were presented as log2-transformed TPM values as we described before.41 Kaplan-Meier (non-parametric) and Cox (semi-parametric) regression survival analysis were conducted using IBM SPSS statistics software. For survival analysis, patients were grouped into high or low based on median log2 gene expression. For multivariate analysis, Cox proportional hazards regression model was constructed by incorporating variate, which gave p ≤ 0.05 using log rank test from the univariate Kaplan-Meier analysis. A forward stepwise Cox regression was applied in which variables with stepwise probability of ≥0.05 were included in the model, while variables with probability of ≥0.1 were excluded. Once signatures were derived, a combination score of all genes in the signature was constructed by calculating the sum score for each gene (1 for high and 0 for low) and subsequently dividing the cohort into high or low, based on the sum score before subjecting to Kaplan-Meier analysis. The log rank test was used to compare the outcome between expression groups. The Cox proportional hazards multiple regression model was used to identify the independent prognostic factors against other confounding variables, such as tumor size (continuous), age (old versus young), intrinsic subtype (basal versus others), and treatment (PTX and radiotherapy). Statistical analyses to compare specific gene expression and graphing were performed using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA).
TNBC cell culture
Human BC cell lines BT-549 and MDA-MB-231 were maintained in RPMI 1640 medium (ATCC modification; catalog number A1049101) and DMEM (Dulbecco’s modified Eagle’s medium), respectively. Both were supplemented with D-glucose 4,500 mg/L, 2–4 mM L-glutamine, 10% fetal bovine serum, and 1× penicillin-streptomycin (Pen-Strep) (all purchased from GIBCO-Invitrogen, Waltham, MA, USA). Cells were grown as monolayers at 37°C in humidified CO2 (5%) incubator.
siRNA transfection
The scrambled siRNA control and ON-TARGETplus SMARTpool siRNA targeting human BYSL, FDPS, ENO1, MED20, MRPL9, MRPL37, NDUFB11, PMVK, MYC, and GSTP1 were purchased from Dharmacon (Lafayette, CO, USA). Transfection was performed using a reverse transfection approach as previously described.42 In brief, siRNA at a final concentration of 30 nM was diluted in 50 μL of Opti-MEM (11058-021; GIBCO, Carlsbad, CA, USA), and 1 μl of Lipofectamine 2000 (catalog no. 52758; Invitrogen) was diluted in 50 μl of OPTI-MEM. The diluted siRNA and Lipofectamine 2000 were mixed together and incubated at room temperature for 20 min. Twenty microliters of transfection mixture was added to the tissue culture plate, and subsequently 10,000 cells in 60 μl transfection medium (complete DMEM without Pen-Strep) were added to each well. Twenty-four hours later, the transfection cocktail was replaced with complete DMEM.
CFU assay and PTX sensitivity of control and siRNA-transfected MDA-MB-231 and BT-549 cells
Transfected cells were cultured for 48 h and treated with PTX (20 nM). On day 5, cells were fixed with 4% PFA for 5 min followed by washing twice in PBS and stained with crystal violet (0.1% in 10% EtOH) for 10 min at room temperature. The images were taken and compared with appropriate control. Subsequently, plates were air dried at room temperature, and CFUs were quantified by dissolving crystal violet in 5% SDS and measured absorbance at 590 nm. The experiments were repeated twice, and data are represented as mean ± SD from four technical replicas.
Statistical analyses
Statistical analyses and graphing were performed using Microsoft Excel 2016 and GraphPad Prism 8.0 software (GraphPad, San Diego, CA, USA). Two-tailed t test was used for comparative groups. p values ≤0.05 (two-tailed t test) were considered significant. For IPA analyses, a Z score (2.0 ≤ Z ≥ 2.0) was considered significant.
Acknowledgments
This work was supported by Qatar Biomedical Research Institute (QBRI) start-up fund (grant no. QB13 to N.M.A.).
