

Abstract
Ectopic lymphoid follicles (ELFs), resembling germinal centre‐like structures, emerge in a variety of infectious and autoimmune and neoplastic diseases. ELFs can be found in the meninges of around 40% of the investigated progressive multiple sclerosis (MS) post‐mortem brain tissues and are associated with the severity of cortical degeneration and clinical disease progression. Of predominant importance for progressive neuronal damage during the progressive MS phase appears to be meningeal inflammation, comprising diffuse meningeal infiltrates, B‐cell aggregates and compartmentalized ELFs. However, the absence of a uniform definition of ELFs impedes reproducible and comparable neuropathological research in this field. In this review article, we will first highlight historical aspects and milestones around the discovery of ELFs in the meninges of progressive MS patients. In the next step, we discuss how animal models may contribute to an understanding of the mechanisms underlying ELF formation. Finally, we summarize challenges in investigating ELFs and propose potential directions for future research.
Keywords: autoimmunity, B Cell, neurodegeneration, neuroinflammation, T follicular helper cell
ELFs are frequently found in the meninges of the deep sulci in about 40% of investigated progressive MS tissues. The typical structure of organized ELFs resembles the architecture of germinal centres in secondary lymphoid organs. In addition to compartmentalized B‐ and T‐cell zones, ELFs also feature specialized TFH cells, which are in close contact to B cells (predominantly CD27+ memory B cells), as well as follicular dendritic cells (FDC), which are essential for B‐cell differentiation and activation.
![]()
Abbreviations
- 2D2 mice
C57BL/6 2D2MOG35–55‐specific mice
- ATAMS
Atacicept in multiple sclerosis
- BAFF
B‐cell‐activating factor
- BBB
Blood–brain barrier
- BCR
B‐cell receptor
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- CXCL
Chemokine (C‐X‐C motif) ligand
- CXCR
Chemokine (C‐X‐C motif) receptor
- EAE
Experimental autoimmune encephalomyelitis
- ELFs
Ectopic lymphoid follicles
- EMA
European Medicines Agency
- FDA
U.S. Food and Drug Administration
- FDC
Follicular dendritic cells
- FOXP3
Forkhead box protein 3
- IFN‐γ
Interferon gamma
- Ig
Immunoglobulin
- IL
Interleukin
- MBP
Myelin basic protein
- MOG
Myelin oligodendrocyte protein
- MS
Multiple sclerosis
- NFATc1
Cytoplasmic nuclear factor of activated T cells 1
- PLP
Proteolipid protein
- RORγt
Retinoic acid receptor‐related orphan receptor‐γt
- RRMS
Relapsing–remitting multiple sclerosis
- S1Pr
Sphingosine 1‐phosphate receptor
- SPMS
Secondary progressive multiple sclerosis
- TCR
T‐cell receptor
- TFH
Follicular T‐helper cells
- TFR cells
FOXP3+ regulatory follicular T cells
- Th mice
MOG‐specific Ig heavy‐chain knock‐in mice
- TNF
Tumour necrosis factor
PATHOLOGY OF PROGRESSIVE MS AND B‐CELL TARGETED THERAPY
Multiple sclerosis (MS) is an inflammatory demyelinating and neurodegenerative disease of the central nervous system (CNS), characterized by inflammatory plaques in the white matter, demyelination and axonal damage. Clinically, most patients initially present with a relapsing–remitting disease course (RRMS), which is characterized by the appearance of recurrent neurological symptoms and subsequent partial or complete recovery. After 10–15 years, 85% of the patients enter the secondary progressive MS disease stage (SPMS), in which there is a progressive accumulation of disability over time. Around 10% of all patients initially present with a deterioration of symptoms without classical relapses and remissions from the disease onset, called primary progressive multiple sclerosis (PPMS).
At the pathological level, the progressive MS disease courses (i.e. SPMS and PPMS) display a set of key histopathological and clinical characteristics, including (i) the presence of ectopic lymphoid follicles (ELFs) in the meninges (in around 40% of SPMS patients) [1], (ii) grey matter demyelination and brain atrophy [2, 3], (iii) diffuse white matter damage [3], (iv) profound oxidative injury [4, 5], (v) progressive worsening of motor and/or cognitive functions [6], (vi) moderate parenchymal immune cell infiltration [7] and (vii) a relative preservation of the blood–brain barrier (BBB) integrity, especially when compared to the focal yet severe BBB integrity loss during RRMS [8, 9]. While RRMS is understood to be an autoimmune‐mediated and inflammation‐driven disease, the influence of T‐cell‐driven inflammatory processes on disease progression and neurodegeneration in progressive MS is controversially discussed.
Various therapeutic agents are currently available for the symptom control of RRMS. In 2008, Hauser et al. demonstrated that the use of rituximab, a chimeric CD20+ B‐cell‐depleting monoclonal antibody, can significantly reduce disease activity and lesion formation in RRMS [10]. The rapid and marked effect of a B‐cell‐depleting therapy on MS disease activity has drawn attention to B cells and T‐cell / B‐cell interactions in RRMS. Of note, increased clinical disease activity was observed in the randomized, placebo‐controlled, double‐blind, phase 2 trial ‘atacicept in multiple sclerosis’ (ATAMS) in RRMS patients treated with atacicept, a recombinant fusion protein that suppresses B‐cell function and antibody production of plasma cells [11], highlighting a complex yet important function of B cells and plasma cells during MS.
Drugs aiming to delay the progression of disability during progressive MS are exceptionally scarce [6], which reflects the elusive pathological mechanisms underlying MS disease progression. Currently, only two drugs (i.e. siponimod [Mayzent®] and ocrelizumab [Ocrevus®]) have been approved for the treatment of progressive MS [6, 12, 13, 14, 15]. In 2017, the B‐cell‐depleting drug ocrelizumab, a second‐generation humanized anti‐CD20 antibody, was approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of PPMS, which has become a milestone in progressive MS therapy [13]. Although the mode of action of ocrelizumab in progressive MS is yet to be fully elucidated, the success of a B‐cell‐depleting therapy in progressive MS emphasizes essential roles of B cells in disease progression during progressive MS [16, 17].
Drugs targeting B‐cell epitopes are not used exclusively in the treatment of MS. For instance, the use of rituximab has been reported to ameliorate symptoms of patients with Susac's syndrome [18, 19], which is a rare cerebral microangiopathy, mainly driven by cytotoxic CD8+ T cells causing an endotheliopathy, which leads to a progressive demyelination of the CNS white matter with subsequent neurological deficits [20]. Another disease, which is as well characterized by high numbers of intracerebral CD8+ T cells is Rasmussen's encephalitis, causing a severe inflammatory response strictly limited to one cerebral hemisphere [21]. Therapeutic attempts with monoclonal anti‐CD20 antibodies (i.e. rituximab) have been reported to improve clinical symptoms in cases of Rasmussen's encephalitis, although the number of cytotoxic T cells significantly exceeds the number of B cells [22, 23]. These two examples suggest close interactions between B cells and T cells, in particular CD8+ cytotoxic T cells. Of note, MS is as well a CD8+ T‐cell‐dominated disease [24, 25]. A presumed involvement of B cells in progressive MS is mainly based on three evidences: (i) the presence of ELFs containing B cells in the meninges of around 40% of SPMS patients, which is associated with a more severe cortical pathology and disease progression [1, 26, 27], (ii) increased levels of immunoglobulins in the cerebrospinal fluid (CSF) of RRMS and progressive MS patients [28] and (iii) clinical alleviation of disease progression in progressive MS by B‐cell‐depleting therapies [13, 29, 30].
In this review, we first summarize neuropathological evidence for the presence of ELFs in the meninges of progressive MS patients (Table 1). Then, we review animal studies, which have been performed to shed light on the mechanisms involved during ELF formation in the meninges (Table 2), and briefly discuss how clinical and pathological features of progressive MS can be modelled in different animal models (Table 3). Finally, we aim to point out challenges and provide possible directions for future studies addressing the relevance of ELFs for MS disease progression.
TABLE 1.
Main studies on meningeal inflammation and ELFs in progressive MS
| Year | MS type and patient numbers | Tissue samples | Main technique | Key finding(s) on ELFs | Ref |
|---|---|---|---|---|---|
| 2004 | 3 SPMS and 2 PPMS | Brain, spinal cord | IHC/IF |
|
[26] |
| 2007 | 29 SPMS and 7 PPMS | Brain | IHC/IF |
|
[1] |
| 2007 | 13 SPMS and 3 PPMS | Brain, CSF | IHC/IF, PCR, ISH |
|
[31] |
| 2009 | 12 SPMS and 7 PPMS | Brain | IHC |
|
[57] |
| 2010 | 7 SPMS | Brain | Microarray, PCR |
|
[46] |
| 2010 | 37 SPMS | Brain | IHC/IF |
|
[54] |
| 2010 | 7 SPMS | Brain, CSF | IHC, B cell repertoire analysis |
|
[58] |
| 2011 | 123 SPMS | Brain | IHC/IF |
|
[27] |
| 2012 | 26 PPMS | Brain | IHC |
|
[56] |
| 2013 | 12 SPMS (6 ELFs‐positive and 6 ELFs‐negative) | Brain, CSF | IHC/IF |
|
[62] |
| 2015 | 5 SPMS or PPMS | Brain | High‐throughput antigen arrays |
|
[59] |
| 2016 | 10 SPMS | Brain | IHC/IF |
|
[65] |
| 2018 | 11 SPMS | Brain | IHC/IF, digital PCR |
|
[53] |
| 2018 | 21 SPMS/PPMS, 12 acute MS# | Brain | IHC/IF, ISH |
|
[122] |
| 2020 | 22 SPMS, 11 PPMS | Brain, spinal cord | IHC/IF |
|
[66] |
| 2020 | 22 SPMS | Brain, spinal cord | IHC/IF |
|
[55] |
Abbreviations: #acute MS, MS cases that were diagnosed and died before the advent of disease‐modifying therapies; ↑, upregulation; IF, immunofluorescence; IFN‐γ, interferon gamma; Ig, immunoglobulin; IHC, immunohistochemistry; ISH, in situ hybridization; PCR, polymerase chain reaction; RORγt, retinoic acid receptor‐related orphan receptor γt; TNF, tumour necrosis factor.
