

Abstract
The G-type nerve agents, sarin (GB), soman (GD) and cyclosarin (GF) are among the most toxic compounds known. Much progress has been made in evolving the enzyme phosphotriesterase (PTE) from Pseudomonas diminuta for the decontamination of the G-agents, however the extreme toxicity of the G-agents makes the use of substrate analogs necessary. Typical analogs utilize a chromogenic leaving group to facilitate high-throughput screening, and substitution of an O-methyl for the P-methyl group found in the G-agents, in an effort to reduce toxicity. To date there has been no systematic evaluation of the effects of these substitutions on catalytic activity, and the presumed reduction in toxicity has not been tested. A series of 21 G-agent analogs, including all combinations of O-methyl, p-nitrophenyl and thiophosphate substitutions, has been synthesized and evaluated for their ability to unveil the stereoselectivity and catalytic activity of PTE variants against the authentic G-type nerve agents. The potential toxicity of these analogs was evaluated by measuring the rate of inactivation of acetylcholinesterase (AChE). All of the substitutions reduced inactivation of AChE by more than 100-fold, with the most effective being the thiophosphate analogs, which reduced the rate of inactivation by about 4 to 5 orders of magnitude. The analogs were found to reliably predict changes in catalytic activity and stereoselectivity of the PTE variants, and led to the identification of the BHR-30 variant, which has no apparent stereoselectivity against GD and a kcat/Km of 1.4 × 10⁶, making it the most efficient enzyme for GD decontamination reported to date.
Graphical Abstract
Introduction
One of the major challenges in the directed evolution of enzyme-catalyzed reactions is the identification of an appropriate substrate that enables the selection of variants with enhanced catalytic activity. Modest sized enzyme libraries generally contain thousands to tens of thousands of variants (1, 2). Screening a library for improved catalytic activity often requires high-throughput selection techniques, making the use of the actual target compound impractical in many cases. Substrate analogs are often used to overcome this obstacle by facilitating rapid screening, but identifying the most appropriate analog that reports reliably on the desired reaction over a broad range of enzyme variants is a major challenge. This obstacle becomes greater as the activity of the enzyme increases with each round of selection becoming more and more discriminating for the reaction screened relative to the desired reaction (3, 4). This often results in the latter rounds of evolution producing variants with substantial increases in activity for the analog, but not the target compound (3, 5, 6).
These difficulties are exacerbated by toxicity and stereoselectivity in the case of the organophosphate nerve agents. These compounds are among the most toxic compounds known, making their use undesirable in most situations. The G-type nerve agents, sarin (GB, 1), cycolosarin (GF, 9), and soman (GD, 17), contain a fluoride leaving group attached to a chiral methylphosphonate center. The toxicity of the G-agents is dependent on the stereochemistry of the phosphorus center with the (SP)-isomers being more than 1000-fold more toxic than the (RP)-isomers (7, 8). To reliably target G-type nerve agents, substrate analogs must accurately predict both the hydrolytic activity of the enzyme variants, as well as their stereoselectivity, and at the same time reduce the toxicity to levels appropriate for large-scale screening.
The bacterial enzyme phosphotriesterase (PTE) from Pseudomonas diminuta exhibits strong catalytic activity for hydrolysis of the G-type nerve agents, but unfortunately, the wild-type enzyme has a strong stereoselective preference for the less toxic (RP)-isomers of the G-type nerve agents (6, 9). Directed evolution of PTE for G-agent decontamination has been successful, resulting in the identification of the H257Y/L303T (YT) variant, which exhibits the appropriate stereoselectivity for hydrolysis of the most toxic G-agents and a catalytic efficiency >106 M−1 s−1 for hydrolysis of GB (6). YT has now been adapted for in vivo prophylactic use, where it is able to protect test animals against repeated exposures of GB over 7 days (10). However, the activity against one of the more toxic stereoisomers of GD remains an order of magnitude lower than that observed for the hydrolysis of GB.
Some work with PTE has been done using low concentrations of authentic nerve agents with very good results, but the toxicity and security concerns of the G-agents makes this practically impossible for most efforts and thus requires the use of analogs (5, 11, 12). Efforts to evolve PTE for the enhanced hydrolysis of a phosphorus-fluorine bond without concern for the stereochemistry of the phosphorus center using the achiral analog diisopropylfluorophosphate failed to yield variants that were significantly enhanced against the authentic G-agents (13). Most efforts to evolve PTE for G-agent decontamination have utilized chromogenic aromatic leaving group analogs, with or without a chiral phosphate center including both P-methyl and O-methyl analogs, which are believed to have reduced toxicity (5, 6, 9, 14, 15). Unfortunately, these efforts have often led to variants with dramatically enhanced activity for the analogs, but weak or no enhancement for the authentic G-agents.
To date, there has not been a systematic evaluation of G-agent analogs to determine which chemical moieties are most important for predicting catalytic activity against the authentic agents. Analogs often utilize aromatic leaving groups such as p-nitrophenol or coumarin coupled with O-methyl esters, rather than the P-methyl functional group in the authentic agents in an effort to reduce toxicity. However, these perceived advantages are only made by analogy to insecticides (16). No significant effort has been made to evaluate the ability of these compounds to inactivate acetyl cholinesterase. In this work, a series of G-agent analogs was created to systematically alter the structure of the authentic nerve agents with an O-methyl ester vs. P-methyl phosphonate, p-nitrophenol vs. fluoride leaving group, and substitution of the phosphoryl oxygen for sulfur. This set of 21-compounds includes all possible combinations of one, two, and three alterations to the authentic nerve agents.
These analogs have been evaluated with a set of known PTE variants with differing degrees of catalytic activity and stereoselectivity against the authentic G-agents, and the ability of these analogs to inactivate the neural enzyme acetylcholine esterase was evaluated. Substitution of the methyl phosphonate moiety with an O-methyl group was least effective at reducing inactivation (102- to 103-fold) of acetylcholine esterase. Substitution with a thiophosphate derivative or a p-nitrophenyl leaving group reduced the rate of inactivation 103 to 106-fold. Combinations of these substitutions were further reduced, but the effects were not additive. The best reporter for catalytic activity on the authentic nerve agents was dependent on the agent. The thiophosphonate analog (2) was the best reporter for GB (1), while the activity against GF (9) and GD (17) was best represented by the O-methyl analogs 11 and 19. Surprisingly, the variant BHR-30 demonstrated little selectivity against authentic GD, hydrolyzing all 4-isomers with a single exponential phase with a kcat/Km of 1.4 × 10⁶ M−1 s−1, which is the highest reported activity against (RCSP)-GD to date.
Materials and Methods
Materials.
General lab supplies were purchased from VWR International. Growth media were obtained from RPI Corporation. Chemicals were from Sigma Aldrich. Acetylcholine esterase, type VI-S, and acetylthiocholine chloride were from Sigma Aldrich. Racemic versions of GB (1), GF (9), and GD (17) were Chemical Agent Analytical Reference Materials synthesized at the Aberdeen Proving Ground. Purity of compound 1 was greater than 97% by gas chromatography and 31P NMR spectroscopy. Compound 17 was >95% pure as determined by 1H and 31P NMR spectroscopy. Compound 9 was 95% pure as determined by gas chromatography using thermal conductivity detection. Compounds 7, 15 and 16 were prepared as previously described (17, 18) at Texas A&M University and were greater than 95% pure based on 1H and 31P NMR spectroscopy. Compounds 2-6, 8, 10-14, and 18-24 (see below) were synthesized at Texas A&M University. The purity of all of these compounds was >95% based on 1H and 31P NMR spectroscopy. The structures of all compounds are provided in Schemes 1 and 2. These compounds are toxic and should be handled with the appropriate precautions.
Scheme 1:
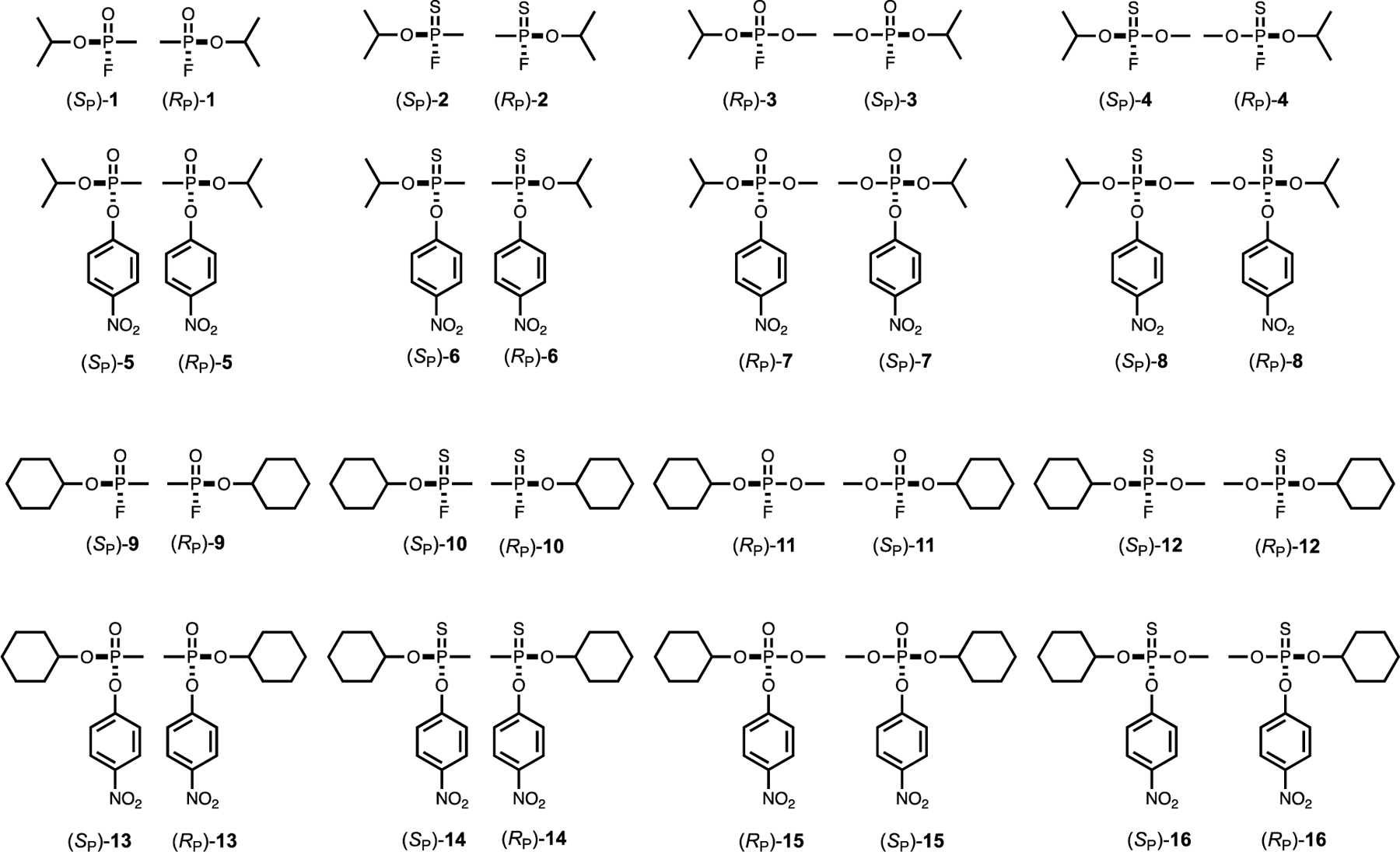
Structures of analogs of sarin (1) and cyclosarin (9) used in this investigation.
