

Summary
Asymmetric dimethylarginine (ADMA) is an endogenous nitric oxide synthase (NOS) inhibitor/uncoupler inducing vascular pathology. Vascular pathology is an important factor for the development and progression of CNS pathology of MS, yet the role of ADMA in MS remains elusive. Patients with multiple sclerosis (MS) are reported to have elevated blood levels of ADMA, and mice with experimental autoimmune encephalomyelitis (EAE, an animal model of MS) generated by auto‐immunization of myelin oligodendrocyte glycoprotein (MOG) and blood–brain barrier (BBB) disruption by pertussis toxin also had increased blood ADMA levels in parallel with induction of clinical disease. To explore the role of ADMA in EAE pathogenesis, EAE mice were treated with a daily dose of ADMA. It is of special interest that ADMA treatment enhanced the BBB disruption in EAE mice and exacerbated the clinical and CNS disease of EAE. ADMA treatment also induced the BBB disruption and EAE disease in MOG‐immunized mice even without pertussis toxin treatment, suggesting the role of ADMA in BBB dysfunction in EAE. T‐cell polarization studies also documented that ADMA treatment promotes TH1‐ and TH17‐mediated immune responses but without affecting Treg‐mediated immune response in EAE mice as well as in in vitro T‐cell culture. Taken together, these data, for the first time, document the vascular and immunopathogenic roles of ADMA in EAE, thus pointing to the potential of ADMA‐mediated mechanism as a new target of potential therapy for MS.
Keywords: Asymmetric dimethylarginine (ADMA), blood–brain barrier (BBB), experimental autoimmune encephalomyelitis (EAE), multiple sclerosis, myelin, T helper lymphocytes, TH1, TH17, vascular pathology
In EAE mice, the blood level of ADMA, an endogenous inhibitor/uncoupler of nitric oxide synthases, is increased in parallel to clinical disease. ADMA induces EAE disease in MOG‐immunized mice by disrupting BBB and by enhancing TH1/TH17‐mediated autoimmune responses. Overall, these data document the vascular and immunopathogenic roles of ADMA in EAE.
![]()
Abbreviations
- ADMA
asymmetric dimethylarginine
- AdoHcyase
S‐adenosylhomocysteine hydrolase
- Arg
arginine
- AUC
area under the curve
- BBB
blood–brain barrier
- CFA
complete Freund's adjuvant
- CNS
central nervous system
- CVD
cardiovascular disease
- DDAH
dimethylarginine dimethylamino‐hydrolases
- EAE
experimental autoimmune encephalomyelitis
- eNOS
endothelial nitric oxide synthase
- Fig
figure
- GSH
glutathione
- GSNO
S‐nitrosoglutathione
- H&E
haemotoxylin and eosin
- Hcy
homocysteine
- iNOS
inducible nitric oxide synthase
- LFB
Luxol fast blue
- MBP
myelin basic protein
- MMP
matrix metalloprotease
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NOS
nitric oxide synthase
- ONOOˉ
peroxynitrite
- PTX
pertussis toxin
- SAH
S‐adenosylhomocysteine
- SAM
S‐adenosylmethionine
- TH1
type 1 T helper cell
- TH17
type 17 T helper cell
- Treg
regulatory T cell
- Tyr
tyrosine
- VEGF
vascular endothelial growth factor
INTRODUCTION
Multiple sclerosis (MS) is the most common debilitating neurologic disease affecting individuals in their most productive years. The disease is induced by peripheral activation of myelin‐specific autoreactive lymphocytes and their CNS infiltration across the blood–brain barrier (BBB) leading to encephalitogenic demyelination. Accordingly, immune modulation has been the major target for Food and Drug Administration (FDA)‐approved disease‐modifying drugs for MS but with limited efficacies as CNS disease progression continues.
Nitric oxide (NO) has been shown to have diverse actions in the mammalian vascular and immune systems. NO exerts its vascular effects principally by activating soluble guanylyl cyclase, which increases the intracellular concentration of cGMP. Alternatively, NO also exerts its action via the formation of secondary redox metabolites, such as peroxynitrite (ONOOˉ) or S‐nitrosoglutathione (GSNO). ONOOˉ is generated by the reaction between NO and superoxide anion (•O2ˉ) under pathological conditions [1]. ONOOˉ is the most powerful oxidative and nitrosative agent that has been implicated in CNS pathogenesis of MS and its animal model, experimental autoimmune encephalomyelitis (EAE) [2]. On the other hand, GSNO is a physiological NO carrier molecule generated by the reaction between NO and glutathione (GSH).
GSNO by itself does not release free NO efficiently; rather, it exerts its biological regulation/activity via modifying protein thiols, a process termed S‐nitrosylation [1]. GSNO‐induced S‐nitrosylation is now known to participate in various cell signalling processes for the maintenance of vascular haemodynamics [3] and anti‐inflammatory processes [4, 5]. Our laboratory has documented that GSNO‐mediated S‐nitrosylation mechanism modulates proinflammatory processes via inhibiting NF‐κB and STAT3 pathways [6, 7]. We also reported that the GSNO‐mediated mechanism regulates TH17 vs. Treg cell balance and their CNS infiltration under EAE conditions [5, 8, 9, 10, 11]. More recently, we reported that the GSNO‐mediated mechanism also regulates endothelial actin cytoskeleton dynamics and protects BBB against the extravasation of peripheral immune cells [5, 11], thereby documenting NO metabolism as a potential target for MS/EAE therapy.
NO is synthesized by three isoforms of NO synthase (NOS): neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS). Under physiological conditions, NOS catalyses the oxidation of L‐Arg to produce NO. Under the pathological conditions, however, NOS is uncoupled from its cofactor (tetrahydrobiopterin) and/or substrate (L‐Arg) and produces more •O2ˉ and less NO, thus increasing ONOOˉ generation, a process known as ‘NOS uncoupling’ [12]. NOS is uncoupled by endogenous NOS inhibitor, asymmetric dimethylarginine (ADMA), a by‐product of L‐Arg metabolism that competes with L‐Arg for all isoforms of NOS [13, 14]. Recently, a growing body of evidence points to ADMA as a risk factor for vascular dysfunction based on the effect of ADMA on eNOS inhibition and uncoupling [15]. Elevated ADMA levels have been reported in a variety of cardiovascular diseases (CVD), and ADMA infusion has been shown to induce pathologies associated with CVD and stroke [15]. Patients with MS are known to have significantly increased levels of ADMA in the blood [16]. Accordingly, MS patients are known to have a very high incidence of CVD and ischaemic stroke [17, 18]. However, the role of ADMA in the MS/EAE pathogenesis is not understood at present.
The majority of ADMA (>80%) is cleared by its enzymatic degradation by dimethylarginine dimethylamino‐hydrolases (DDAH‐1 and DDHA‐2), and the rest is excreted through the renal system [15]. Sulfhydryl groups (cysteines) at the active sites of DDAHs are essential for their activities [19]. Various studies reported that oxidative stress or reactive thiols, such as L‐homocysteine (HCy), inhibit DDAH activities for ADMA degradation by modifying the sulfhydryl groups in the active sites [20, 21, 22]. It is of interest that MS patients have elevated plasma levels of HCy (hyperhomocysteinaemia) [23, 24, 25, 26, 27] as well as ADMA [16], thus suggesting the potential role of impaired ADMA catabolism in MS/EAE pathogenesis. Based on these studies, here, we investigated the role of ADMA homeostasis and its participation in autoimmune pathogenesis using an animal model of EAE.