Author contributions
R.V. performed experiments and wrote the manuscript; N.M.A. conceived the study, obtained funding, performed bioinformatics analysis, and finalized the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.09.002.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2015. CA Cancer J. Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Paik S., Shak S., Tang G., Kim C., Baker J., Cronin M., Baehner F.L., Walker M.G., Watson D., Park T. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 3.Ellsworth R.E., Blackburn H.L., Shriver C.D., Soon-Shiong P., Ellsworth D.L. Molecular heterogeneity in breast cancer: State of the science and implications for patient care. Semin. Cell Dev. Biol. 2017;64:65–72. doi: 10.1016/j.semcdb.2016.08.025. [DOI] [PubMed] [Google Scholar]
- 4.Hamam R., Hamam D., Alsaleh K.A., Kassem M., Zaher W., Alfayez M., Aldahmash A., Alajez N.M. Circulating microRNAs in breast cancer: novel diagnostic and prognostic biomarkers. Cell Death Dis. 2017;8:e3045. doi: 10.1038/cddis.2017.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sikov W.M., Berry D.A., Perou C.M., Singh B., Cirrincione C.T., Tolaney S.M., Kuzma C.S., Pluard T.J., Somlo G., Port E.R. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance) J. Clin. Oncol. 2015;33:13–21. doi: 10.1200/JCO.2014.57.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guarneri V., Broglio K., Kau S.W., Cristofanilli M., Buzdar A.U., Valero V., Buchholz T., Meric F., Middleton L., Hortobagyi G.N., Gonzalez-Angulo A.M. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J. Clin. Oncol. 2006;24:1037–1044. doi: 10.1200/JCO.2005.02.6914. [DOI] [PubMed] [Google Scholar]
- 7.Liedtke C., Mazouni C., Hess K.R., André F., Tordai A., Mejia J.A., Symmans W.F., Gonzalez-Angulo A.M., Hennessy B., Green M. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 8.Bianchini G., Balko J.M., Mayer I.A., Sanders M.E., Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig D.W., O’Shaughnessy J.A., Kiefer J.A., Aldrich J., Sinari S., Moses T.M., Wong S., Dinh J., Christoforides A., Blum J.L. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013;12:104–116. doi: 10.1158/1535-7163.MCT-12-0781. [DOI] [PubMed] [Google Scholar]
- 10.Kendrick H., Regan J.L., Magnay F.A., Grigoriadis A., Mitsopoulos C., Zvelebil M., Smalley M.J. Transcriptome analysis of mammary epithelial subpopulations identifies novel determinants of lineage commitment and cell fate. BMC Genomics. 2008;9:591. doi: 10.1186/1471-2164-9-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koren S., Bentires-Alj M. Breast Tumor Heterogeneity: Source of Fitness, Hurdle for Therapy. Mol. Cell. 2015;60:537–546. doi: 10.1016/j.molcel.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 12.Gupta P.B., Fillmore C.M., Jiang G., Shapira S.D., Tao K., Kuperwasser C., Lander E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 13.Kreso A., Dick J.E. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Nowell P.C. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 15.Bissell M.J., Hines W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell. 2018;173:879–893.e13. doi: 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vishnubalaji R., Sasidharan Nair V., Ouararhni K., Elkord E., Alajez N.M. Integrated Transcriptome and Pathway Analyses Revealed Multiple Activated Pathways in Breast Cancer. Front. Oncol. 2019;9:910. doi: 10.3389/fonc.2019.00910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M. Defining a Cancer Dependency Map. Cell. 2017;170:564–576.e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 20.Masuda H., Baggerly K.A., Wang Y., Zhang Y., Gonzalez-Angulo A.M., Meric-Bernstam F., Valero V., Lehmann B.D., Pietenpol J.A., Hortobagyi G.N. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013;19:5533–5540. doi: 10.1158/1078-0432.CCR-13-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denkert C., Loibl S., Noske A., Roller M., Müller B.M., Komor M., Budczies J., Darb-Esfahani S., Kronenwett R., Hanusch C. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010;28:105–113. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- 22.García-Teijido P., Cabal M.L., Fernández I.P., Pérez Y.F. Tumor-Infiltrating Lymphocytes in Triple Negative Breast Cancer: The Future of Immune Targeting. Clin. Med. Insights Oncol. 2016;10(Suppl 1):31–39. doi: 10.4137/CMO.S34540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ladoire S., Mignot G., Dabakuyo S., Arnould L., Apetoh L., Rébé C., Coudert B., Martin F., Bizollon M.H., Vanoli A. In situ immune response after neoadjuvant chemotherapy for breast cancer predicts survival. J. Pathol. 2011;224:389–400. doi: 10.1002/path.2866. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y., Su H., Shen X., Du J., Zhang X., Zhao Y. The immunological function of CD52 and its targeting in organ transplantation. Inflamm. Res. 2017;66:571–578. doi: 10.1007/s00011-017-1032-8. [DOI] [PubMed] [Google Scholar]