TABLE 2.
Main studies on meningeal inflammation and ELFs in MS‐related animal models
| Year | Model(s) used | Key finding(s) on ELFs | Subsidiary findings | Ref |
|---|---|---|---|---|
| 1990s | Lewis rat immunized with MBP |
|
|
[83, 84] |
| 2003 | 2D2 mice |
|
|
[81] |
| 2004 |
PLP‐EAE ABH‐EAE |
|
|
[85] |
| 2006 | PLP‐EAE |
→ inhibits the formation of ELFs |
|
[90] |
| 2006 | 2D2×Th |
|
|
[91] |
| 2006 | MP4‐EAE |
|
|
[71] |
| 2009 | Adoptive transfer of TH1, TH2, TH9, TH17 from 2D2×Th |
|
|
[92] |
| 2011 | Adoptive transfer of TH17 from 2D2×Th |
|
|
[93] |
| 2012 | MP4‐EAE |
|
|
[33] |
| 2013 | Crossbred of transgenic MOG‐specific B‐cell receptor mice and Th mice |
|
|
[95] |
| 2015 | Injection of IFN‐γ and TNF into the subarachnoid space of Dark Agouti rats preimmunized with MOG |
|
|
[62] |
| 2015 | 2D2×Th |
|
|
[94] |
| 2015 | PLP‐EAE |
|
→ GC formation in NP‐KLH mice↓ |
[97] |
| 2016 | 2D2×Th |
|
|
[34] |
| 2016 | 2D2×Th |
|
|
[106] |
| 2018 |
MOG‐EAE Adoptive transfer EAE model |
|
|
[107, 108] |
| 2018 | Conditional knockout of PD‐L1 in CD11c+ dendritic cells together with adoptive transfer EAE |
|
|
[109] |
| 2019 | MP4‐EAE |
|
|
[96] |
Abbreviations: ↑, upregulation/ increased; ↓, reduced; 2D2×Th, a spontaneous EAE model derived from the crossbred of TCR transgenic mice (C57BL/6 2D2 MOG35–55‐specific, referred to as 2D2 mice) and MOG‐specific Ig heavy‐chain knock‐in mice (referred to as Th mice); ABH‐EAE, Biozzi ABH mice immunized with spinal cord homogenate developing a disease course with relapsing–remitting episodes and secondary progressive disability; GC, germinal centre; IHC, immunohistochemistry; MOG‐EAE, C57BL/6 mice immunized with MOG35‐55 peptide developing a monophasic chronic disease course; MP4‐EAE, C57BL/6 mice immunized with MPB‐PLP fusion protein (MP4) to induce a B‐cell‐dependent pathology; n.a., not applicable; NP‐KLH, 4‐hydroxy‐3‐nitrophenyl acetyl hapten conjugated to keyhole limpet haemocyanin; PD‐L1, programmed death ligand 1; PLP‐EAE, SJL mice immunized with PLP139–151 peptide developing a relapsing–remitting disease course; TFH, follicular T‐helper cells; TH, T‐helper cells.
TABLE 3.
Correlation between MS‐related animal models and clinical/pathological features of progressive MS
| MS‐related animal model | Hypothesized driving force for disease progression | Clinical/pathological features of progressive MS | ||||||
|---|---|---|---|---|---|---|---|---|
| Presence of ELFs | Grey matter demyelination and/or brain atrophy | Diffuse white matter damage | Profound oxidative injury | Progressive worsening of motor/cognitive functions | Moderate parenchymal immune cell infiltration | Moderate BBB integrity loss | ||
| MOG‐EAE | Inflammation | Moderate | Severe | |||||
| PLP‐EAE | Inflammation | Moderate | Severe | |||||
| MP4‐EAE | Inflammation | Moderate | Severe | |||||
| ABH‐EAE | Inflammation | Severe | ||||||
| 2D2×Th | Inflammation | Moderate | Severe | |||||
| Cup/EAE | Neurodegeneration | Unknown | ||||||
| Cup | Neurodegeneration | |||||||
Characteristics can be studied (green) or cannot be studied (red).
MOG‐EAE: C57BL/6 mice immunized with MOG35‐55 peptides develop a monophasic, chronic disease course.
PLP‐EAE: SJL mice immunized with PLP139–151 peptides develop a relapsing–remitting disease course.
MP4‐EAE: C57BL/6 mice immunized with an MPB‐PLP fusion protein (MP4) induces a B‐cell‐dependent pathology.
ABH‐EAE: Biozzi ABH mice immunized with spinal cord homogenate develop a disease course with relapsing–remitting episodes and a secondary progressive disability.
2D2×Th: A spontaneous EAE model derived from the crossbreeding of TCR transgenic mice (C57BL/6 2D2 MOG35–55‐specific) and MOG‐specific Ig heavy‐chain knock‐in mice (referred to as Th mice).
Cup/EAE: MOG‐EAE mice predisposed to a 3‐week cuprizone intoxication and 2‐week normal chow develop inflammatory demyelination in the forebrain.
Cup: C57BL/6 mice intoxicated with cuprizone, a reagent inducing apoptosis in mature oligodendrocytes, developing innate immune activation within the CNS followed by demyelination.
TERMINOLOGY OF ELFS IN MS
A variety of terms have been used to describe ELFs in the meninges of progressive MS patients, including meningeal B‐cell follicles, meningeal ectopic lymphoid follicle‐like structures, meningeal ectopic lymphoid tissues, meningeal tertiary lymphoid tissue or ectopic B‐cell follicles [1, 26, 31, 32, 33, 34]. In this article, we consistently use the abbreviation ‘ELFs’ to refer to the above‐mentioned structures as ectopic lymphoid follicles. In the related literature, the fundamental definition of ELFs ranges from ‘meningeal foci’ (e.g. >10 inflammatory cells) to germinal centre‐like structures consisting of stromal/follicular dendritic cells, T cells and B cells, as well as plasma cells. To make a clear distinction between ELFs and meningeal inflammation, we adopt the definition of ELFs formulated by Aloisi et al.: large B‐cell aggregates localized in the subarachnoid space, mainly inside the cerebral sulci, that display several germinal centre‐like features (i.e. presence of stromal/follicular dendritic cells expressing CXCL13, B‐cell proliferation, expression of activation‐induced cytidine deaminase and plasma cell differentiation) but lack the typical structure of lymphoid follicles with a germinal centre and a mantle zone and contain mainly memory B cells [1, 26, 32] (Figure 1).
FIGURE 1.
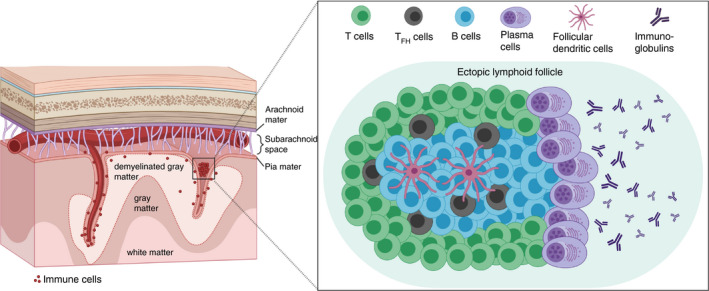
Schematic illustration of the architecture of ectopic lymphoid follicles (ELFs) in the CNS of progressive multiple sclerosis patients. ELFs are frequently found in the meninges of the deep sulci in about 40% of investigated progressive MS tissues. The typical structure of organized ELFs resembles the architecture of germinal centres in secondary lymphoid organs. In addition to compartmentalized B‐ and T‐cell zones, ELFs also feature specialized TFH cells, which are in close contact to B cells (predominantly CD27+ memory B cells), as well as follicular dendritic cells (FDC), which are essential for B‐cell differentiation and activation. B cells that experienced a first T‐cell‐dependent and a second FDC‐ or TFH cell‐supported antigen contact can mature into immunoglobulin‐producing plasma cells. The immunoglobulins, if directed against CNS‐specific antigens, could play an important role during disease progression in progressive MS
NEUROPATHOLOGICAL EVIDENCE OF ELFS IN PROGRESSIVE MS
In some studies, the presence of ELFs in SPMS patients' meninges is positively correlated with disease progression [1, 26, 27]. However, it remains largely unknown to what extent ELFs contribute to the progression of MS and what the underlying pathophysiological mechanisms are. Many conclusions drawn about ELFs in MS are based on the analysis of post‐mortem brain tissues of progressive MS cohorts including some RRMS and undetermined MS cases. In this review, we chronologically summarize neuropathological studies to provide a historical perspective (summarized in Table 1).