MATERIALS AND METHODS
EAE induction and clinical score assessment
C57BL/6 mice were purchased from Jackson Laboratory (Stock no: 000664, Bar Harbor, ME). Mice were supplied with food and water ad libitum and kept in ventilated cages in a specific pathogen‐free animal care facility maintained by the Medical University of South Carolina throughout the entire study. They were housed at controlled temperature (22°C), humidity (45–55%) and 12‐h light/dark cycle. All animal studies were reviewed and approved by the Medical University of South Carolina's Institutional Animal Care and Use Committee (IACUC) (AR # 2019‐00761). The mice were randomly assigned to 0, 7, 11, 19 and 44 days of post‐immunization or control group, vehicle‐treated EAE group and ADMA‐treated EAE group. The induction of EAE and vehicle/ADMA treatment was blinded to the personnel performing the experiments. EAE was induced by the subcutaneous injection of female C57BL/6 mice (8–12 weeks old) with myelin oligodendrocyte glycoprotein (MOG)35‐55 peptide emulsified in the complete Freund's adjuvant (CFA) as described in the kit instruction (Hook Laboratories, Lawrence, MA). Two hundred nanograms of pertussis toxin was injected intraperitoneally on days 0 and 1. Mice were weighed and assessed for clinical signs every day starting from day 0 through the day of experiment termination. Alternatively, MOG‐immunized mice received daily ADMA (50 mg/kg body weight /day/ip; Sigma‐Aldrich, St. Louis, MO, USA) starting day 0 and ending day 10 post‐immunization instead of PTX treatments. EAE score was evaluated as follows: 0 = no clinical signs of disease; 0·5 = partial tail paralysis; 1 = limp tail or waddling gait with tail tonicity; 2 = waddling gait with limp tail (ataxia); 2·5 = ataxia with partial limb paralysis; 3 = full paralysis of one limb; 3·5 = full paralysis of one limb with partial paralysis of second limb; 4 = full paralysis of two limbs; 4·5 = moribund stage; 5 = death. EAE mice were treated with daily doses of a vehicle (100 μl of 10% DMSO in saline) or ADMA (50 mg/Kg body weight/day/ip) starting the disease onset (day 11 of post‐immunization).
ELISA for serum ADMA, L‐Arg, VEGF and HCy
Serum levels of ADMA, L‐arginine, VEGF and HCy were determined with commercial enzyme‐linked immunosorbent assay (ELISA) kits according to the manufacturers’ instructions (MyBioSource, San Diego, CA, USA, Cat# mbs705936 for ADMA, mbs726317 for L‐arginine, mbs165329 for VEGF and mbs7252797 for HCy).
Assay of DDAH activity
DDAH activity was measured as reported previously [28]. Briefly, kidneys and livers were homogenized in sodium phosphate buffer containing 0·1% Triton X‐100, preincubated with urease (100 U/ml) for elimination urea. The samples were mixed with 1 mM ADMA, incubated at 37°C and treated with the sample volume of 4% sulfosalicylic acid to stop the reaction and followed by centrifugation at 3000 g for 10 min. The supernatant was mixed with an equal amount of colour mixture (1 part of oxime reagent: 2 parts of antipyrine/H2SO4) and incubated for 110 min at 60°C in the dark. The reaction was stopped by cooling samples at 20°C for 10 min, and the levels of citrulline were measured at 466 nm.
Histological and electron microscopic analysis
Control mice, EAE mice treated with vehicle or ADMA were anaesthetized and perfused first with saline and then 4% paraformaldehyde as described previously [29]. Tissue samples were paraffin‐embedded and transversely sectioned (4 μm thick). Haematoxylin and eosin (H&E) staining was performed to assess the infiltration of mononuclear cells, and Luxol fast blue (LFB) staining was performed to evaluate the status of myelin (demyelination) in spinal cord sections. Pictures were taken using BX‐60 Olympus Microscope (Olympus, Tokyo, Japan). Electron microscopy was performed as we described previously [30]. Animals were anaesthetized and perfused with 10 ml of normal saline containing 0·1% sodium nitrite followed by 15 ml of a mixture of 4% paraformaldehyde and 2% glutaraldehyde in 0·1 M phosphate buffer, pH 7·4. Then, the spinal cords were fixed in the same fixative (above) and post‐fixed with 1% osmium tetroxide‐1·5% ferrocyanide for 2 h in the dark, then dehydrated and embedded in Epon LX 112 resin. Semi‐thin sections, approximately 1 μm thick, were cut and stained with toluidine blue. Ultrathin sections (70 nm thick) were stained with uranyl acetate and lead citrate and examined by electron microscopy.
Western analysis: Western blot analysis
Western immunoblot analysis was performed by a standard method using 50μg of cell lysates. Following the SDS–PAGE electrophoresis, proteins were transferred from the gel onto the polyvinylidene fluoride membrane (GE Healthcare Life Sciences, Marlborough, MA, USA). Membranes were blocked with non‐fat dry milk (Santa Cruz Biotechnology) and incubated with a primary antibody specific to myelin basic protein (MBP; Santa Cruz Biotech, Delaware Avenue, CA), Claudin‐1 (Invitrogen‐Thermo Fisher Inc., Cat# 37–4900), β‐actin (Cell Signaling Technology, Danvers, MA), β‐tubulin (Cell Signaling Technology) or MMP‐9 (Cell Signaling Technology). Following washing, the membranes were incubated with horseradish peroxidase‐conjugated secondary antibody (Jackson Immunoresearch Lab, West Grove, PA), washed and then incubated with ECL reagent (Amersham Life Science, Pittsburgh, PA), and exposed to Amersham Hyperfilm ECL film. The density of specific bands was analysed by ImageJ (NIH) software.
Evaluation of BBB disruption by Evans blue (EB) extravasation
BBB leakage was assessed as previously described from our laboratory [31, 32]. The mice received 100 μl of a 5% solution of EB in saline administered intravenously. At 24 h, cardiac perfusion was performed under deep anaesthesia with 200 ml of saline to clear the brain/spinal cord circulation of EB. The brain and spinal cord were removed and sliced. The tissues were homogenized in 750 μl of N, N‐dimethylformamide (DMF) and centrifuged at 10,000 ×g for 25 min, and EB content in the supernatant was fluorometrically analysed (λex 620 nm, λem 680 nm).
Fluorescence flow cytometry analysis of splenic TH1, TH17 and Treg cells and their cytokine production
Fluorescence flow cytometry analysis for each subset of CD4+ T cells (TH1, TH17, CD4+/CD25+/FOXP3+ Treg and CD4+/CD25+/FOXP3− Treg) was performed as reported previously with some modification [9]. Briefly, control and EAE mice were killed for the collection of the spleens. Following the preparation of single‐cell suspension, red blood cells were lysed with Pharma lyse buffer (BD Pharmingen) and the remaining cells were washed with RPMI 1640. CD4+ T cells were purified with CD4 microbeads (Miltenyi Biotech) then resuspended with complete RPMI‐media in 12‐well plates (5 × 106 cells/2 ml per well) containing MOG peptide (25 μg/ml) for 48 h Following the centrifugation, the resulting supernatants were collected for ELISA for CD4+ subset‐specific cytokines. The cell pellets were washed with cell staining solution (Ebioscience, Waltham, MA, USA) and stained with a fluorescence‐labelled antibody specific to IFN‐γ for TH1, IL‐17/IL‐23R for TH17 and CD25 and FOXP3+ for Treg (Ebioscience). The cells were counted and analysed using a Beckman Coulter instrument (Beckman Coulter, Inc., Brea, CA, USA). ELISA assay was performed for analysis of CD4+ T‐cell subset‐specific cytokines (IFN‐γ, IL‐17 and IL‐10) released from cultured lymphocytes. The ELISA assay kits for IFN‐γ, IL‐17, GM‐CSF and IL‐10 were purchased from R&D Systems (Minneapolis, MN, USA).
For in vitro T‐cell differentiation assay, naive CD4+ T cells were isolated from naïve C57BL/6 mice as described above. The CD4+ T cells (2 × 105 cells/well) suspended in RPMI‐complete media were pretreated with ADMA (200 µM) and then stimulated with plate‐bound anti‐CD3ε antibody and soluble anti‐CD28 antibody and IL‐2 (20 U/ml) in the presence or absence of TH1 (10 ng/ml IL‐12 and 1 ug/ml anti‐IL‐4 mAb) or TH17 (30ng/ml IL‐6, 2ng/ml TGF‐β, 5ng/ml IL‐23, 1ug/ml anti‐IFN‐γ mAb, 1ug/ml anti‐IL‐4 mAb) polarization as described previously [8]. The cells were collected at 36–96 h, and the numbers of TH1, TH17, and FOXP3+ Treg cells and the levels of IFN‐γ, GM‐CSF, IL‐17a and IL‐10 in the cell culture media were analysed by fluorescence flow cytometric analysis and ELISA as described above. For fluorescence flow cytometric analysis, the cells were treated with phorbol myristate acetate (PMA: 50 ng/ml), ionomycin (1 μg/ml) and 1× Brefeldin A for 5 h before the analysis.
