

Abstract
An exclusive feature of dendritic cells (DCs) is their capacity to present exogenous antigens by MHC class I molecules, called cross‐presentation. Here, we show that protein antigen can be conserved in mature murine DCs for several days in a lysosome‐like storage compartment, distinct from MHC class II and early endosomal compartments, as an internal source for the supply of MHC class I ligands. Using two different uptake routes via Fcγ receptors and C‐type lectin receptors, we could show that antigens were routed towards the same endolysosomal compartments after 48 h. The antigen‐containing compartments lacked co‐expression of molecules involved in MHC class I processing and presentation including TAP and proteasome subunits as shown by single‐cell imaging flow cytometry. Moreover, we observed the absence of cathepsin S but selective co‐localization of active cathepsin X with protein antigen in the storage compartments. This indicates cathepsin S‐independent antigen degradation and a novel but yet undefined role for cathepsin X in antigen processing and cross‐presentation by DCs. In summary, our data suggest that these antigen‐containing compartments in DCs can conserve protein antigens from different uptake routes and contribute to long‐lasting antigen cross‐presentation.
Keywords: antigen cross‐presentation, cathepsin, C‐type lectin receptors, dendritic cells, Fc receptors
Antigens taken up by dendritic cells via Fcγ receptors or C‐type lectin receptor MGL1 are routed towards the same endolysosomal compartments, which express LAMP1 and active cathepsin X. These compartments can conserve antigen for several days and serve as an antigen depot for prolonged antigen cross‐presentation to CD8+ T cells.
Abbreviations
- BMDCs
bone marrow‐derived dendritic cells
- CLRs
C‐type lectin receptors
- CTL
cytotoxic T‐lymphocyte
- DCs
dendritic cells
- DIC
differential interference contrast
- ER
endoplasmic reticulum
- FcγRs
Fcγ receptors
- IRAP
insulin‐regulated aminopeptidase
- Lex
LewisX
- OVA IC
OVA‐IgG immune complexes
- qABP
quenched activity‐based probe
- TAP
transporter associated with antigen processing
INTRODUCTION
Dendritic cells (DCs) are specialized antigen‐presenting cells due to their exclusive ability to cross‐present tumour antigens to initiate CD8+ T‐cell response [1, 2, 3]. DCs scan the peripheral tissue and take up antigens they encounter through receptors, such as Fcγ receptors (FcγRs) and C‐type lectin receptors (CLRs). Upon antigen internalization and DC activation, DCs migrate towards the draining lymph node where they can present antigens to T cells. Two main intracellular pathways for antigen cross‐presentation in DCs have been proposed: the vacuolar and the cytosolic pathways. Antigen processing through the vacuolar pathway is mainly dependent on antigen degradation in endosomes, probably by proteases such as cathepsin S, but independent of transporter associated with antigen processing (TAP) and the proteasome [4]. Thus, antigen processing and loading on MHCI probably occur in endocytic compartments. In the cytosolic pathway, exogenous antigens in endosomal vesicles are transported in the cell cytosol and degraded by the proteasome. The peptides that are generated by the proteasome are then transported by TAP to the endoplasmic reticulum (ER) where they are loaded on MHCI molecules [5, 6, 7]. In addition, it has been shown that some proteasome‐generated peptides can be transported back into endocytic compartments where they are trimmed by insulin regulated aminopeptidase (IRAP) and directly loaded on MHCI molecules [8].
We have previously reported that protein antigen can be stored in DCs for several days, which facilitates prolonged cytotoxic T‐lymphocyte (CTL) cross‐priming capacity [9, 10]. Moreover, we have shown that DCs in vivo can store antigen up to a week, which contributes to sustained antigen cross‐presentation to T cells in vivo and ex vivo [11]. Both cDC1 and cDC2 subsets contribute to prolonged antigen presentation capacity in MHC class I (cDC1) and MHC class II and I (cDC2) in vivo. Furthermore, we have shown that autophagy can regulate long‐term cross‐presentation by degrading antigen stored in DCs [12]. Sustaining antigen cross‐presentation capacity is important since it may take up to a few days for DCs to migrate from the infection site to the lymph nodes to encounter T cells. Moreover, the turnover rate of surface MHCI molecules is shorter compared to MHCII, most MHCI–peptide complexes disappear from the cell surface within 24 h [9]. Therefore, prolonged antigen storage in DCs and sustained supply of newly synthesized MHCI–peptide complexes is beneficial to ensure efficient T‐cell cross‐priming [13].
Here, we analysed the routing of antigens targeted to FcγRs and C‐type lectin receptor MGL1. Our results revealed that protein antigens targeted to FcγRs or MGL1 end up in the same storage compartment. We characterized the compartments where antigen is stored in DCs by using immunofluorescent staining of several proteins related to the endosomal trafficking and antigen processing pathways. The antigen‐containing compartments are LAMP1 positive, distinct from early endosomal or MHCI/ MHCII loading compartments. Moreover, the lack of cathepsin S indicates that this cathepsin is not involved in antigen degradation in these storage compartments. Interestingly, the increased presence of cathepsin X in the storage compartments in time suggests there might be a distinct but yet unidentified function of cathepsin X in DCs. Our data show that DC can conserve antigens derived from different uptake routes, which suggests a common role of these compartments in sustaining antigen cross‐presentation.
MATERIALS AND METHODS
Cells
Bone marrow‐derived dendritic cells (BMDCs) from C57BL/6 mice were generated in the presence of 30% R1 supernatant from NIH3T3 fibroblasts transfected with GM‐CSF for 10 days. The D1 dendritic cells, a long‐term growth factor‐dependent immature splenic DC line derived from C57BL/6 (BL/6) mice, were kindly provided by P. Ricciardi‐Castagnoli (University of Milano‐Bicocca, Italy) and cultured as described [14]. CD8+ T cells (CD8+/ CD45.1+) were purified (Mouse CD8 T Lymphocyte Enrichment Set, BD Biosciences) from the spleen of OTI mice (CD8+ T cell transgenic mice expressing a TCR recognizing the OVA‐derived Kb presented epitope SIINFEKL) that were bred and kept at the LUMC animal facility under SPF conditions. B3Z is a CD8+ T‐cell hybridoma specific for SIINFEKL‐H2‐Kb MHCI molecules and expresses LacZ upon activation.
DC antigen presentation
OVA‐IgG immune complexes (OVA IC) were formed by incubating 0.25 µg/ml OVA (native protein, Worthington Biochemical and 75 µg/ml anti‐OVA IgG (rabbit polyclonal, LSBio) for 30 min at 37°C. Immature D1 DCs were incubated with OVA IC or 16 µg/ml OVA‐LeX [10] for 2 h, extensively washed with culture medium and chased for the indicated periods. CFSE labelled purified CD8+ T cells (CD8+/ CD45.1+) from OTI mice were added, and T‐cell proliferation was measured 3 days later by flow cytometry.