Detection and typical chemokine expression of ELFs in SPMS
Although immune cell infiltration in MS has been studied since the 1970s and remains a major focus of MS research [35, 36, 37, 38], it was not until 2004 when Serafini et al. [26] reported the existence of ELFs containing CD20+ B cells, CD3+ T cells, CD138+ plasma cells and a network of CD21+CD35+ follicular dendritic cells producing chemokine (C‐X‐C motif) ligand 13 (CXCL13) in the cerebral meninges of 2 of 3 SPMS patients. No ELFs were found in RRMS (1 patient investigated), PPMS (2 patients investigated) or a non‐neurological control (1 patient investigated) [26]. Of note, chemokines, such as CXCL12 and CXCL13, have been shown to regulate B‐cell migration and germinal centre organization in secondary lymphoid tissues [39]. In 2006, another group investigated the expression of CXCL12 and CXCL13 in CSF samples from a cohort of 30 RRMS, 8 PPMS and 14 SPMS together with 14 non‐inflammatory neurological disease patients. The CSF of the majority of SPMS and PPMS patients showed increased levels of CXCL12 when compared to non‐inflammatory neurological disease patients but little or no detectable CXCL13 [40]. In the majority of RRMS patients, CXCL12 and CXCL13 levels were both elevated compared with non‐inflammatory neurological disease patients and the detected levels correlated positively with intrathecal immunoglobulin production and the presence of B cells in the CNS. The low levels of CXCL13 in the CSF of SPMS and PPMS patients were further verified in a larger cohort of 40 SPMS and 24 PPMS patients together with 14 healthy controls [41]. Based on these results, although CXCL13‐producing follicular dendritic cells were previously observed in ELFs, so far there is no convincing evidence that ELFs are linked to the levels of CXCL13 in the CSF of progressive MS patients. Levels of CXCL13 in the CSF of RRMS patients and other inflammatory neurological disease patients can provide information about the extent of inflammation in the CNS. However, activated macrophages act as an additional source of CXCL13 [40, 42, 43] and can be found in high densities in the CNS of RRMS patients [44]. Thus, elevated levels of CXCL13 do not necessarily indicate a possible presence of CXCL13‐producing follicular dendritic cells or the existence of ELFs in the meninges of RRMS patients.
Association between ELFs and disease progression in MS
The first study to investigate a possible clinical impact of the presence of ELFs in progressive MS was published in 2007. Magliozzi et al. [1] reported that in 41.4% of SPMS patients, but neither in PPMS nor in control patients, ELFs can be found. These ELFs were mainly located in the subarachnoid space of the meninges entering the cerebral sulci, adjacent to large subpial cortical lesions. Most importantly, the presence of ELFs positively correlated with an early disease onset and severe cortical demyelination in the investigated cohort of SPMS patient [1]. By using in situ hybridization, Serafini et al. found that ELFs in the meninges of SPMS patients are major sites of Epstein–Barr virus infection (15 of 22 patients investigated) [31]. Epstein–Barr virus is a human pathogenic B‐lymphotropic DNA herpesvirus that is strongly associated with MS prevalence based on large‐scale sero‐epidemiological studies showing higher anti‐Epstein–Barr virus antibody titres in MS patients compared with control individuals paralleled by a higher risk of developing MS after Epstein–Barr virus‐induced infectious mononucleosis [45]. At sites of major accumulations of Epstein–Barr virus‐infected B cells, CD8+ T cells expressing interferon gamma (IFNγ) were also observed, suggesting a proinflammatory cytotoxic environment [31]. Although not in the focus of this review, it is pertinent to point out that a latent Epstein–Barr virus infection, although still controversial [46, 47, 48, 49, 50], might partly contribute to the sustained B‐cell dysregulation in MS [32, 51, 52, 53].
Additional evidence demonstrating an association between the presence of ELFs and cortical pathology in SPMS was published in 2010, when Magliozzi et al. observed that the presence of ELFs was positively correlated with the severity of neuronal loss in superficial cortical layers in SPMS patients. In the investigated post‐mortem brain tissues, SPMS cases with ELFs showed a more severe neuronal degeneration in cortical layers I‐IV compared with SPMS brains without ELFs, where neuronal loss was equally distributed across all cortical layers [54]. Reali et al. recently reported that the presence of ELFs located in meninges of the forebrain, accompanied by a diffuse B‐cell‐dominated inflammation of spinal cord meninges, is associated with ongoing spinal cord pathology in SPMS patients [55]. Although ELFs were only found in the meninges of SPMS but not PPMS patients, Choi et al. [56] showed that diffuse meningeal and perivascular infiltrates consisting of CD3+ T cells and CD20+ B cells can also be found in PPMS patients. Moreover, the extent of meningeal inflammation was positively correlated with the disease severity in PPMS.
Challenges in the detection of ELFs in SPMS
The successful detection of ELFs depends on (i) extensive tissue sampling due to the heterogeneous and sparse distribution of ELFs per se in SPMS patients, and (ii) optimal preservation of meningeal structures during the preparation and embedding procedures of the investigated brain tissues [32].
Not all studies were able to demonstrate the presence of ELFs in the meninges of cases with progressive MS. Based on a cohort investigating 7 PPMS and 12 SPMS patients, Kooi et al. found significant meningeal inflammation, composed mainly of CD3+ T cells, CD20+ B cells, CD68+ macrophages, granzyme B+ activated cytotoxic T cells and DC‐SIGN+ (im)mature dendritic cells, but no ELFs were detected in this study. In addition, the extent of meningeal inflammation was not found to be associated with the extent of subpial cortical demyelination [57]. Torkildsen et al. found massive upregulation of immunoglobulin (Ig)‐related genes in normal‐appearing grey matter and grey matter lesions of MS patients. They also reported the presence of small numbers of CD3+ T cells, CD20+ B cells and CD138+ plasma cells in the meninges but, in line with the results by Kooi et al., did not observe ELFs in a cohort of 7 SPMS patients [46].
In 2011, Howell et al. [27] conducted an extensive sampling of brain tissues from 123 SPMS patients and found that ELFs could be observed in around 40% of the investigated SPMS cases. A widespread distribution throughout the forebrain was observed, but most frequently, ELFs were found in the deep sulci of the temporal, cingulate, insula and frontal cortex. Moreover, subpial cortical lesions were found both adjacent and distant to ELFs. The presence of ELFs was again positively correlated with the extent of diffuse meningeal inflammation, microglial activation and grey matter demyelination. Clinically, the presence of ELFs positively correlated with more pronounced clinical symptoms and neurological deficits in progressive MS patients [27]. Lovato et al. reported that the majority of the expanded B‐cell clones isolated from ELFs are also present in the CNS parenchyma of progressive MS brain samples [58]. These findings suggest a close association between the occurrence of ELFs in the subarachnoid space and the existence of associated lymphocytic infiltrates in the CNS white and grey matter of progressive MS patients. However, intrathecally produced immunoglobulins in general, as well as immunoglobulins produced by plasma cells originating from ELFs in the meninges, might not display high antigen specificity against CNS‐derived antigens and could therefore be of minor pathological relevance [59].
Insights into the formation of ELFs in SPMS
Since the identification of ELFs in the large cohort of SPMS patients by Howell et al. in 2011 [27], a substantial research focus in this field has shifted aiming to understand how ELFs form and finding potential inhibitors for ELF formation as a prospective therapeutic option to treat progressive MS. Possible methods to evaluate the formation of ELFs, or even to measure the effect of drugs on ELFs, could be done by monitoring ELF‐specific cells in the peripheral blood or the CSF of patients [60].
In 2013, Christensen et al. [61] reported higher frequencies of ICOS+CXCR5+CD4+ follicular T‐helper (TFH) cells, IL23R+CD4+ TH17 cells, DC‐SIGN+ and CD83+ activated B cells, as well as CD27HighCD38High plasmablasts in peripheral blood samples of SPMS patients. They emphasized that elevated levels of ELF‐associated cells can be detected in the periphery and suggest a possible link between systemic inflammation and disease progression [61]. In the same study, higher frequencies of ICOS+ TFH cells together with an increased expression of genes associated with TFH‐ and B‐cell activation in the CSF were additionally found in RRMS cases.
Gardner et al. found elevated levels of the proinflammatory cytokines IFN‐γ and tumour necrosis factor (TNF) in the meninges of SPMS cases with ELFs. Beyond, it was elegantly demonstrated in that study that the application of IFN‐γ and TNF into the subarachnoid space leads to cortical demyelination and inflammation in preimmunized Dark Agouti rats, supporting the hypothesis that proinflammatory molecules, produced in the meninges, play a major role in cortical demyelination in MS [62]. While this study investigated the mechanisms of cortical damage by proinflammatory cytokines, no implications for the possible formation of ELFs in the meninges were drawn. In 2016, two studies identified IFN‐γ signalling as a crucial pathway in regulating autoimmune B‐cell and TFH cell responses [63], as well as spontaneous germinal centre formation [64], emphasizing a possible role of IFN‐γ during ELF formation in progressive MS.