Statistical analysis
Statistical analysis was performed with GraphPad Prism (version 8.2: GraphPad Software, Inc.). Values are expressed as mean ±standard deviation (SD). All data were subjected to Shapiro–Wilk normality test, and then, the group means were statistically compared by ordinary one‐way ANOVA with Tukey's multiple comparisons test. Comparisons among means of two groups were made with a two‐tailed unpaired t test. Results were considered statistically significant if p < 0·05.
RESULT
EAE increases the blood ratio of ADMA vs. L‐Arg with reduction of DDAH activity
Our laboratory has recently reported that ADMA induces endothelial/vascular dysfunction by interacting with VEGF [33]. Based on these studies, we analysed blood levels of ADMA and VEGF in EAE mice. For induction of EAE, female C57BL/6 mice were treated with complete Freund's adjuvant (CFA) and myelin oligodendrocyte glycoprotein peptide (MOG) with/without pertussis toxin (PTX) treatment. Following the induction of EAE, we analysed blood (serum) levels ADMA and VEGF on day 0 (control), before the onset of the EAE disease (day 7 post‐immunization), at the disease onset (day 11 post‐immunization), at the peak of disease (day 19 post‐immunization) and during the chronic disease phase (day 44 post‐immunization). Figure 1A‐i, A‐iii shows that ADMA levels were significantly increased on the day of disease onset (day 11 post‐immunization) and stayed at high levels till day 44 post‐immunization. The EAE mice also had increased blood VEGF levels even before the onset of disease (day 7 post‐immunization) and which was decreased during the chronic phase of EAE disease (day 44 post‐immunization) (Figure 1A‐ii, ‐iii).
FIGURE 1.
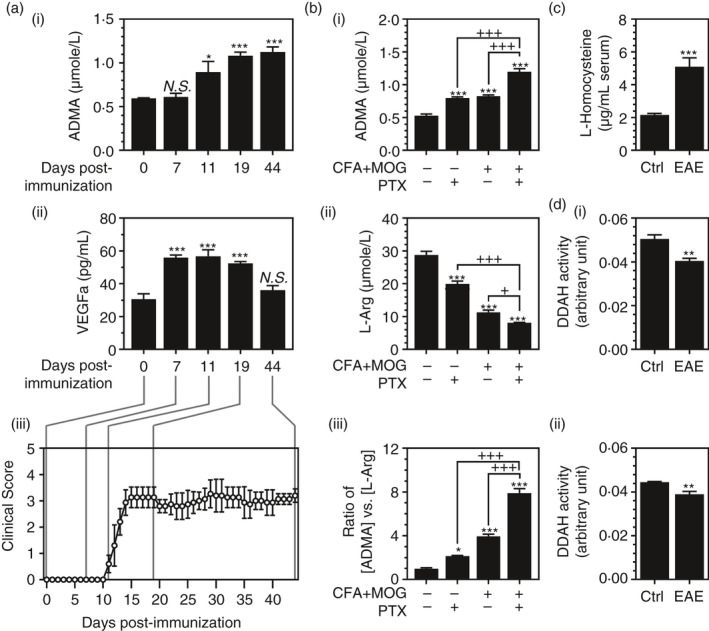
ADMA is elevated in the blood of EAE animals: Female C57BL/6 mice were treated with complete Freund's adjuvant (CFA), myelin oligodendrocyte glycoprotein peptide (MOG) and pertussis toxin (PTX) for induction of EAE; then, blood (serum) levels ADMA (A‐i) and VEGF (A‐ii) were analysed in control (0‐day post‐immunization) and EAE mice before the onset of the disease (day 7 post‐immunization), on the day of disease onset (day 11 of post‐immunization), at the peak of disease (day 19 post‐immunization), and during the chronic disease phase (day 44 of post‐immunization) (A‐iii). On the day of disease onset (day 11 post‐immunization), blood levels of ADMA (B‐i), L‐arginine (L‐Arg) (B‐ii) and the ratio of [ADMA] vs. [L‐Arg] (B‐iii) were analysed in the mice immunized with CFA+MOG with or without PTX treatment. In EAE mice (CFA+MOG+PTX), the blood levels of L‐homocysteine (C) and DDAH activities in the kidney and liver (D‐i and ii) were analysed on the day of disease onset (Day 11 post‐immunization). Data represent mean ±standard error mean (SEM) from n = 6 animals. N.S. (not significant: p ≥ 0·05); *p ≤ 0·05, **p ≤ 0·01, ***p ≤ 0·001 vs. control. +p ≤ 0·05, ++p ≤ 0·01, +++p ≤ 0·001 vs. as indicated. Six animals were used for mice for each group
On the day of disease onset (day 11 post‐immunization), the animals treated with PTX or CFA+MOG or PTX+CFA+MOG were analysed for blood levels of ADMA, L‐Arg and the ratio of ADMA vs. L‐Arg (Figure 1B). The blood levels of ADMA were increased by treatment either with CFA+MOG treatments or with PTX treatment (Figure 1B‐i) while the blood levels of L‐Arg were reduced by these treatments (Figure 1B‐ii). A combination of CFA+MOG and PTX further increased the blood levels of AMDA and further reduced the blood levels of L‐Arg (Figure 1B‐i, ii). Accordingly, the ratio of blood levels of ADMA vs. L‐Arg was increased either by CFA+MOG or by PTX treatment and further increased by their combination (Figure 1B‐iii).
In the body, the majority of ADMA (>80%) is known to be cleared by its enzymatic degradation by DDAH in the liver and kidney by DDAH [15, 34, 35, 36]. DDAH is a redox‐sensitive enzyme [19] whose activity is affected by reactive thiols, such as L‐homocysteine (HCy) [20, 21, 22]. Similar to the MS patients [23, 24, 25, 26, 27], we observed that the blood levels of HCy were greatly increased in EAE animals (CFA+MOG+PTX) (Figure 1C). Accordingly, we observed significant reductions in DDAH activities in the kidney and liver (Figure 1D‐I, ii). Taken together, these data indicate the potential role of hyperhomocysteinaemia in the inhibition of DDAH‐mediated ADMA catabolism and the increased accumulation of blood ADMA in EAE.
ADMA treatment exacerbates clinical EAE disease
Next, we investigated the effect of ADMA overburden on clinical EAE disease. Recently, we have reported that systemic treatment of C57BL/6 mice with a daily dose of ADMA (50mg/kg/day/ip) increases the levels of ADMA in the blood [33]. Based on these data, we treated EAE mice with the same daily dose of ADMA starting on the day of disease onset (day 11 post‐immunization). Figure 2A shows that the treatment of EAE mice with ADMA had no significant effect till day 14 post‐immunization. However, it significantly increased the disease starting day 15 post‐immunization. The areas under the curves (AUC) of the daily clinical scores (Figure 2B) show that ADMA significantly increased the overall EAE disease symptom.
FIGURE 2.