Scheme 2:
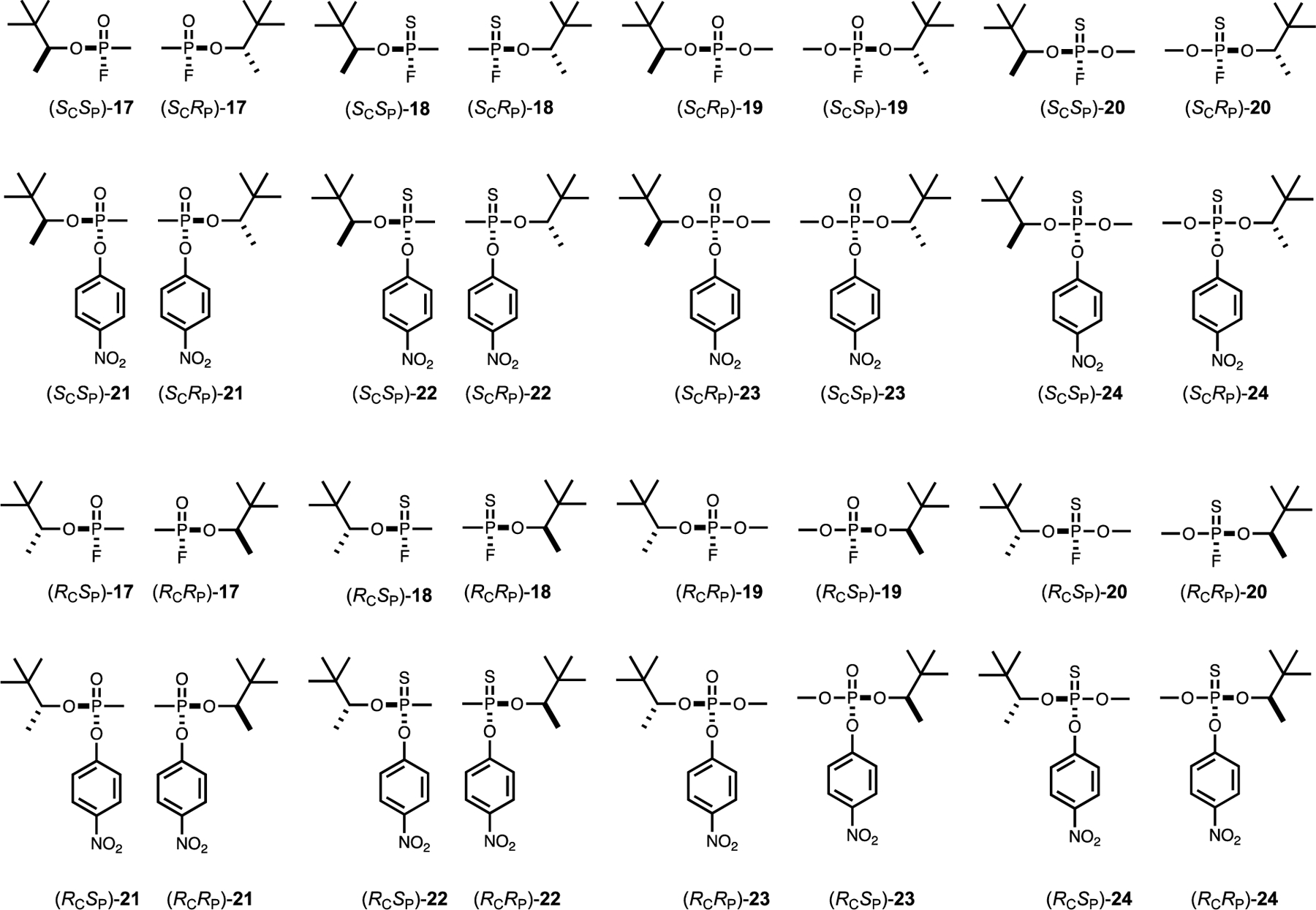
Structures of analogs of soman (17) used in this investigation.
Synthesis of (R)-Pinacolyl Alcohol.
A mixture of (R)-mandelic acid (10.0 g, 65.7 mmol) and (R/S)-pinacolyl alcohol (43.0 g, 420 mmol) was heated with stirring at 80 °C. Dry HCl gas was bubbled into the reaction mixture for 4 h while maintaining the temperature at 80 °C. The reaction was cooled to room temperature and the excess alcohol removed under vacuum. The resulting solid product (50:50 mixture of (R)-mandelate (R)-pinacolyl ester and (R)-mandelate (S)-pinacolyl ester) was recrystallized from MeOH/water (1.4:1). The crystalline product was further purified by five recrystallizations using the same solvent mixture. Purity of the (R)-mandelate (R)-pinacolyl ester diastereomer was monitored by 1H NMR spectroscopy to obtain 3.7 g (48% yield, 97% ee) of the (R,R) product. (R)-mandelic acid (R)-pinacolyl ester: 1H NMR (400 MHz, CDCl3) δ 7.46–7.42 (m, 2H), 7.39 – 7.29 (m, 3H), 5.13 (d, J = 5.2 Hz, 1H), 4.73 (q, J = 6.4 Hz, 1H), 3.61 (d, J = 5.2 Hz, 1H), 1.21 (d, J = 6.4 Hz, 3H), 0.65 (s, 9H).
Sodium hydroxide (10 mL of a 2.5 N solution) was added to 3.0 g (12.7 mmol) of the (R)-mandelate (R)-pinacolyl ester and the mixture was refluxed for 2 h. After cooling, the (R)-pinacolyl alcohol was extracted with ethyl ether (4 × 25 mL). The ether extracts were combined, dried over Na2SO4, and carefully evaporated (at low vacuum) to remove solvent and leave 0.72 g (45%) of the (R)-pinacolyl alcohol as a colorless liquid.
Synthesis of (S)-Pinacolyl Alcohol.
A mixture of (S)-mandelic acid (10.0 g, 65.7 mmol) and (R/S)-pinacolyl alcohol (43.0 g, 420 mmol) was heated with stirring at 80 °C. Dry HCl (g) was bubbled into the reaction mixture for 4 h at the same temperature. The reaction mixture was cooled to room temperature, and the excess alcohol was removed under vacuo resulting in a solid product. The solid product (50:50 mixture of (S)-mandelate R-pinacolyl ester and (S)-mandelate (S)-pinacolyl ester) was crystalized from MeOH/water (1.4:1). The crystalline product was further recrystallized 8 times using the same solvent mixture. Purity of (S)-mandelate (S)-pinacolyl ester diastereomer was monitored by 1H NMR spectroscopy to obtain 3.3 g (43% yield, 97% ee) of the (S, S) product. (S)-mandelate (S)-pinacolyl ester: 1H NMR (400 MHz, CDCl3) δ 7.46–7.42 (m, 2H), 7.39 – 7.29 (m, 3H), 5.13 (d, J = 5.2 Hz, 1H), 4.73 (q, J = 6.4 Hz, 1H), 3.61 (d, J = 5.2 Hz, 1H), 1.21 (d, J = 6.4 Hz, 3H), 0.65 (s, 9H).
Sodium hydroxide (10 mL of a 2.5 M solution) was added to 3.0 g (12.7 mmol) of (S)-mandelate (S)-pinacolyl ester (97% ee) and the mixture was refluxed for 2 h. After cooling, (S)-pinacolyl alcohol was extracted using ethyl ether (4 × 25 mL). The ether extracts were combined, dried over Na2SO4, and carefully evaporated (at low vacuum) to remove solvent and leave 0.72 g (45%) of (S)-pinacolyl alcohol as a colorless liquid.
Synthesis of O-methyl, p-Nitrophenyl Phosphate Analogs.
A solution of the appropriate alcohol (4.0 mmol, 1.0 equiv) in anhydrous diethyl ether (10 mL), was chilled to −78 °C and 2.0 mL of LDA (2.0 M in THF, 4.0 mmol, 1.0 equiv) was added. The reaction mixture was stirred for 30 min and transferred via cannula to another flask at −78 °C that contained methyl dichlorophosphate (0.47 mL, 4.0 mmol, 85%, 1.0 equiv) in diethyl ether (20 mL). The cooling bath was removed, and the reaction allowed to warm to ambient temperature (23 °C). After stirring for 5 h, p-nitrophenol (0.56 g, 4.0 mmol, 1.0 equiv) was added at room temperature, followed by the addition of dry triethylamine (0.56 mL, 4.0 mmol, 1.0 equiv). The yellow mixture was stirred for 16 h and then filtered. After concentration, the residue was purified by silica gel column chromatography (hexanes/ethyl acetate mixtures) to give phosphate products as colorless oils (33 to 50% yields).