In 2016, Serafini et al. described the presence of interleukin‐17 (IL‐17)‐releasing and retinoic acid receptor‐related orphan receptor‐γt (RORγt)‐positive cells in ELFs of SPMS patients, suggesting that these cells might play critical roles during lymphoid neogenesis in the meninges of progressive MS patients [65]. In 2019, Bell et al. investigated a cohort of 11 PPMS and 22 SPMS patients together with 2 Parkinson's disease controls and 13 healthy controls. B‐cell‐enriched inflammatory infiltrates could be observed in the meninges and the parenchyma of the investigated SPMS cases. However, substantial B‐cell aggregates that meet the definition of ELFs containing CXCL13+ cells were not detected in the investigated cohort of PPMS and SPMS patients. Abundant CXCR5+ cells and cytoplasmic nuclear factor of activated T‐cell‐positive (NFATc1+) cells, enriched CD3+CD27+ memory cells and CD4+CD69+ tissue‐resident cells were, however, identified in the inflammatory meningeal and perivascular infiltrates [66]. Simultaneously, forkhead box protein 3+ (FOXP3+) regulatory T cells were almost entirely absent in the CNS of the investigated patients, which led the authors to conclude that the uncontrolled humoral immune response might allow and promote the formation of CNS‐specific autoantibodies in the investigated infiltrates. However, conclusions about the relevance of ELFs in MS are, in the study by Bell et al., not supported by histological evidence of ELFs in the meninges and are therefore of limited applicability.
Little is known about the cellular dynamics during the formation of ELFs. With regard to the heterogeneous results and descriptions of ELFs in the related literature, a dynamic transition from loose B‐cell aggregates to highly organized and compartmentalized ELFs in the meninges of progressive MS patients seems very likely [26]. The series of events leading to the formation of ELFs is still not known. Two possible cascades are principally conceivable: (i) ELFs arise primarily in the subarachnoid space and contribute to a secondary cortical pathology via cytokine release and immunoglobulin production, or (ii) a primary cortical pathology triggers the formation of ELFs in the meninges via the release of chemotactic substances. The exact mechanism behind the formation of ELFs remains unknown, yet the sequence of events is of great importance for understanding the relevance of ELFs in the context of pathological processes driving disease progression in SPMS patients. In addition, future studies are required to demonstrate the extent to which changes in ELF‐specific cells in the peripheral blood of patients or chemokine levels in the serum or CSF reflect possible changes in the microenvironment of ELFs.
INSIGHTS INTO ELFS FROM ANIMAL STUDIES
The presence of ELFs emerges as one of the hallmarks of SPMS pathology and the amelioration of ELF formation could provide effective new treatment options for progressive MS patients. Animal studies using gene editing or/and pharmacological treatment might support our understanding of how ELFs form and how their formation might be inhibited (summarized in Table 2). In this section of the article, we first describe the animal models used to investigate different clinical and pathological features of progressive MS. Then, we discuss how animal studies provide insights into the development and possible inhibition of ELF formation. Finally, we discuss how emerging knowledge of follicular T‐helper cells (TFH) may improve our understanding of ELF formation in MS.
The experimental autoimmune encephalomyelitis model: many variants for a complex human disease
The experimental autoimmune encephalomyelitis (EAE) model, the most frequently used animal model in MS‐related research, has contributed significantly to the development of several new therapeutic agents for RRMS [67, 68]. In brief, EAE can be induced by active immunization with CNS‐related antigens, for example CNS homogenate, proteins/peptides of myelin basic protein (MBP), proteolipid protein (PLP) or myelin oligodendrocyte protein (MOG), together with (in)complete Freund's adjuvant and pertussis toxin [69]. Certain aspects of CNS inflammation and ensuing neurological symptoms can be studied in a variety of different EAE models [70]. For example, C57BL/6 mice immunized with MOG35‐55 peptides (MOG‐EAE) develop a monophasic and chronic disease course. SJL mice immunized with PLP139–151 peptides (PLP‐EAE) develop a relapsing–remitting disease course. C57BL/6 mice immunized with MPB‐PLP fusion proteins (so‐called MP4‐EAE) are characterized by a strongly B‐cell‐dependent EAE pathology. Biozzi AB/H mice immunized with spinal cord homogenate (ABH‐EAE) exhibit a disease stage with relapsing–remitting episodes followed by secondary disease progression [71, 72, 73, 74]. As recently established by our group, MOG‐EAE induced in mice initially intoxicated with cuprizone (so‐called Cup/EAE model) [75, 76, 77], a chemical compound inducing apoptosis of mature oligodendrocytes, develop multifocal, inflammatory and demyelinating lesions in the forebrain and spinal cord. [78].
Alternatively, EAE can also be induced passively by the adoptive transfer of encephalitogenic lymphocytes, where T cells are isolated from myelin protein/peptide‐primed donors, ex vivo‐restimulated with encephalitogenic peptides and injected into immunocompetent or immunodeficient recipient mice [79]. Furthermore, transgenic mice developing spontaneous EAE (e.g. SJL 5B6 PLP139–151‐specific or C57BL/6 2D2 MOG35–55‐specific T‐cell receptor (TCR) transgenic mice) are available and allow the study of myelin‐specific T cells [80, 81].
It must be noted that there is no perfect animal model for MS, which is a disease described purely in humans with a still unknown disease aetiology, both in RRMS and in progressive disease phases. Most notably, there is an extreme paucity of models for progressive MS, mostly due to the controversial and largely unknown roles of inflammatory processes in disease progression in SPMS and PPMS [82]. Different animal models only reflect distinct features of MS instead of its entire complexity [72]. Therefore, we try to summarize how clinical and pathological features of progressive MS can be modelled in different animal models (i.e. MOG‐EAE, PLP‐EAE, MP4‐EAE, ABH‐EAE, 2D2×Th spontaneous EAE, Cup/EAE and the Cuprizone model, summarized in Table 3).
Insights into the formation of ELFs in different EAE models
The use of a consistent definition for ELFs is essential in both human pathological and experimental animal studies. For a most appropriate comparison of ELF‐related observations in animal models and in human pathological studies, we here again use the definition of Aloisi et al. given in chapter 2 of this manuscript to distinguish ELFs from meningeal inflammation. One exception must be added in this regard for studies on ELFs in rodent brains: as rodent brains do not have gyri and sulci, the localization of ELFs in rodent brains does not apply as a criterion.
Since the 1990s, inflammatory meningeal infiltrates were observed in the spinal cord of rats with actively induced EAE [83, 84]. In 2003, inflammatory meningeal infiltrates were also found in a model of spontaneous autoimmune optic neuritis using TCR transgenic mice [81]. In 2004, meningeal infiltrates resembling ELFs, together with an intracerebral upregulation of CXCL13 and B‐cell‐activating factor (BAFF), were found in models of relapsing–remitting and chronic relapsing EAE, namely in PLP139‐151‐induced EAE in SJL mice and in Biozzi AB/H mice immunized with spinal cord homogenate [85]. In subsequent studies by the same group, it was investigated whether inhibition of lymphoid tissue neogenesis influenced the formation of ELFs and the severity of EAE symptoms in mice. The signalling pathway via the lymphotoxin β receptor on reticular stromal cells plays an important role in the formation of new lymphoid tissue [86]. Activated T and B cells express lymphotoxin‐α1β2 [87] and, by binding to the lymphotoxin β receptor, regulate the induction of lymphoid chemokines such as CCL21 and CXCL13, which regulate the trafficking of lymphocytes within lymphoid tissues [88] and, additionally, contribute to the differentiation of follicular dendritic cells [89]. In 2006, Columba‐Cabezas et al. showed that blocking lymphoid tissue organization by treating mice with a lymphotoxin beta receptor‐Ig fusion protein does not only inhibit the formation of ELFs and reduces T‐ and B‐cell infiltration, but also ameliorates clinical EAE signs, emphasizing critical roles of ELFs for the development of PLP139‐151‐induced EAE in SJL mice [90]. Meanwhile, ELFs were observed in the spinal cord and optic nerves in the spontaneous ‘2D2×Th’‐EAE model, derived from the crossbreeding of TCR transgenic mice (C57BL/6 2D2 MOG35–55‐specific, referred to as 2D2 mice) and MOG‐specific Ig heavy‐chain knock‐in mice (referred to as Th mice) [80, 81, 91].