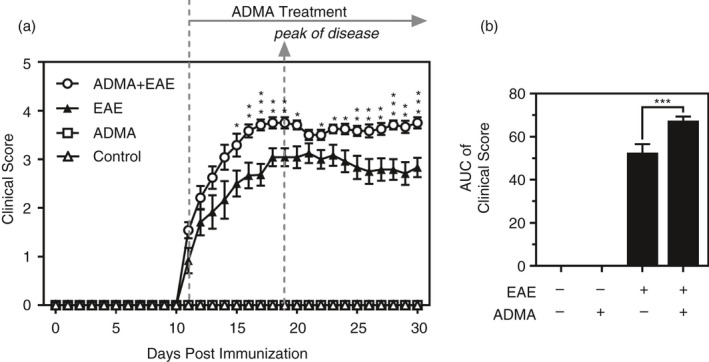
ADMA treatment exacerbates EAE disease: Female C57BL/6 mice were treated with complete Freund's adjuvant (CFA), myelin oligodendrocyte glycoprotein (MOG) and pertussis toxin (PTX) for induction of EAE and daily clinical scores of control, EAE and ADMA‐treated EAE mice (A) and the areas under the curves as overall disease severity (B) were analysed. Data represent mean ± standard error mean (SEM) from n = 12 animals. *p ≤ 0·05, **p ≤ 0·01, ***p ≤ 0·001 vs. EAE mice
ADMA treatment increases myelin and axonal pathologies in the spinal cord of EAE animals
Next, we investigated the effect of ADMA overburden in EAE‐induced demyelination and axonal degeneration in the spinal cord. LFB staining in Figure 3A shows that EAE mice had decreased myelin content as compared to the control mice and which was further decreased with ADMA treatment. Accordingly, Western analysis of myelin basic protein (MBP) in Figure 3B shows that ADMA treatment significantly enhanced the EAE‐induced reduction in MBP levels in the spinal cord. Electron microscope study of the spinal cord for ultrastructural conformation of myelin and axonal loss (Figure 3C) shows that ADMA treatment also enhanced the loss of myelin ultrastructure as shown by the loss of doughnut‐like structure of myelin sheet. ADMA treatment also increased the loss of axons in the spinal cord of EAE mice as shown by Bielschowsky silver staining (Figure 3D) and β‐tubulin (axonal marker) Western analysis (Figure 3E). Overall, these data document that ADMA overburden augmented EAE‐induced demyelination and axonal loss.
FIGURE 3.
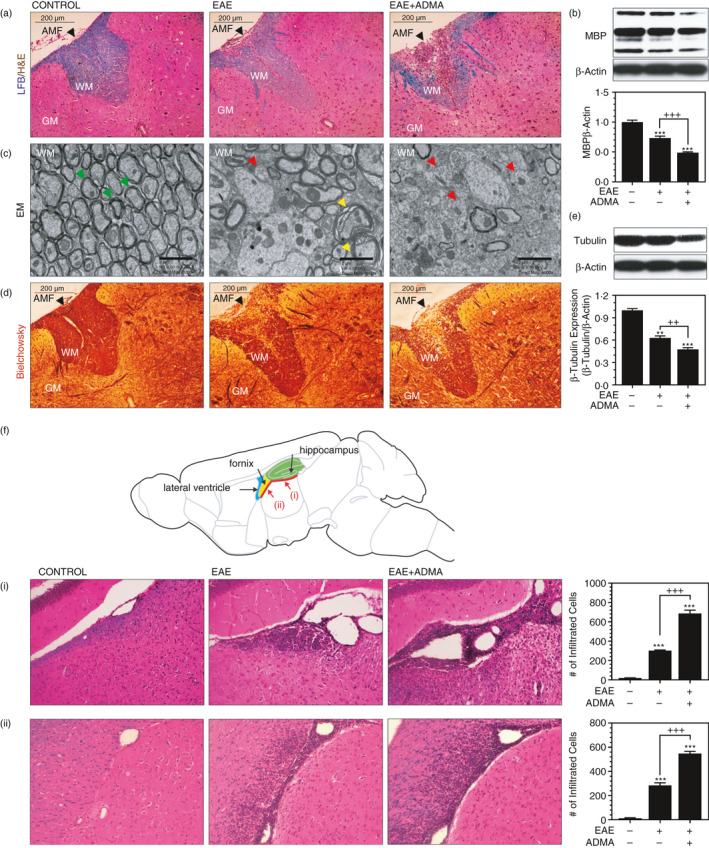
ADMA treatment increases myelin and axonal pathologies in EAE mice: Female C57BL/6 mice were treated with complete Freund's adjuvant (CFA), myelin oligodendrocyte glycoprotein (MOG) and pertussis toxin (PTX) for induction of EAE. On day 30 of post‐immunization, lumbar spinal cord sections (A, C and D) and brain sections (F) were stained by Luxol fast blue (LFB) for visualization of myelin (blue colour) and hematoxylin and eosin (H&E) for mononuclear cells (dark brown dots) (A and F), electron microscope (EM) for visualization of axon‐myelination ultrastructure (C) and Bielschowsky silver staining for visualization of axonal pathology (D). In the EM study, the green arrowhead indicates intact axon–myelin structure; the yellow arrowhead indicates demyelinating axon; the red arrowhead indicates demyelinated axon. Demyelination and axonal loss were also analysed by Western analysis for myelin basic protein (MBP) (B) and β‐tubulin (E) using the spinal cord tissue lysates. β‐Actin was used for the internal loading standard. The bar graphs represent mean ± standard error mean (SEM) from n = 6 animals. *p ≤ 0·05, **p ≤ 0·01, ***p ≤ 0·001 vs. control; +p ≤ 0·05, ++p ≤ 0·01, +++p ≤ 0·001 vs. as indicated. AMF (anterior median fissure), WM (white matter), GM (grey matter)
EAE mice are known to exhibit widespread brain lesions under long‐term and chronic disease conditions (e.g. day 66 post‐immunization) [37]. We did not observe such widespread brain lesions during the acute disease conditions (day 30 post‐immunization), but we observed localized lesions in certain brain areas. Haematoxylin and eosin (H&E) staining in Figure 3F shows that EAE mice had increased inflammatory infiltration near the leptomeninges underneath the hippocampal formation (Figure 3F‐i) and fornix (Figure 3F‐ii) and which were further increased by ADMA treatment.
ADMA treatment enhances BBB dysfunction in EAE animals and increases the CNS infiltration of peripheral effector T cells
Recently, our laboratory has reported that ADMA enhances endothelial/vascular dysfunction under neuropathological conditions [33]. To investigate the effect of ADMA overburden on BBB functions and integrity in EAE animals, we performed Evans blue extravasation assay. We observed that daily ADMA treatment increased Evans blue extravasation into the CNS (brain and spinal cord) in EAE mice (Figure 4A‐i). However, ADMA by itself had relatively little effect on Evans blue extravasation in the control mice (Figure 4A‐i). Matrix metalloprotease‐9 (MMP‐9) is known to play a key role in BBB dysfunction in MS and EAE [38, 39]. We observed that EAE mice had increased expression of MMP‐9 in the spinal cord of EAE animals and which was further increased by daily ADMA treatment (Figure 4A‐ii). Claudin‐5 is one of the BBB tight junction proteins [40] which is decreased during EAE disease [41]. MMP‐9 is known to degrade claudin‐5 [42]. Accordingly, we observed that the claudin‐5 level was decreased in the spinal cord of EAE mice and ADMA treatment further reduced it (Figure 4A‐iii).
FIGURE 4.