O-Methyl, O-(p-nitrophenyl), O-(S)-pinacolyl phosphate (23): 1H NMR (400 MHz, CDCl3) δ 8.24 – 8.22 (m, 2H), 7.40 – 7.37 (m, 2H), 4.40–4.32 (m, 1H), 3.88 (d, J = 11.0 Hz, 1.5 H), 3.88 (d, J = 11.0 Hz, 1.5 H), 3.87 (d, J = 11.0 Hz, 1.5 H), 1.35 (d, J = 6.4 Hz, 1.5 H), 1.27 (d, J = 6.4 Hz, 1.5 H), 0.95 (s, 4.5 H), 0.91 (s, 4.5 H). 31P NMR (160 MHz, CDCl3) δ −6.47 (s), −6.28 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO6P: 318.1106, found: 318.1125.
O-Methyl, O-(p-nitrophenyl), O-(R)-pinacolyl phosphate (23): 1H NMR (400 MHz, CDCl3) δ 8.24 – 8.22 (m, 2H), 7.40 – 7.37 (m, 2H), 4.40–4.32 (m, 1H), 3.88 (d, J = 11.0 Hz, 1.5 H), 3.88 (d, J = 11.0 Hz, 1.5 H), 3.87 (d, J = 11.0 Hz, 1.5 H), 1.34 (d, J = 6.4 Hz, 1.5 H), 1.27 (d, J = 6.4 Hz, 1.5 H), 0.94 (s, 4.5 H), 0.90 (s, 4.5 H). 31P NMR (160 MHz, CDCl3) δ −6.48 (s), −6.29 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO6P: 318.1106, found: 318.1125.
General Procedure for Synthesis of p-Nitrophenyl Methylphosphonate Analogs.
A solution of the appropriate alcohol (4 mmol, 1.0 equiv) in anhydrous diethyl ether (10 mL), was chilled to −78 °C and 2.0 mL of LDA (2.0 M in THF, 4.0 mmol, 1.0 equiv) was added. The mixture was stirred for 30 min and transferred via cannula to another flask at −78 °C that contained methyl phosphonate dichloride (0.36 mL, 4.0 mmol, 98%, 1.0 equiv) in diethyl ether (25 mL). The cooling bath was removed and the reaction was allowed to warm to ambient temperature (23 °C). After stirring for 6 h, p-nitrophenol (0.55 g, 4.0 mmol, 1.0 equiv) was added at room temperature followed by the addition of dry triethylamine (0.62 mL, 4.4 mmol, 1.1 equiv). The yellow mixture was stirred for 16 h and then filtered. After concentration, the residue was purified by silica gel column chromatography (hexanes/ethyl acetate mixtures) to give the phosphonate product as a colorless oil (40 to 58% yields).
Methyl O-(p-nitrophenyl), O-isopropyl phosphonate ((RP/SP)-5): 1H NMR (400 MHz, CDCl3) δ 8.29 – 8.22 (m, 2H), 7.44 – 7.38 (m, 2H), 4.92–4.80 (m, 1H), 1.69 (d, J = 17.6 Hz, 3H), 1.39 (d, J = 6.4 Hz, 3H), 1.30 (d, J = 6.4 Hz, 3H). 31P NMR (160 MHz, CDCl3) δ 27.02 (s). HRMS (ESI+) m/z [M+H]+ calc. for C10H15NO5P: 260.0688, found: 260.0681.
Methyl O-(p-nitrophenyl), O-cyclohexyl phosphonate ((RP/SP)-13): 1H NMR (400 MHz, CDCl3) δ 8.29 – 8.22 (m, 2H), 7.44 – 7.38 (m, 2H), 4.62–4.52 (m, 1H), 2.03–1.95 (m, 1H), 1.91–1.81 (m, 1H), 1.80–1.70 (m, 2H), 1.71 (d, J = 17.3 Hz, 3H), 1.66–1.44 (m, 3H), 1.43–1.21 (m, 3H). 31P NMR (160 MHz, CDCl3) δ 27.02 (s). HRMS (ESI+) m/z [M+H]+ calc. for C10H19NO5P: 300.1001, found: 300.0992.
Methyl O-(p-nitrophenyl), O-(S)-pinacolyl phosphonate ((SCRP)-21 + (SCSP)-21): 1H NMR (400 MHz, CDCl3) δ 8.27 – 8.22 (m, 2H), 7.46 – 7.39 (m, 2H), 4.46–4.34 (m, 1H), 1.70 (d, J = 17.6 Hz, 2H), 1.71 (d, J = 17.6 Hz, 1H), 1.36 (d, J = 6.4 Hz, 2H), 1.19 (d, J = 6.4 Hz, 1H), 0.96 (s, 3H), 0.90 (s, 6H). 31P NMR (160 MHz, CDCl3) δ 27.69 (s), 26.88 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO5P: 302.1157, found: 302.1148.
Methyl O-(p-nitrophenyl), O-(R)-pinacolyl phosphonate ((RCRP)-21 + (RCSP)-21): 1H NMR (400 MHz, CDCl3) δ 8.27 – 8.24 (m, 2H), 7.44 – 7.41 (m, 2H), 4.40–4.46–4.35 (m, 1H), 1.70 (d, J = 17.6 Hz, 1.8 H), 1.71 (d, J = 17.6 Hz, 1.2 H), 1.37 (d, J = 6.4 Hz, 1.8 H), 1.19 (d, J = 6.4 Hz, 1.2 H), 0.96 (s, 3.6 H), 0.91 (s, 5.4 H). 31P NMR (160 MHz, CDCl3) δ 27.28 (s), 26.87 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO5P: 302.1157, found: 302.1148.
General Procedure for Synthesis of Thiophosphates 8, 16, and 24.
Lawesson’s reagent (0.800 g, 1.98 mmol, 1.1 eq) was added to a stirred solution of the appropriate phosphates 7, 15, 23 (1.8 mmol, 1 equiv) in anhydrous toluene (25 mL) and the mixture was heated at 105 °C for 42 h. The reaction was cooled, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate, 15:1 to 10:1) to give the thiophosphate product as a colorless oil (35 to 40 % yields).
O-Methyl-, O-(p-nitrophenyl), O-isopropyl thiophosphate ((RP/SP)-8): 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 8.7 Hz, 2H), 7.39–7.33 (m, 2H), 4.96 – 4.85 (m, 1H), 3.88 (d, J = 14.0 Hz, 3H), 1.41 (d, J1 = 6.2 Hz, 3H) 1.38 (d, J = 6.2 Hz, 3H). 31P NMR (160 MHz, CDCl3) δ + 62.52 (s). HRMS (ESI+) m/z [M+H]+ calc. for C10H14NO5PS: 292.0409, found: 292.0395.
O-Methyl-, O-(p-nitrophenyl), O-(S)-pinacolyl thiophosphate ((SCRP)-24 + (SCSP)-24): 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 8.6 Hz, 2H), 7.36 – 7.32 (m, 2H), 4.48–4.40 (m, 1H), 3.86 (d, J = 14.0 Hz, 1.5 H), 3.85 (d, J = 14.0 Hz, 1.5 H), 1.32 (d, J = 6.4 Hz, 1.5 H), 1.27 (d, J = 6.4 Hz, 1.5 H), 0.96 (s, 4.5 H), 0.93 (s, 4.5 H). 31P NMR (160 MHz, CDCl3) δ +63.46 (s), +63.16 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO5PS: 334.0878, found: 334.0870.
O-Methyl, O-(p-nitrophenyl), O-(R)-pinacolyl thiophosphate ((RCRP)-24 + (RCSP)-24): 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 8.6 Hz, 2H), 7.36 – 7.32 (m, 2H), 4.48–4.40 (m, 1H), 3.86 (d, J = 14.0 Hz, 1.5 H), 3.85 (d, J = 14.0 Hz, 1.5 H), 1.32 (d, J = 6.4 Hz, 1.5 H), 1.27 (d, J = 6.4 Hz, 1.5 H), 0.96 (s, 4.5 H), 0.93 (s, 4.5 H). 31P NMR (160 MHz, CDCl3) δ +63.45 (s), +63.16 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO5PS: 334.0878, found: 334.0868.
General Synthesis of p-Nitrophenyl Thiophosphonate Analogs.
Lawesson’s reagent (0.996 g, 2.39 mmol, 1.5 eq) was added to a stirred solution appropriate methyl phosphonate (5, 13, 21) (1.6 mmol, 1 equiv) in anhydrous toluene (25 mL) and the mixture was heated at 100 °C for 4 h. The reaction was cooled, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate, 8:1) to give the methyl thiophosphonate products as colorless oils (45 to 60% yields).
Methyl O-(p-nitrophenyl), O-isopropyl thiophosphonate ((RP/SP)-6). 1H NMR (400 MHz, CDCl3) δ 8.29 – 8.22 (m, 2H), 7.38 – 7.33 (m, 2H), 5.00–4.88 (m, 1H), 2.04 (d, J = 15.4 Hz, 3H), 1.38 (d, J = 6.2 Hz, 3H), 1.31 (d, J = 6.2 Hz, 3H). 31P NMR (160 MHz, CDCl3) δ 92.45 (s). HRMS (ESI+) m/z [M+H]+ calc. for C10H15NO4PS: 276.0459, found: 276.0453.