In 2009, using adoptive transfer of specific subsets of T‐helper cells (i.e. TH1, TH2, TH9, TH17) from 2D2 mice, Jäger et al. found that only TH17 cells cultured with IL‐23 were able to induce the formation of ELFs in the recipient mice [92]. Furthermore, in 2011, the researchers proved that the development of ELFs induced by TH17 cells in the 2D2×Th model is partly dependent on IL‐17 and the cell membrane molecule podoplanin, which is also crucial for the development of secondary lymphoid structures [93]. Lee et al. found that Il21r and Il23r deletion reduces EAE disease severity and abolishes the number of ELFs, respectively, in the 2D2×Th spontaneous EAE model [94]. In 2013, another group reported that the induction of spontaneous opticospinal EAE by the crossbreeding of transgenic MOG‐specific B‐cell receptor (BCR) mice and MOG‐specific Ig heavy‐chain knock‐in mice leads to the development of neurological symptoms closely resembling pathological characteristics of the human Devic's disease with inflammatory lesion development primarily in the optic nerve and the spinal cord. Using this animal model, Molnarfi et al. were able to detect ELFs in transgenic mice, highlighting the important role of B cells in the formation of ELFs [95].
In 2016, Lehmann‐Horn et al. further described that ELFs exhibit characteristics of germinal centre‐like activity in the 2D2×Th spontaneous EAE model. They could detect an increased expression of activation‐induced cytidine deaminase in the ELFs, which is required for somatic hypermutation and class switch recombination [34]. In 2012, Kuerten et al. proved that the formation of B‐cell aggregates and ELFs in the cerebellum can be induced using the MP4‐EAE model, which is a strongly B‐cell‐dependent MS model [33]. Further investigations by the same group showed that CD3+CD5+CD4+RORγt+ TH17 cells rather than CD3−CD5−CD4+RORγt+ lymphoid tissue inducer cells might contribute to the formation of ELFs in the MP4‐EAE model, emphasizing the importance of TH17 cells in the formation of ELFs [96].
As previously mentioned, CXCL13 plays a crucial role in B‐cell migration and germinal centre organization in lymphoid tissues. In 2015, Klimatcheva et al. developed a human monoclonal anti‐CXCL13 antibody (i.e. MAb 5261) and elegantly demonstrated that the antibody could inhibit germinal centre formation induced by 4‐hydroxy‐3‐nitrophenyl acetyl hapten conjugated to keyhole limpet haemocyanin. Moreover, the novel antibody reduced the clinical severity in both actively induced PLP139‐151‐EAE and passively induced EAE models with adoptive transfer of TH17 cells in SJL mice. However, a possible influence on the formation of ELFs was not investigated [97]. In Figure 2, we schematically summarize the EAE models where the formation of ELFs or a meningeal inflammation can be studied.
FIGURE 2.
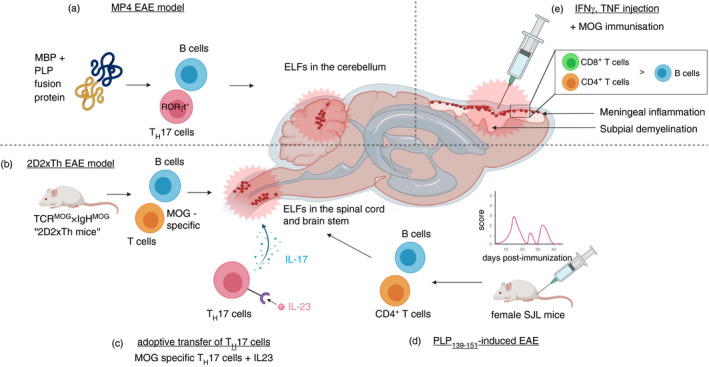
Schematic representation of selected experimental autoimmune encephalomyelitis (EAE) models that allow the study of ELFs or meningeal inflammation in rodents. In the B‐cell‐dependent MP4‐EAE model (A), formation of ELFs in the cerebellum can be observed after immunization with the MBP‐PLP fusion protein MP4. Retinoic acid receptor‐related orphan receptor‐γt positive (RORγt+) TH17 cells, in addition to B cells, are thought to play a crucial role during the formation of ELFs in MP4‐immunized mice. In the spontaneous ‘2D2xTh EAE model’ (B), where T‐cell receptor (TCR) transgenic mice (C57BL/6 2D2 MOG35–55‐specific, referred to as 2D2 mice) are crossbred with MOG‐specific Ig heavy‐chain knock‐in mice (referred to as Th mice), ELFs develop spontaneously in the spinal cord. MOG‐specific TH17 cells isolated from 2D2 mice and cultured in vitro with interleukin‐23 (IL‐23) can induce formation of ELFs in the spinal cord of recipient mice (C). In the PLP139‐151 induced relapsing–remitting EAE model, ELFs can be found in the brain stem and spinal cord of SJL mice (D). Injection of proinflammatory cytokines (i.e. interferon gamma [IFN‐γ], tumour necrosis factor [TNF]) directly into the subarachnoid space leads to the induction of subpial demyelination and meningeal inflammation (predominantly CD4+ and CD8+ T cells, less B cells) in Dark Agouti rats when preimmunized with MOG peptides (E)
Insights into ELFs from TFH cells
TFH cells, a subtype of CD4+ T cells showing a CD44hiCD62LlowCCR7lowPSGL1lowCXCR5hiICOShiPD‐1hi phenotype, are crucial for humoral immunity and significantly involved in controlling the formation and the cellular reactions in germinal centres [98, 99, 100, 101]. As illustrated in Figure 3, the interaction between TFH cells and B cells in the germinal centres is critical for B‐cell differentiation into antibody‐producing plasma cells and memory B cells [98, 102, 103]. The dysregulation of TFH cells is thought to play an important role in the development of a broad range of autoimmune diseases (e.g. systemic lupus erythematosus, rheumatoid arthritis and Sjögren's syndrome) [104]. Fonseca et al. found that the frequency of activated PD‐1+ICOS+ TFH cells is positively correlated with disease activity in patients with primary Sjögren's syndrome [105]. Furthermore, in the same study, Fonseca et al. demonstrated that the ratio of FOXP3+ regulatory follicular T cells (TFR cells) to TFH cells in the blood of patients is not only a reliable predictor for the presence of primary Sjögren's syndrome, but also correctly indicates the occurrence of ELFs in the salivary glands of investigated patients. While TFH cells are important promoters of B‐cell development and activation, TFR cells limit the formation of germinal centres. The relevance of TFH cells and, in particular, regulatory TFR cells in the pathogenesis and formation of ELFs in MS is largely unknown. In 2016, Varrin‐Doyer et al. demonstrated that the immunomodulatory drug laquinimod is able to significantly reduce the number of TFH and B‐cell aggregates in ELFs (mainly located in the spinal cord meninges) in the 2D2×Th spontaneous EAE model [106]. In 2018, several studies claimed that TFH cells most likely maintain but do not induce the formation of ELFs in spinal cord meninges using actively induced MOG‐EAE and passively induced adoptive transfer EAE models [107, 108]. Moreover, conditional knockout mice lacking the programmed death ligand 1 in CD11c+ dendritic cells showed an inhibited differentiation of TFH cells and an increased number of inflammatory foci in the meninges and the parenchyma using passively induced adoptive transfer EAE [109].
FIGURE 3.
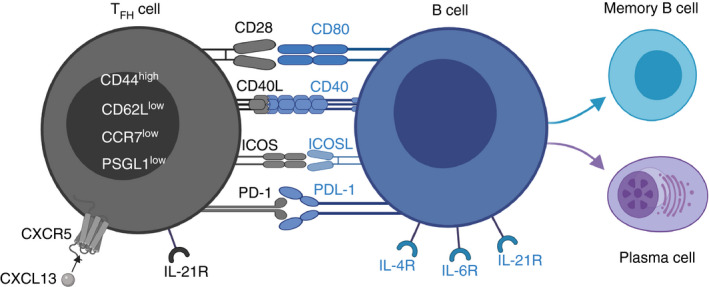
Schematic illustration depicting the interactions between TFH cells and B cells. Specialized CD4+ T‐helper cells (TFH cells), displaying a CD44hiCD62LlowCCR7lowPSGL1lowCXCR5hiICOShiPD‐1hi phenotype, are crucial during B‐cell differentiation. TFH cells are suggested to play important roles during formation and/or maintenance of ELFs
There are several challenges in studying the roles of TFH cells during formation and/or ELF maintenance: firstly, it is difficult to identify TFH cells due to the lack of lineage‐specifying cytokines [110, 111] and their dynamic plasticity, which is shaped largely by the respective microenvironment [98]. Secondly, although TFH cells are suggested to play critical roles during the formation of germinal centres, blocking key regulators of TFH cells (e.g. CXCR5, BCL‐6) does not completely inhibit the function of germinal centres [98]. Beyond, there are substantial differences between in vivo and in vitro studies investigating TFH cells, and molecules regulating the development of TFH cells such as BCL‐6, ICOS, CXCL13, CXCR5, IL‐21/IL‐21R, PD‐1/PD‐L1 have opposite roles during EAE development [94, 109, 112, 113, 114, 115], possibly reflecting the heterogeneity of TFH cells in different MS‐related animal models. Finally, these molecules are not specific to TFH cells and their inhibition can affect a variety of other cells and cellular interactions.