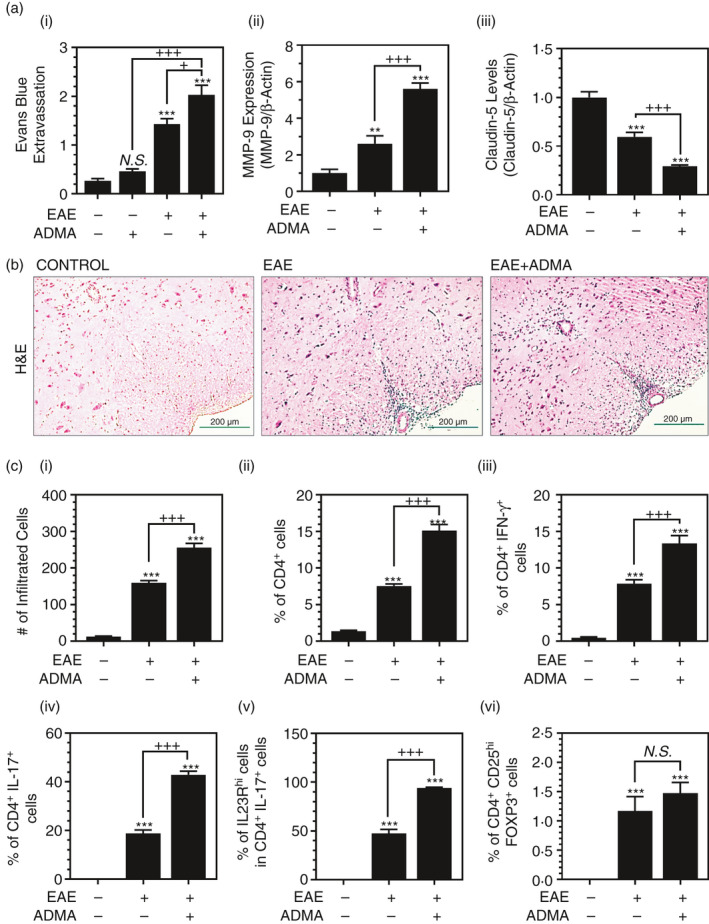
ADMA treatment induces BBB dysfunction and increases CNS infiltration of lymphocytes in EAE mice: Female C57BL/6 mice were treated with complete Freund's adjuvant (CFA), myelin oligodendrocyte glycoprotein (MOG) and pertussis toxin (PTX) for induction of EAE with/without daily exogenous ADMA treatment. At the peak of disease (day 19 post‐immunization), the effect of ADMA on EAE‐induced BBB dysfunction, such as Evans blue extravasation into the CNS (brain and spinal cord) (A‐i), and the spinal cord expression levels of MMP‐9 (A‐ii) and claudin‐5 (A‐iii) were analysed. At the peak of disease, infiltration of mononuclear cells into the lumbar area of the spinal cord was visualized as H&E staining (B). In addition, the numbers of infiltrated mononuclear cells (C‐i) and % of CD4+ T cells in CNS infiltrated mononuclear cells (C‐ii), % of IFN‐γ+ TH1 cells (C‐iii) and IL‐17+ TH17 cells (C‐iv) in CD4+ T cells, % of IL‐23Rhi cells in TH17 cells (C‐v), and % of CD25hiFOXP3+ Treg cells in CD4+ T cells (C‐vi) were measured by fluorescent flow cytometry. Please see Figure S1 for the representative flow cytometry dot plots. Data represent mean ± standard error mean (SEM) from n = 6 animals. N.S. (not significant: p ≥ 0·05); *p ≤ 0·05, **p ≤ 0·01, ***p ≤ 0·001 vs. control; +p ≤ 0·05, ++p ≤ 0·01, +++p ≤ 0·001 vs. as indicated
Next, we investigated the effect of ADMA overburden in the extravasation of mononuclear cells into the CNS. H&E staining of the lumbar spinal cord sections (Figure 4B) and counting of infiltrated mononuclear cells (Figure 4C‐i) show that daily ADMA treatment increased the extravasation of mononuclear cells in the lumbar area of the spinal cord in EAE mice.
The activation and differentiation of myelin‐reactive CD4+ T cells into effector (TH1 and TH17) and regulatory (Tregs) subsets at the peripheral tissues, and their subsequent infiltration into the CNS are decisive events in the pathogenesis of MS and EAE [43]. To investigate the effect of ADMA overburden on the CNS infiltration of peripheral CD4+ T cells, total T cells (CD3+) were isolated from the spinal cord and the per cent of CD4+ T cells was analysed by fluorescence flow cytometry. Figure 4C‐ii shows that ADMA treatment enhanced the EAE‐induced CNS infiltration of CD4+ T cells. ADMA treatment also increased the CNS infiltrations of TH1 (CD4+ IFN‐γ+) and TH17 (CD4+ IL‐17+) cells (Figure 4C‐iii, iv). IL‐23R is one of the signature genes for the development of pathogenic TH17 cells [44]. We observed that daily ADMA treatment of EAE mice also increased IL‐23Rhi TH17 cells (Figure 4C‐v), thus suggesting the role of ADMA in the induction of pathogenic TH17 cells. However, daily ADMA treatment of EAE mice did not significantly affect CNS infiltration of Treg (CD4+ CD25hi FOXP3+) cells (Figure 4C‐vi). These data indicate the potential role of ADMA in the dysfunction of BBB and the augment of peripheral effector T‐cell (TH1 and TH17) infiltration into the CNS under EAE conditions.
ADMA treatment induces autoreactive effector T‐cell immune responses in the spleen
Next, we investigated the potential role of ADMA in the development of autoreactive effector and regulatory T cells in the spleen. For it, CD4+ T cells, purified from spleens of naïve, EAE and ADMA‐treated EAE mice on day 30 of post‐immunization, were cultured ex vivo, and the per cent of CD4+ IFN‐γ+ (TH1), CD4+ IL‐17+ (TH17) and CD4+ CD25+ FOXP3+ (Treg) were analysed. Figure 5A‐i and ii shows that EAE animals had increased proportions of splenic TH1 and TH17 cells and which were further increased by daily ADMA treatment. EAE animals had decreased numbers of Treg cells in the spleens compared to naïve mice, and ADMA treatment did not affect these decreases (Figure 5A‐iii).
FIGURE 5.
ADMA treatment enhances TH1 and TH17 polarization: Female C57BL/6 mice were treated with complete Freund's adjuvant (CFA), myelin oligodendrocyte glycoprotein (MOG), and pertussis toxin (PTX) for induction of EAE with or without daily ADMA treatment starting the day of disease onset. At the peak of disease (day 19 post‐immunization), % of IFN‐γ+ TH1 cells (A‐i), IL‐17+ TH17 cells (A‐ii) and CD25hi FOXP3+ Treg cells (A‐iii) in the splenic CD4+ cells of control, EAE and ADMA‐treated EAE mice were analysed by fluorescence flow cytometry, and their secretion of IFN‐γ (B‐i), IL‐17a (B‐ii), GM‐CSF (B‐iii) and IL‐10 (B‐iv) was analysed by ELISA. Please see Figure S2 for the representative flow cytometry dot plots. The bar graphs on panels A and B represent mean ± standard error mean (SEM) from n = 4 animals. In vitro‐cultured naïve splenic CD4+ T cells were activated by IL‐2 (5 ng/ml), anti‐CD3 mAb (2 μg/ml) and anti‐CD28 mAb (1 μg/ml) under unpolarized conditions in the presence or absence of ADMA (400 μM) and % of IFN‐γ+ TH1 cells (C‐i), IL‐17+ TH17 cells (C‐ii) and CD25+ FOXP3+ Treg cells were analysed by fluorescence flow cytometry. In vitro‐cultured naïve splenic CD4+ T cells were activated by IL‐2 and anti‐CD3 and anti‐CD28 mAbs under TH1 (10 ng/ml IL‐12 and 1 ug/ml anti‐IL‐4 mAb)‐ or TH17 (30ng/ml IL‐6, 2ng/ml TGF‐β, 5ng/ml IL‐23, 1ug/ml anti‐IFN‐γ mAb, 1ug/ml anti‐IL‐4 mAb)‐polarized conditions in the presence or absence of ADMA and % of IFN‐γ+ TH1 cells (D‐i) and IL‐17+ TH17 cells (D‐ii) in CD4+ T cells, and % of IL‐23Rhi cells in TH17 cells (D‐iii) were analysed by fluorescence flow cytometry. Please see Figure S3 for the representative flow cytometry dot plots. Under the same experimental conditions, T‐cell secretions of IFN‐γ and GM‐CSF under TH1‐polarized conditions (E‐i and ii) and secretions of IL‐17a and GM‐CSF under TH17‐polarized conditions (E‐iii and iv) were analysed by ELISA. The bar graphs on panels C, D and E represent mean ± standard error mean (SEM) from n = 3 independent cell cultures. N.S. (not significant: p ≥ 0·05); *p ≤ 0·05, **p ≤ 0·01, ***p ≤ 0·001 vs. control; +p ≤ 0·05, ++p ≤ 0·01, +++p ≤ 0·001 vs. as indicated
Similar to the proportions of CD4+ T‐cell subsets, splenic CD4+ T cells isolated from EAE animals produced increased levels of proinflammatory cytokines, such as IFN‐γ (Figure 5B‐i), IL‐17a (Figure 5B‐ii) and GM‐CSF (Figure 5B‐iii), and which were further increased by ADMA treatment. However, ADMA treatment did not affect EAE‐induced reduction in IL‐10 production in splenic CD4+ T cells (Figure 5B‐iv).