Methyl, O-(p-nitrophenyl), O-cyclohexyl thiophosphonate ((RP/SP)-14). 1H NMR (400 MHz, CDCl3) δ 8.29 – 8.22 (m, 2H), 7.38 – 7.31 (m, 2H), 4.72–4.60 (m, 1H), 2.06 (d, J = 15.5 Hz, 3H), 2.00–1.92 (m, 1H), 1.89–1.71 (m, 3H), 1.61–1.46 (m, 3H), 1.45–1.24 (m, 3H). 31P NMR (160 MHz, CDCl3) δ 92.55 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H19NO4PS: 316.0772, found: 316.0765
Methyl, O-(p-nitrophenyl), O-(S)-pinacolyl thiophosphonate ((SCRP)-22 + (SCSP)-22). 1H NMR (400 MHz, CDCl3) δ 8.27 – 8.20 (m, 2H), 7.40 – 7.32 (m, 2H), 4.56 – 4.45 (m, 1H), 2.04 (d, J = 15.4 Hz, 0.8H), 2.03 (d, J = 15.4 Hz, 2.2H), 1.33 (d, J = 6.4 Hz, 0.8H), 1.16 (d, J = 6.4 Hz, 2.2H), 0.96 (s, 6.6H), 0.92 (s, 2.4 H). 31P NMR (160 MHz, CDCl3) δ 93.83 (s), 92.18 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO4PS: 318.0929, found: 318.0917
Methyl, O-(p-nitrophenyl), O-(R)-pinacolyl thiophosphonate ((RCRP)-22 + (RCSP)-22). 1H NMR (400 MHz, CDCl3) δ 8.27 – 8.24 (m, 2H), 7.38 – 7.35 (m, 2H), 4.56 – 4.44 (m, 1H), 2.05 (d, J = 15.4 Hz, 1.15 H), 2.04 (d, J = 15.4 Hz, 1.85 H), 1.34 (d, J = 6.4 Hz, 1.15 H), 1.17 (d, J = 6.4 Hz, 1.85 H), 0.97 (s, 5.55 H), 0.93 (s, 3.45 H). 31P NMR (160 MHz, CDCl3) δ 93.83 (s), 92.18 (s). HRMS (ESI+) m/z [M+H]+ calc. for C13H21NO4PS: 318.0929, found: 318.0922
Synthesis of Phosphorus-Fluorine Analogs.
Approximately 2.0 mg of the relevant p-nitrophenyl analog was dissolved in 2.0 mL of DMF in a 1-dram screw cap vial. Solid CsF (~10 mg) was added and the mixture stirred until the reaction was complete. Progress of the reaction was monitored by diluting 10 µL samples into 1.0 mL 50 mM HEPES/KOH buffer (pH 8.5) and recording the absorbance at 400 nm. An appropriate variant of PTE capable of hydrolyzing the p-nitrophenyl analog was added and the absorbance recorded to determine if any of the p-nitrophenyl analog remained. Once the reaction was complete the liquid was transferred to a clean test tube and ~200 mg of dry Dowex-1X4 chloride (Sigma Aldrich) was added. The reaction mixture was allowed to incubate with intermittent mixing for 30 min. The Dowex 1X4 resin was removed by filtration through a plastic syringe fitted with a plastic frit (Torviq). Quantitative conversion of the p-nitrophenyl analog to the fluoridate analog was demonstrate by 19F and 31P NMR spectroscopy.
Expression and Purification of Phosphotriesterase Variants.
The high expression PTE variant A80V/K185R/I274N (VRN) and the additional variants demonstrating unique stereochemical preferences, G60A, H257Y/L303T (YT), BHR-30, and BHR-72 were expressed and purified as previously described (1, 9, 19). All variants were expressed with the addition of 1.0 mM CoCl2 in the growth medium, purified, and stored with 100 µM CoCl2.
Measurement of Kinetic Constants.
Kinetic measurements for the hydrolysis of analogs containing a p-nitrophenyl leaving group were determined using total hydrolysis reactions by following the release of p-nitrophenol (E400 = 17,000 M−1 cm−1) in a 1.0 mL volume reaction (50 mM CHES, pH 9.0, and 0.5% DMF) with ~40–60 µM racemic substrate at 30 oC. Substrates were dissolved in DMF prior to use. Reactions were initiated by the addition of appropriately diluted enzyme and the absorbance at 400 nm recorded as a function of time in a 1-cm cuvette using a Spectramax 384 Plus UV-vis spectrophotometer (Molecular Devices). For variants which showed large differences in the rate of hydrolysis of each isomer, the hydrolysis of the first isomer was initiated at a low enzyme concentration until complete, followed by the addition of a second aliquot of enzyme at a higher concentration to catalyze the hydrolysis of the slower isomer. Reactions were conducted in triplicate and data reported as the average ± the error of mean.
Hydrolyses of analogs containing the fluoride leaving group were followed by the release of fluoride using a Mettler-Toledo SevenMulti meter equipped with a fluoride selective electrode and recorded using LabX direct Software. Reactions were 5 mL total volume in 50 mM HEPES (pH 8.0) with 1% DMF and 50 to 100 µM substrate. Substrates were prepared in DMF as described above and used without further purification. Reactions were initiated by the addition of appropriately diluted enzyme. Variant and substrate combinations showing large differences in the rate of hydrolysis of the two isomers were followed initially at a low enzyme concentration to allow for the complete hydrolysis of the first isomer, followed by addition of a second aliquot at higher enzyme concentration to catalyze the hydrolysis of the second isomer. To ensure sufficient responsiveness of the electrode, all reactions were followed in triplicate with at least two different enzyme concentrations. Data are reported as the average ± error in mean.
Hydrolysis of GB (1), GD (17), and GF (9) were followed in total hydrolysis reactions by fluoride release using a fluoride selective electrode connected to an Accumet XL250 ion selective meter (Thermo Fisher). Reactions were 10 mL total volume with 50 mM bis-tris-propane (pH 7.2) and 100 µM substrate. Reactions were initiated by the addition of appropriately diluted enzyme and followed until completion.
Acetylcholine Esterase Inactivation Assays.
Acetylcholine esterase (AChE) was dissolved in 50 mM HEPES (pH 8.0) and catalytic activity determined by an assay with 1.0 mM acetylthiolcholine (50 mM HEPES, pH 8.0, and 0.3 mM DTNB). AChE was subsequently diluted to 0.8 U/mL (1 U = 1 µmol/min) in 50 mM HEPES (pH 8.0). Inhibitors were dissolved in pure DMF. AChE (190 µL) was mixed with 10 µL of diluted inhibitor. An initial test was conducted to determine maximal solubility of inhibitor and/or concentration which resulted in 70% to 90% inhibition in 10 min. A subsequent dilution series was prepared for each inhibitor resulting in 7 inhibitor concentrations and a control containing only DMF for each inhibition test. Remaining activity was measured at 1-to-3-minute intervals over 30 min using a rapid dilution method by removing 5-µL samples and diluting into an activity assay (250 µL total volume, 50 mM HEPES (pH 8.0), 1.0 mM acetylthiocholine, and 0.3 mM DTNB). Remaining activity was followed by release of the thiol product by addition of DTNB (ΔE412 = 12,500 M−1 cm−1). Assays were conducted in a 96-well format using a Spectramax 384 Plus 96-well plate reader.
Data Analysis.
Data from the total hydrolysis reactions followed by the absorbance of p-nitrophenol were converted to fraction hydrolyzed according to eqn. 1 where At is absorbance at time (t), Ao is the initial absorbance, and Af is final absorbance. Analogous procedures were used for reactions followed by fluoride release by substituting the initial, final, and intermediate concentrations of free fluoride. The fraction hydrolyzed was plotted as a function of time and fit to eqn. 2 for curves displaying a single exponential or eqn. 3 for curves displaying two exponentials. For each reaction, kcat/Km was calculated from the exponential rates according to eqn. 4 where a1 and a2 are the magnitude of the exponential phases, k1 and k2 are the exponential rate constants, t is time and [E] is concentration of enzyme used in the assay.
| (1) |
| (2) |
| (3) |
| (4) |
All assays for acetylcholinesterase activity were conducted using 1.0 mM acetylthiocholine. At each time point an uninhibited sample, which was treated in exactly the same manner as the inhibition tests, was included. The observed change in absorbance (ΔA/min) was converted to fractional activity according to eqn. 5, where ΔAt is the rate of change in absorbance after enzyme is incubated for time t in the presence of inhibitor and ΔAo is the rate of change in absorbance after enzyme is incubated in the absence of the inhibitor for time t. The natural log of the fractional activity is plotted as a function of time yielding a linear relationship with an intercept of zero and the slope of negative kapp according to eqn. 6. Replotting kapp as a function of inhibitor concentration yields a linear plot at low inhibitor concentration with the slope equal to the bimolecular inactivation constant kinact/KI (M−1 min−1) according to eqn. 7 (20).
| (5) |
| (6) |
| (7) |
Results
Synthesis of Substrates.
All of the analogs were initially synthesized as the p-nitrophenyl phosphate and phosphonate analogs (5, 7, 13, 15, 21 and 23) with yields of 30% - 60%. Portions of these compounds were subsequently converted to the p-nitrophenyl thiophosphate or thiophosphonate analogs (6, 8, 14,16, 22, and 24) by reaction with Lawesson’s reagent with 40% - 60% yields. Conversion of the p-nitrophenyl analogs with solid CsF in DMF resulted in a quantitative transformation of the p-nitrophenyl analogs to the corresponding fluorophosphate (3, 11, and 19), fluorothiophosphate (4, 12, and 20), or fluorothiophosphonate substrates (2, 10, and 18). The structures of all compounds are illustrated in Schemes 1 and 2.
Selection of Variants.
To evaluate the analogs, a set of PTE variants with known stereoselective properties was selected. The wild-type-like high-expression variant VRN (A80V/K185R/I274N) was used as the wild-type enzyme and is known to prefer the less toxic (RP)-isomers of the G-type nerve agents (9, 21). The variant G60A shows a much stronger stereoselectivity for the (RP)-isomers of the nerve agents and analogs (6, 9). The variant YT (H257Y/L303T) demonstrates a reversed stereoselectivity from the wild-type enzyme (6). Two additional variants, BHR-30 (A80V/F132C/K185R/H254G/I274T) and BHR-72 (A80V/I106A/F132C/K185R/H254G/I274N) were selected for characterization due to the demonstration of reduced stereoselectivity with other nerve agents and analogs (1).