Confocal microscopy
OVA‐IgG immune complexes (OVA IC) were formed by incubating 1 µg/ml OVA (Alex Fluor 488 or 647 conjugated; Life Technologies) and 300 µg/ml anti‐OVA IgG (rabbit polyclonal, LSBio) for 30 min at 37°C. BMDCs were incubated with OVA IC (Alexa Fluor 488, or 647 conjugated) or OVA‐LeX (DyLight 488) for the indicated time points. Cells were washed and transferred to glass bottom dishes (MatTek Corporation, Ashland, USA), fixed with 4% formaldehyde (Merck) and permeabilized with 0.5% saponin. Cells were then incubated with one of the following primary antibodies: goat anti‐EEA‐1 (C‐15, Santa Cruz Biotechnology), LAMP‐1 (CD107a, Alexa Fluor 647, BioLegend), mouse anti‐Rab5 (D11, Santa Cruz Biotechnology), mouse anti‐Rab7 (Rab7‐117, Sigma‐Aldrich), rat anti‐MHCI ((H‐2Db, ER‐HR 52, Abcam), rat anti‐MHCII (I‐A/I‐E, M5/114.15.2, BioLegend), goat anti‐TAP1 (B‐8, Santa Cruz Biotechnology) and goat anti‐PA28β (L‐19, Santa Cruz Biotechnology). The following secondary antibodies were used: goat anti‐mouse IgG (H + L) Alexa Fluor 647, rabbit anti‐goat IgG (H + L) Alexa Fluor 647 and donkey anti‐rat IgG (H + L) Alexa Fluor 647 (all from Thermo Fisher Scientific). For the cathepsin experiments, BMDCS were incubated with OVA IC (Alexa Fluor 488) for the indicated time points. Cells were fixed and permeabilized as described before and then incubated for 30 min with 1 µM cysteine cathepsin activity‐based probe BMV109 [15]. The cathepsin probe covalently reacts with the active site of Cathepsin X, B, S and L. Once the probe binds to the target enzyme, the quencher is removed and the probe emits the fluorophore Cy5. Co‐staining was performed with the following primary antibodies: mouse anti‐cathepsin S (E‐3, Santa Cruz Biotechnology) and polyclonal goat anti‐cathepsin X/Z/P (R&D systems). The following secondary antibodies were used: goat anti‐mouse IgG A568 (H + L, Thermo Fisher Scientific) and rabbit anti‐goat A568 (H + L, Thermo Fisher). The cells were imaged using Leica SP5 STED confocal microscope with a 63× objective lens. Differential interference contrast (DIC) was additionally used to image cell contrast. Images were acquired in 10x magnification and processed with ImageJ software.
Imaging flow cytometry
BMDCs were incubated with OVA IC (Alexa Fluor 647) for the indicated time points, fixed with 4% formaldehyde (Merck) and permeabilized with 0.5% Saponin. Cells were stained with primary antibodies as described under the confocal microscopy section and subsequently stained with the following secondary antibodies: goat anti‐mouse IgG F(ab’)2 Alexa Fluor 488, donkey anti‐rat IgG (H + L) Alexa Fluor 488 (both from Thermo Fisher Scientific) and rabbit anti‐goat IgG Cy3 (Jackson ImmunoResearch). Cells were acquired on an ImageStream X100 (Amnis) imaging flow cytometer. A minimum of 15000 cells was measured per sample at a flow rate ranging between 50 and 100 cells/s at 60× magnification, and the analysis was performed using the IDEAS v6.1 software (Amnis). Cells were first gated based on the Gradient RMS (brightfield) feature and then further gated based on area vs aspect ratio intensity (both on brightfield). The first gating identified the cells, which appeared in focus (Figure S2A), while the second gating excluded doublets and other cells than BMDCs (Figure S2B). Co‐localization between OVA IC and the markers was calculated on double positive cells (Figure S2C) using the bright detail co‐localization feature (Figure S2D) as reported elsewhere [16].
Electron microscopy
BMDCs were pulsed with OVA IC (Alexa Fluor 488) for 15 min or pulsed for 2 h and chased for 48 h. Cells were fixed for 2 h in PHEM buffer containing 2% paraformaldehyde and 0.2% glutaraldehyde. The cells were rinsed in PBS and pelleted in pre‐warmed PBS containing 12% gelatin. The cell pellet was prepared for cryo sectioning and immunogold labelling as described elsewhere [17]. Briefly, small blocks were cut form the cold cell pellet, which were infiltrated in 2.3 M sucrose in phosphate buffer for 3 h. The cryo‐protected samples were mounted on an aluminium pin, plunged in liquid nitrogen and sectioned with a cryo ultramicrotome (Leica, Vienna) using a diamond knife (Diatome, Biel). Ultra‐thin sections (70 nm) were attached to a formvar and carbon‐coated copper EM grid and labelled with anti‐Alexa 488 antibody (Invitrogen) and protein A gold (10 nm, CMC, Utrecht) and embedded in 2% methylcellulose in water containing 0.6% uranyl acetate and subsequently air‐dried. The sections were imaged in a Tecnai 12 transmission electron microscope (Thermo Fisher) operating at 120 kV equipped with a 4K Eagle camera (Thermo Fisher).
Fluorescent SDS‐PAGE
BMDCs were left untreated or were incubated with OVA IC for 24 and 48 h. Residual immune complexes were removed by washing with culture medium. Antigen‐presenting cell subsets (macrophages, cDC1, cDC2 and pDC) were isolated from spleens of C57BL/6 mice and sorted on BD FACSAria II SORP (BD Biosciences) based on markers CD11b, CD11c, CD8α, Ly6C, B220 and F4/80 as described earlier [11]. Subsequently, cells were incubated with 1 µM quenched activity‐based probe BMV109 for 1 h at 37°C. After washing with PBS, total cell lysates were prepared and proteins were separated by SDS‐PAGE (200.000 cells/lane) on a 15% polyacrylamide gel. Cy5‐labelled cathepsins were measured directly in the gel with the Typhoon Imager.
RESULTS
Storage of antigen–antibody complexes in dendritic cells for prolonged T‐cell activation
We have studied the uptake kinetics before of OVA bound to OVA‐specific IgG antibodies (OVA immune complexes, OVA IC) and showed that the uptake of OVA IC is a 1000‐fold more efficient than soluble OVA [9]. Moreover, we could show that antibody‐bound antigen is conserved in DCs for several days, whereas soluble OVA is not detectable anymore after one day [9]. In order to study the kinetics of antigen cross‐presentation, DCs were pulsed with OVA IC for 2 h and chased for 2, 24, 48 or 72 h. Antigen‐specific T‐cell activation was detected already after 2 h and T‐cell proliferation remained high in time (Figure 1a). DCs that were chased for 72 h were still able to induce ~90% T‐cell proliferation (Figure 1a).
FIGURE 1.
