

Abstract
Loss of the B2M gene is associated with tumour immune escape and resistance to immunotherapy. However, genetic alterations of the B2M gene are rare. We performed an integrative analysis of the mutational and transcriptional profiles of large cohorts of non‐small‐cell lung cancer (NSCLC) patients and found that epigenetic downregulation of B2M is common. B2M‐low tumours exhibit a suppressive immune microenvironment characterized by reduced infiltration of immune cells of various lineages; in B2M‐high tumours, more T and natural killer cells are present, but their activities are constrained by immune checkpoint molecules, indicating the diverse mechanisms of immune evasion. High levels of B2M mRNA, but not PD‐L1, are correlated with an enhanced response to PD‐1‐based immunotherapy, suggesting its value for immunotherapy response prediction in solid tumours. Notably, a high tumour mutation burden (TMB) is associated with low B2M expression, which may explain the poor predictive value of the TMB in some situations. In syngeneic mouse models, genetic ablation of B2M in tumour cells causes resistance to PD‐1‐based immunotherapy, and B2M knockdown also diminishes the therapeutic efficacy. Moreover, forced expression of B2M in tumour models improves the response to immunotherapy, suggesting that B2M levels have significant impacts on treatment outcomes. Finally, we provide insight into the roles of transcription factors and KRAS mutations in B2M expression and the anticancer immune response. In conclusion, genetic and epigenetic regulation of B2M fundamentally shapes the NSCLC immune microenvironment and may determine the response to checkpoint blockade‐based immunotherapy.
Keywords: B2M, immunotherapy, lung cancer, transcriptional regulation, tumour microenvironment
Anti‐PD‐1 immunotherapy has significantly improved the survival of patients with lung cancer, but drug resistance greatly limited its long‐term benefits. We found that a defective B2M gene expression is the underlying mechanism of the resistance, and we proposed some potential strategies to improve the efficacy of anit‐PD‐1 immunotherapy and overcome the resistance.

Abbreviations
- APM
antigen processing machinery
- DEGs
differentially expressed genes
- FACS
fluorescence activated cell sorting
- GSEA
gene set enrichment analysis
- ICB
immune checkpoint blockade
- LUAD
lung adenocarcinomas
- MFIs
mean fluorescence intensities
- NSCLC
non‐small‐cell lung cancer
- TCRs
T‐cell receptors
- TFs
transcription factors
- TMB
tumor mutation burden
INTRODUCTION
The clinical use of immune checkpoint blockade (ICB), including PD‐1 and PD‐L1 antibodies, has significantly improved the overall survival of patients with advanced lung cancer [1, 2]. However, innate and acquired resistance to ICB treatment are common in lung cancer, and the mechanisms of resistance are not fully understood, which not only hinders the selection of patients but also limits the long‐term benefits of treatments [3, 4, 5]. The critical role of the B2M gene in immunotherapy resistance was recognized when a genetic deletion of B2M was identified in a melanoma patient who acquired resistance to a PD‐1 inhibitor [6]. B2M, also known as β2‐microglobulin, is the β chain of MHC class I molecules and an essential component in MHC‐I antigen processing and presentation [7]. Peptides from neoantigens bind to MHC‐I molecules composed of MHC‐I α chains (HLA‐I) and B2M, and then, the complexes are presented on the cell surface and recognized by the T‐cell receptors (TCRs) of CD8+ T cells [8]. Therefore, peptide–MHC‐I complexes cannot be successfully formed and presented to stimulate T cells in the absence of B2M, leading to resistance to PD‐1‐based immunotherapy [6, 8].
B2M mutations were also identified in two lung cancer patients resistant to PD‐1 inhibitors [9]. However, overall, no recurrent mutations in genes involved in antigen processing and presentation, including B2M, were identified in the relatively small cohort of patients [9]. Lee et al. found that downregulation of MHC‐I, rather than complete loss of MHC‐I molecule expression, was closely associated with resistance to PD‐1 inhibitors in melanoma patients [10]. In a cohort of lung cancer patients treated with PD‐1 or PD‐L1 antibodies, the antigen processing machinery (APM) scores, which are generated by utilizing the overall expression of eight genes, including B2M, CALR and PSME1, were significantly higher in responders than in nonresponders [11]. These results suggest that epigenetic downregulation of genes related to antigen processing and presentation may lead to innate and acquired resistance to lung cancer immunotherapy.
B2M is required for the formation of MHC‐I complexes comprised of all types of HLA‐I molecules [12], so it might be the limiting factor that determines the density of the peptide–MHC‐I complexes on the cell membrane, which determines the strength of the so‐called ‘first signalling’ for the activation of CD8+ T cells [13, 14]. In addition, peptide–MHC‐I complexes on the cell membrane of tumour cells are targets for CD8+ cytotoxic T cells (CTLs). Therefore, it is not surprising that tumour clones with B2M mutations have a survival advantage under the pressure of CTL attack. Since B2M is critical for anticancer immunity and genetic mutations in B2M are not commonly seen in lung cancers, we reasoned that the epigenetic downregulation of B2M may play a role in lung tumorigenesis and immunotherapy resistance. In the present study, we found that B2M expression is frequently suppressed in lung cancers. Our study suggests that genetic and epigenetic regulation of B2M fundamentally shapes the immune landscape of lung adenocarcinomas (LUADs) and determines the response to immunotherapy.
MATERIALS AND METHODS
Bioinformatics analysis
Data sources and genetic alteration analysis
The publicly available LUAD data set was downloaded from The Cancer Genome Atlas (TCGA) Data Portal and corrected by the R package ‘limma’. The LUAD dataset GSE116959 was downloaded from the NCBI Gene Expression Omnibus (GEO) database, and the LUAD data set from the International Cancer Genome Consortium (ICGC) database was downloaded from the University of California, Santa Cruz (UCSC) Xena platform. Protein expression data were downloaded from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database. B2M copy number variation (CNV) and mutation were analysed by the cBioPortal tool. The tumour mutation burden (TMB) score of a tumour sample was calculated as follows: the total number of mutations/human genome size 38 MB.
Gene expression and survival analysis
The analysis of differentially expressed genes (DEGs) between tumour and normal tissues in the abovementioned data sets was conducted by using the ‘Wilcox_test’ function in R, and DEGs with a p‐value < 0·05 were considered significant. Gene set enrichment analysis (GSEA) of DEGs was conducted with GSEA 4.0 software. The R package ‘clusterProfiler’ was used to conduct Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) enrichment analysis. Gene sets with nominal p ≤ 0·05 and false discovery rate ≤0·25 were considered significantly enriched. The R package ‘ggplot2’ was used to draw plots. Survival analysis was conducted with the R packages ‘survival’ and ‘survminer’. Survival time was compared by using Kaplan–Meier curves, and a log‐rank p‐value <0·05 was considered to indicate a significant difference.
Tumour microenvironment analysis
The tumour microenvironment score was calculated by the R package ‘estimate’. The R packages ‘GSVA’ and ‘GSEABase’ were used to perform single sample gene set enrichment analysis (ssGSEA) [15, 16]. Samples were divided into two groups based on the median B2M expression, and the R code provided by CIBERSORT was used to estimate the infiltration of various immune cells [17]. The immune cell infiltration between groups was compared with the ‘Wilcox_test’ function in R. The Tumor IMmune Estimation Resource (TIMER) tool was used to estimate the relationship between the levels of chemokine expression and various immune cells [18].
Transcription factor analysis
The correlation between transcription factors (TFs) and B2M was analysed by a correlation test, and the Spearman correlation coefficient was used to indicate a correlation. A p‐value <0·05 was considered statistically significant. TF binding sites on targeted genes were predicted using the ConTra V3 server [19] and the Cistrome tool [20].
Cell lines and cell culture
The human lung cancer cell lines H358, H23 and H1975 and the mouse colorectal cancer cell lines MC38 and CT26 were obtained from the American Type Tissue Culture Collection (ATCC). The human normal lung epithelial cell line BEAS‐2B and the human lymphoma cell line NK92 were purchased from FuHeng Biology. NK‐92 cells were cultured in α‐minimum essential medium supplemented with 2 mM l‐glutamine, 1·5 g/l sodium bicarbonate, 0·2 mM inositol, 0·1 mM 2‐mercaptoethanol, 0·02 mM folic acid, 100–200 U/ml recombinant IL‐2, 12·5% horse serum, 12·5% foetal bovine serum (FBS) and 100–200 U/ml recombinant IL‐2. H358, H23, H1975 and CT26 cells were cultured in RPMI‐1640, and MC38 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were grown at 37°C in an incubator with 5% CO2. The cells passed mycoplasma testing on a regular basis, and their identity was regularly confirmed by SNP testing.