Next, we investigated the effects of ADMA on the differentiation of naïve CD4+ T cells under in vitro stimulatory conditions. For it, splenic naïve CD4+ T cells were stimulated in vitro by treatment with anti‐CD3 and CD28 mAbs and IL‐2 under non‐polarized conditions in the presence or absence of ADMA treatment (Figure 5C). ADMA treatment had no obvious effect on the differentiation of CD4+ T cells under unstimulatory conditions but it enhanced the TH1 and TH17 differentiation under stimulatory conditions (Figure 5C‐i, C‐ii). In contrast to ex vivo‐cultured splenic CD4+ T cells from EAE animals (Figure 5A‐iii), in vitro stimulation of naïve CD4+ T cells increased the numbers of Treg cells (Figure 5C‐iii). However, ADMA treatment did not affect these changes.
Next, we investigated the effects of ADMA on the polarization of TH1 or TH17 cells under each polarized condition. For this, the stimulated CD4+ T cells were further polarized with IL‐12 and anti‐IL‐4 mAb for TH1 cells or IL‐6, TGF‐β, anti‐IFN‐γ mAb and anti‐IL‐4 mAb for TH17 cells in the presence/absence of ADMA. Figure 5D‐i, D‐ii shows that ADMA treatment increased TH1 and TH17 polarization under each polarized condition. However, ADMA treatment had no obvious effect on IL‐23R expression in TH17 cells (Figure 5D‐iii). ADMA treatment increased IFN‐γ and IL‐17a productions under TH1‐ and TH17‐polarized conditions, respectively (Figure 5E‐i and E‐iii). However, ADMA treatment did not affect GM‐CSF release of T cells under both TH1‐ and TH17‐polarized conditions (Figure 5E‐ii, E‐iv). Taken together, these data document the pathogenic role of ADMA in CD4+ T cell‐mediated autoimmune response in addition to its role in the induction of BBB disruption and peripheral immune cell extravasation.
ADMA treatment during the early phase of EAE induces BBB disruption and CNS infiltration of inflammatory T cells in the absence of PTX
Current EAE models are based on peripheral immunization of animals with myelin–antigen (e.g. MOG) and CFA. However, the myelin immunization by itself is not enough for developing clinical EAE disease due to a lack of CNS infiltration of immunocytes [45]. Accordingly, we observed no significant induction of EAE disease in CFA+MOG‐treated group (Figure 6A‐i, ii) as well as no significant extravasation of Evans blue dye into the CNS (brain and spinal cord) in this group (Figure 6B). In addition, we observed no obvious myelin pathology and mononuclear cell infiltration into the spinal cord in CFA+MOG‐treated group (Figure 6C‐i, ii). Pertussis toxin (PTX) has been widely used to induce BBB pathology for facilitating the CNS infiltration of peripheral immunocytes and thus induction of EAE disease [45]. Accordingly, we observed that PTX treatment in addition to CFA+MOG treatment induced the extravasation of Evans blue dye into the CNS (Figure 6B) and the infiltration of mononuclear cells into the spinal cord (Figure 6C‐ii) that parallel with the clinical EAE disease induction (Figure 6A‐i, ii).
FIGURE 6.
ADMA treatment induces EAE disease in MOG‐immunized mice in the absence of PTX: Female C57BL/6 mice were immunized with complete Freund's adjuvant (CFA) and myelin oligodendrocyte glycoprotein (MOG) with or without pertussis toxin (PTX) or ADMA treatment. PTX was treated on days 0 and 1 of post‐immunization. ADMA (50mg/kg) was treated daily starting day 0 and ending day 10 post‐immunization. Following immunization, daily clinical scores (A‐i) and overall disease severity (AUC: area under the curve) (A‐ii) were analysed. The line and bar graphs on panel A represent mean ± standard error mean (SEM) from n = 8 animals. On day 28 post‐immunization, CNS extravasation of Evans blue (B), mononuclear cell infiltration (H&E: hematoxylin and eosin staining) and myelin status (LFB: Luxol fast blue staining) in the lumbar area of spinal cord sections (C‐i), and the number of mononuclear cells in the spinal cord (C‐ii) were analysed. Under the same experimental conditions, % of CD4+ T cells in CNS‐infiltrated mononuclear cells (D‐i), % of IFN‐γ+ TH1 cells (D‐ii), and IL‐17+ TH17 cells (D‐iii) in CNS infiltrated CD4+ T cells, and % of IL‐23R+ cells in CNS infiltrated CD4+ IL‐17+ TH17 cells (D‐iv) were analysed by fluorescence flow cytometry. Please see Figure S4 for the representative flow cytometry dot plots. The bar graphs on panels B, C and D represent mean ± standard error mean (SEM) from n = 6 animals. N.S. (not significant: p ≥ 0·05); *p ≤ 0·05, **p ≤ 0·01, ***p ≤ 0·001 vs. control; +p ≤ 0·05, ++p ≤ 0·01, +++p ≤ 0·001 vs. as indicated
Next, we examined whether daily ADMA treatment during the early phase of EAE induction (day 0 to day 10 post‐immunization) can substitute the role of PTX for induction of BBB disruption and mononuclear cell infiltration into the spinal cord and the clinical EAE disease. ADMA treatment of MOG‐immunized mice (CFA+MOG) induced the CNS extravasation of Evans blue dye (Figure 6B) and the spinal cord infiltration of immunocytes (Figure 6B‐ii) without PTX treatment and accordingly induced the spinal cord myelin pathology (Figure 6C‐i) and clinical disease of EAE (Figure 6A‐i, ii). We also observed that daily ADMA treatment of MOG‐immunized mice (CFA+MOG) also increased the CNS infiltration of CD4+ T cells (Figure 6D‐i), such as TH1 (Figure 6D‐ii) and TH17 (total and IL‐23R+; Figure 6D‐iii,d, D‐iv), which were comparable to the levels observed in PTX treated MOG‐immunized mice. Taken together, these data document that ADMA induces BBB pathology, thus facilitating CNS infiltration of peripheral immunocytes for the development of EAE.
DISCUSSION
In the present study, we investigated the potential role of ADMA in the vascular and immune pathogenesis of EAE. EAE animals had an increased blood level of HCy and reduced DDAH activities in the kidney and liver and thus elevation of blood levels of ADMA (Figure 1). Overburden of blood AMDA by daily treatment of ADMA [33] caused exacerbation of clinical EAE disease (Figure 2) with increased loss of myelin and axons in the spinal cord (Figure 3) as well as increased BBB disruption and mononuclear cell infiltration into the CNS (Figure 4). ADMA treatment also promoted TH1‐ and TH17‐mediated immune responses in the spleen but without affecting Treg‐mediated immune response under EAE conditions (Figure 5). In addition, ADMA was also able to induce EAE disease in MOG‐immunized mice by substituting PTX for BBB disruption and CNS infiltration of mononuclear cells (Figure 6). Taken together, these data, for the first time, document the role of ADMA in vascular and immune pathologies of ADMA and thus pointing to the potential of ADMA‐mediated mechanisms in the clinical disease of EAE and MS.
A growing body of evidence points to ADMA as a risk factor for vascular dysfunction [15]. Elevated ADMA levels have been reported in a variety of cardiovascular diseases (CVD) and ADMA infusion has been shown to induce pathologies associated with CVD and stroke [15]. MS patients are known to have increased blood levels of ADMA [16]. Accordingly, a meta‐analysis showed that people with MS have an increased risk of developing stroke [17, 18, 46]. For instance, in a population‐based cohort study including more than 8,000 patients, MS patients have 85% more heart attacks, 71% more strokes and 97% more heart failure as compared to age and sex‐matched healthy controls [18]. These studies suggest mechanistic interaction between the MS pathogenesis and CVD/stroke and the potential role of ADMA in these interactions. In the EAE model, whether the elevated blood ADMA levels induce cardiovascular dysfunction is not known at present. However, we observed that treatment of MOG‐immunized mice with a daily dose of exogenous ADMA before the onset of the disease induced the EAE disease without PTX treatment for BBB disruption (Figure 6A). Moreover, daily ADMA treatment during the effector phase of EAE disease further worsened the EAE disease severity (Figure 2). Accordingly, the induction or worsening of EAE disease by daily ADMA treatment involved cerebrovascular dysfunction as observed by increased extravasation of Evans blue dye and inflammatory mononuclear cells into the CNS (Figures 4A, B and 6B, C). Recently, we also reported that systemic overburden of ADMA in healthy C57BL/6 mice, by daily treatment with the same dose of ADMA (50 mg/kg/day), caused elevation of mean arterial pressure but without any obvious effects on the cerebrovascular function as observed by no changes in the extravasation of Evans blue dye into the CNS [11]. These data suggest that systemic ADMA overburden by itself has no major effect on cerebrovascular function under healthy conditions, but it enhances cerebrovascular dysfunction under EAE disease conditions and thus worsening the disease.