Kinetic Characterization of Analogs.
The use of a low concentration total hydrolysis assay allows for the determination of kcat/Km for both isomers of the analogs in a single reaction. In some cases, such as the hydrolysis of 5 by YT, the time course of the reaction proceeds as an obvious 2-exponential process with the magnitude of each phase being approximately equal and the rate for the first phase being 9-fold faster than the second (Figure 1a). In other cases, the difference in catalytic rates for the two isomers is sufficiently large that one isomer is effectively consumed before hydrolysis of the other has proceeded. For example, Figure 1b illustrates the hydrolysis of racemic 3 by G60A. The first isomer is effectively consumed in 500 s by 1.4 nM G60A while the second isomer has not been hydrolyzed to a substantial degree. The hydrolysis of the second isomer was achieved by addition of 70 nM G60A, allowing the two exponential rates to be calculated from a single experiment. This methodology also allows for the assignment of stereoselectivity by comparison to G60A. G60A has a strong and well-known stereoselectivity with chiral phosphorus centers, and conditions can be established under which it will effectively hydrolyze only a single isomer (9, 22). As demonstrated in Figure 1c, once a variant has hydrolyzed half of a racemic mixture, the addition of G60A will either have no effect (same selectivity), or as seen when YT was used to hydrolyzed the initial isomer of 3, G60A will result in the rapid hydrolysis of the other isomer (opposite selectivity).
Figure 1.
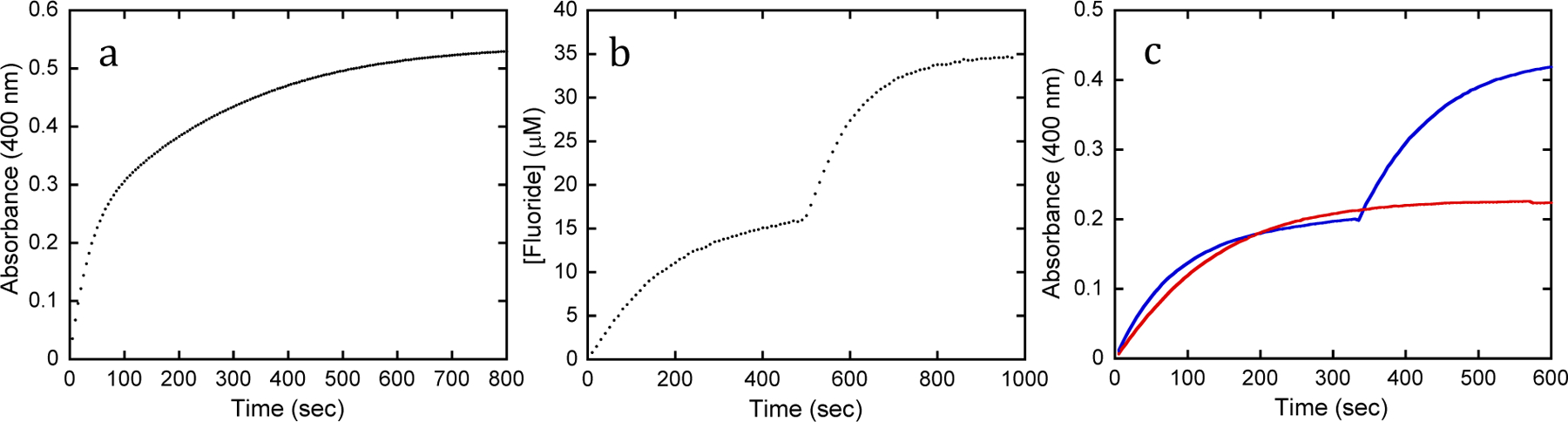
Kinetic time courses for PTE variants with G-agent analogs. (a) Complete hydrolysis of 30 µM (RP/SP)-5 by 40 nM YT showing biphasic hydrolysis of the two enantiomers. (b) Hydrolysis of 35 µM (RP/SP)-3 by G60A. First isomer is hydrolyzed after the addition of 1.4 nM G60A, while the second isomer is hydrolyzed by the subsequent addition of 70 nM G60A. (c) Determination of stereoselectivity of YT with the mixture of the (SCSP)- and (SCRP)-diastereomers of 21. Red trace is for hydrolysis of (SCRP)-21 in the mixture by 0.35 µM G60A. The blue curve was initiated by the addition 4.0 µM YT, which hydrolyzes only one isomer. The other isomer is hydrolyzed after the addition of 0.35 µM G60A, demonstrating the preference of YT to be (SCSP)-21.
Effects of Substrate Substitutions on Activity of Wild-Type PTE.
Replacement of one of the substituents to the phosphorus center in the compounds tested for activity in this investigation resulted in changes in the magnitude of the kinetic constants, which depended on the specific substrate being considered. For the wild-type enzyme with the analogs for GB (SP/RP)-1, substitution of the phosphoryl oxygen with sulfur in (RP)-2 has relatively little effect and hydrolysis proceeds at nearly the same rate as with (RP)-1 (Figure 2a). Wild-type PTE exhibits no stereoselectivity against GB ((SP)-1 and (RP)-1) but substitution with sulfur results in a 15-fold preference for hydrolysis of the (RP)-isomer of 2. Substitution of an O-methyl ester for the methyl phosphonate moiety increased the catalytic rate of wild-type PTE for both isomers of 3 compared to 1 with no selectivity observed. Addition of the methyl substitution to the thiol analog in compound 4 did not further alter the activity of the wild-type enzyme. Substitution of the leaving group for p-nitrophenyl with compound 5 resulted in a substantial increase in rate with (RP)-5 but little effect on the rate of (SP)-5, resulting in more than an order of magnitude preference for the (RP)-enantiomer. Combination of the sulfur substitution with the p-nitrophenyl leaving group resulted in slightly diminished activity against (SP)-6 and slightly increased activity against (RP)-6, compared to the p-nitrophenyl alone ((SP)-5 and (RP)-5). Conversely, the combination of the O-methyl with the p-nitrophenyl substitution in 7 resulted in increases in the activity for both isomers compared to the p-nitrophenyl alone (5). Combination of all three substitutions in 8 resulted in activity intermittent between the O-methyl ((SP)-7) and thiol ((RP)-6) for the preferred isomers (please note that the absolute relative orientation of constituents is identical in (SP)-7 and (RP)-6 with the change in priority between the P-methyl and O-methyl resulting in a change in the SP and RP designations, with the same being true for compounds 3, 7, 11, 15, 19 and 23). The inclusion of all three substitutions substantially decreased the activity of the wild-type enzyme for the hydrolysis of (SP)-8, compared to all of the p-nitrophenyl analogs ((RP)-3, (SP)-5, (SP)-6, and (RP)-7). These relationships are shown graphically in Figure 2a.
Figure 2.
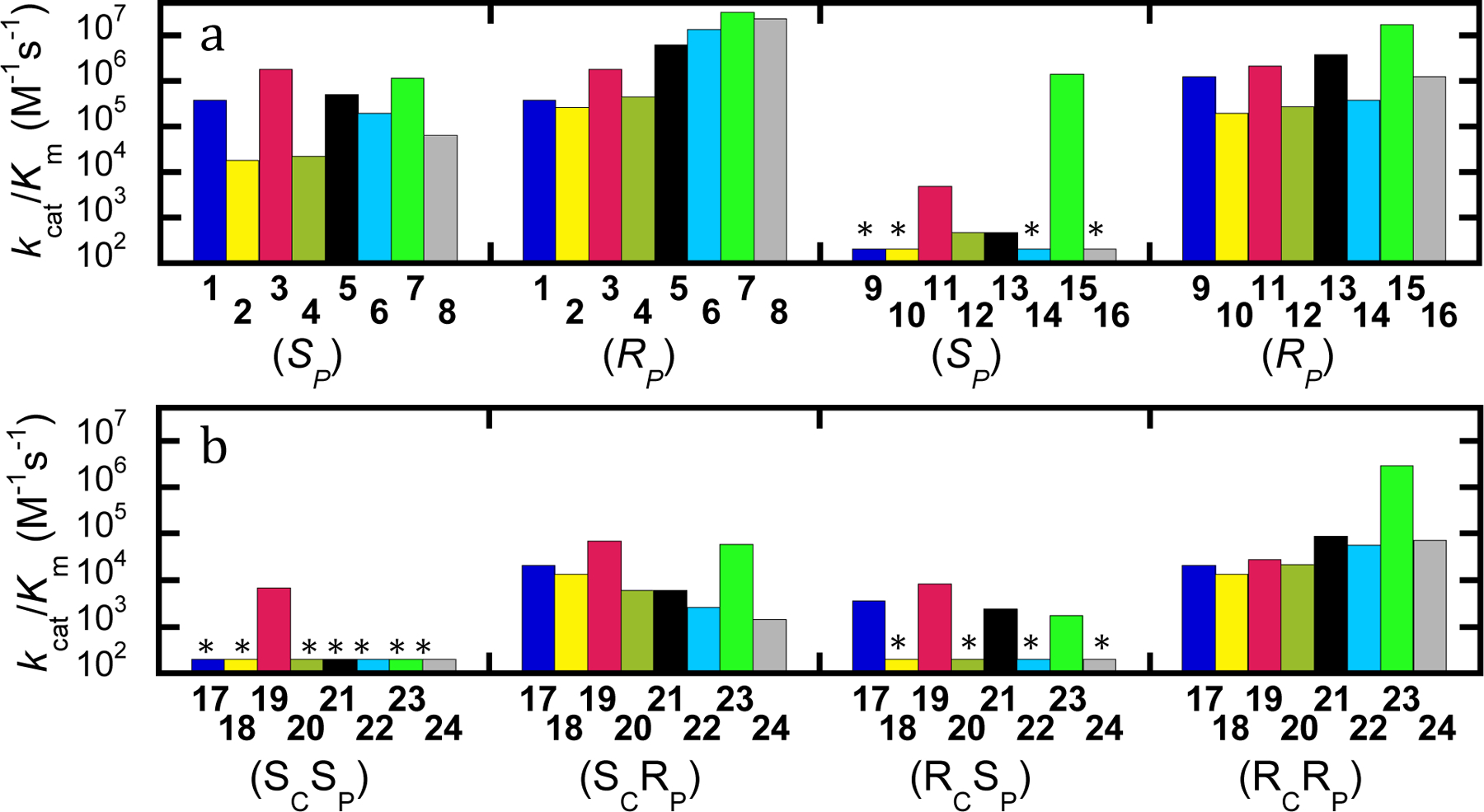
(A) Activity of wild-type PTE with analogs of G-agents. GB (1-8) and GF (9-16) analogs. (B) Activity of wild-type PTE with analog of GD analogs (17-24). Compounds are given by number with isomer given below. Asterisk indicates compound with insufficient activity to be quantified. Estimates of the upper limit of kcat/Km for these compounds are provided in Tables 1 and 2.