Animals and in vivo studies
C57BL/6, BALB/c and Balb/c‐nu mice at 6–8 weeks of age were purchased from the laboratory animal center of Nantong University. All mice were bred and housed in specific pathogen‐free facilities at the laboratory animal center of Nantong University. MC38 or CT26 cells (3 × 105) were subcutaneously injected into C57BL/6 or BALB/c mice, respectively, and when the tumour volume reached ~100 mm3, the mice were treated with phosphate‐buffered saline (PBS) or anti‐PD‐L1 every 3 days 3–4 times via intraperitoneal injection. Tumours were measured every 3 days by callipers, and tumour volume was calculated as (length×width2)/2. For the xenograft assay, Balb/c‐nu mice were injected subcutaneously with 5 × 106 H358 cells. When the tumours reached ~100 mm3 in size, the mice were treated with AMG510 by oral garage (10 mg/kg, daily). These studies were performed in compliance with an approved protocol and the institutional guidelines of the Ethical Committee of Nantong University.
Antibodies and chemicals
The source and identifier of antibodies used are as follows: antibodies against human STAT5A, p‐ERK1/2, ERK1/2, PD‐L1 and CD8A were purchased from Abcam; human anti‐pSTAT5A antibody was purchased from CST; antibodies against human GAPDH, B2M and β‐catenin were purchased from Proteintech. For fluorescence‐activated cell sorting (FACS) analysis, antibodies against PE‐Cy7‐CD3, PE‐CD8, FITC‐CD4 and APC‐Cy7‐CD45 were purchased from Thermo Fisher; antibodies against PerCP‐B2M were purchased from BD Bioscience; and antibodies against APC‐mouse H‐2Kb/2Db and APC‐mouse B2M were purchased from Biolegend. The STAT5A inhibitor SH454, KRAS inhibitor ARS1620 and FGFR1 inhibitor AZD4547 were purchased from Selleck. The KRAS inhibitor AMG510 was purchased from Abmole. The MEK inhibitor PD0325901 was purchased from MCE.
Plasmids and virus production
The lentiviral vector pLVX‐IRES‐Puro‐mB2M was used to overexpress mouse B2M. The CRISPR/Cas9 technique was used to generate B2M knockout mouse cell lines using the pLentiCRISPR‐GFP‐v2 vector as previously described [21]. The selected B2M single‐guide RNA (sgRNA) sequence was 5′‐CTGGTGCTTGTCTCACTGAC‐3′, and the control nontargeting sgRNA sequence was 5′‐GCTTTCACGGAGGTTCGACG‐3′. The pSLenti‐U6‐mB2M‐shRNA‐EGFP‐F2A‐Puro‐WPRE vector was used to knockdown mouse B2M. The selected B2M‐shRNA sequence for in vivo experiments was 5′‐ACGGTGATTCTAATCATCTTAA‐3′, and the control nontargeting shRNA sequence was 5′‐CACAGGTTGGTGGTGCAAGTGA‐3′. Human KRASG12V was introduced by the pLenti‐CMV‐KrasG12V‐Hygro lentiviral vector. All plasmids were generated by modifying the vector backbones in the laboratory of the Department of Immunology, School of Medicine, Nantong University. The lentivirus was prepared using HEK293T cells as previously described [21].
Western blotting
Western blotting was performed using whole‐cell lysates. Briefly, aliquots of total protein (20–50 μg/lane) were electrophoresed on 10% SDS‐polyacrylamide gradient gels and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were incubated at 4°C overnight with primary antibodies against p‐ERK, ERK, p‐STAT5A, STAT5A, B2M, GAPDH or β‐catenin. After being rinsed in wash buffer, the membranes were incubated with a horseradish peroxidase‐conjugated secondary antibody diluted at 1:10,000, and the signal was visualized with SuperSignal West Dura reagents (Thermo Fisher).
Real‐time PCR
RNA was extracted using the RNeasy Mini Kit (Qiagen), and cDNA was synthesized using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher). Real‐time PCR was performed with the ABI StepOnePlus system (Thermo Fisher) and iTaq Universal SYBR Green Supermix (Bio‐Rad). For data analysis, the method was used to calculate the fold changes. GAPDH expression was considered to be unaffected under our treatment conditions and was used as a reference gene. The primer sequences used for real‐time PCR were as follows (5′‐3′): B2M, forward, AAGCAGCATCATGGAGGTTTG; reverse, GAGCTACCTGTGGAGCAACC. GAPDH, forward, GAAGGTGAAGGTCGGAGTC; reverse, GAAGATGGTGATGGGATTTC. Each experiment was run in triplicate, and the error bars represent the range of the fold changes calculated from three or four independent experiments.
Flow cytometry analysis by FACS
The cultured cells were harvested and washed, and a single‐cell suspension was prepared in flow cytometry staining buffer (PBS supplemented with 2% FBS and 0·1% sodium azide). In some in vivo experiments, tumours were collected from euthanized mice and dissociated into a single‐cell suspension by using the GentleMAX™ Dissociator (Miltenyi Biotec). Primary antibodies with the appropriate fluorescent labels were added to 106 cells resuspended in 100 μl of staining buffer and then incubated at 4°C for 30 min. After staining, the cells were washed with staining buffer three times and analysed with an ACEA NovoCyte flow cytometer (ACEA Biosciences).
Immunohistochemistry (IHC)
Serial 5‐μm sections were cut from the tissue blocks, deparaffinized in xylene and hydrated in a graded series of alcohol. The slides were then immersed in citrate unmasking solution (10×) (CST) in a pressure cooker for 10 min to retrieve the antigen. After inactivating endogenous peroxidase activity with 3% H2O2, the slides were incubated with primary antibodies against B2M, PD‐L1 or CD8A at a 1:100 dilution overnight at 4°C in a humidified chamber. For detection, the slides were treated with SignalStain Boost Detection reagents from CST according to the manufacturer’s instructions, stained with 3,3'‐diaminobenzidine (DAB) for 3–5 min and counterstained with haematoxylin for 5–15 s. Finally, the slides were dehydrated and mounted. All pictures were obtained using a Zeiss microscope (Observer Z1). The scores for B2M and PD‐L1 expression in LUAD samples were obtained with an IHC profiler [22]. The staining intensities of B2M or PD‐L1 (0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining) for 5 randomly selected high‐power fields (20×) in each tumour sample were evaluated, and the final scores were calculated by summing the 5 scores of each sample and then were categorized into three groups: Low (<5), Middle (between 5 and 10) and High (>10). CD8+ T cell numbers were obtained by averaging the numbers of CD8A‐positive stained cells in 5 randomly selected 20× fields in each LUAD sample.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 7 software (GraphPad Software). In general, values were plotted as the mean ± standard deviation (SD). The means between independent groups were compared by Student’s t‐test (2 groups) or Kruskal‐Wallis one‐way ANOVA (3 or more groups) with pairwise comparisons, and differences with p < 0·05 were considered statistically significant.
RESULTS
B2M expression is downregulated in lung cancer
Analysis of the TCGA database showed that 1·41% of LUADs harbour genetic alterations in the B2M gene, among which only deep deletion significantly reduced the levels of B2M transcription (Figure S1A,B). In addition, the overall survival times between patients with mutant or wild‐type B2M were not significantly different (Figure S1C), suggesting that genetic alteration of B2M is rare in resectable primary LUAD tumours and that these tumours evade immune attack mainly through other mechanisms. Analysis of LUAD transcriptome data (RNA‐seq results) revealed that B2M expression was significantly reduced in tumour tissues compared with adjacent normal tissues (Figure 1a,b). Similar results were obtained from the other two datasets from the GEO (GSE116959) and ICGC databases (Figure 1c,d). Further analysis of the CPTAC database indicated that B2M protein levels were lower in LUAD tissues than in normal lung tissues (Figure 1e). We then performed IHC staining for B2M in surgically resected LUAD tissues and adjacent normal tissues, and the results demonstrated that tumours exhibited lower B2M expression levels than normal tissues that universally express B2M at medium or high levels (Figure 1f,g). These results suggest that B2M is downregulated in LUAD primarily through epigenetic mechanisms.
FIGURE 1.