PERSPECTIVES
Inflammatory infiltrates consisting of macrophages, predominantly CD8+ T cells, and relatively few B cells, are found in the CNS of RRMS patients. At the progressive stage (i.e. SPMS), the presence of ELFs in the meninges, diffuse inflammatory CD8+ T‐cell and B‐cell infiltrates in the parenchyma, gliosis, diffuse myelin destruction and axonal injury are evident in and around the demyelinating lesions [82, 116]. Investigations on ELFs hold the potential to significantly contribute to the research of new therapeutic approaches acting on the irreversible disease progression in progressive MS. In this section, we try to point out the next challenges in investigating ELFs and potential research directions.
Paucity of standards: one cannot measure the distance without a ruler
As mentioned earlier, the presence of ELFs in progressive MS patients is still being discussed by some authors. The successful investigation of ELFs requires extensive sampling and well‐preserved meningeal structures during the tissue preparation process due to the sparse and heterogeneous ELF distribution [32]. ELFs are most frequently observed in the deep sulci of the brain, perhaps because meningeal structures within the sulci are especially well‐preserved during tissue preparation. As mentioned already in the introduction, ELFs were and still are mistakenly termed ‘meningeal inflammation’ in the related literature, although the criteria for ELFs according to previously postulated definitions were not met. Therefore, different definitions of ELFs complicate the comparison of different possible treatment options. Moreover, the paucity of progressive MS animal models additionally limits any comparison between ELFs found in animals and in progressive MS tissue. In general, animal models are not suitable for the study of aetiological factors of MS, but, in the context of ELF formation and resolution, might only be useful to investigate possible new ELF‐disrupting drugs. Furthermore, neuroanatomical differences between rodent and human brains could limit the relevance of in vivo findings related to ELF formation and resolution.
Little is known about the formation of ELFs in progressive MS
Most of the current studies on inhibition of ELF formation are based on the hypothesis that germinal centre‐like structures (i.e. ELFs) could, in principle, be eradicated by application of reagents that inhibit the formation of germinal centres. Specific biomarkers for the detection of ELFs in progressive MS remain absent. Furthermore, little is known about why ELFs exist in a variety of autoimmune diseases (e.g. rheumatoid arthritis, Sjögren's syndrome and uveitis) [117, 118, 119, 120, 121]. Recent studies indicate that ELFs might also contribute to disease progression during early stages of MS pathology. Bevan et al. detected meningeal inflammation and ELFs in 4 acute MS cases. The investigated acute MS cases were diagnosed and died before the advent of disease‐modifying therapies [122]. Large B‐cell aggregates were also observed in the meninges of investigated RRMS cases or cases of clinically isolated syndrome, before a formal diagnosis of MS was even made. However, necessary immunohistochemical labellings were not performed to conclusively assess whether the structures found resemble ELFs [123].
Based on neuropathological studies, many findings indicate that ELFs and meningeal inflammation are associated with disease progression (i.e. cognitive and motor disability). However, it remains largely unknown how immune cells are recruited to the meninges in the first place and how exactly they contribute to the disease progression. It remains intriguing whether the observed meningeal infiltrations and the formation of ELFs might be associated with the presence of meningeal lymphatic vessels [124, 125].
Possible research directions
Due to the localization of ELFs and the vulnerability of the meninges during the tissue embedding process, it would be fundamentally important to preserve the histological entirety of the meninges along with careful consideration of the tissue preparation procedure. It would be as well beneficial to evaluate the stability of ELFs over time and whether they undergo spontaneous resolution depending on the disease activity or during the course of different MS disease stages. The rapid development of intravital imaging, for example 2‐photon laser scanning microscopy and tissue transparency technology, could provide opportunities for more accurate evaluations of meningeal inflammation or ELFs in the future [126, 127]. In 2020, the approval of siponimod by the FDA and EMA for SPMS treatment has brought moderate benefits in alleviating the clinical disease progression. Siponimod is a novel sphingosine 1‐phosphate receptor (S1Pr) modulator that selectively binds to S1Pr1 and S1Pr5. However, it remains largely unknown whether siponimod ameliorates clinical progression mainly by inhibiting peripheral immune cell recruitment or by alleviating neurodegeneration in the CNS. Therefore, it would be intriguing to study whether siponimod influences the formation and/or resolution of ELFs. As mentioned in the introduction of this review, the mode of action of anti‐CD20 antibody therapies (e.g. ocrelizumab) is yet to be fully elucidated. The moderate success of B‐cell‐depleting therapies in progressive MS and RRMS suggests an important role of B cells during disease progression. However, B cells in the CNS parenchyma and in meningeal ELFs can presumably not be reached and selectively inhibited by anti‐CD20 antibodies due to the largely preserved blood–brain barrier in SPMS patients. Different EAE models in which the formation of ELFs could be observed may be used to investigate more auspicious drugs targeting immune cell differentiation or lymphoid neogenesis in the CNS. New therapeutic approaches with, for example, small‐molecule drugs that can trespass the blood–brain barrier might allow the specific inhibition of B‐cell differentiation, B‐cell / T‐cell interactions in ELFs or even the formation of ELFs in the first place. This harbours promising potential for a protective effect on chronic inflammation and progressive neurodegeneration in SPMS patients.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
JZ and HK wrote the manuscript. MK and WH revised parts of the manuscript. All authors contributed to the article and approved the submitted version.
ACKNOWLEDGEMENTS
We thank Prof. Hai Qi from the Tsinghua University for the fruitful discussion about follicular T‐helper cells. We also acknowledge the suggestions and the help of Prof. Francesca Aloisi, Department of Neuroscience, Istituto Superiore di Sanità, Rome, Italy. All figures were created using BioRender https://biorender.com. Open Access funding enabled and organized by Projekt DEAL.
Zhan J, Kipp M, Han W, Kaddatz H. Ectopic lymphoid follicles in progressive multiple sclerosis: From patients to animal models. Immunology. 2021;164:450–466. 10.1111/imm.13395
Funding information
JZ is financially supported by the China Scholarship Council for a living stipend (CSC201706010354)
REFERENCES
- 1. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, et al. Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(Pt 4):1089–104. [DOI] [PubMed] [Google Scholar]
- 2. Bermel RA, Bakshi R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol. 2006;5:158–70. [DOI] [PubMed] [Google Scholar]
- 3. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(Pt 11):2705–12. [DOI] [PubMed] [Google Scholar]
- 4. Mao P, Reddy PH. Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta. 2010;1802:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lassmann H, van Horssen J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim Biophys Acta. 2016;1862:506–10. [DOI] [PubMed] [Google Scholar]
- 6. Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019;18:905–22. [DOI] [PubMed] [Google Scholar]
- 7. Frischer JM, Bramow S, Dal‐Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(Pt 5):1175–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood‐brain barrier leakage and active demyelination. J Pathol. 2003;201:319–27. [DOI] [PubMed] [Google Scholar]
- 9. Leech S, Kirk J, Plumb J, McQuaid S. Persistent endothelial abnormalities and blood‐brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol. 2007;33:86–98. [DOI] [PubMed] [Google Scholar]
- 10. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med. 2008;358:676–88. [DOI] [PubMed] [Google Scholar]
- 11. Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurol. 2014;13:353–63. [DOI] [PubMed] [Google Scholar]
- 12. Kappos L, Bar‐Or A, Cree BAC, Fox RJ, Giovannoni G, Gold R, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‐blind, randomised, phase 3 study. Lancet. 2018;391:1263–73. [DOI] [PubMed] [Google Scholar]
- 13. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar‐Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376:209–20. [DOI] [PubMed] [Google Scholar]
- 14. Kipp M. Does siponimod exert direct effects in the central nervous system? Cells. 2020;9:1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Correale J, Gaitan MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 2017;140:527–46. [DOI] [PubMed] [Google Scholar]
- 16. Moreno Torres I, García‐Merino A. Anti‐CD20 monoclonal antibodies in multiple sclerosis. Expert Rev Neurother. 2017;17:359–71. [DOI] [PubMed] [Google Scholar]
- 17. Klein C, Lammens A, Schäfer W, Georges G, Schwaiger M, Mössner E, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gertner E, Rosenbloom MH. Susac syndrome with prominent dermatological findings and a prompt response to intravenous immunoglobulin, steroids, and rituximab: a case report. J Med Case Rep. 2016;10:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Monferrer‐Adsuara C, Remolí‐Sargues L, Hernández‐Bel L, Gracia‐García A, Hernández‐Garfella ML, Cervera‐Taulet E. Rituximab in the treatment of Susac's syndrome: report of a case. Eur J Ophthalmol. 2020;1120672120924545. [DOI] [PubMed] [Google Scholar]