The effects of group substitutions tended to be less dramatic with the GF (9) analogs. Wild-type PTE has a strong stereoselectivity for (RP)-9, with the activity using (SP)-9 not being quantifiable. Substitution with sulfur reduced the activity for the preferred isomer by nearly an order of magnitude compared to (RP)-9, whether alone ((SP)-10) or in combination with the O-methyl ((RP)-12), or p-nitrophenyl ((SP)-14) substitutions. Replacement of the P-methyl group for an O-methyl ((SP)-11) or fluoride for p-nitrophenyl ((RP)-13) had only modest effects on the activity of the wild-type enzyme compared to (RP)-9. However, the combination of the O-methyl and p-nitrophenyl groups resulted in activity for the preferred enantiomer ((SP)-15) increased ~10-fold. When the sulfur was substituted as in 16 the activity for the preferred isomer dropped to levels similar that of the original compound ((RP)-9). The marginal activity with the slow isomers prevented quantification of the activity of the wild-type enzyme with the (SP)-isomers of GF (9), and the analogs 10, 14, and 16. However, it can be determined that the activity is greater than 1000-fold reduced relative to the (RP)-isomers. Inclusion of the O-methyl group rather than the P-methyl allowed the quantification of the activity against the slower isomer both as the single substitution ((RP)-11) or in combination with the sulfur substitution ((SP)-12) as did the substitution of the fluoride leaving group for p-nitrophenyl ((SP)-13). The combination of the O-methyl and p-nitrophenyl substitutions ((RP)-15) dramatically improved the activity with the wild-type enzyme, with greater than 1000-fold higher activity compared to (SP)-9. Unfortunately, the dramatic improvement was reversed when the sulfur substitution was included with the O-methyl and p-nitrophenyl leaving group modifications in (SP)-16. These relationships are represented graphically in Figure 2a.
GD (17) contains a carbon stereocenter in addition to the chiral phosphorus center. As was seen with GF (9) there is very strong stereoselectivity for the hydrolysis of GD. With the (SC)-stereocenter there is more than 200-fold selectivity for the (RP)-isomer. Substitution of the phosphoryl oxygen with sulfur reduced the activity for the preferred isomer ((SCRP)-18) modestly, while substitution of the leaving group for p-nitrophenyl (SCRP)-21 or combination of the sulfur and O-methyl group substitutions ((SCSP)-20) further reduced the activity. Substitution of the O-methyl for the P-methyl alone (SCSP)-19 or in combination with the p-nitrophenyl leaving group (SCSP)-23 significantly improved activity compared to (SCRP)-17. Inclusion of the p-nitrophenyl leaving group with the thiophosphate center dramatically reduced activity with (SCRP)-24, or without (SCRP)-22, the O-methyl substitution compared to (SCRP)-17. Against the slower isomers with the (SC)-stereocenter, the activity found to be 100 to 1000-fold reduced (kcat/Km < 200 M−1 s−1), but the only analog which had quantifiable activity was (SCRP)-19 where the substitution of the P-methyl with the O-methyl improves the activity to 6.8 × 10⁴ and gives only 10-fold selectivity compared to (SCSP)-19. These relationships are represented graphically in Figure 2b.
With the RC-stereocenter the effects were substantially different for the two isomers. For the preferred isomer the inclusion of the sulfur substitution ((RCRP)-18), the O-methyl ((RCSP)-19), or the p-nitrophenyl leaving group ((RCRP)-21) had little effect. Similarly, the combination of the sulfur with O-methyl ((RCRP)-20) or the p-nitrophenyl leaving group ((RCRP)-22) only modestly altered the activity compared to (RCRP)-17. Combination of the O-methyl and the p-nitrophenyl group in (RCSP)-23 was the only preferred isomer to show significantly higher activity than (RCRP)-17. However, when the sulfur substitution was included in (RCRP)-24, the activity was diminished to levels similar to (RCRP)-17. For the slower isomers the inclusion of the thiophosphate reduced activity below quantifiable levels for all combination ((RCSP)-18, (RCSP)-20, (RCSP)-22, and (RCSP)-24). Substitution of the O-methyl ((RCRP)-19), or the p-nitrophenyl leaving group ((RCSP)-21) and the combination ((RCRP)-23) only had minimal effects on the activity of the wild-type enzyme compared to (RCSP)-17. These relationships are represented graphically in Figure 2b. The kinetic constants for the hydrolysis of the substrates are provided in Tables 1 and 2.
Table 1.
Kinetic constants for inactivation of AChEa, and for hydrolysis of compounds 1-16 using mutants of phosphotriesterase.b
| Compound | Inactivation rate constant kinact/KI, (M−1 min−1) | wild-type | G60A | YT kcat/Km (M−1 s−1) | BHR-30 | BHR-72 |
|---|---|---|---|---|---|---|
| (SP)-1 | (1.4 × 10⁷)c | 3.7 × 105 | 4.7 × 104 | 5.8 × 106 | 2.4 × 106 | 3.9 × 105 |
| (RP)-1 | 3.7 × 105 | 3.5 × 105 | <1 × 104 | 2.4 × 106 | 2.6 × 106 | |
| (SP)-2 | 2.8 × 103 | 1.7 × 104 | <1 × 102 | 4.8 × 104 | 4.0 × 104 | 3.6 × 104 |
| (RP)-2 | 2.5 × 105 | 5.6 × 105 | 4.8 × 104 | 4.0 × 104 | 3.6 × 104 | |
| (RP)-3 | 1.0 × 104 | 1.7 × 106 | 2.6 × 105 | 1.4 × 106 | 4.7 × 106 | 1.7 × 106 |
| (SP)-3 | 1.7 × 106 | 5.7 × 106 | 1.2 × 105 | 4.7 × 106 | 1.7 × 106 | |
| (SP)-4 | 1.8 × 102 | 2.2 × 104 | <1 × 103 | 2.0 × 104 | 1.6 × 104 | 4.1 × 104 |
| (RP)-4 | 4.3 × 105 | 1.8 × 106 | 2.0 × 104 | 6.0 × 104 | 4.1 × 104 | |
| (SP)-5 | 4.4 × 103 | 4.8 × 105 | 3.0 × 104 | 8.1 × 105 | 4.9 × 105 | 3.6 × 105 |
| (RP)-5 | 6.0 × 106 | 4.2 × 106 | 9.0 × 104 | 4.9 × 105 | 3.6 × 105 | |
| (SP)-6 | NOd | 1.9 × 105 | 1.6 × 103 | 2.0 × 105 | 4.5 × 104 | 1.8 × 105 |
| (RP)-6 | 1.3 × 107 | 2.0 × 107 | 2.0 × 105 | 2.9 × 105 | 1.8 × 105 | |
| (RP)-7 | 1.2 × 103 | 1.1 × 106 | 1.4 × 105 | 1.1 × 106 | 3.9 × 105 | 4.7 × 105 |
| (SP)-7 | 3.1 × 107 | 5.6 × 107 | 1.1 × 106 | 8.7 × 106 | 3.8 × 106 | |
| (SP)-8 | NO | 6.3 × 104 | <1 × 103 | 2.8 × 104 | 3.5 × 104 | 5.6 × 104 |
| (RP)-8 | 2.2 × 107 | 4.0 × 107 | 2.8 × 104 | 2.4 × 106 | 3.6 × 105 | |
| (SP)-9 | (2.6 × 10⁸)c | <1 × 104 | <1 × 103 | 5.4 × 105 | 1.2 × 105 | <1 × 10³ |
| (RP)-9 | 1.2 × 106 | 8.8 × 10⁵ | <1 × 10³ | 1.3 × 106 | 3.0 × 105 | |
| (SP)-10 | 3.2 × 103 | <1 × 102 | <1 ×102 | 9.5 × 102 | <1.5 × 102 | 4.0 × 103 |
| (RP)-10 | 1.9 × 105 | 2.2 × 105 | 9.4 × 103 | 1.1 × 105 | 2.8 × 104 | |
| (RP)-11 | 3.0 × 105 | 4.8 × 103 | <1 × 103 | 3.9 × 105 | 3.3 × 104 | 1.4 × 105 |
| (SP)-11 | 2.1 × 106 | 2.9 × 106 | 3.9 × 105 | 6.0 × 106 | 1.8 × 106 | |
| (SP)-12 | 3.1 × 103 | 4.6 × 102 | <1 × 103 | <1 × 102 | 1.1 × 103 | 4.2 × 103 |
| (RP)-12 | 2.6 × 105 | 5.3 × 105 | 8.4 × 103 | 2.0 × 105 | 1.2 × 105 | |
| (SP)-13 | 5.1 X 104 | 4.6 × 102 | <6 × 102 | 1.7 × 105 | 8.8 × 102 | 4.5 × 104 |
| (RP)-13 | 3.7 × 106 | 3.0 × 106 | 1.7 × 105 | 9.9 × 105 | 4.0 × 105 | |
| (SP)-14 | NO | <2 × 102 | <6 × 102 | 2.6 × 103 | <2 × 102 | 3.8 × 103 |
| (RP)-14 | 3.6 × 105 | 6.0 × 105 | 2.7 × 104 | 1.7 × 105 | 1.0 × 105 | |
| (RP)-15 | 8.7 × 103 | 1.4 × 106 | <1 × 103 | 6.1 × 104 | 1.2 × 104 | 4.8 × 104 |
| (SP)-15 | 1.7 × 107 | 1.9 × 107 | 2.3 × 105 | 6.3 × 106 | 4.1 × 106 | |
| (SP)-16 | 4.4 × 102 | < 2 × 10² | <1 × 103 | <1 × 10² | <2 × 102 | 9.7 × 103 |
| (RP)-16 | 1.2 × 106 | 1.5 × 106 | 3.0 × 103 | 2.7 × 105 | 5.7 × 104 |
Table 2.