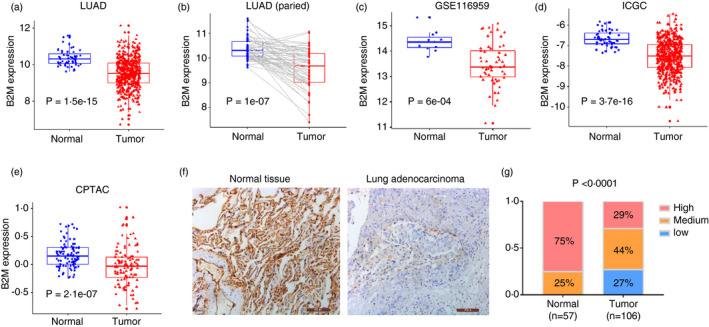
B2M downregulation in LUAD. B2M expression in LUAD tissues (a) or their paired adjacent normal lung tissues (b). (c–d) B2M expression in LUAD tissues compared with adjacent normal tissues in GSE116959 and ICGC data sets. (e) Protein levels of B2M in LUAD tissues compared with adjacent normal tissues in the CPTAC data set. (f) Representative images of IHC staining for B2M in LUAD tissues and adjacent normal lung tissues. (g) Quantification of the results in Figure (f)
The association between B2M expression and the immune landscape of lung cancer
The LUAD tumours were classified into either B2M ‐high tumours (B2Mhi) or B2M ‐low (B2Mlo) tumours, and DEGs between the two groups were identified and analysed with the GSEA program. As shown in Figure 2a, among the ten most enriched pathways, several immune‐related pathways, including the antigen processing and presentation pathway, were upregulated in B2Mhi tumours. Then, GO analysis of enriched biological process (BP), molecular function (MF), and cellular component (CC) terms and KEGG pathways was conducted with 255 immune‐related DEGs between B2Mhi and B2Mlo tumours. As shown in Figure S2A, functions related to T and natural killer (NK) cells were highly enriched, suggesting that B2M levels are closely associated with immune infiltration. Consistently, B2Mhi tumours exhibited higher stromal scores and immune scores according to the CIBERSORT program [17] (Figure 2b,c). Next, the LUAD tumours were divided into high, moderate and low immune infiltration groups (immunity‐H, immunity‐M and immunity‐L, respectively) by the ssGSEA algorithm [15, 16]. We found that B2M levels and immune infiltration levels were significantly different among the three groups, with the highest B2M levels seen in the immune‐H group and the lowest B2M levels seen in the immune‐L group (Figure 2d, Figure S2B).
FIGURE 2.
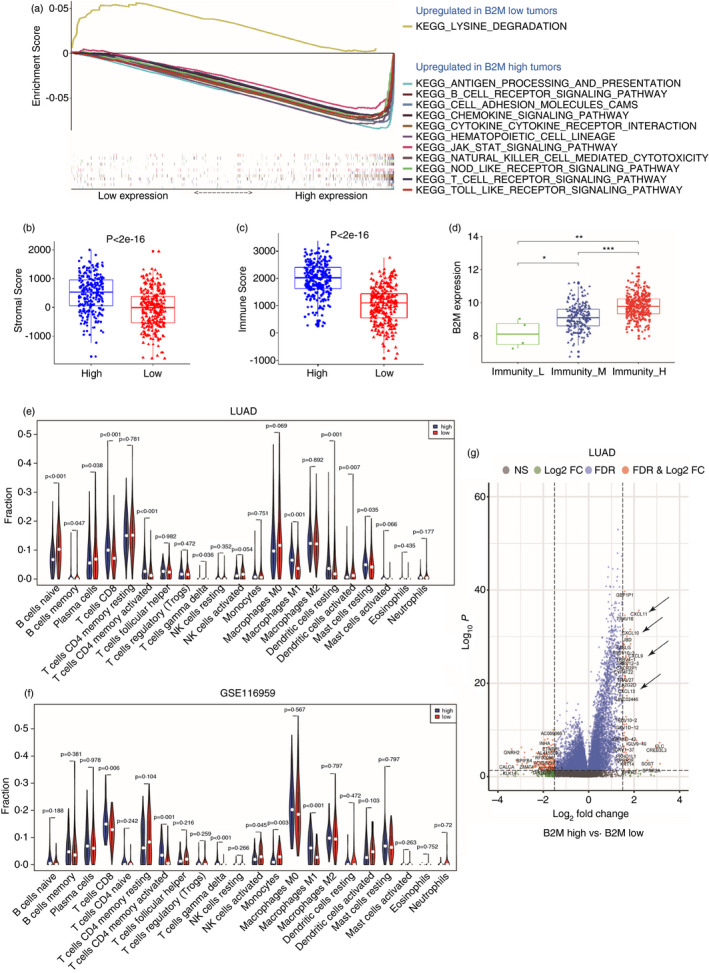
B2M levels affect the immune landscape of lung cancer. (a) GSEA of B2Mhi LUAD tumours compared with B2Mlo tumours. (b, c) Stromal scores and immune scores in B2Mhi LUAD tumours vs. B2Mlo tumours. (d) B2M expression in the LUAD tumours of immunity‐H, ‐M, or ‐L. (e,f) CIBERSORT analysis of the fractions of infiltrated immune cells in the LUAD and GSE116959 datasets. (g) Differentially expressed immune‐related genes in LUAD B2Mhi tumours vs. B2Mlo tumours
We further analysed the fractions of various immune cell types in the LUAD tumours and found that B2Mhi tumours contained more CD8+ T cells (Figure 2e) than B2Mlo tumours, and we obtained similar results in the GSE116959 data set (Figure 2f). In addition, both data sets showed that B2Mhi tumours contained more activated CD4+ T cells than B2Mlo tumours (Figure 2e). We reasoned that increased chemokines in B2Mhi tumours might drive enhanced T‐cell recruitment. Indeed, CCL5, CXCL9, CXCL10, CXCL11 and CXCL13 were upregulated in B2Mhi tumours and may play critical roles in recruiting T cells (Figure 2g and Figure S3) [23, 24, 25, 26].
B2M influences the infiltration and function of T and NK cells
Many TCR genes were highly expressed in B2Mhi tumours (Figure S4A), indicating increased infiltration of CD8+ T cells (Figure 2e,f); however, B2Mhi tumours also expressed higher levels of immune checkpoint molecules, including PDCD1 (PD‐1), LAYN and HAVCR2 (Figure S4C–E), all of which are reportedly related to exhausted T cells [27, 28]. Therefore, CD8+ T cells might be recruited and transiently activated, but their cytotoxic activity is suppressed by various mechanisms in the tumour microenvironment, including mechanisms involving those checkpoint molecules. This may also explain why the increased level of T cells in B2Mhi tumours did not translate into improved survival in lung cancer patients with this tumour type (Figure S4F).
In addition, compared to B2Mlo tumours, B2Mhi tumours have greater infiltration of NK cells, as indicated by the positive association between B2M and NKP46 or NKP30, two natural cytotoxicity receptors expressed explicitly by NK cells (Figure S5A,B) [29], which is consistent with the increased expression of NK cell‐recruiting chemokines, such as CCL5, in B2Mhi tumours (Figure S3) [30]. However, B2Mhi tumours also have higher expression levels of CD96 and TIGIT, two checkpoint molecules related to the inhibition of NK cell activity (Figure S5C,D) [31]. We also found a strong correlation between B2M and HLA‐I molecules, implying that they may share some common regulatory mechanisms (Figure S5E). Since HLA‐I expression on normal cells is thought to be critical for self‐tolerance [32], we reasoned that low B2M expression might stimulate NK cell cytotoxic activity in lung cancers. Indeed, the fraction of activated NK cells tended to increase in B2Mlo tumours in the LUAD (p = 0·054) and GSE116959 (p = 0·045) data sets (Figure 2e,f). Together, the results suggest that B2Mhi tumours recruit more NK cells, but the suppressive tumour microenvironment limits their antitumour activities. In B2Mlo tumours, even though NK cells seem to be activated locally due to low HLA‐I expression on tumour cells, their abundance is low. Therefore, through various mechanisms, lung cancers can evade NK cell‐mediated immune attack.
Value of B2M levels in predicting the response to immunotherapy
B2M is critical for tumour antigen presentation and cognate CD8+ T cell activation. We have shown that T‐cell infiltration is high in B2Mhi tumours, but immune checkpoint molecules, including PD‐L1, may suppress their cytotoxic function. We wondered whether B2M levels could predict the response to PD‐1 inhibition. Since no public RNA‐seq data on lung cancer immunotherapy are currently available, we analysed a cohort of melanoma patients treated with PD‐1 antibodies and found that pretreatment B2M levels are higher in patients with a complete response (CR) than in those with a partial response (PR) or progressive disease (PD) (Figure 3a) [33]. PD‐L1 mRNA levels were comparable among the three groups (Figure 3b), implying that B2M levels, but not PD‐L1 mRNA levels, might have predictive value in PD‐1‐based immunotherapy.
FIGURE 3.