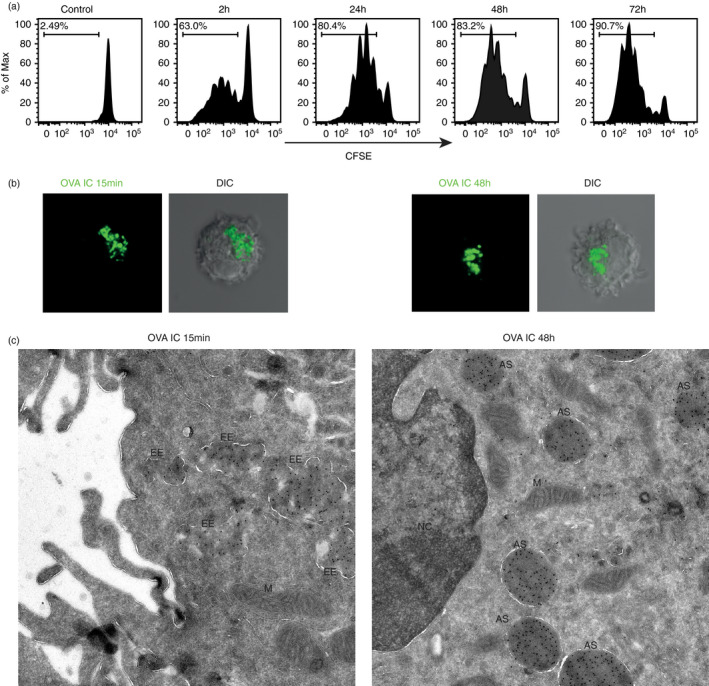
Antigen storage in dendritic cells. DCs were pulsed with OVA IC for 2 h, washed and chased for 2, 24, 48 or 72 h. CFSE‐labelled CD8+ OTI T cells were added, and T‐cell proliferation was analysed by flow cytometry (a). DCs were pulsed with OVA IC (Alexa Fluor 488) for 15 min (b, left panel) or pulsed with OVA IC for 2 h and chased for 48 h (b, right panel). OVA IC uptake and presence in DCs were imaged by confocal microscopy, and differential interference contrast (DIC) was additionally used to image cell contrast. Immuno‐electron microscopy images of DCs after 15‐min OVA IC (Alexa Fluor 488) pulse (c, left) and DCs pulsed with OVA IC for 2 h and chased for 48 h (c, right). Sections were labelled with immunogold for Alexa488 with 10‐nm gold particle size. EE = early endosome, M = mitochondria, AS = antigen storage compartment, NC = nucleus
We further characterized internal antigen storage sources with the use of fluorescently labelled OVA IC. Large amounts of OVA IC were already taken up by DCs after only 15‐min pulse, visualized by confocal microscopy (Figure 1b, left panel). Interestingly, DCs that were pulsed with OVA IC for 2 h, washed and chased for 48 h still contain extensive amounts of OVA IC (Figure 1b, right panel). These OVA IC are located perinuclear in a condensed fashion. More detailed visualization of the uptake of OVA IC was done by 10‐nm gold particle labelling for electron microscopy. After 15 min OVA IC pulse in DCs, many gold beads were detected in cloud shaped compartments (Figure 1c, left). Strikingly, 48 h after pulse‐loading DCs with OVA IC, large spherical structures filled with gold beads were visualized near the nucleus (Figure 1c, right). These results confirmed once more that antigen can be stored in DCs for several days in compartments proportionate to endosomal structures.
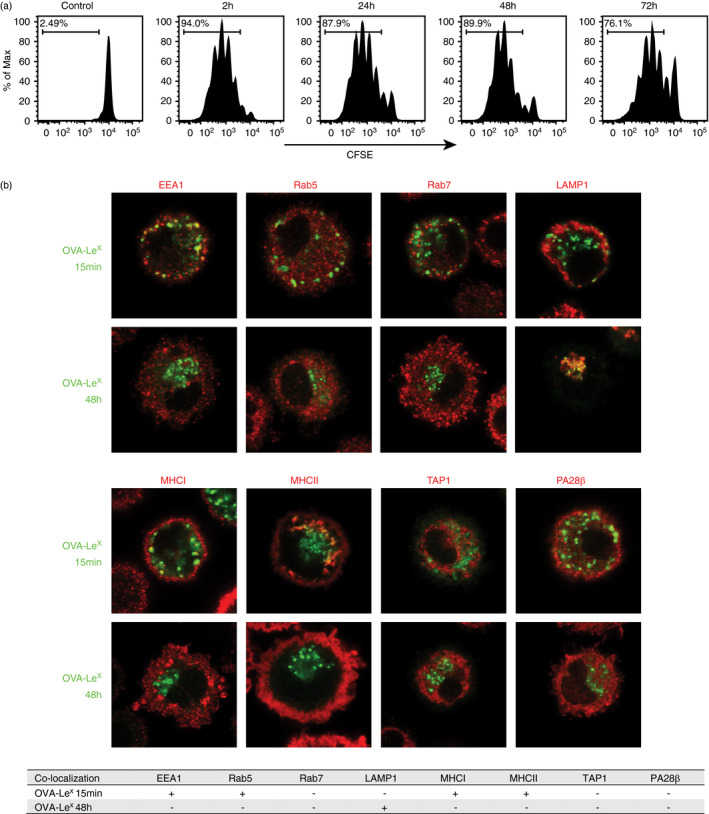
Antigen–antibody complexes are stored in LAMP1 positive compartments in dendritic cells
We further characterized the storage compartments by performing co‐staining with antibodies against several members of the endosomal trafficking pathways with confocal microscopy. After the initial 15‐min uptake of OVA IC by DCs, OVA IC were taken up in EEA1‐ and Rab5‐positive compartments (Figure 2, Figure S1). However, after 2‐h pulse and 48‐h chase, OVA IC were not found in EEA1 or Rab5 compartments. Rab7, which is a marker for more matured endosomes, did not show any co‐localization with OVA IC after 15 min or 48 h. The lysosomal marker LAMP1 was spread out through the cell cytosol after 15 min, and no co‐localization was found with OVA IC. However, a clear perinuclear reorganization of LAMP1 was detected after 48 h and co‐localization with OVA IC was observed. MHCI and MHCII positive compartments in the cell cytosol were detected at 15‐min time point, and co‐localization with OVA IC was observed (Figure 2, Figure S1). As described before, targeting FcγRs on DCs with OVA IC induced strong DC maturation, including an increased expression of MHCI and MHCII [9]. Indeed, 48 h after antigen pulse, most MHCI and MHCII were detected on the cell surface, and not co‐localizing anymore with OVA IC (Figure 2, Figure S1). Since we have shown before that antigens from the storage compartments are cross‐presented in a TAP‐ and proteasome‐dependent pathway [9], antibody staining for TAP1 and PA28β was performed. The proteasome activator PA28β subunit is known to be strongly upregulated in maturing dendritic cells [18]. However, neither TAP1 nor PA28β co‐localized with OVA IC at any measured time point (Figure 2, Figure S1).