Kinetic constants for inactivation of AChEa, and for the hydrolysis of compounds 17-24 using mutants of phosphotriesterase.b
| Compound | Inactivation rate constant kinact/KI, (M−1 min−1) | wild-type | G60A | YT kcat/Km (M−1 s−1) | BHR-30 | BHR-72 |
|---|---|---|---|---|---|---|
| (SCSP)-17 | (1.8 × 10⁸)c | <1 × 102 | <3 × 10¹ | 3.9 × 105 | 1.4 × 106 | 4.1 × 104 |
| (SCRP)-17 | 2.1 × 104 | 1.7 × 104 | <1 × 103 | 1.4 × 106 | 4.1 × 104 | |
| (SCSP)-18 | 3.9 × 103 | <1 × 102 | <1.5 × 102 | 2.8 × 103 | <1.5 × 102 | <1 × 102 |
| (SCRP)-18 | 1.3 × 104 | 4.3 × 104 | 2.8 × 103 | 3.3 × 103 | 3.9 × 103 | |
| (SCRP)-19 | 8.6 × 105 | 6.8 × 103 | <1 × 103 | 1.3 × 105 | 3.4 × 104 | 2.5 × 104 |
| (SCSP)-19 | 7.0 × 104 | 4.1 × 105 | <1 × 103 | 5.7 × 105 | 2.5 × 104 | |
| (SCSP)-20 | 1.2 × 103 | <1 × 103 | <1 × 103 | <1 × 102 | <1 × 103 | <1.0 × 103 |
| (SCRP)-20 | 5.9 × 103 | 2.4 × 104 | <1 × 102 | 4.5 × 103 | 4.0 × 103 | |
| (SCSP)-21 | 3.3 × 101 | <2 × 102 | <6 × 102 | 3.7 × 103 | <2 × 102 | <2 × 102 |
| (SCRP)-21 | 5.9 × 103 | 3.2 × 104 | <2 × 102 | 1.4 × 104 | 3.9 × 103 | |
| (SCSP)-22 | NOd | <2 × 102 | <6 × 102 | 2.2 × 102 | <2 × 102 | <2 102 |
| (SCRP)-22 | 2.6 × 103 | 8.3 × 103 | 2.2 × 102 | 7.3 × 102 | 4.5 × 102 | |
| (SCRP)-23 | 2.2 × 102 | <5 × 102 | <1 × 103 | 1.4 × 103 | <3 × 102 | <3.0 × 102 |
| (SCSP)-23 | 5.8 × 104 | 7.3 × 106 | <3 × 102 | 3.3 × 105 | 1.0 × 105 | |
| (SCSP)-24 | NO | <2 × 102 | <1 × 103 | <3 × 102 | <3 × 102 | <3 × 102 |
| (SCRP)-24 | 1.4 × 103 | 2.3 × 104 | <3 × 102 | 1.7 × 103 | 9.1 × 102 | |
| (RCSP)-17 | (2.8 × 10⁸)c | 3.6 × 103 | <3 × 10¹ | 3.9 × 105 | 1.4 × 106 | 4.1 × 104 |
| (RCRP)-17 | 2.1 × 104 | 6.0 × 10⁴ | 8.4 × 104 | 1.4 × 106 | 4.1 × 104 | |
| (RCSP)-18 | 1.0 × 103 | <1 × 102 | <1.5 × 102 | 1.8 × 103 | 1.1 × 103 | 2.4 × 103 |
| (RCRP)-18 | 1.3 × 104 | 3.9 × 104 | 1.8 × 103 | 5.0 × 103 | 2.4 × 103 | |
| (RCRP)-19 | 2.9 105 | 8.2 × 103 | <1 × 103 | 1.0 × 105 | 9.5 × 104 | 1.5 × 104 |
| (RCSP)-19 | 2.7 × 104 | 4.5 × 105 | 3.1 × 103 | 5.8 × 105 | 6.3 × 104 | |
| (RCSP)-20 | 4.4 × 102 | <1 × 103 | <1 × 103 | <1 × 102 | <1 × 103 | <5 × 103 |
| (RCRP)-20 | 2.2 × 104 | 2.6 × 105 | <1 × 102 | 2.2 × 104 | 9.6 × 103 | |
| (RCSP)-21 | 3.2 × 102 | 2.4 × 103 | <6 × 102 | 2.0 × 104 | 3.0 × 103 | 5.2 × 103 |
| (RCRP)-21 | 8.9 × 104 | 2.2 × 105 | 6.9 × 102 | 5.5 × 104 | 2.1 × 104 | |
| (RCSP)-22 | NO | <2 × 102 | <6 × 102 | <2 × 102 | <2 × 102 | <2 × 102 |
| (RCRP)-22 | 5.7 × 104 | 1.5 × 105 | 2.6 × 102 | 6.0 × 103 | 2.1 × 103 | |
| (RCRP)-23 | 8.5 × 102 | 1.7 × 103 | <1 × 103 | 2.6 × 103 | 5.9 × 102 | 8.5 × 102 |
| (RCSP)-23 | 2.9 × 106 | 1.0 × 107 | 2.6 × 103 | 6.1 × 105 | 5.0 × 105 | |
| (RCSP)-24 | NO | <5 × 102 | <1 × 103 | <3 × 102 | <3 × 102 | <5 × 102 |
| (RCRP)-24 | 7.2 × 104 | 2.2 × 106 | <3 × 102 | 2.8 × 104 | 7.1 × 103 |
Utility of Analogs in Predicting Variant Activity.
Testing the analogs with variants with differing activity and stereoselectivity against the G-type nerve agents has enabled an evaluation of this set of analogs for their ability to predict the change in the activity with the actual nerve agents. With GB (1) the wild-type enzyme shows no stereoselectivity. The variant G60A exhibits a significantly reduced activity toward the SP-isomer resulting in significant selectivity for (RP)-1. The YT variant shows strong and reversed stereoselectivity for GB (1) with activity increased an order of magnitude for (SP)-1 and diminished more than an order of magnitude for (RP)-1 giving this variant a strong selectivity for hydrolysis of the (SP)-isomer. The variant BHR-30 shows no specificity, but increased activity for GB (1) compared to wild-type enzyme, while BHR-72 shows a significant increase in activity for the (RP)-isomer resulting in a ~7-fold preference for hydrolysis of (RP)-1. These relationships are displayed graphically in Figure 3a.
Figure 3.
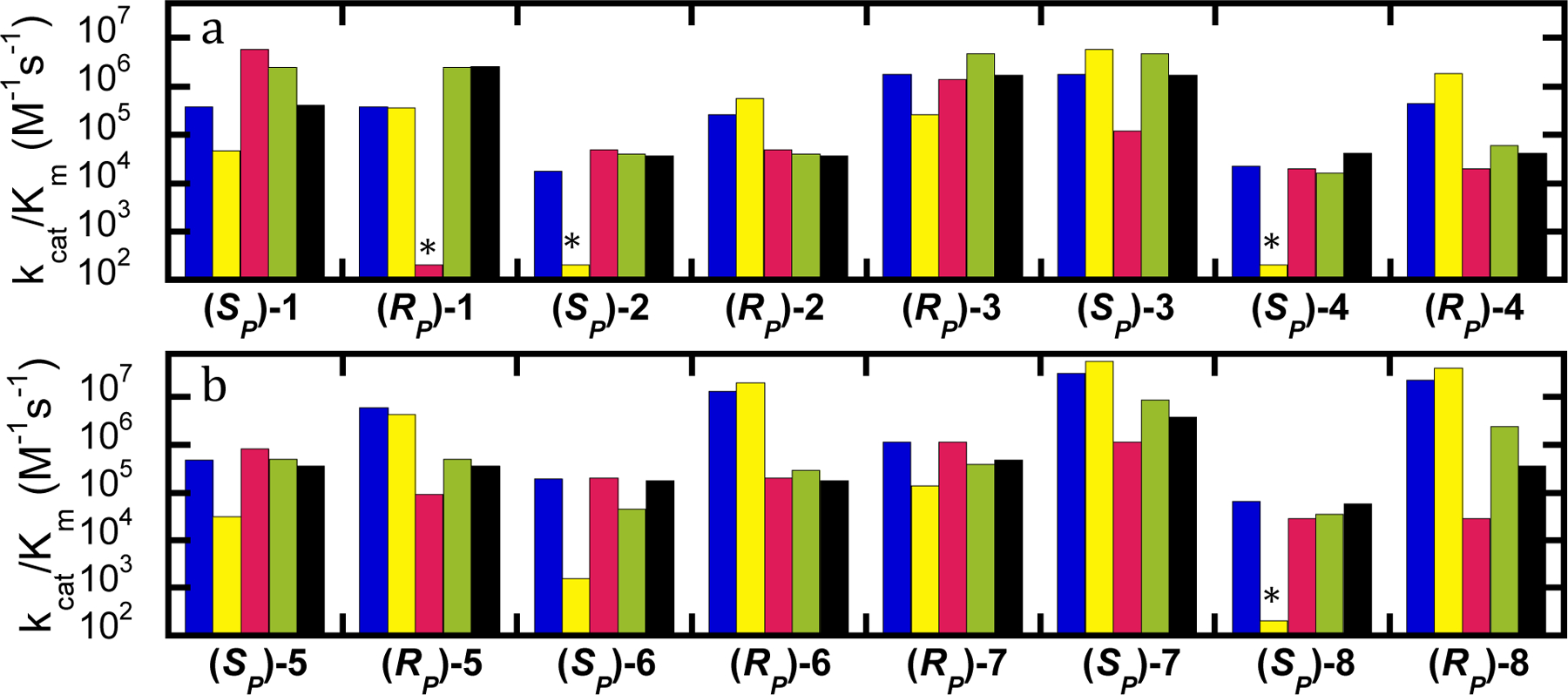
Values of kcat/Km for hydrolysis of GB (1) and analogs (2-8) by variants of PTE. Wild-type PTE (blue), G60A (yellow), YT (red), BHR-30 (green), and BHR-72 (black). (A) Analogs with a fluoride leaving group. (B) Analogs with a p-nitrophenyl leaving group. Variants without quantifiable activity are marked with an asterisk, and the detection limit for the assay is provided in Table 1.