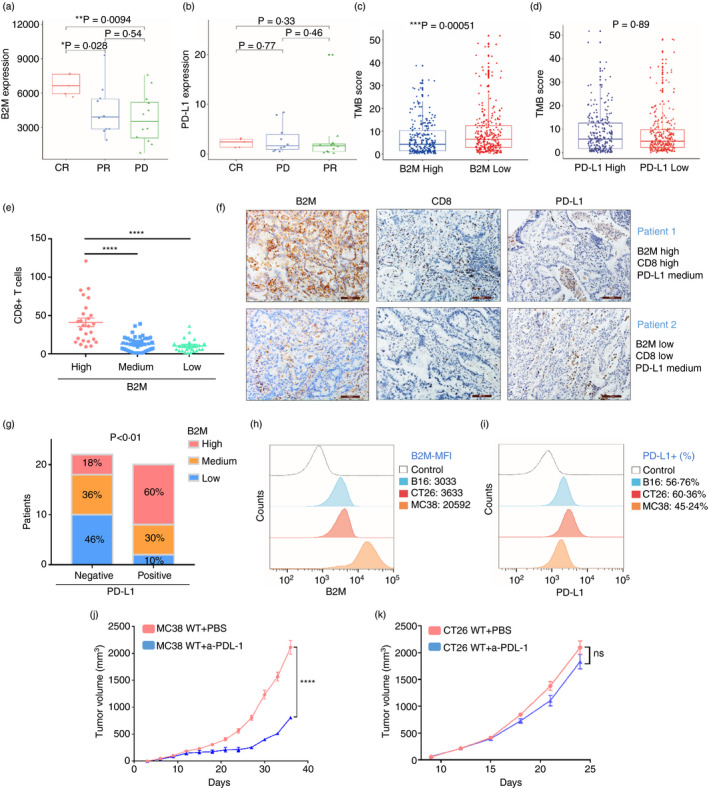
Value of B2M levels in predicting the response to immunotherapy. (a,b) Levels of B2M or PD‐L1mRNA in melanoma patients treated with PD‐1 inhibition. (c,d) TMB scores in the LUAD tumours divided by the levels of B2M or PD‐L1. E, The average numbers of CD8+ T cells in five random images of IHC staining for CD8A in LUAD tissues. (f) The stacked histogram shows the numbers of patients with different levels of B2M and PD‐L1 based on IHC staining. (g) Representative images of LUAD tumours from two patients. (h–i) FACS analysis of B2M and PD‐L1 expression in MC38, B16, and CT26 cells. (j–k) Tumour volumes of subcutaneously implanted MC38 or CT26 tumours treated with α‐PD‐L1 or PBS as a control (*p < 0·05, **p < 0·01, ***p < 0·001, ****p < 0·0001 vs. control)
Tumour mutation burden has been linked to the response to PD‐1 inhibition, as high TMB tumours likely produce more neoantigens [34]. Surprisingly, we found that TMB scores were lower in B2Mhi LUAD tumours than in B2Mlo LUAD tumours, whereas they were not different between tumours expressing high or low levels of PD‐L1 (Figure 3c,d), suggesting that lung tumours with a high TMB may downregulate B2M expression to escape T‐cell attack. We speculate that some lung tumours with high TMB and significant T‐cell infiltration could still fail to respond to PD‐1 blockade immunotherapy due to low B2M expression and insufficient neoantigen presentation.
Next, we performed IHC staining for PD‐L1, B2M and CD8A in LUAD specimens (n = 85) and found that tumours with high B2M expression contained more CD8+ T cells than tumours with moderate or low B2M expression. However, no significant difference was found between tumours with moderate and low levels of B2M, suggesting that high B2M expression might be a prerequisite for an optimal T‐cell response (Figure 3e). Figure 3f shows the representative IHC images from two patients; high T‐cell infiltration was found in tumours from patient 1 with high B2M expression but not in tumours from patient 2 with low B2M, although tumours from both patients expressed moderate levels of PD‐L1. Of patients with positive staining of PD‐L1 (≥1%) in tumour tissues, who might be considered suitable for PD‐1 inhibition in the clinic [35], ~40% expressed low to medium levels of B2M in tumours (Figure 3g). These patients may not benefit from the treatment due to defective CD8+ T‐cell recruitment and cytotoxic activity.
MC38 cells are murine colon carcinoma cells, and syngeneic mice subcutaneously transplanted with these cells are widely used in preclinical immunotherapy studies because they are sensitive to PD‐1 inhibition [36]. On the other hand, the CT26 murine colon carcinoma model [37, 38, 39] and the B16 murine melanoma model [40, 41, 42] are reportedly less sensitive to PD‐1 or PD‐L1 blockade. Interestingly, we found that MC38 cells expressed higher levels of B2M than the other two lines, as indicated by the mean fluorescence intensities of B2M expression in the FACS results (Figure 3h). In contrast, the PD‐L1 levels were comparable among the three cell lines (Figure 3i). We then established MC38 or CT26 tumour models and treated them with anti‐PD‐L1 antibody, and in line with previous studies, we found that the treatment effectively suppressed the growth of MC38 tumours (Figure 3j). However, in the CT26 models, anti‐PD‐L1 treatment only induced a partial response and did not inhibit tumour growth significantly compared to the PBS‐treated control (Figure 3k). Together, these results suggest that high B2M expression is associated with the response to PD‐1 inhibition.
Modulation of B2M expression can determine the outcome of immunotherapy
To directly assess the impact of B2M expression on the response to PD‐1 inhibition, we first checked whether the genetic loss of B2M causes resistance to immunotherapy. B2M deletion was achieved with the CRISPR technique by using a pLenti‐CRISPR‐GPF‐v2 vector as previously described [21]. FACS analysis demonstrated that transduction was very effective, as the cell populations infected by the lentivirus that expressed B2M ‐sgRNA or control sgRNA, referred to as MC38‐sg B2M or MC38‐sgNS cells, respectively, were mostly positive for GFP expression (~90%) (Figure 4a). As expected, ~70% of MC38‐sgB2M cells lost B2M and MHC class I (MHC‐I) expression (Figure 4a and Figure S6A).
FIGURE 4.
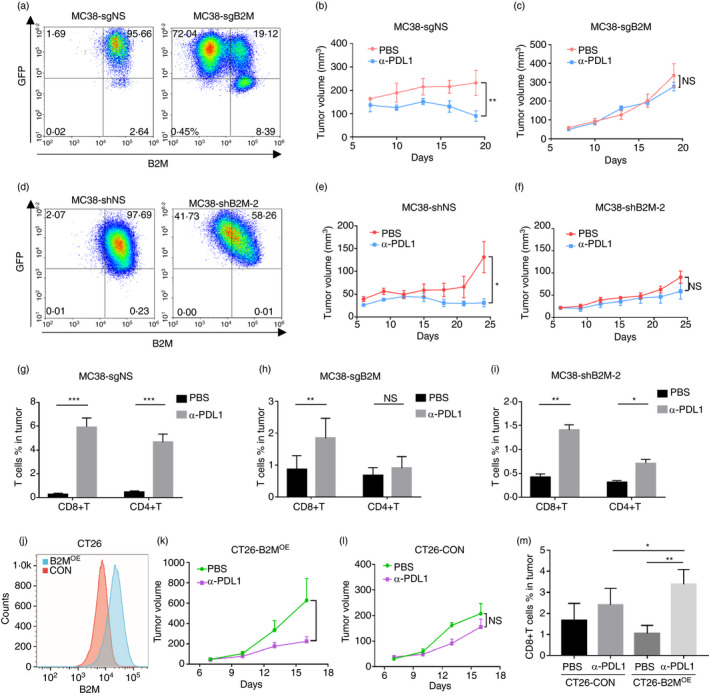
Modulation of B2M expression can determine the outcome of immunotherapy. (a) FACS analysis validated the loss of B2M expression on the cell surface in a proportion of cells infected with lentivirus carrying pLenti‐CRISPR‐mB2M‐sgRNA‐GFP (MC38‐sgB2M); cells infected with lentivirus carrying a nonspecific sgRNA (MC38‐sgNS) were used as a control. (b,c) Subcutaneously implanted MC38‐sgNS or MC38‐sgB2M tumours were treated with α‐PD‐L1 or PBS as a control. (d) FACS analysis validated B2M knockdown in MC38 cells (MC38‐shB2M) infected with lentivirus shRNA. (e,f) Mice bearing MC38‐shB2M or MC38‐shNS control tumours were treated with α‐PD‐L1 or PBS. (g–i) FACS analysis of T cells in single‐cell suspensions from MC38‐sgNS, MC38‐sgB2M, and MC38‐sgB2M‐2 tumours. (j) FACS analysis validated the overexpression of B2M in CT26 cells (B2MOE) compared to control cells (CON). (k–l) Subcutaneously implanted B2MOE or CON tumours were treated with anti‐PD‐L1 or PBS. (m) FACS analysis of T cells in B2MOE or CON tumours (*p < 0·05, **p < 0·01, ***p < 0·001, ****p < 0·0001 vs. control)
We reasoned that it is not necessary to use a pure B2M−/− population to test whether deletion of the B2M gene can cause resistance to anti‐PD‐L1 treatment; therefore, MC38‐sg B2M or MC38‐sgNS cells without sorting were subcutaneously injected into C57BL/6 mice, and when tumours reached approximately 100 mm3, the mice were treated by intraperitoneal injection of mouse anti‐PD‐L1 (α‐PD‐L1) as previously described [43]. While MC38‐sgNS tumours responded well to the treatment, MC38‐sgB2M tumours were resistant to the therapy (Figure 4b,c). These results suggest that intact B2M expression and MHC class I antigen presentation are required for the response to ICB treatment. Next, B2M knockdown MC38 cells (MC38‐shB2M) were generated by shRNA interference (Figure 4d and Figure S7A). Knocking down B2M suppressed MHC class I expression in MC38 cells (Figure S7B), and the MC38‐shB2M tumours grew similarly to the control MC38‐shNS tumours in the absence of treatment (Figure S7C). Importantly, treatment with α‐PD‐L1 induced the regression of MC38‐shNS tumours, but the effects diminished in MC38‐shB2M tumours (Figure 4e,f), indicating that B2M expression is critical for the optimal response to PD‐1 inhibition. We collected MC38‐sgNS tumours after treatment and prepared a single‐cell suspension for FACS analysis. The results demonstrated that the numbers of CD8+ and CD4+ T cells in the α‐PD‐L1‐treated tumours increased by ~10‐fold compared with those in the tumours treated with PBS (Figure 4g). In contrast, the increase in T‐cell infiltration upon treatment was much less significant in B2M knockout or knockdown tumours (Figure 4h,i).