FIGURE 2.
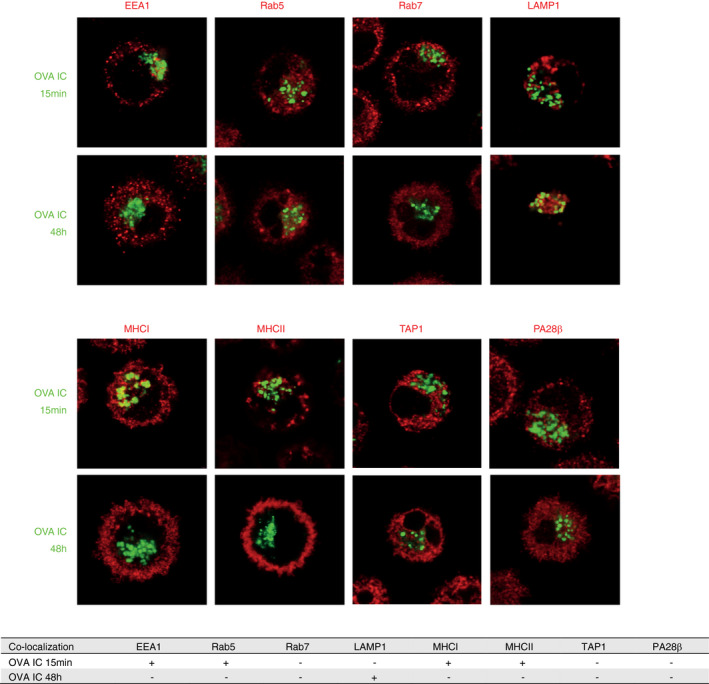
Characterization of the antigen storage compartments in DCs upon FcγR targeting. DCs were pulsed with OVA IC (Alexa Fluor 488, green) for 15 min or pulsed with OVA IC for 2 h and chased for 48 h. OVA IC presence in DCs was imaged by confocal microscopy, and DIC was used to image cell contrast. Specific antibodies against EEA1, Rab5, Rab7, LAMP1, MHCI, MHCII, TAP1 and PA28β (red) were used and analysed for co‐localization with OVA IC. Co‐localization between OVA IC and the antibodies is summarized in a table, ‘+’ indicates co‐localization, ‘‐’ indicates no co‐localization
In order to follow the co‐localization kinetics of OVA IC after uptake by DCs, we used imaging flow cytometry, a method that allows high‐throughput image analysis of cells in flow with near‐confocal resolution to analyse intracellular routing of fluorescently labelled OVA IC (Figure S2). OVA IC enters EEA1‐positive compartments after DC uptake, but the co‐localization score decreased swiftly within 15 min (Figure 3). One hour after the initial antigen pulse, most OVA IC were not present in EEA1 positive compartments. Similar kinetics was observed for Rab5, OVA IC enters Rab5‐positive compartments, but after 1 h, the co‐localization score between Rab5 and OVA IC had diminished. In line with the confocal analysis, Rab7 did not show any remarkable co‐localization with OVA IC, although a slight increase could be observed after 48 h. OVA IC was strongly co‐localizing with LAMP1 after 24 and 48 h, confirming our previous confocal data. MHCI and MHCII co‐localized with OVA IC at early time points (10 min, 1 h), but the co‐localization score was decreased after 24 and 48 h. The co‐localization kinetics for both TAP1 and PA28β with OVA IC did not change in time.
FIGURE 3.
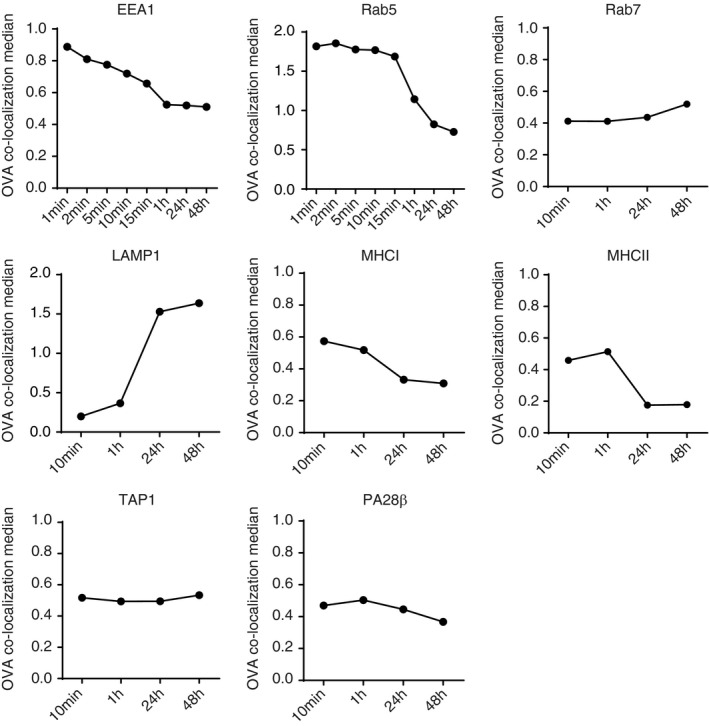
Characterization of the antigen storage compartments in DCs with imaging flow cytometry. DCs were pulsed with OVA IC (Alexa Fluor 647 labelled OVA) for 1, 2, 5, 10, 15 min or 1 h (different for each antibody), and pulsed for 2 h and chased for 24 and 48 h. Co‐localization between OVA IC and EEA1, Rab5, Rab7, LAMP1, MHCI, MHCII, TAP1 and PA28β was analysed by imaging flow cytometry
Taken together, we observed that OVA IC enter the early endosomal pathway after uptake by DCs. Besides, OVA IC are present in endosomal compartments that contain MHCI and MHCII at early time points. However, at later time points (24 and 48 h), OVA IC are stored in LAMP1‐positive compartments, distinct from early endosomal and MHCII loading compartments. Since TAP1 and PA28β were not detected in the antigen storage compartments, but shown before to be crucial in antigen processing and cross‐presentation [9, 18], this suggests that antigen is translocated from the compartments into the cell cytosol for further processing.