The eNOS‐produced ONOOˉ (nitrotyrosine) is reported to participate in the disruption of endothelial actin dynamics (F‐actin stress fibre formation) and cerebrovascular dysfunction under EAE conditions [11]. Recently, we also described the role of ADMA in brain endothelial barrier dysfunction using in vitro human brain microvessel endothelial cell (hBMVEC) culture model [11]. In hBMVECs, ADMA by itself did not induce the endothelial F‐actin stress fibre formation and the barrier disruption. However, ADMA treatment in the presence of VEGF induced the endothelial F‐actin stress fibre formation and the barrier disruption by inducing eNOS activity for the production of ONOOˉ [11]. In EAE mice, blood VEGF levels began to elevate before the disease onset (day 7 post‐immunization) and blood ADMA levels began to increase on the day of the disease onset (day 11 post‐immunization) (Figure 1A), thereby suggesting a potential role of ADMA and VEGF interaction in BBB dysfunction leading to CNS infiltration of immune cells causing CNS disease of EAE. Moreover, these data also explain why ADMA had no obvious effect on BBB integrity in healthy mice (Figure 4C) having low levels of blood VEGF. It should be of interest to investigate the potential role of VEGF as a second vascular stressor in ADMA‐mediated EAE pathogenesis.
In the body, ADMA can be excreted through the renal system but the majority of ADMA (>80%) is known to be cleared by its enzymatic degradation by DDAH in the liver and kidney [34, 35, 36]. DDAH activity is reported to be inhibited by oxidative stress, such as reactive oxygen species (ROS) and/or HCy [47, 48]. It is of interest to note that MS patients have elevated plasma levels of HCy (hyperhomocysteinaemia) [23, 24, 25, 26, 27] and meta‐analysis reported hyperhomocysteinaemia as a strong and modifiable risk factor for MS [49]. Accordingly, we observed increased blood levels of HCy (Figure 1C) and decreased DDAH activities in the kidney and liver in EAE animals (Figure 1D). These studies suggest that hyperhomocysteinaemia plays a role in the elevation of blood ADMA levels. At present, the mechanism underlying the induction of hyperhomocysteinaemia under MS and EAE conditions is not fully understood. HCy is produced during the methylation process, in which S‐adenosylmethionine (SAM), a methyl donor, is converted to S‐adenosylhomocysteine (SAH) which is hydrolysed to HCy by SAH hydrolase (AdoHcyase) [50]. Expression of AdoHcyase is reported to be elevated by IFN‐γ [51], thus suggesting that TH1‐mediated immune responses potentially participate in the induction of AdoHcyase expression and thus increased production of HCy and ADMA during the course of EAE/MS. In support, pharmacological inhibitors of AdoHcyase were reported to inhibit ADMA accumulation [52] as well as EAE disease [53]. Therefore, these studies suggest a role for the HCy‐DDAH‐ADMA axis in the development and progression of EAE/MS pathogenesis.
Taken together, these data, for the first time, document the ADMA‐mediated mechanisms in vascular (BBB dysfunction) and immune (induction of effector functions of TH1 and TH17 cells) dysfunctions and thus the clinical diseases of EAE and MS and secondly HCy‐DDAH‐ADMA axis as a potential target for vascular and immune pathogenesis of EAE and MS.
CONFLICT OF INTERESTS
Authors declare no competing interests.
AUTHOR CONTRIBUTIONS
I.S. designed the study, analysed data and co‐wrote the manuscript. J.K. performed experiments and edited the manuscript. N.S. performed experiments and analysed data. S.C., S.M.T.I., A.K.S and M.K. performed the experiments. J.W. designed the study, conducted experiments, analysed data and co‐wrote the manuscript.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Singh I, Kim J, Saxena N, Choi S, Islam SMT, Singh AK, et al. Vascular and immunopathological role of Asymmetric Dimethylarginine (ADMA) in Experimental Autoimmune Encephalomyelitis. Immunology. 2021;164:602–616. 10.1111/imm.13396
Funding information
This work was supported in part by the U.S. Department of Veterans Affairs (BX002829) and the National Institutes of Health (NS037766).
Contributor Information
Inderjit Singh, Email: singhi@musc.edu.
Jeseong Won, Email: wonj@musc.edu.
REFERENCES
- 1. Gaston BM, Carver J, Doctor A, Palmer LA. S‐nitrosylation signaling in cell biology. Mol Interv. 2003;3:253–63. [DOI] [PubMed] [Google Scholar]
- 2. Witherick J, Wilkins A, Scolding N, Kemp K. Mechanisms of oxidative damage in multiple sclerosis and a cell therapy approach to treatment. Autoimmune Dis. 2010;2011:164608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haldar SM, Stamler JS. S‐nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest. 2013;123:101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Won JS, Kim J, Annamalai B, Shunmugavel A, Singh I, Singh AK. Protective role of S‐nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J Alzheimers Dis. 2013;34:621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prasad R, Giri S, Nath N, Singh I, Singh AK. GSNO attenuates EAE disease by S‐nitrosylation‐mediated modulation of endothelial‐monocyte interactions. Glia. 2007;55:65–77. [DOI] [PubMed] [Google Scholar]
- 6. Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, et al. S‐Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab. 2005;25:177–92. [DOI] [PubMed] [Google Scholar]
- 7. Kim J, Won JS, Singh AK, Sharma AK, Singh I. STAT3 regulation by S‐nitrosylation: implication for inflammatory disease. Antioxid Redox Signal. 2014;20:2514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nath N, Morinaga O, Singh I. S‐nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol. 2010;5:240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saxena N, Won J, Choi S, Singh AK, Singh I. S‐nitrosoglutathione reductase (GSNOR) inhibitor as an immune modulator in experimental autoimmune encephalomyelitis. Free Radic Biol Med. 2018;121:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh I, Nath N, Saxena N, Singh AK, Won JS. Regulation of IL‐10 and IL‐17 mediated experimental autoimmune encephalomyelitis by S‐nitrosoglutathione. Immunobiology. 2018;223:549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choi S, Saxena N, Dhammu T, Khan M, Singh AK, Singh I, et al. Regulation of endothelial barrier integrity by redox‐dependent nitric oxide signaling: Implication in traumatic and inflammatory brain injuries. Nitric Oxide. 2019;83:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo S, Lei H, Qin H, Xia Y. Molecular mechanisms of endothelial NO synthase uncoupling. Curr Pharm Des. 2014;20:3548–53. [DOI] [PubMed] [Google Scholar]
- 13. Sibal L, Agarwal SC, Home PD, Boger RH. The Role of Asymmetric Dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr Cardiol Rev. 2010;6:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forstermann U, Schmidt HH, Pollock JS, Sheng H, Mitchell JA, Warner TD, et al. Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochem Pharmacol. 1991;42:1849–57. [DOI] [PubMed] [Google Scholar]
- 15. Visser M, Paulus WJ, Vermeulen MA, Richir MC, Davids M, Wisselink W, et al. The role of asymmetric dimethylarginine and arginine in the failing heart and its vasculature. Eur J Heart Fail. 2010;12:1274–81. [DOI] [PubMed] [Google Scholar]
- 16. Haghikia A, Kayacelebi AA, Beckmann B, Hanff E, Gold R, Haghikia A, et al. Serum and cerebrospinal fluid concentrations of homoarginine, arginine, asymmetric and symmetric dimethylarginine, nitrite and nitrate in patients with multiple sclerosis and neuromyelitis optica. Amino Acids. 2015;47:1837–45. [DOI] [PubMed] [Google Scholar]