Interestingly, although the implanted MC38‐sgB2M cells were a mixed population, in which ~70% were negative for B2M and MHC‐I expression (Figure 4a and Figure S6A), FACS analysis with the single‐cell suspension from MC38‐sgB2M tumours at the end of experiments indicated that most of the cells (~90%) became negative for both B2M and MHC‐I, regardless of treatment with PBS or anti‐PD‐L1, whereas the small population positive for MHC‐I predominantly consisted of immune cells because most of them expressed CD45 (Figure S6B). We also performed FACS analysis with a single‐cell suspension from MC38‐sgNS tumours harvested at the end of experiments treated with PBS or anti‐PD‐L1, and we found that they remained positive for MHC class I expression (Figure S6C). These results imply that the immune reaction in vivo can drive the enrichment of B2M‐negative tumour cells.
To test whether we can improve the efficacy of immunotherapy by upregulating B2M expression, we established tumours with CT26 cells overexpressing B2 M (CT26‐B2MOE) (Figure 4j). We found that α‐PD‐L1 treatment significantly suppressed the growth of CT26‐B2MOE cells (Figure 4k), whereas the control tumours (CT26‐CON) were resistant to the treatment (Figure 4l). FACS analysis demonstrated that α‐PD‐L1 treatment increased the number of CD8+ T cells in the CT26‐B2MOE tumours almost by ~5‐fold (Figure 4m), and after treatment, the B2MOE tumours exhibited more CD8+ T cells than control tumours (Figure 4m).
Regulatory mechanisms of B2M expression in lung cancer
To explore the regulatory mechanisms of B2M expression, we searched for TFs that are positively or negatively correlated with B2M expression in LUAD tumours (Figure 5a). IRF1 and STAT1 are two TFs known to mediate IFN‐γ‐induced B2M expression [44, 45, 46]. STAT5A was also positively associated with B2M expression in LUAD tumours, and analyses of several datasets consistently showed that LUAD tissues expressed lower levels of STAT5A than normal tissues (Figure S5F). Potential binding sites for STAT5A on the B2M promoter were also identified (Figure 5a), suggesting that it may directly regulate B2M gene expression. H1975 lung cancer cells highly expressed STAT5A and B2M, and treatment with SH454, a STAT5 inhibitor, suppressed B2M expression, as demonstrated by Western blotting and real‐time PCR (Figure 5b,c). IL‐2 activates STAT5, and the NK cell line NK92 is routinely cultured in media supplemented with IL‐2. As expected, NK92 cells exhibited high STAT5 and p‐STAT5 expression, and treatment with SH454 decreased p‐STAT5 and B2M expression (Figure 5d,e). Collectively, these results suggest that STAT5A may participate in the regulation of B2M in lung cancer cells.
FIGURE 5.
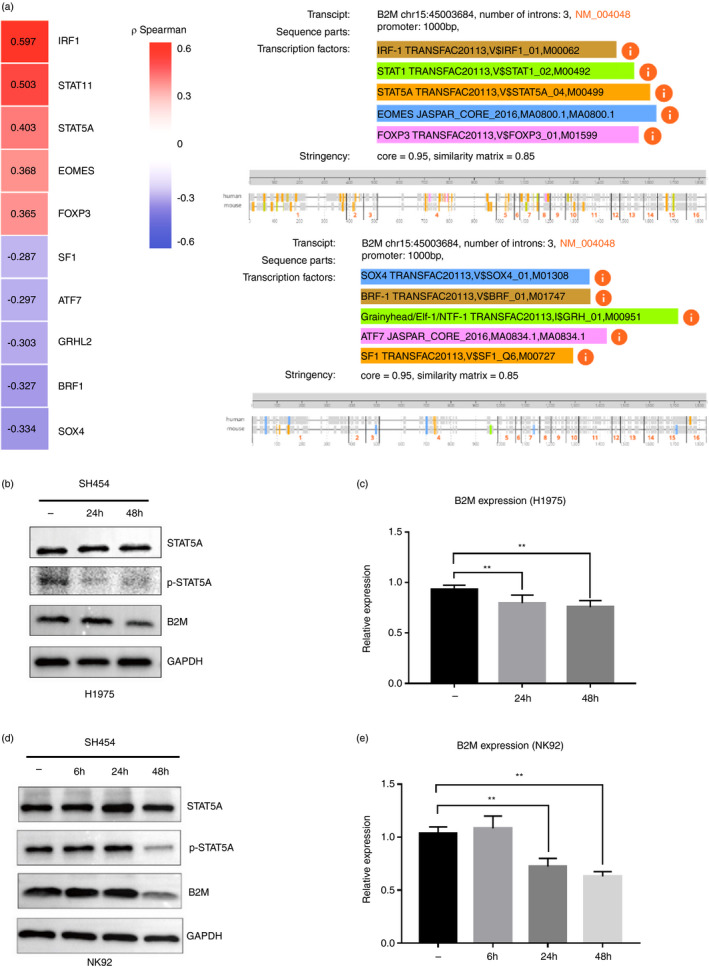
STAT5 may regulate B2M expression in lung cancer. (a) The TFs that are positively (red) or negatively (blue) correlated with B2M expression in the LUAD dataset (left panel) and their potential binding sites on the B2M promoter region (right panel). (b–e) H1975 lung cancer cells or NK92 cells were treated with the STAT5 inhibitor SH454 for 24 and 48 h, and the levels of STAT5, p‐STAT5, and B2M were assessed by Western blotting and real‐time PCR; GAPDH was used as an internal control (**p < 0·01 vs. control)
We also investigated whether B2M downregulation is associated with lung cancer‐related mutations. Among the top 10 frequently mutated genes in LUAD (Figure 6a), B2M exhibited lower expression in tumours harbouring mutations in TTN, MUC16, LRP18, KRAS and XIPR2 than in their wild‐type counterparts, with the most significant difference seen between KRAS‐mutant and KRAS wild‐type tumours (Figure 6b). H358 and H23 are human lung cancer cell lines that harbour the KRASG12C mutation, and treatment with ARS‐1620, a specific KRASG12C inhibitor, suppressed their growth (Figure 7A). We treated H358 cells with AMG510 (a KRASG12C inhibitor in clinical trials) for 24 h, and FACS analysis indicated increased B2M expression on the cell membranes of H358 and H23 cells (Figure 7b,c). Western blotting demonstrated that B2M levels increased while pERK levels decreased upon ARS‐1620 treatment in H358 cells (Figure 7d). Partial recovery of pERK expression was observed 48 h after KRASG12C inhibition (Figure 7d), which might result from feedback‐related upregulation of FGFR1 expression (unpublished data). We next treated H358 and H23 cells with AMG510 (Figure 7e,f), ARS1620 (Figure S8), ARS1620+AZD4547 (an FGFR1 inhibitor, Figure S8) or PD0325901 (a MEK inhibitor, Figure S8), and FACS analysis and real‐time PCR demonstrated that all treatments for 24 h enhanced the expression of B2M in both H358 and H23 cells.
FIGURE 6.