Antigen storage compartments contain cathepsin X but no cathepsin S
Since the antigen‐containing compartments can be characterized as lysosomal‐like compartments, we investigated the role of lysosomal proteases, such as cysteine cathepsins. The role of cathepsin S in antigen degradation has been described for the vacuolar pathway in DC cross‐presentation [4]. Whether cysteine cathepsins play a role in endosome‐to‐cytosol pathway is not excluded. Therefore, we investigated the presence of proteolytically active cysteine cathepsins in antigen‐containing compartments in DCs with the use of the cysteine cathepsin quenched activity‐based probe (qABP) BMV109 [15]. This intrinsically dark qABP reacts with active cathepsin X, B, S and L to yield a fluorescent Cy5 conjugate. Lysates of naïve DCs exposed to 1 µM BMV109 for 1 h were run on 15% PAGE gel and scanned for Cy5 fluorescence to show the presence of active cathepsin X, B, S and L, with cathepsin S being the most dominantly labelled (Figure 4a, left lane). Interestingly, after 2‐h OVA IC pulse and 24‐h chase, the amount of cathepsin X was increased compared to immature DCs (Figure 4a, right lane). We continued to investigate cathepsin S and X with the use of confocal microscopy to analyse their presence in the antigen‐positive compartments. BMV109 labelling showed partial co‐localization with antibody‐stained cathepsin S in naïve DCs (Figure 4b, Figure S3A). DCs that were pulsed with fluorescently labelled OVA IC for 30 min revealed co‐localization of OVA IC with BMV109 labelling, but not with cathepsin S (Figure 4b, Figure S3A). After 2‐h OVA IC pulse and 24‐h chase, co‐localization between OVA IC and BMV109 labelling was more distinct, but no co‐localization was observed with cathepsin S (Figure 4b, Figure S3A). A more detailed kinetic experiment following co‐localization of OVA IC with cysteine cathepsin activity and cathepsin X in time revealed that BMV109 labelling and cathepsin X were distributed in the cell cytosol in naïve DCs, but became more clustered to a central location 3 h after OVA IC uptake (Figure 4c, Figure S3B). Moreover, after 30‐min, 1‐h or 2‐h OVA IC pulse, OVA IC were co‐localizing with BMV109 fluorescence but not with cathepsin X (Figure 4c, Figure S3B). After 3‐h OVA IC pulse, co‐localization between OVA IC and BMV109 labelling, and partial co‐localization between OVA IC with cathepsin X was observed. When DCs were pulse‐loaded with OVA IC for 2 h and chased for 24 h, both the BMV109 fluorescence and cathepsin X were co‐localizing with OVA IC (Figure 4c, Figure S3B). These results indicate that the antigen storage compartments in DCs contain cathepsin X and possibly other yet unidentified cathepsins, such as cathepsins B or L. We further investigated the presence of Cathepsin X in DCs in vivo, and we observed differences in expression levels in antigen‐presenting cell subsets sorted from C57BL/6 spleens (Figure S3C). Remarkable is that CD8α+ DCs, generally known as the most potent antigen cross‐presenting subset [11, 19, 20, 21], expressed the highest levels of active cathepsin X.
FIGURE 4.
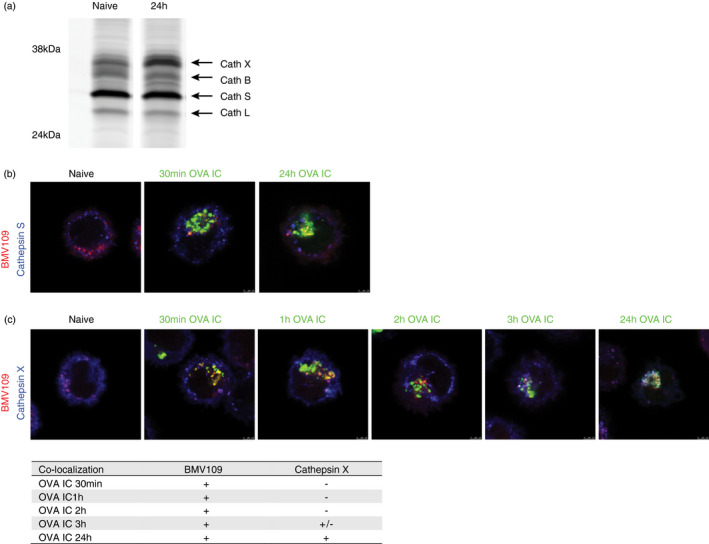
The presence of cathepsins in the antigen storage compartments in DCs. Naïve DCs or DCs pulsed with OVA IC for 2 h and chased for 24 h were run on a 15% PAGE gel. Quenched activity‐based probe BMV109 was used to stain active cathepsin X, B, S and L, indicated by arrows (a). Co‐localization between BMV109 (red) and specific antibody against cathepsin S (blue) was analysed in naïve DCs with confocal microscopy. DCs were pulsed with OVA IC (Alexa Fluor 488) for 30 min or pulsed for 2 h and chased for 24 h. Co‐localization between OVA IC (green), cathepsin S (blue) and BMV109 (red) was analysed by confocal microscopy (b). Naïve DCs, DCs pulsed with OVA IC (Alexa Fluor 488) for 30 min, 1, 2, 3 h or DCs pulsed for 2 h and chased for 24 h were stained with cathepsin X antibody and BMV109. Co‐localization between OVA IC (green), cathepsin X (blue) and BMV109 (red) was analysed by confocal microscopy. Co‐localization between OVA IC, BMV109, cathepsin X and cathepsin S is summarized in a table, ‘+’ indicates co‐localization, ‘+/−’ indicates partial co‐localization, and ‘‐’ indicates no co‐localization (c)
Antigen targeted to MGL1 is stored in LAMP1‐positive compartments in DCs
We extended our antigen uptake studies by specific targeting to C‐type lectin receptor MGL1. LeX is a carbohydrate ligand of the MGL1 receptor. Conjugation of LeX to OVA alters its routing from mannose receptor to MGL1 receptor, which results in prolonged cross‐presentation to CD8+ T cells [10]. We pulsed DCs with OVA‐LeX for 2 h and chased for 2, 24, 48 or 72 h. Similar to OVA IC, OVA‐LeX showed sustained antigen cross‐presentation to T cells, even after 72 h pulse, DCs were able to induce ~75% T‐cell proliferation (Figure 5a). To characterize the routing of the antigen, DCs were pulsed with fluorescently labelled OVA‐LeX for 15 min or pulsed for 2 h and chased for 48 h. Co‐staining between OVA‐LeX and several members of the endosomal trafficking and processing pathways was performed and analysed by confocal microscopy. After 15‐min pulse, OVA‐LeX was distributed through the cell cytosol and co‐localization with EEA1 and partially with Rab5 was observed (Figure 5b, Figure S4). After 48 h, OVA‐LeX was redistributed in a more compact manner located near the nucleus and not co‐localizing with either EEA1 or Rab5. No co‐localization between OVA‐LeX and Rab7 was observed at the measured time points (Figure 5b, Figure S4). Since we observed co‐localization of OVA IC with LAMP1 after 48 h, one could speculate that OVA‐LeX also ends up in similar LAMP1‐positive compartments. Indeed, we observed an overlap of OVA‐LeX with LAMP1 after 48 h, whereas no co‐localization was observed after 15‐min antigen pulse (Figure 5b, Figure S4). Interestingly, OVA‐LeX was present in intracellular MHCI‐ and MHCII‐positive compartments during the early stage of antigen uptake, but after 48‐h chase both MHCI and MHCII were mainly expressed on the cell surface and not co‐localizing with OVA‐LeX (Figure 5b, Figure S4). Both TAP1 and PA28β did not co‐localize with OVA‐LeX regardless of the measured time points (Figure 5b, Figure S4). These results indicate that OVA‐LeX is following a similar uptake route as FcγR‐targeted OVA IC and both receptor‐mediated targeting routes towards the same LAMP1‐positive endolysosomal compartments.