- 20. Gross CC, Meyer C, Bhatia U, Yshii L, Kleffner I, Bauer J, et al. CD8(+) T cell‐mediated endotheliopathy is a targetable mechanism of neuro‐inflammation in Susac syndrome. Nat Commun. 2019;10:5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider‐Hohendorf T, Mohan H, Bien CG, Breuer J, Becker A, Görlich D, et al. CD8(+) T‐cell pathogenicity in Rasmussen encephalitis elucidated by large‐scale T‐cell receptor sequencing. Nat Commun. 2016;7:11153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laxer K, Wilfong A, Morris G, Andermann F. Study of rituximab to treat chronic focal encephalitis: 1.277. Epilepsia. 2008;49:121. [Google Scholar]
- 23. Thilo B, Stingele R, Knudsen K, Boor R, Bien CG, Deuschl G, et al. A case of Rasmussen encephalitis treated with rituximab. Nat Rev Neurol. 2009;5:458–62. [DOI] [PubMed] [Google Scholar]
- 24. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gay FW, Drye TJ, Dick GW, Esiri MM. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain. 1997;120(Pt 8):1461–83. [DOI] [PubMed] [Google Scholar]
- 26. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B‐cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134(Pt 9):2755–71. [DOI] [PubMed] [Google Scholar]
- 28. Archelos JJ, Storch MK, Hartung HP. The role of B cells and autoantibodies in multiple sclerosis. Ann Neurol. 2000;47:694–706. [PubMed] [Google Scholar]
- 29. Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing‐remitting multiple sclerosis: a phase 2 study. Neurology. 2014;82:573–81. [DOI] [PubMed] [Google Scholar]
- 30. Kappos L, Li D, Calabresi PA, O'Connor P, Bar‐Or A, Barkhof F, et al. Ocrelizumab in relapsing‐remitting multiple sclerosis: a phase 2, randomised, placebo‐controlled, multicentre trial. Lancet. 2011;378:1779–87. [DOI] [PubMed] [Google Scholar]
- 31. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein‐Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aloisi F, Serafini B, Magliozzi R, Howell OW, Reynolds R. Detection of Epstein‐Barr virus and B‐cell follicles in the multiple sclerosis brain: what you find depends on how and where you look. Brain. 2010;133(Pt 12):e157. [DOI] [PubMed] [Google Scholar]
- 33. Kuerten S, Schickel A, Kerkloh C, Recks MS, Addicks K, Ruddle NH, et al. Tertiary lymphoid organ development coincides with determinant spreading of the myelin‐specific T cell response. Acta Neuropathol. 2012;124:861–73. [DOI] [PubMed] [Google Scholar]
- 34. Lehmann‐Horn K, Wang SZ, Sagan SA, Zamvil SS, von Budingen HC. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight. 2016;1:e87234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prineas JW. Multiple sclerosis: presence of lymphatic capillaries and lymphoid tissue in the brain and spinal cord. Science. 1979;203:1123–5. [DOI] [PubMed] [Google Scholar]
- 36. Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest. 1978;38:409–21. [PubMed] [Google Scholar]
- 37. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. 1983;62(1–3):219–32. [DOI] [PubMed] [Google Scholar]
- 38. Guseo A, Jellinger K. The significance of perivascular infiltrations in multiple sclerosis. J Neurol. 1975;211:51–60. [DOI] [PubMed] [Google Scholar]
- 39. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–52. [DOI] [PubMed] [Google Scholar]
- 40. Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisakk P, Ransohoff RM, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up‐regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129(Pt 1):200–11. [DOI] [PubMed] [Google Scholar]
- 41. Khademi M, Kockum I, Andersson ML, Iacobaeus E, Brundin L, Sellebjerg F, et al. Cerebrospinal fluid CXCL13 in multiple sclerosis: a suggestive prognostic marker for the disease course. Mult Scler. 2011;17:335–43. [DOI] [PubMed] [Google Scholar]
- 42. Narayan K, Dail D, Li L, Cadavid D, Amrute S, Fitzgerald‐Bocarsly P, et al. The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann Neurol. 2005;57:813–23. [DOI] [PubMed] [Google Scholar]
- 43. Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P. Monocyte‐like and mature macrophages produce CXCL13 (B cell‐attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood. 2004;104:3021–7. [DOI] [PubMed] [Google Scholar]
- 44. Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. 2009;66:739–53. [DOI] [PubMed] [Google Scholar]
- 45. Krone B, Oeffner F, Grange JM. Is the risk of multiple sclerosis related to the ‘biography’ of the immune system? J Neurol. 2009;256:1052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Torkildsen O, Stansberg C, Angelskar SM, Kooi EJ, Geurts JJ, van der Valk P, et al. Upregulation of immunoglobulin‐related genes in cortical sections from multiple sclerosis patients. Brain Pathol. 2010;20:720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, et al. Epstein‐Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain. 2009;132(Pt 12):3318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Opsahl ML, Kennedy PG. An attempt to investigate the presence of Epstein Barr virus in multiple sclerosis and normal control brain tissue. J Neurol. 2007;254:425–30. [DOI] [PubMed] [Google Scholar]
- 49. Morre SA, van Beek J, De Groot CJ, Killestein J, Meijer CJ, Polman CH, et al. Is Epstein‐Barr virus present in the CNS of patients with MS? Neurology. 2001;56:692. [DOI] [PubMed] [Google Scholar]
- 50. Hilton DA, Love S, Fletcher A, Pringle JH. Absence of Epstein‐Barr virus RNA in multiple sclerosis as assessed by in situ hybridisation. J Neurol Neurosurg Psychiatry. 1994;57:975–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jaquiery E, Jilek S, Schluep M, Meylan P, Lysandropoulos A, Pantaleo G, et al. Intrathecal immune responses to EBV in early MS. Eur J Immunol. 2010;40:878–87. [DOI] [PubMed] [Google Scholar]
- 52. Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV‐specific CD8+ T‐cell response in patients with early multiple sclerosis. Brain. 2008;131(Pt 7):1712–21. [DOI] [PubMed] [Google Scholar]
- 53. Veroni C, Serafini B, Rosicarelli B, Fagnani C, Aloisi F. Transcriptional profile and Epstein‐Barr virus infection status of laser‐cut immune infiltrates from the brain of patients with progressive multiple sclerosis. J Neuroinflammation. 2018;15:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–93. [DOI] [PubMed] [Google Scholar]
- 55. Reali C, Magliozzi R, Roncaroli F, Nicholas R, Howell OW, Reynolds R. B cell rich meningeal inflammation associates with increased spinal cord pathology in multiple sclerosis. Brain Pathol. 2020;30:779–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choi SR, Howell OW, Carassiti D, Magliozzi R, Gveric D, Muraro PA, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain. 2012;135(Pt 10):2925–37. [DOI] [PubMed] [Google Scholar]
- 57. Kooi EJ, Geurts JJ, van Horssen J, Bo L, van der Valk P. Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol. 2009;68:1021–8. [DOI] [PubMed] [Google Scholar]
- 58. Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, et al. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain. 2011;134(Pt 2):534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Willis SN, Stathopoulos P, Chastre A, Compton SD, Hafler DA, O'Connor KC. Investigating the antigen specificity of multiple sclerosis central nervous system‐derived immunoglobulins. Front Immunol. 2015;6:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fan X, Lin C, Han J, Jiang X, Zhu J, Jin T. Follicular helper CD4+ T cells in human neuroautoimmune diseases and their animal models. Mediators Inflamm. 2015;2015:638968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Romme Christensen J, Bornsen L, Ratzer R, Piehl F, Khademi M, Olsson T, et al. Systemic inflammation in progressive multiple sclerosis involves follicular T‐helper, Th17‐ and activated B‐cells and correlates with progression. PLoS ONE. 2013;8:e57820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gardner C, Magliozzi R, Durrenberger PF, Howell OW, Rundle J, Reynolds R. Cortical grey matter demyelination can be induced by elevated pro‐inflammatory cytokines in the subarachnoid space of MOG‐immunized rats. Brain. 2013;136(Pt 12):3596–608. [DOI] [PubMed] [Google Scholar]
- 63. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN‐γ receptor signaling promotes autoimmune germinal centers via cell‐intrinsic induction of BCL‐6. J Exp Med. 2016;213:733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, et al. IFN‐γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. 2016;213:715–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serafini B, Rosicarelli B, Veroni C, Zhou L, Reali C, Aloisi F. RORgammat expression and lymphoid neogenesis in the brain of patients with secondary progressive multiple sclerosis. J Neuropathol Exp Neurol. 2016;75:877–88. [DOI] [PubMed] [Google Scholar]
- 66. Bell L, Lenhart A, Rosenwald A, Monoranu CM, Berberich‐Siebelt F. Lymphoid aggregates in the CNS of progressive multiple sclerosis patients lack regulatory T cells. Front Immunol. 2019;10:3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nutma E, Willison H, Martino G, Amor S. Neuroimmunology ‐ the past, present and future. Clin Exp Immunol. 2019;197:278–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Olitsky PK, Yager RH. Experimental disseminated encephalomyelitis in white mice. J Exp Med. 1949;90:213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1810–9. [DOI] [PubMed] [Google Scholar]
- 70. Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol. 2014;122:173–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kuerten S, Lichtenegger FS, Faas S, Angelov DN, Tary‐Lehmann M, Lehmann PV. MBP‐PLP fusion protein‐induced EAE in C57BL/6 mice. J Neuroimmunol. 2006;177(1–2):99–111. [DOI] [PubMed] [Google Scholar]