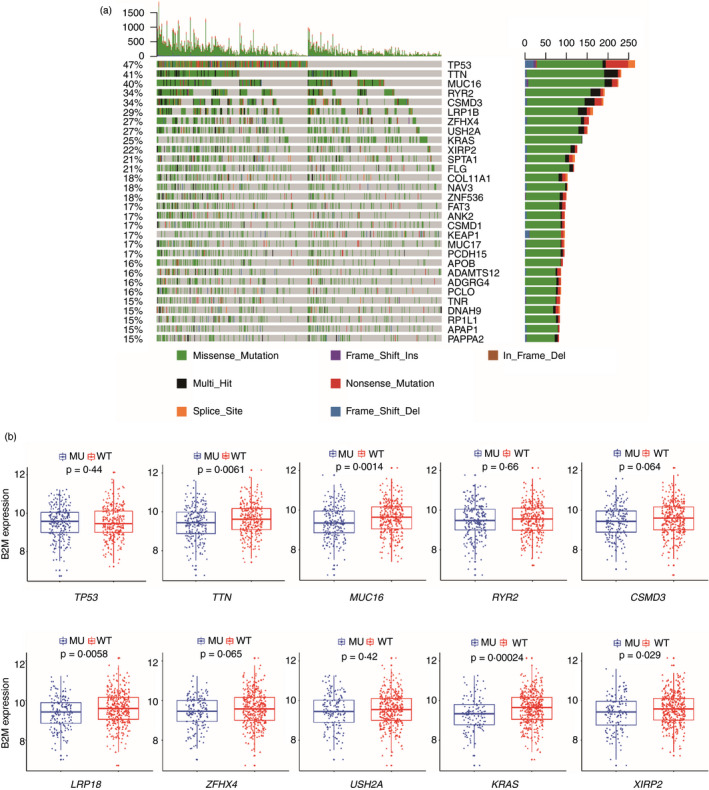
The association between B2M expression and recurrent mutations in lung cancer. (a) Recurrent mutations and their alteration profiles in the LUAD data set. (b) The levels of B2M in tumours with or without the indicated mutations in the LUAD data set
FIGURE 7.
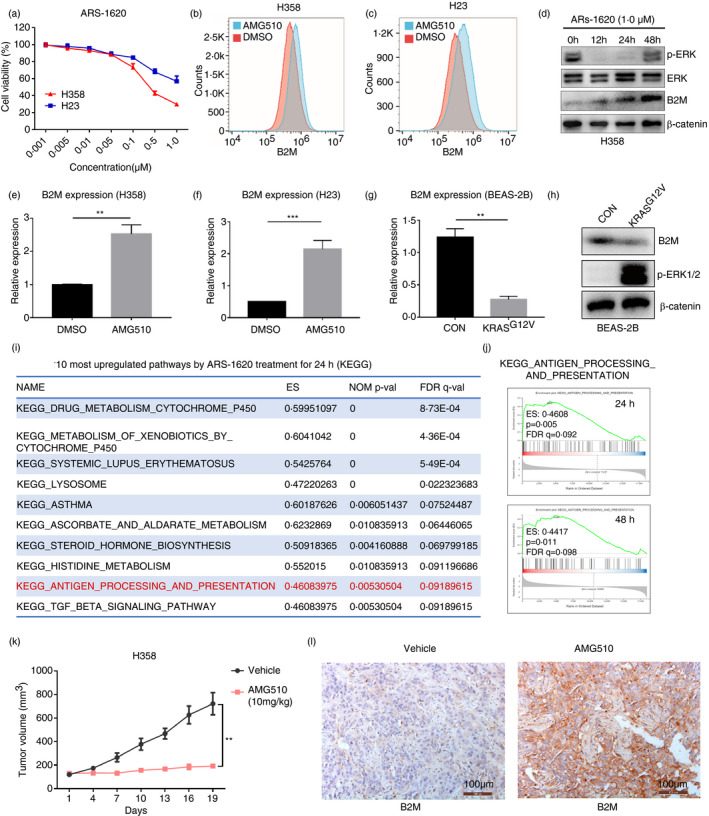
KRAS mutations suppress B2M expression in lung cancer. (a) Cell viability assay for H358 and H23 cells treated with ARS‐1620 at various concentrations for 96 h. (b,c) FACS analysis of B2M on the surface of H23 and H358 cells treated with AMG510 for 24 h. (d) Western blot demonstrating the levels of p‐ERK, ERK and B2M, with β‐catenin as a loading control. (e,f) Real‐time PCR results indicating B2M transcription upon AMG510 treatment for 24 h in H358 and H23 cells. (g) Mutant KRASG12V was introduced into the BEAS‐2B normal bronchial epithelial cell line, and B2M transcription was assessed by real‐time PCR. (h) Western blot showing the reduced B2M in BEAS‐2B cells overexpressing KRASG12V. (i–j) GSEA of the RNA‐seq results and the top 10 upregulated pathways upon treatment with ARS‐1620. (k) Tumour volumes of H358 xenografts in mice treated with AMG510 or vehicle (n = 5). (l) IHC staining for B2M in H358 xenografts treated with AMG510 or control (**p < 0·01 vs. control)
Moreover, forced expression of KRASG12V in BEAS‐2B normal lung epithelial cells suppressed B2M expression, as indicated by Western blotting and real‐time PCR (Figure 7g,h). Previously, we conducted RNA‐seq to study the effects of KRASG12C inhibition on H358 cells (available in the GEO database, GSE164326). In the present study, we performed GSEA with the RNA‐seq data and found that the antigen processing and presentation pathway was one of the ten most upregulated pathways after ARS‐1620 treatment for 24 h or 48 h (Figure 7i,j, and Figure S8D). Next, we implanted H358 cells subcutaneously into immunocompromised mice, and when tumours reached ~100 mm3, the mice were treated with AMG510 by oral gavage. AMG510 significantly suppressed the growth of H358 tumours (Figure 7k), and IHC staining demonstrated a marked increase in B2M expression in AMG510‐treated tumours compared to control tumours (Figure 7l). Together, these data suggest that KRAS mutations suppress B2M expression, which may contribute to the immune evasion of KRAS‐mutant lung cancer.
DISCUSSION
Although they occur infrequently, B2M mutations may be involved in resistance to PD‐1‐based immunotherapy in melanoma and lung cancer patients [6, 8]. Here, we show that B2M knockout in otherwise sensitive MC38 tumours led to resistance to PD‐1 inhibition and that B2M knockdown also diminished the therapeutic effects of immunotherapy. We found that B2M expression is markedly reduced in lung tumour tissues compared to normal tissues and that only ~30% of tumours exhibit high levels of B2M. These results suggest that not only genetic mutations but also epigenetic dysregulation of the B2M gene could be responsible for resistance to lung cancer immunotherapy.
The tumour microenvironment plays an essential role in lung cancer development [47, 48]. Immune effector cells, such as CD8+ T and NK cells, are either prevented from entering the tumour or inhibited functionally within the tumour milieu, which contributes to immune escape [47]. These scenarios are manifested in tumours expressing either high or low levels of B2M. We showed that B2M expression has a significant impact on the immune landscape in LUAD; B2Mhi tumours feature enhanced infiltration of immune cells, including CD8+ T and NK cells, but their cytotoxic functions are suppressed, probably due to increased expression of immune checkpoint molecules such as PD‐L1. On the other hand, B2Mlo tumours fail to recruit sufficient CD8+ T and NK cells, even though these tumours might be sensitive to NK‐mediated cytotoxicity. Therefore, established tumours evade immune surveillance and even take advantage of immune cell‐derived factors to promote their growth and metastasis [49].
The protumorigenic immune microenvironment could be corrected by PD‐1 inhibition if it is the PD/PD‐L1 interaction that restrains T cell function. However, for the immune system to launch an effective antitumour response upon PD‐1 inhibition, tumour cells need to be antigenic and recognizable (i.e. they need to express targetable neoantigen peptides in the context of MHC‐I molecules). We demonstrated that the downregulation of B2M diminished the responsiveness to PD‐1 inhibition in otherwise sensitive tumour models, whereas B2M overexpression sensitized resistant tumour models to the treatment, suggesting that B2M levels have a significant impact on the therapeutic efficacy of PD‐1 inhibition, probably through modulating the antigenicity of tumour cells. This may explain why PD‐L1 expression is not always predictive of the response to PD‐1 inhibition in the clinic [50, 51].
TMB is also linked to tumour cell antigenicity [50]. However, even in solid tumours that generally exhibit a high TMB, such as melanomas and LUADs, a potential neoantigen reservoir cannot be realized if antigen processing and presentation are defective. Interestingly, we found that the TMB scores in LUADs were higher in B2Mlo tumours than in B2Mhi tumours (Figure 3c), suggesting that the antigenicity of these tumours might be restrained by the downregulation of B2M, although the mechanisms are not clear at the moment; nevertheless, this implies that lung cancers may co‐opt those mechanisms to evade T‐cell attack induced by PD‐1 inhibition. Indeed, a CR to PD‐1 inhibition was more often seen in melanoma patients whose tumours expressed high levels of B2M (Figure 3a).