- 25.Osterborg A., Dyer M.J., Bunjes D., Pangalis G.A., Bastion Y., Catovsky D., Mellstedt H. Phase II multicenter study of human CD52 antibody in previously treated chronic lymphocytic leukemia. European Study Group of CAMPATH-1H Treatment in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 1997;15:1567–1574. doi: 10.1200/JCO.1997.15.4.1567. [DOI] [PubMed] [Google Scholar]
- 26.Hale G., Jacobs P., Wood L., Fibbe W.E., Barge R., Novitzky N., Toit C., Abrahams L., Thomas V., Bunjes D. CD52 antibodies for prevention of graft-versus-host disease and graft rejection following transplantation of allogeneic peripheral blood stem cells. Bone Marrow Transplant. 2000;26:69–76. doi: 10.1038/sj.bmt.1702477. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H., Lu H., Xiang L., Bullen J.W., Zhang C., Samanta D., Gilkes D.M., He J., Semenza G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA. 2015;112:E6215–E6223. doi: 10.1073/pnas.1520032112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan J., Shi X., Chen C., He H., Liu L., Wu J., Yan H. High expression of CD47 in triple negative breast cancer is associated with epithelial-mesenchymal transition and poor prognosis. Oncol. Lett. 2019;18:3249–3255. doi: 10.3892/ol.2019.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skinner H.D., Giri U., Yang L., Woo S.H., Story M.D., Pickering C.R., Byers L.A., Williams M.D., El-Naggar A., Wang J. Proteomic Profiling Identifies PTK2/FAK as a Driver of Radioresistance in HPV-negative Head and Neck Cancer. Clin. Cancer Res. 2016;22:4643–4650. doi: 10.1158/1078-0432.CCR-15-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z., Zhang F., He J., Wu P., Tay L.W.R., Cai M., Nian W., Weng Y., Qin L., Chang J.T. Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev. Cell. 2017;43:186–197.e7. doi: 10.1016/j.devcel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian Y., Chai S., Liang Z., Wang Y., Zhou Y., Xu X., Zhang C., Zhang M., Si J., Huang F. KIF5B-RET fusion kinase promotes cell growth by multilevel activation of STAT3 in lung cancer. Mol. Cancer. 2014;13:176. doi: 10.1186/1476-4598-13-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chua C.Y., Liu Y., Granberg K.J., Hu L., Haapasalo H., Annala M.J., Cogdell D.E., Verploegen M., Moore L.M., Fuller G.N. IGFBP2 potentiates nuclear EGFR-STAT3 signaling. Oncogene. 2016;35:738–747. doi: 10.1038/onc.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sawada T., Hilhorst R., Rangarajan S., Yoshida M., Tanabe Y., Tamura K., Kinoshita T., Shimoyama T., van Beuningen R., Ruijtenbeek R. Inactive immune pathways in triple negative breast cancers that showed resistance to neoadjuvant chemotherapy as inferred from kinase activity profiles. Oncotarget. 2018;9:34229–34239. doi: 10.18632/oncotarget.26026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang B., Jiang H., Zhou T., Ma N., Liu W., Wang Y., Zuo L. Expression of ICOSL is associated with decreased survival in invasive breast cancer. PeerJ. 2019;7:e6903. doi: 10.7717/peerj.6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H.L., Wang P., Lu M.Z., Zhang S.D., Zheng L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019;17:4487–4493. doi: 10.3892/ol.2019.10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leinonen R., Sugawara H., Shumway M., International Nucleotide Sequence Database Collaboration The sequence read archive. Nucleic Acids Res. 2011;39:D19–D21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkatasubramanian M., Chetal K., Schnell D.J., Atluri G., Salomonis N. Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF. Bioinformatics. 2020;36:3773–3780. doi: 10.1093/bioinformatics/btaa201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaath H., Vishnubalaji R., Elango R., Khattak S., Alajez N.M. Single-cell long noncoding RNA (lncRNA) transcriptome implicates MALAT1 in triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy. Cell Death Discov. 2021;7:23. doi: 10.1038/s41420-020-00383-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zambon A.C., Gaj S., Ho I., Hanspers K., Vranizan K., Evelo C.T., Conklin B.R., Pico A.R., Salomonis N. GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics. 2012;28:2209–2210. doi: 10.1093/bioinformatics/bts366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvano S.E., Xiao W., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Inflamm and Host Response to Injury Large Scale Collab. Res. Program A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 41.Elango R., Vishnubalaji R., Shaath H., Alajez N.M. Molecular subtyping and functional validation of TTK, TPX2, UBE2C, and LRP8 in sensitivity of TNBC to paclitaxel. Mol. Ther. Methods Clin. Dev. 2021;20:601–614. doi: 10.1016/j.omtm.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vishnubalaji R., Elango R., Al-Toub M., Manikandan M., Al-Rikabi A., Harkness L., Ditzel N., Atteya M., Hamam R., Alfayez M. Neoplastic Transformation of Human Mesenchymal Stromal Cells Mediated via LIN28B. Sci. Rep. 2019;9:8101. doi: 10.1038/s41598-019-44536-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.