FIGURE 5.
Characterization of the antigen storage compartments in DCs upon MGL1 targeting. DCs were pulsed with OVA‐LeX for 2 h, washed and chased for 2, 24, 48 or 72 h. CFSE‐labelled OTI CD8+ T cells were added, and T‐cell proliferation read‐out was analysed by flow cytometry (a). DCs were pulsed with OVA‐LeX (DyLight 488) for 15 min or pulsed with OVA‐LeX for 2 h and chased for 48 h. OVA‐LeX presence in DCs was imaged by confocal microscopy and specific antibodies against EEA1, Rab5, Rab7, LAMP1, MHCI, MHCII, TAP1 and PA28β (red) were used and analysed for co‐localization with OVA‐LeX (green). Co‐localization between OVA‐LeX and the antibodies is summarized in a table, ‘+’ indicates co‐localization, and ‘−’ indicates no co‐localization (b)
Antigen uptake by FcγRs and MGL1 routes the antigen to the same storage compartments
To investigate the intracellular fate of antigen taken up by different DC uptake receptors, we investigated antigen targeting via FcγRs and the C‐type lectin receptor MGL1, simultaneously. Here, we pulsed DCs with fluorescently labelled OVA IC and OVA‐LeX and followed the antigen in time. Fifteen minutes after antigen pulse, great amounts of both OVA IC and OVA‐LeX were already taken up by DCs (Figure 6). Punctuated hotspots of OVA IC and OVA‐LeX were spread through the cell cytosol and showed partial co‐localization. After 1 h, OVA IC and OVA‐LeX were more compactly distributed in the cell cytosol compared to 15 min, still showing partial co‐localization (Figure 6). More strikingly, after 2‐h pulse loading DCs and 24‐h or 48‐h chase, both OVA IC and OVA‐LeX appeared as concentrated hotspots near the nucleus of DCs (Figure 6). Co‐localization analysis showed complete overlap of both OVA IC and OVA‐LeX, indicating the antigens were present in the same compartments (Figure 6). Moreover, even subsequently internalized antigens were targeted to the same compartments in DCs. DCs that were first pulse‐loaded with OVA IC for 24 h followed by 2 h OVA‐LeX,or the other way around, showed co‐localization of both antigens even when internalized 24 h later (Figure S5). These results demonstrate that targeting antigen to DCs through FcγRs and MGL1 routes the antigen to the same storage compartments at later time points.
FIGURE 6.
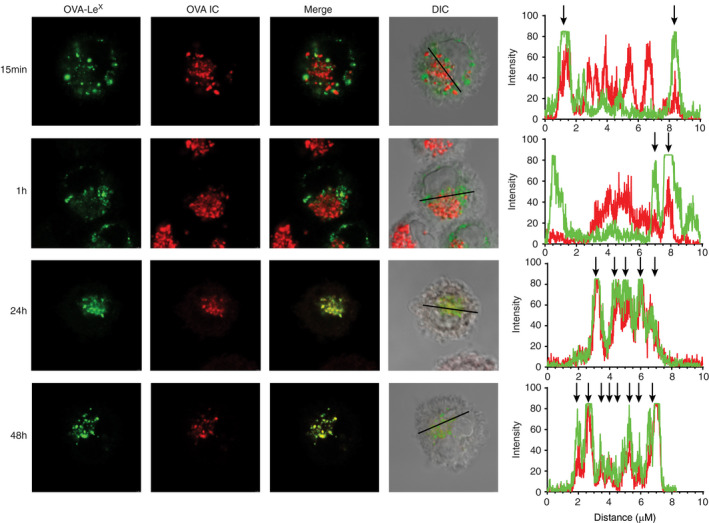
Antigens targeted to FcγRs and MGL1 on DCs end up in the same compartments. DCs were pulsed with OVA IC (Alexa Fluor 647) or OVA‐LeX (DyLight 488) for 15 min and 1 h, or pulsed for 2 h and chased for 24 or 48 h. Co‐localization between OVA IC (red) and OVA‐LeX (green) was visualized by confocal microscopy and DIC was used to image cell contrast. Histograms for each fluorophore were created for a selected area (indicated by a line on the image) and overlays were made with the ImageJ software. Arrows indicate co‐localization between OVA IC (red) and OVA‐LeX (green)
DISCUSSION
We have previously described that antigen targeted to either FcγRs or C‐type lectin receptor MGL1 greatly enhanced sustained antigen cross‐presentation by dendritic cells (DCs) [9, 10, 11]. The presence of antigen storage compartments in DCs where antigen could be conserved for several days, and thereby contributing to prolonged antigen cross‐presentation, had been described before by our group [9]. In the current study, we have further characterized the antigen storage compartments in DCs by tracking protein antigens targeted to FcγRs and C‐type lectin receptor MGL1. Using fluorescently labelled antibody‐bound OVA immune complexes (OVA IC, FcγR targeting) and OVA‐LeX (MGL1 targeting), we observed that antigen from both uptake routes end up in the same compartments in DCs. Several markers of the endosomal trafficking and antigen processing pathways were investigated at different time points and detected by confocal microscopy. We found similar endosomal trafficking pathways of antigen targeted to FcγRs compared to MGL1. We observed that antigen targeted to both receptors were efficiently taken up by DCs already after 15 min. The antigen first entered early endosomal compartments characterized by the presence of EEA1 and Rab5. Co‐localization with MHCI and MHCII was also detected after 15 min antigen uptake, suggesting a route for early antigen presentation. However, when DCs were pulse loaded with antigen for 2 h and chased for 48 h, antigen was stored in LAMP1‐positive compartments, distinct from EEA1, Rab5, or MHCI/ MHCII loading compartments. Moreover, TAP1 or the proteasomal activator PA28β were not present in the storage compartments. We have shown before that antigen cross‐presentation from the storage compartments is TAP1 and proteasome dependent, suggesting that antigen is translocated from the storage compartments into the cytosol for further processing before loading on MHCI [9]. It has been reported that in the endosome‐to‐cytosol pathway antigens need to be transported from endosomal compartments into the cytosol where they are degraded by the proteasome [6, 7]. After proteasomal degradation, these derived peptides are further transported by TAP into the ER. However, it has also been reported that some proteasome‐generated peptides may be transported back into endocytic compartments and trimmed by IRAP and then directly loaded on MHCI molecules [8].