In LUAD tumours, high B2M is associated with enhanced CD8+ T cell infiltration. In general, the transcription of B2M is positively correlated with that of PD‐L1 (Figure S4B), probably because various cytokines stimulate the transcription of both genes in inflamed tumours. However, differences exist in terms of the regulatory mechanisms of the two genes. For example, KRAS mutation was reported to enhance PD‐L1 expression [52], whereas our results demonstrated that KRAS mutation reduced B2M expression. In addition, B2M expression is stable under hypoxic and normoxic conditions, but PD‐L1 is a direct target of HIF‐1α and upregulated by hypoxia [53]. Future studies are warranted to evaluate the predictive and prognostic value of the combined evaluation of B2M and PD‐L1 in immunotherapy.
Our studies revealed that several TFs might be involved in regulating B2M expression; two of these TFs, IRF‐1 and STAT1, are activated by IFN‐γ [54, 55]. IFN‐γ stimulates the expression of a group of immune‐related genes, including PD‐L1, B2M, MHC class I molecules and MHC class II molecules [44, 54, 56, 57]. Interestingly, JAK2 mutation was also identified in melanoma patients who developed resistance to PD‐1 inhibition, suggesting that disruption of IFN‐γ signalling might enable tumour cells to escape T‐cell attack [58, 59]. We speculate that IFN‐γ signalling is critical for the upregulation of PD‐L1 expression by tumour cells, which thereby suppresses CTL activity [60, 61]; however, this effect can be overcome by PD‐1 inhibition. Under such a scenario, JAK2 mutation and defective IFN‐γ‐induced B2M expression could still lead to resistance to PD‐1 inhibition.
STAT5 may also regulate B2M expression in LUAD (Figure 5a). Analysis of public databases indicated that lung cancer exhibits reduced STAT5 expression (Figure S5F), which may contribute to the downregulation of B2M. STAT5 can be activated by a wide variety of cytokines and growth factors [62]. In H1975 cells that express high levels of STAT5, B2M is downregulated by STAT5 inhibition, and similar effects have been seen in NK92 cell lines with constitutively active STAT5. In several leukaemias and some solid tumours, STAT5 has been reported to be activated and to contribute to the survival and proliferation of malignant cells [63], and targeting STAT5 is thought to be a promising anticancer strategy [62]. We propose that this strategy needs to be carefully evaluated in lung cancer, as STAT5 inhibition may suppress B2M expression and impair antitumour immune responses.
Genetic alterations in KRAS occur in 20%–30% of LUADs and are key drivers of lung tumorigenesis [64]. We revealed that KRAS mutations suppress B2M expression in LUAD, probably through abnormal activation of the downstream MAPK pathway. Our results are in line with previous studies showing that MEK/BRAF inhibition enhances HLA‐I antigen presentation and improves the therapeutic efficacy of PD‐1 inhibitors [65, 66, 67]. However, MEK inhibition may also suppress the activation of naive T cells [68]. Therefore, the time and duration of MEK inhibitor application must be carefully evaluated before MEK inhibitors are added to any combination therapies [69]. The newly developed KRASG12C inhibitors specifically target tumour cells and have shown promising results in clinical trials [70, 71]. We speculate that, in combination with PD‐1 inhibitors, KRASG12C inhibitors provide a unique opportunity to treat patients with KRASG12C‐expressing lung cancers. Consistently, a recent study showed that targeting KRASG12C with AMG510 stimulated antitumour immunity and had synergistic effects with PD‐1 inhibitors in mouse models, but the mechanisms were not very clear [43], and our study suggests that KRASG12C inhibition might induce B2M upregulation and thus contribute to improved therapeutic efficacy.
In conclusion, we demonstrated that genetic and epigenetic dysregulation of the B2M gene significantly impacts the immune landscape and suppresses anticancer immunity in LUADs. Moreover, we uncovered several mechanisms of B2M downregulation and provided strategies for restoring its expression, which may help overcome resistance to immunotherapy in patients with lung cancer.
CONFLICT OF INTEREST
The authors declare that they have no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
X.Z. performed the conception and design; Y.Z., YJ.C., YQ.C., H.H., and C.J. involved in data acquisition; X.Z., Y.Z., YQ.C., and YJ.C. analysed the data and interpreted the data; X.Z. wrote manuscript; X.Z. involved in funding acquisition.
ETHICAL APPROVAL
These studies were performed in compliance with an approved protocol and the institutional guidelines of the Ethical Committee of Nantong University.
Supporting information
Fig S1‐S8
ACKNOWLEDGMENTS
We thank Mr. Xu Wang at laboratory animal center of Nantong University for his support in animal studies.
Zhao Y, Cao Y, Chen Y, Wu L, Hang H, Jiang C, et al. B2M gene expression shapes the immune landscape of lung adenocarcinoma and determines the response to immunotherapy. Immunology. 2021;164:507–523. 10.1111/imm.13384
Yu Zhao and Yuejiao Cao Co‐first authors.
Funding information
This work was supported by the National Natural Science Foundation of China (Grant number 81771681 to XZ), the Jiangsu Six Elite Units Foundation (Grant number WSW‐058 to XZ), the Nantong Science and Technology project (Grant number XG202008‐5 to XZ) and the Beijing Xisike Clinical Oncology Research Foundation (Grant number Y‐XD2019‐130 to XZ).
REFERENCES
- 1. Gandhi L, Rodriguez‐Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non‐small‐cell lung cancer. N Engl J Med. 2018;378:2078–92. [DOI] [PubMed] [Google Scholar]
- 2. Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five‐year follow‐up of nivolumab in previously treated advanced non‐small‐cell lung cancer: results from the CA209‐003 study. J Clin Oncol. 2018;36:1675–84. [DOI] [PubMed] [Google Scholar]
- 3. Aspeslagh S, Chabanon RM, Champiat S, Postel‐Vinay S. Understanding genetic determinants of resistance to immune checkpoint blockers. Semin Cancer Biol. 2020;65:123–39. [DOI] [PubMed] [Google Scholar]
- 4. Hamilton G, Rath B. Immunotherapy for small cell lung cancer: mechanisms of resistance. Expert Opin Biol Ther. 2019;19:423–32. [DOI] [PubMed] [Google Scholar]
- 5. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zaretsky JM, Garcia‐Diaz A, Shin DS, Escuin‐Ordinas H, Hugo W, Hu‐Lieskovan S, et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med. 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. 2017;2:e000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pereira C, Gimenez‐Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, et al. Genomic profiling of patient‐derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin Cancer Res. 2017;23:3203–13. [DOI] [PubMed] [Google Scholar]
- 10. Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, et al. Transcriptional downregulation of MHC class I and melanoma de‐ differentiation in resistance to PD‐1 inhibition. Nat Commun. 2020;11:1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson JC, Davis C, Deshpande C, Hwang WT, Jeffries S, Huang A, et al. Gene signature of antigen processing and presentation machinery predicts response to checkpoint blockade in non‐small cell lung cancer (NSCLC) and melanoma. J Immunother Cancer. 2020;8:e000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bern MD, Parikh BA, Yang L, Beckman DL, Poursine‐Laurent J, Yokoyama WM. Inducible down‐regulation of MHC class I results in natural killer cell tolerance. J Exp Med. 2019;216:99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Azuma M. Co‐signal molecules in T‐cell activation : historical overview and perspective. Adv Exp Med Biol. 2019;1189:3–23. [DOI] [PubMed] [Google Scholar]
- 14. Hashimoto‐Tane A, Saito T. Dynamic regulation of TCR‐microclusters and the microsynapse for T cell activation. Front Immunol. 2016;7:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: A web server for comprehensive analysis of tumor‐infiltrating immune cells. Cancer Res. 2017;77:e108–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kreft L, Soete A, Hulpiau P, Botzki A, Saeys Y, De Bleser P. ConTra v3: a tool to identify transcription factor binding sites across species, update 2017. Nucleic Acids Res. 2017;45:W490–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li S, Wan C, Zheng R, Fan J, Dong X, Meyer CA, et al. Cistrome‐GO: a web server for functional enrichment analysis of transcription factor ChIP‐seq peaks. Nucleic Acids Res. 2019;47:W206–W211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou X, Updegraff BL, Guo Y, Peyton M, Girard L, Larsen JE, et al. PROTOCADHERIN 7 Acts through SET and PP2A to potentiate MAPK signaling by EGFR and KRAS during lung tumorigenesis. Cancer Res. 2017;77:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014;9:e96801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, et al. Macrophage‐derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin Cancer Res. 2020;26:487–504. [DOI] [PubMed] [Google Scholar]