- 72. Kipp M, Nyamoya S, Hochstrasser T, Amor S. Multiple sclerosis animal models: a clinical and histopathological perspective. Brain Pathol. 2017;27:123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Peferoen LA, Breur M, van de Berg S, Peferoen‐Baert R, Boddeke EH, van der Valk P, et al. Ageing and recurrent episodes of neuroinflammation promote progressive experimental autoimmune encephalomyelitis in Biozzi ABH mice. Immunology. 2016;149:146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Al‐Izki S, Pryce G, O'Neill JK, Butter C, Giovannoni G, Amor S, et al. Practical guide to the induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. Mult Scler Relat Disord. 2012;1:29–38. [DOI] [PubMed] [Google Scholar]
- 75. Zhan J, Mann T, Joost S, Behrangi N, Frank M, Kipp M. The Cuprizone model: dos and do nots. Cells. 2020;9:843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fischbach F, Nedelcu J, Leopold P, Zhan J, Clarner T, Nellessen L, et al. Cuprizone‐induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage‐inducible transcript 3. Glia. 2019;67:263–76. [DOI] [PubMed] [Google Scholar]
- 77. Ruther BJ, Scheld M, Dreymueller D, Clarner T, Kress E, Brandenburg LO, et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia. 2017;65:1900–13. [DOI] [PubMed] [Google Scholar]
- 78. Storch MK, Stefferl A, Brehm U, Weissert R, Wallstrom E, Kerschensteiner M, et al. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998;8:681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1952–60. [DOI] [PubMed] [Google Scholar]
- 80. Waldner H, Whitters MJ, Sobel RA, Collins M, Kuchroo VK. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein‐specific T cell receptor. Proc Natl Acad Sci USA. 2000;97:3412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein‐specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545–58. [DOI] [PubMed] [Google Scholar]
- 83. Tsuchida M, Hanawa H, Hirahara H, Watanabe H, Matsumoto Y, Sekikawa H, et al. Identification of CD4‐ CD8‐ alpha beta T cells in the subarachnoid space of rats with experimental autoimmune encephalomyelitis. A possible route by which effector cells invade the lesions. Immunology. 1994;81:420–7. [PMC free article] [PubMed] [Google Scholar]
- 84. Shin T, Kojima T, Tanuma N, Ishihara Y, Matsumoto Y. The subarachnoid space as a site for precursor T cell proliferation and effector T cell selection in experimental autoimmune encephalomyelitis. J Neuroimmunol. 1995;56:171–8. [DOI] [PubMed] [Google Scholar]
- 85. Magliozzi R, Columba‐Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle‐like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148(1–2):11–23. [DOI] [PubMed] [Google Scholar]
- 86. Gonzalez M, Mackay F, Browning JL, Kosco‐Vilbois MH, Noelle RJ. The sequential role of lymphotoxin and B cells in the development of splenic follicles. J Exp Med. 1998;187:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, et al. Characterization of lymphotoxin‐alpha beta complexes on the surface of mouse lymphocytes. J Immunol. 1997;159:3288–98. [PubMed] [Google Scholar]
- 88. Cupedo T, Mebius RE. Role of chemokines in the development of secondary and tertiary lymphoid tissues. Semin Immunol. 2003;15:243–8. [DOI] [PubMed] [Google Scholar]
- 89. Tumanov A, Kuprash D, Lagarkova M, Grivennikov S, Abe K, Shakhov A, et al. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity. 2002;17:239–50. [DOI] [PubMed] [Google Scholar]
- 90. Columba‐Cabezas S, Griguoli M, Rosicarelli B, Magliozzi R, Ria F, Serafini B, et al. Suppression of established experimental autoimmune encephalomyelitis and formation of meningeal lymphoid follicles by lymphotoxin beta receptor‐Ig fusion protein. J Neuroimmunol. 2006;179(1–2):76–86. [DOI] [PubMed] [Google Scholar]
- 91. Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein‐specific T and B cells cooperate to induce a Devic‐like disease in mice. J Clin Invest. 2006;116:2393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35:986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee Y, Mitsdoerffer M, Xiao S, Gu G, Sobel RA, Kuchroo VK. IL‐21R signaling is critical for induction of spontaneous experimental autoimmune encephalomyelitis. J Clin Invest. 2015;125:4011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Molnarfi N, Schulze‐Topphoff U, Weber MS, Patarroyo JC, Prod'homme T, Varrin‐Doyer M, et al. MHC class II‐dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin‐specific antibodies. J Exp Med. 2013;210:2921–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Schropp V, Rohde J, Rovituso DM, Jabari S, Bharti R, Kuerten S. Contribution of LTi and TH17 cells to B cell aggregate formation in the central nervous system in a mouse model of multiple sclerosis. J Neuroinflammation. 2019;16:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Klimatcheva E, Pandina T, Reilly C, Torno S, Bussler H, Scrivens M, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Qi H. T follicular helper cells in space‐time. Nat Rev Immunol. 2016;16:612–25. [DOI] [PubMed] [Google Scholar]
- 99. Schmitt N. Role of T follicular helper cells in multiple sclerosis. J Nat Sci. 2015;1:e139. [PMC free article] [PubMed] [Google Scholar]
- 100. Quinn JL, Axtell RC. Emerging role of follicular T helper cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Int J Mol Sci. 2018;19:3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin‐21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity. 2016;45:471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hamel KM, Liarski VM, Clark MR. Germinal center B‐cells. Autoimmunity. 2012;45:333–47. [DOI] [PubMed] [Google Scholar]
- 104. Gensous N, Charrier M, Duluc D, Contin‐Bordes C, Truchetet ME, Lazaro E, et al. T follicular helper cells in autoimmune disorders. Front Immunol. 2018;9:1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fonseca VR, Romao VC, Agua‐Doce A, Santos M, Lopez‐Presa D, Ferreira AC, et al. The ratio of blood t follicular regulatory cells to T follicular helper cells marks ectopic lymphoid structure formation while activated follicular helper T cells indicate disease activity in primary Sjogren's syndrome. Arthritis Rheumatol. 2018;70:774–84. [DOI] [PubMed] [Google Scholar]
- 106. Varrin‐Doyer M, Pekarek KL, Spencer CM, Bernard CC, Sobel RA, Cree BA, et al. Treatment of spontaneous EAE by laquinimod reduces Tfh, B cell aggregates, and disease progression. Neurol Neuroimmunol Neuroinflamm. 2016;3:e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Guo J, Zhao C, Wu F, Tao L, Zhang C, Zhao D, et al. T follicular helper‐like cells are involved in the pathogenesis of experimental autoimmune encephalomyelitis. Front Immunol. 2018;9:944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Quinn JL, Kumar G, Agasing A, Ko RM, Axtell RC. Role of TFH cells in promoting T helper 17‐induced neuroinflammation. Front Immunol. 2018;9:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, Sharpe AH. Dendritic cell PD‐L1 limits autoimmunity and follicular T cell differentiation and function. J Immunol. 2018;200:2592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rottman JB, Smith T, Tonra JR, Ganley K, Bloom T, Silva R, et al. The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat Immunol. 2001;2:605–11. [DOI] [PubMed] [Google Scholar]
- 113. Li Q, Zhou L, Wang L, Li S, Xu G, Gu H, et al. Bcl6 modulates innate immunity by controlling macrophage activity and plays critical role in experimental autoimmune encephalomyelitis. Eur J Immunol. 2020;50:525–36. [DOI] [PubMed] [Google Scholar]
- 114. Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting edge: IL‐21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. 2008;180:7097–101. [DOI] [PubMed] [Google Scholar]
- 115. Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, et al. PD‐1/PD‐L1, but not PD‐1/PD‐L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;182(1–2):124–34. [DOI] [PubMed] [Google Scholar]
- 116. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183–93. [DOI] [PubMed] [Google Scholar]
- 117. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid‐like structures in infection, cancer and autoimmunity. Nat Rev Immunol. 2014;14:447–62. [DOI] [PubMed] [Google Scholar]
- 118. Corsiero E, Nerviani A, Bombardieri M, Pitzalis C. Ectopic lymphoid structures: powerhouse of autoimmunity. Front Immunol. 2016;7:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Jones GW, Jones SA. Ectopic lymphoid follicles: inducible centres for generating antigen‐specific immune responses within tissues. Immunology. 2016;147:141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Epps SJ, Coplin N, Luthert PJ, Dick AD, Coupland SE, Nicholson LB. Features of ectopic lymphoid‐like structures in human uveitis. Exp Eye Res. 2020;191:107901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kielczewski JL, Horai R, Jittayasothorn Y, Chan CC, Caspi RR. Tertiary lymphoid tissue forms in retinas of mice with spontaneous autoimmune uveitis and has consequences on visual function. J Immunol. 2016;196:1013–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bevan RJ, Evans R, Griffiths L, Watkins LM, Rees MI, Magliozzi R, et al. Meningeal inflammation and cortical demyelination in acute multiple sclerosis. Ann Neurol. 2018;84:829–42. [DOI] [PubMed] [Google Scholar]
- 123. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rossi B, Constantin G. Live imaging of immune responses in experimental models of multiple sclerosis. Front Immunol. 2016;7:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cai R, Pan C, Ghasemigharagoz A, Todorov MI, Forstera B, Zhao S, et al. Panoptic imaging of transparent mice reveals whole‐body neuronal projections and skull‐meninges connections. Nat Neurosci. 2019;22:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]