Lysosomal proteases, such as cathepsin S, are essential for the generation of antigenic peptides for MHCII antigen presentation, Ii degradation and dissociation from MHCII [22, 23]. Besides, cathepsin S, B, H and L redistribute from lysosomes to MHCII‐containing endosomal antigen‐processing compartments upon DC activation [24]. The role of cathepsin S in antigen degradation has been described for the vacuolar pathway in DC cross‐presentation [4]. Whether cathepsins play a role in endosome‐to‐cytosol antigen cross‐presentation pathway is not reported yet. Here, we show that the antigen storage compartments containing OVA IC lack cathepsin S activity, but do possess cathepsin X activity, as determined by qABP (BMV109) labelling of cysteine cathepsin activity in combination with immunofluorescence staining. We observed co‐localization of cysteine cathepsin activity with OVA IC already after 30 min uptake. However, staining with antibodies against cathepsin S or cathepsin X did not show any co‐localization with OVA IC after 30 min uptake. The storage compartments where antigen is conserved during later stage were cathepsin S negative but contained cathepsin X; however, the presence of other cathepsins could not be ruled out. Antigen degradation in the storage compartments could be slower due to the lack of cathepsin S. Rapid antigen degradation of internalized antigen is assumed to be negatively affecting cross‐presentation outcome. It has been demonstrated that DCs expressed lower levels of lysosomal proteases compared to macrophages [25]. Expression of cathepsin L, S, D and B in DC phagosomes was reduced compared to macrophages, which could impair phagolysosomal degradation and sustained antigen stability in DCs. Cathepsin X is predominantly found in monocytes, macrophages and dendritic cells [26]. It has been described that cathepsin X played a role in β2 integrin activation in DCs, which was crucial for effective antigen presentation and initiation of T‐cell immune response [27]. Obermajer et al[28] showed that during DC maturation, cathepsin X translocated to the plasma membrane and enabled Mac‐1 activation and cell adhesion. Moreover, cathepsin X redistributed from the membrane to the perinuclear region in mature DCs, which coincided with de‐adhesion of DCs, cell cluster formation and acquisition of the mature phenotype. Importantly, they showed that inhibition of cathepsin X in DCs reduced cell surface expression of co‐stimulatory molecules, abolished cytokine production, diminished DC migration and decreased stimulation of CD4+ T cells. In contrast to other cathepsins, cathepsin X is an exopeptidase and mainly acting as carboxypeptidase [29, 30]. In our previous study [11] and reported by Kamphorst et al[31], it is shown that different spleen‐derived DC subsets have differential antigen presentation capacity in MHC class II and I. We have studied the presence of active cathepsins in DC subsets purified from spleens and observed high expression of cathepsin X in the CD8α+ cDC1 dendritic cell subset known as the most potent cross‐presenting DC subset. This suggests a possible role for cathepsin X in pre‐processing antigen for MHCI loading; however, additional research is needed to determine the role of cathepsin X in DC cross‐presentation.
Since the turnover rate of surface MHCI molecules is shorter compared to MHCII, most MHCI–peptide complexes disappear from the cell surface within 24 h [9]. The migration of DCs after antigen encountering to the T‐cell zones might take up to several days; therefore, prolonged antigen storage in DCs and sustained supply of newly synthesized MHCI–peptide complexes is beneficial to ensure efficient T‐cell cross‐priming.[13] The current study was performed with in vitro‐cultured BMDCs; however, the existence and relevance of antigen storage compartments in vivo were confirmed by our previous work where we showed sustained antigen presence in different DC subsets and prolonged cross‐presentation in vivo [11]. The presence of storage compartments was visible after isolation of both CD8α+ and CD8α− DC subsets after in vivo antigen uptake, showing similar punctuated endosomal structures. These DC subsets could efficiently present antigen to CD8+ and CD4+ T cells, respectively. Taken together, our studies indicate the importance of antigen storage compartments in DCs in vitro as well as in vivo.
Next to direct antigen uptake, processing and presentation, also carry‐over of antigen from one DC type to another is currently accepted as a feasible model of cross‐presentation in vivo [32, 33, 34] as the role of multiple DC subsets in T cell priming become more evident [35, 36, 37, 38]. In this two‐step priming model, naïve CD4+ and CD8+ T cells are activated by different DC populations. The activated CD8+ T cells recruit lymph node‐resident XCR1+ DCs, which receive cross‐presented antigen from the DCs that carried out the first priming step [39, 40]. The XCR1+ DCs interact with both activated CD4+ and CD8+ T cells and thereby inducing optimal signals for CD8+ T cell differentiation into cytotoxic T lymphocytes (CTLs) and memory CTLs. We speculate that antigen storage compartments may play a central role in these processes between cell types as well since prolonged antigen storage and presentation by DCs would be beneficial for this multi‐step T cell priming model.
Here, we show that antigen targeted to different uptake receptors on DCs can result in the same antigen routing and ultimately be conserved in LAMP1‐positive endolysosomal storage compartments. The absence of cathepsin S and core components of the antigen processing and loading machineries in these compartments suggest that the main function of the compartments is to store antigen for sustained antigen presentation, which is crucial to elicit prolonged efficient T cell priming, although the possibility of antigen pre‐processing by cathepsin X could be an interesting additional function of these compartments that requires future research. The current understanding of the intracellular pathways underlying DC cross‐presentation reveals a complex molecular and subcellular interplay.
CONFLICT OF INTERESTS
There are no competing interests.
AUTHOR CONTRIBUTIONS
Each author acknowledges that he/she has contributed in a substantial way to the work described in the manuscript and its preparation.
ETHICAL APPROVAL
The experiments conducted on animals were conducted in accordance with the legislation and guidelines of the Netherlands.
Supporting information
Fig S1‐S5
ACKNOWLEDGEMENTS
N.I.H., M.G.M.C. and F.O. designed the study. N.I.H., M.G.M.C., J.J.G.V. and E.B. carried out research experiments. N.I.H., M.G.M.C., J.J.G.V., E.B., A.J.K, M.V., Y.v.K. and F.O. analysed and interpreted the data. N.I.H. and F.O. wrote the manuscript.