- 24. Li L, Yang L, Cheng S, Fan Z, Shen Z, Xue W, et al. Lung adenocarcinoma‐intrinsic GBE1 signaling inhibits anti‐tumor immunity. Mol Cancer. 2019;18:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol. 2014;178:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu Rev Med. 2019;70:437–50. [DOI] [PubMed] [Google Scholar]
- 27. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion'. Nat Rev Immunol. 2019;19:665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol. 2020;20:128–36. [DOI] [PubMed] [Google Scholar]
- 29. Pockley AG, Vaupel P, Multhoff G. NK cell‐based therapeutics for lung cancer. Expert Opin Biol Ther. 2020;20:23–33. [DOI] [PubMed] [Google Scholar]
- 30. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5‐dependent manner. Proc Natl Acad Sci USA. 2017;114:E9271–E9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22:5183–8. [DOI] [PubMed] [Google Scholar]
- 32. Sivori S, Della Chiesa M, Carlomagno S, Quatrini L, Munari E, Vacca P, et al. Inhibitory receptors and checkpoints in human NK cells, implications for the immunotherapy of cancer. Front Immunol. 2020;11:2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu‐Lieskovan S, et al. Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell. 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu S, Stein JE, Rimm DL, Wang DW, Bell JM, Johnson DB, et al. Comparison of biomarker modalities for predicting response to PD‐1/PD‐L1 checkpoint blockade: a systematic review and meta‐analysis. JAMA Oncol. 2019;5:1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miyazawa T, Marushima H, Saji H, Kojima K, Hoshikawa M, Takagi M, et al. PD‐L1 expression in non‐small‐cell lung cancer including various adenocarcinoma subtypes. Ann Thorac Cardiovasc Surg. 2019;25:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li N, Tang J, Yang J, Zhu B, Wang X, Luo Y, et al. Tumor perfusion enhancement by ultrasound stimulated microbubbles potentiates PD‐L1 blockade of MC38 colon cancer in mice. Cancer Lett. 2021;498:121–9. [DOI] [PubMed] [Google Scholar]
- 37. Johnston RJ, Comps‐Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–37. [DOI] [PubMed] [Google Scholar]
- 38. Cai X, Wei B, Li L, Chen X, Liu W, Cui J, et al. Apatinib enhanced anti‐PD‐1 therapy for colon cancer in mice via promoting PD‐L1 expression. Int Immunopharmacol. 2020;88:106858. [DOI] [PubMed] [Google Scholar]
- 39. De Luca R, Neri D. Potentiation of PD‐L1 blockade with a potency‐matched dual cytokine‐antibody fusion protein leads to cancer eradication in BALB/c‐derived tumors but not in other mouse strains. Cancer Immunol Immunother. 2018;67:1381–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oba T, Long MD, Keler T, Marsh HC, Minderman H, Abrams SI, et al. Overcoming primary and acquired resistance to anti‐PD‐L1 therapy by induction and activation of tumor‐residing cDC1s. Nat Commun. 2020;11:5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie Z, Ikegami T, Ago Y, Okada N, Tachibana M. Valproic acid attenuates CCR2‐dependent tumor infiltration of monocytic myeloid‐derived suppressor cells, limiting tumor progression. Oncoimmunology. 2020;9:1734268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Waaler J, Mygland L, Tveita A, Strand MF, Solberg NT, Olsen PA, et al. Tankyrase inhibition sensitizes melanoma to PD‐1 immune checkpoint blockade in syngeneic mouse models. Commun Biol. 2020;3:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti‐tumour immunity. Nature. 2019;575:217–23. [DOI] [PubMed] [Google Scholar]
- 44. Neerincx A, Castro W, Guarda G, Kufer TA. NLRC5, at the heart of antigen presentation. Front Immunol. 2013;4:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sznarkowska A, Mikac S, Pilch M. MHC Class I regulation: the origin perspective. Cancers. 2020;12:1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jongsma MLM, Guarda G, Spaapen RM. The regulatory network behind MHC class I expression. Mol Immunol. 2019;113:16–21. [DOI] [PubMed] [Google Scholar]
- 47. Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. 2019;19:9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, et al. The tumor microenvironment regulates sensitivity of murine lung tumors to PD‐1/PD‐L1 antibody blockade. Cancer Immunol Res. 2017;5:767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51:27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD‐1/PD‐L1 inhibitors. Mol Cancer. 2018;17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rizvi H, Sanchez‐Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti‐programmed cell death (PD)‐1 and anti‐programmed death‐ligand 1 (PD‐L1) blockade in patients with non‐small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol. 2018;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS mutation‐induced upregulation of PD‐L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66:1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD‐L1 is a novel direct target of HIF‐1alpha, and its blockade under hypoxia enhanced MDSC‐mediated T cell activation. J Exp Med. 2014;211:781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moon JW, Kong SK, Kim BS, Kim HJ, Lim H, Noh K, et al. IFNgamma induces PD‐L1 overexpression by JAK2/STAT1/IRF‐1 signaling in EBV‐positive gastric carcinoma. Sci Rep. 2017;7:17810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Connett JM, Hunt SR, Hickerson SM, Wu SJ, Doherty GM. Localization of IFN‐gamma‐activated Stat1 and IFN regulatory factors 1 and 2 in breast cancer cells. J Interferon Cytokine Res. 2003;23:621–30. [DOI] [PubMed] [Google Scholar]
- 56. Zhou F. Molecular mechanisms of IFN‐gamma to up‐regulate MHC class I antigen processing and presentation. Int Rev Immunol. 2009;28:239–60. [DOI] [PubMed] [Google Scholar]
- 57. Giroux M, Schmidt M, Descoteaux A. IFN‐gamma‐induced MHC class II expression: transactivation of class II transactivator promoter IV by IFN regulatory factor‐1 is regulated by protein kinase C‐alpha. J Immunol. 2003;171:4187–94. [DOI] [PubMed] [Google Scholar]
- 58. Nguyen TT, Ramsay L, Ahanfeshar‐Adams M, Lajoie M, Schadendorf D, Alain T, et al. Mutations in the IFNgamma‐JAK‐STAT pathway causing resistance to immune checkpoint inhibitors in melanoma increase sensitivity to oncolytic virus treatment. Clin Cancer Res. 2021;27:3432–42. [DOI] [PubMed] [Google Scholar]
- 59. Shin DS, Zaretsky JM, Escuin‐Ordinas H, Garcia‐Diaz A, Hu‐Lieskovan S, Kalbasi A, et al. Primary resistance to PD‐1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD‐L1 expression is mainly regulated by interferon gamma associated with JAK‐STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garcia‐Diaz A, Shin DS, Moreno BH, Saco J, Escuin‐Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Rep. 2017;19:1189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rani A, Murphy JJ. STAT5 in Cancer And Immunity. J Interferon Cytokine Res. 2016;36:226–37. [DOI] [PubMed] [Google Scholar]
- 63. Wittig I, Groner B. Signal transducer and activator of transcription 5 (STAT5), a crucial regulator of immune and cancer cells. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:449–63. [DOI] [PubMed] [Google Scholar]
- 64. Minguet J, Smith KH, Bramlage P. Targeted therapies for treatment of non‐small cell lung cancer – recent advances and future perspectives. Int J Cancer. 2016;138:2549–61. [DOI] [PubMed] [Google Scholar]
- 65. Mimura K, Kua LF, Shiraishi K, Kee Siang L, Shabbir A, Komachi M, et al. Inhibition of mitogen‐activated protein kinase pathway can induce upregulation of human leukocyte antigen class I without PD‐L1‐upregulation in contrast to interferon‐gamma treatment. Cancer Sci. 2014;105:1236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr, Carlino MS, et al. Combined BRAF and MEK inhibition with PD‐1 blockade immunotherapy in BRAF‐mutant melanoma. Nat Med. 2019;25:936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase inhibition promotes T Cell and Anti‐tumor Activity in Combination with PD‐L1 Checkpoint Blockade. Immunity. 2016;44:609–21. [DOI] [PubMed] [Google Scholar]
- 68. Oh YT, Deng J, Yue P, Owonikoko TK, Khuri FR, Sun SY. Inhibition of B‐Raf/MEK/ERK signaling suppresses DR5 expression and impairs response of cancer cells to DR5‐mediated apoptosis and T cell‐induced killing. Oncogene. 2016;35:459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Richmond CS, Vallatharasu Y, Deviley JA, Vos CR, Parsons BM, Kenny PA. Sequential treatment failures in response to BRAF/MEK and immune checkpoint inhibitors mediated by MAP2K2 and B2M mutations in melanoma. Exp Mol Pathol. 2019;110:104260. [DOI] [PubMed] [Google Scholar]
- 70. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS‐mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S8