- 17. Roshanisefat H, Bahmanyar S, Hillert J, Olsson T, Montgomery S. Multiple sclerosis clinical course and cardiovascular disease risk ‐ Swedish cohort study. Eur J Neurol. 2014;21:1353–e88. [DOI] [PubMed] [Google Scholar]
- 18. Jadidi E, Mohammadi M, Moradi T. High risk of cardiovascular diseases after diagnosis of multiple sclerosis. Mult Scler. 2013;19:1336–40. [DOI] [PubMed] [Google Scholar]
- 19. Murray‐Rust J, Leiper J, McAlister M, Phelan J, Tilley S, Santa Maria J, et al. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat Struct Biol. 2001;8:679–83. [DOI] [PubMed] [Google Scholar]
- 20. Stuhlinger MC, Stanger O. Asymmetric dimethyl‐L‐arginine (ADMA): a possible link between homocyst(e)ine and endothelial dysfunction. Curr Drug Metab. 2005;6:3–14. [DOI] [PubMed] [Google Scholar]
- 21. Hong L, Fast W. Inhibition of human dimethylarginine dimethylaminohydrolase‐1 by S‐nitroso‐L‐homocysteine and hydrogen peroxide. Analysis, quantification, and implications for hyperhomocysteinemia. J Biol Chem. 2007;282:34684–92. [DOI] [PubMed] [Google Scholar]
- 22. Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104:2569–75. [DOI] [PubMed] [Google Scholar]
- 23. Zhu Y, He ZY, Liu HN. Meta‐analysis of the relationship between homocysteine, vitamin B(1)(2), folate, and multiple sclerosis. J Clin Neurosci. 2011;18:933–8. [DOI] [PubMed] [Google Scholar]
- 24. Moghaddasi M, Mamarabadi M, Mohebi N, Razjouyan H, Aghaei M. Homocysteine, vitamin B12 and folate levels in Iranian patients with Multiple Sclerosis: a case control study. Clin Neurol Neurosurg. 2013;115:1802–5. [DOI] [PubMed] [Google Scholar]
- 25. Ramsaransing GS, Fokkema MR, Teelken A, Arutjunyan AV, Koch M, De Keyser J. Plasma homocysteine levels in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2006;77:189–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vrethem M, Mattsson E, Hebelka H, Leerbeck K, Osterberg A, Landtblom AM, et al. Increased plasma homocysteine levels without signs of vitamin B12 deficiency in patients with multiple sclerosis assessed by blood and cerebrospinal fluid homocysteine and methylmalonic acid. Mult Scler. 2003;9:239–45. [DOI] [PubMed] [Google Scholar]
- 27. Sahin S, Aksungar FB, Topkaya AE, Yildiz Z, Boru UT, Ayalp S, et al. Increased plasma homocysteine levels in multiple sclerosis. Mult Scler. 2007;13:945–6. [DOI] [PubMed] [Google Scholar]
- 28. Tain YL, Baylis C. Determination of dimethylarginine dimethylaminohydrolase activity in the kidney. Kidney Int. 2007;72:886–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004;172:1273–86. [DOI] [PubMed] [Google Scholar]
- 30. Singh I, Samuvel DJ, Choi S, Saxena N, Singh AK, Won J. Combination therapy of lovastatin and AMP‐activated protein kinase activator improves mitochondrial and peroxisomal functions and clinical disease in experimental autoimmune encephalomyelitis model. Immunology. 2018;154:434–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, et al. Administration of S‐nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khan M, Dhammu TS, Matsuda F, Annamalai B, Dhindsa TS, Singh I, et al. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2016;1630:159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi S, Singh I, Singh AK, Khan M, Won J. Asymmetric dimethylarginine exacerbates cognitive dysfunction associated with cerebrovascular pathology. FASEB J. 2020;34:6808–6823. [DOI] [PubMed] [Google Scholar]
- 34. Siroen MP, van der Sijp JR, Teerlink T, van Schaik C, Nijveldt RJ, van Leeuwen PA. The human liver clears both asymmetric and symmetric dimethylarginine. Hepatology. 2005;41:559–65. [DOI] [PubMed] [Google Scholar]
- 35. Nijveldt RJ, Teerlink T, Siroen MP, van Lambalgen AA, Rauwerda JA, van Leeuwen PA. The liver is an important organ in the metabolism of asymmetrical dimethylarginine (ADMA). Clin Nutr. 2003;22:17–22. [DOI] [PubMed] [Google Scholar]
- 36. Palm F, Onozato ML, Luo Z, Wilcox CS. Dimethylarginine dimethylaminohydrolase (DDAH): expression, regulation, and function in the cardiovascular and renal systems. Am J Physiol Heart Circ Physiol. 2007;293:H3227–45. [DOI] [PubMed] [Google Scholar]
- 37. Hamilton AM, Forkert ND, Yang R, Wu Y, Rogers JA, Yong VW, et al. Central nervous system targeted autoimmunity causes regional atrophy: a 9.4T MRI study of the EAE mouse model of Multiple Sclerosis. Sci Rep. 2019;9:8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liuzzi GM, Trojano M, Fanelli M, Avolio C, Fasano A, Livrea P, et al. Intrathecal synthesis of matrix metalloproteinase‐9 in patients with multiple sclerosis: implication for pathogenesis. Mult Scler. 2002;8:222–8. [DOI] [PubMed] [Google Scholar]
- 39. Opdenakker G, Van Damme J. Probing cytokines, chemokines and matrix metalloproteinases towards better immunotherapies of multiple sclerosis. Cytokine Growth Factor Rev. 2011;22:359–65. [DOI] [PubMed] [Google Scholar]
- 40. Zlokovic BV. The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. [DOI] [PubMed] [Google Scholar]
- 41. Errede M, Girolamo F, Ferrara G, Strippoli M, Morando S, Boldrin V, et al. Blood‐brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2012;71:840–54. [DOI] [PubMed] [Google Scholar]
- 42. Boroujerdi A, Welser‐Alves JV, Milner R. Matrix metalloproteinase‐9 mediates post‐hypoxic vascular pruning of cerebral blood vessels by degrading laminin and claudin‐5. Angiogenesis. 2015;18:255–64. [DOI] [PubMed] [Google Scholar]
- 43. Sonar SA, Lal G. Differentiation and transmigration of CD4 T cells in neuroinflammation and autoimmunity. Front Immunol. 2017;8:1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Linthicum DS, Munoz JJ, Blaskett A. Acute experimental autoimmune encephalomyelitis in mice. I. Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system. Cell Immunol. 1982;73:299–310. [DOI] [PubMed] [Google Scholar]
- 46. Hong Y, Tang HR, Ma M, Chen N, Xie X, He L. Multiple sclerosis and stroke: a systematic review and meta‐analysis. BMC Neurol. 2019;19:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fiedler LR, Wojciak‐Stothard B. The DDAH/ADMA pathway in the control of endothelial cell migration and angiogenesis. Biochem Soc Trans. 2009;37:1243–7. [DOI] [PubMed] [Google Scholar]
- 48. Cooke JP, Ghebremariam YT. DDAH says NO to ADMA. Arterioscler Thromb Vasc Biol. 2011;31:1462–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu Y, Liu HN, Zhang CD, Liang HY. [Meta‐analysis on relationship between hyperhomocysteinemia and multiple sclerosis]. Zhonghua Yi Xue Za Zhi. 2009;89:3055–7. [PubMed] [Google Scholar]
- 50. Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther. 2007;113:50–87. [DOI] [PubMed] [Google Scholar]
- 51. Kominsky DJ, Keely S, MacManus CF, Glover LE, Scully M, Collins CB, et al. An endogenously anti‐inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol. 2011;186:6505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boger RH, Sydow K, Borlak J, Thum T, Lenzen H, Schubert B, et al. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S‐adenosylmethionine‐dependent methyltransferases. Circ Res. 2000;87:99–105. [DOI] [PubMed] [Google Scholar]
- 53. Fu YF, Zhu YN, Ni J, Zhong XG, Tang W, Re YD, et al. A reversible S‐adenosyl‐L‐homocysteine hydrolase inhibitor ameliorates experimental autoimmune encephalomyelitis by inhibiting T cell activation. J Pharmacol Exp Ther. 2006;319:799–808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4