Ho NI, Camps MG, Garcia‐Vallejo JJ, Bos E, Koster AJ, Verdoes M, et al. Distinct antigen uptake receptors route to the same storage compartments for cross‐presentation in dendritic cells. Immunology. 2021;164:494–506. 10.1111/imm.13382
Funding information
This work was financially supported by ZonMW TOP 91211011 to Ferry Ossendorp
REFERENCES
- 1. Fehres CM, Unger WWJ, Garcia‐Vallejo JJ, van Kooyk Y . Understanding the biology of antigen cross‐presentation for the design of vaccines against cancer. Front Immunol. 2014;5:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Embgenbroich M, Burgdorf S. Current concepts of antigen cross‐presentation. Front Immunol. 2018;9:1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ho NI, Huis in ‘t Veld LGM, Raaijmakers TK, Adema GJ. Adjuvants enhancing cross‐presentation by dendritic cells: the key to more effective vaccines? Front Immunol. 2018;9:2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP‐independent MHC class I crosspresentation in vivo. Immunity. 2004;21:155–65. [DOI] [PubMed] [Google Scholar]
- 5. Kovacsovics‐Bankowski M, Rock KL. A phagosome‐to‐cytosol pathway for exogenous antigens presented on MHC class I molecules. Science (80). 1995;267:243–6. [DOI] [PubMed] [Google Scholar]
- 6. Ackerman AL, Kyritsis C, Tampe R, Cresswell P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci USA. 2003;100:12889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Palmowski MJ, Gileadi U, Salio M, Gallimore A, Millrain M, James E, et al. Role of immunoproteasomes in cross‐presentation. J Immunol. 2006;177:983–90. [DOI] [PubMed] [Google Scholar]
- 8. Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, et al. IRAP identifies an endosomal compartment required for MHC class I cross‐presentation. Science (80‐). 2009;325:213–7. [DOI] [PubMed] [Google Scholar]
- 9. Van Montfoort N, Camps MG, Khan S, Filippov DV, Weterings JJ, Griffith JM, et al. Antigen storage compartments in mature dendritic cells facilitate prolonged cytotoxic T lymphocyte cross‐priming capacity. Proc Natl Acad Sci USA. 2009;106:6730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Streng‐Ouwehand I, Ho NI, Litjens M, Kalay H, Boks MA, Cornelissen LAM, et al. Glycan modification of antigen alters its intracellular routing in dendritic cells, promoting priming of T cells. Elife. 2016;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ho NI, Camps MGM, de Haas EFE , Ossendorp F. Sustained cross‐presentation capacity of murine splenic dendritic cell subsets in vivo. Eur J Immunol. 2018;48:1164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ho NI, Camps MGM, Verdoes M, Münz C, Ossendorp F. Autophagy regulates long‐term cross‐presentation by murine dendritic cells. Eur J Immunol. 2021;51:835–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol. 2008;9:282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, et al. Maturation stages of mouse dendritic cells in growth factor‐dependent long‐term cultures. J Exp Med. 1997;185:317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verdoes M, Oresic Bender K, Segal E, van der Linden WA , Syed S, Withana NP, et al. Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. J Am Chem Soc. 2013;135:14726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phadwal K. Analyzing the colocalization of MAP1LC3 and lysosomal markers in primary cells. Cold Spring Harb Protoc. 2015;2015:823–9. [DOI] [PubMed] [Google Scholar]
- 17. Peters PJ, Bos E, Griekspoor A. Cryo‐Immunogold Electron Microscopy. Current Protocols in Cell Biology. 2006;30:1. [DOI] [PubMed] [Google Scholar]
- 18. Ossendorp F, Fu N, Camps M, Granucci F, Gobin SJP, van den Elsen PJ , et al. Differential expression regulation of the α and β subunits of the PA28 proteasome activator in mature dendritic cells. J Immunol. 2005;174:7815–22. [DOI] [PubMed] [Google Scholar]
- 19. den Haan JM , Lehar SM, Bevan MJ. CD8(+) but not CD8(‐) dendritic cells cross‐prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8‐ dendritic cells, but cross‐presented to CD8 T cells by CD8+ dendritic cells. J Immunol. 2001;166:5327–30. [DOI] [PubMed] [Google Scholar]
- 21. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (80‐). 2007;315:107–11. [DOI] [PubMed] [Google Scholar]
- 22. Nakagawa TY, Brissette WH, Lira PD, Griffiths RJ, Petrushova N, Stock J, et al. Impaired invariant chain degradation and antigen presentation and diminished collagen‐induced arthritis in cathepsin S null mice. Immunity. 1999;10:207–17. [DOI] [PubMed] [Google Scholar]
- 23. Shi GP, Villadangos JA, Dranoff G, Small C, Gu L, Haley KJ, et al. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999;10:197–206. [DOI] [PubMed] [Google Scholar]
- 24. Lautwein A, Burster T, Lennon‐Duménil A‐M, Overkleeft HS, Weber E, Kalbacher H, et al. Inflammatory stimuli recruit cathepsin activity to late endosomal compartments in human dendritic cells. Eur J Immunol. 2002;32:3348–57. [DOI] [PubMed] [Google Scholar]
- 25. Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen‐presenting cells determines antigen fate. Science. 2005;307:1630–4. [DOI] [PubMed] [Google Scholar]
- 26. Perišić Nanut M, Sabotič J, Jewett A, Kos J. Cysteine cathepsins as regulators of the cytotoxicity of NK and T cells. Front Immunol. 2014;5:616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kos J, Jevnikar Z, Obermajer N. The role of cathepsin X in cell signaling. Cell Adh Migr. 2009;3:164–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Obermajer N, Svajger U, Bogyo M, Jeras M, Kos J. Maturation of dendritic cells depends on proteolytic cleavage by cathepsin X. J Leukoc Biol. 2008;84:1306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nägler DK, Zhang R, Tam W, Sulea T, Purisima EO, Ménard R. Human cathepsin X: a cysteine protease with unique carboxypeptidase activity. Biochemistry. 1999;38:12648–54. [DOI] [PubMed] [Google Scholar]
- 30. Klemencic I, Carmona AK, Cezari MHS, Juliano MA, Juliano L, Guncar G, et al. Biochemical characterization of human cathepsin X revealed that the enzyme is an exopeptidase, acting as carboxymonopeptidase or carboxydipeptidase. Eur J Biochem. 2000;267:5404–12. [DOI] [PubMed] [Google Scholar]
- 31. Kamphorst AO, Guermonprez P, Dudziak D, Nussenzweig MC. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J Immunol. 2010;185:3426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node‐resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–62. [DOI] [PubMed] [Google Scholar]
- 33. Srivastava S, Ernst JD. Cell‐to‐cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe. 2014;15:741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gurevich I, Feferman T, Milo I, Tal O, Golani O, Drexler I, et al. Active dissemination of cellular antigens by DCs facilitates CD8+ T‐cell priming in lymph nodes. Eur J Immunol. 2017;47:1802–18. [DOI] [PubMed] [Google Scholar]
- 35. Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust anti‐viral immunity requires multiple distinct T cell‐dendritic cell interactions. Cell. 2015;162:1322–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity. 2015;43:554–65. [DOI] [PubMed] [Google Scholar]
- 37. Kitano M, Yamazaki C, Takumi A, Ikeno T, Hemmi H, Takahashi N, et al. Imaging of the cross‐presenting dendritic cell subsets in the skin‐draining lymph node. Proc Natl Acad Sci USA. 2016;113:1044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635–47. [DOI] [PubMed] [Google Scholar]
- 39. Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross‐presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. 2010;207:1273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brewitz A, Eickhoff S, Dähling S, Quast T, Bedoui S, Kroczek RA, et al. CD8 + T cells orchestrate pDC‐XCR1 + dendritic cell spatial and functional cooperativity to optimize priming. Immunity. 2017;46:205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5