All of the analogs predicted the correct enhanced stereoselectivity of G60A and the decrease in activity for hydrolysis of the (SP)-isomer of GB (1). However, compounds 3 and 4 predicted a substantial increase in activity with (RP)-1, which was not seen. Similarly, all of the analogs predicted a decrease in activity of YT with the (RP)-isomer of GB (1), but only 2 and 5 predicted an increase in activity compared to wild-type for the (SP)-isomer, and only 3 predicted the strong selectivity observed with the authentic agent. Likewise, only compound 3 predicted a significant increase in activity against the (RP)-isomer of GB (1) by BHR-30, though all but 7 and 8 correctly predicted the lack of stereoselectivity. Compounds 2 and 3 predicted the observed increase in activity for BHR-30 with the (SP)-isomer of GB (1). Unfortunately, none of the analogs predicted the substantial increase in activity observed with BHR-72 with (RP)-1, although 7 and 8 predicted the correct stereoselectivity and all but 7 predicted little change in the activity of BHR-72 for (SP)-1. While none of the analogs of GB (1) tested were completely accurate at predicting changes in activity for all of the variants with both isomers, (SP)-2 was best at predicting the changes in activity against the actual target (SP)-1. These relationships are displayed graphically in Figure 3.
Against GF (9), wild-type, G60A and BHR-72 show very strong selectivity for hydrolysis of the (RP)-isomer, while the YT variant shows strong selectivity for the (SP)-isomer. BHR-30 exhibits more than an order of magnitude increased activity for the (SP)-isomer and much less stereoselectivity compared to the other variants, though it still has a preference for the (RP)-isomer. All of the analogs tested predicted the correct stereoselectivity for the wild-type enzyme. However, none of the analogs predicted the correct selectivity of the YT variant with GF (9). The sulfur analogs predicted a wild-type-like preference for YT, while the oxygen analogs predicted very little selectivity. Compounds 11, 13, and 14 were able to correctly predict substantially improved activity against the (SP)-isomer of GF (9) by the YT variant, and all of the analogs correctly predicted reduced activity against the (RP)-isomer of GF. Similarly, all of the analogs correctly predicted wild-type like activity of the BHR-30 variant against the (RP)-isomer of GF (9) as well as the observed preference for the (RP)-isomer, but only compounds 11, 12 and 13 predicted the observed improvement against the (SP)-isomer. With BHR-72, all of the analogs predicted a reduction of activity against (RP)-9, but six of the analogs predicted an increase in activity for the SP-isomer, while only 15 correctly predicted the low activity observed with the authentic agent. While none of the analogs were correct with all the variants tested, 11 was best at predicting the correct changes in activity compared to the wild-type enzyme. Compound 11 correctly predicted the stereoselectivity for four of five variants, though the reversed stereoselectivity of YT was not predicted and BHR-72 was incorrectly predicted to have significantly improved activity with (SP)-9. These relationships are shown graphically in Figure 4.
Figure 4.
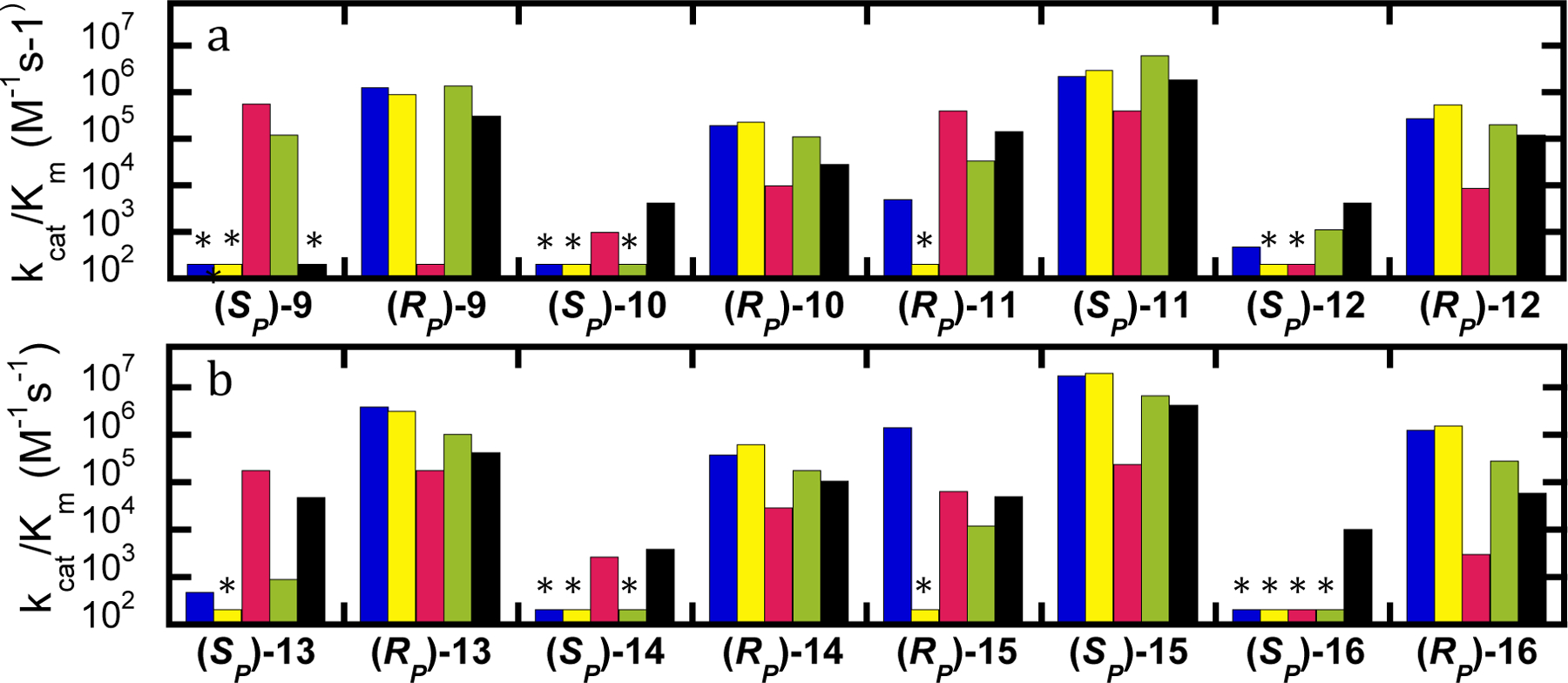
Values of kcat/Km for hydrolysis of GF (9) and analogs (10-16) by variants of PTE. (A) Compounds with fluoride leaving group. (B) Compounds with p-nitrophenyl leaving group. Wild-type PTE (blue), G60A (yellow), YT (red), BHR-30 (green), and BHR-72 (black). Variants without quantifiable activity are marked with an asterisk and the detection limit for the assay is provided in Table 1.
With GD (17) wild-type PTE has reasonable activity against three of the four isomers. The two (RP)-isomers, (SCRP)-17 and (RCRP)-17, are hydrolyzed approximately an order of magnitude faster than the (RCSP)-isomer, and there is only marginal activity observed with the (SCSP)-isomer. The majority of analogs failed to predict this selectivity with compound 21 giving the best result. Unfortunately, the activity with the (SP)-isomers with the sulfur substitution (18, 20, 22, and 24) was too low to differentiate. Compound 19 predicted similar activity for both the (RCSP)- and (SCSP)-isomers. Compound 23 correctly predicted the selectivity between the (RCSP)- and (SCSP)-isomers, but predicted nearly two orders of magnitude selectivity between the (RCRP)- and (SCRP)-isomers. The YT variant shows a dramatic enhancement with the (SP)-isomers of GD (17) as well as a stereochemical preference for the SP-isomers, the least favored isomer being (SCRP)-17. Compounds 18, 19, 21, and 23 predicted the improvement in activity against the (SP)-isomers, but none of the variants correctly predicted the improvement of YT over the wild-type enzyme for (RCRP)-17. Only 19 and 21 were able to predict the correct stereoselectivity for YT. BHR-30 surprisingly hydrolyzed all isomers of GD (17) without significant stereoselectivity, and with a rate substantially improved compared to the other variants. Similarly, BHR-72 did not show significant selectivity with GD (17). The activity of BHR-72 with the (SP)-isomers is significantly enhanced compared to wild-type enzyme, while maintaining wild-type-like activity for the (RP)-isomers. Only 19 was able to accurately predict the high activity of BHR-72 against all isomers, though it did predict a slight decrease with respect to wild-type for hydrolysis of (SCRP)-17, which was not seen. Overall, the best predictor of activity for GD (17) was compound 19 with correct predictions of activity for all but one isomer. While 21 was also a reasonable predicter of activity, this analog showed much smaller changes compared to GD (17), and failed to predict YT having high activity with the (RCRP)-isomer. These relationships are shown graphically in Figure 5. The values of kcat/Km for the hydrolysis of compounds 1–24 by the mutant enzymes G60A, YT, BHR-30 and BHR-72 are provided in Figures S2–S5.
Figure 5.
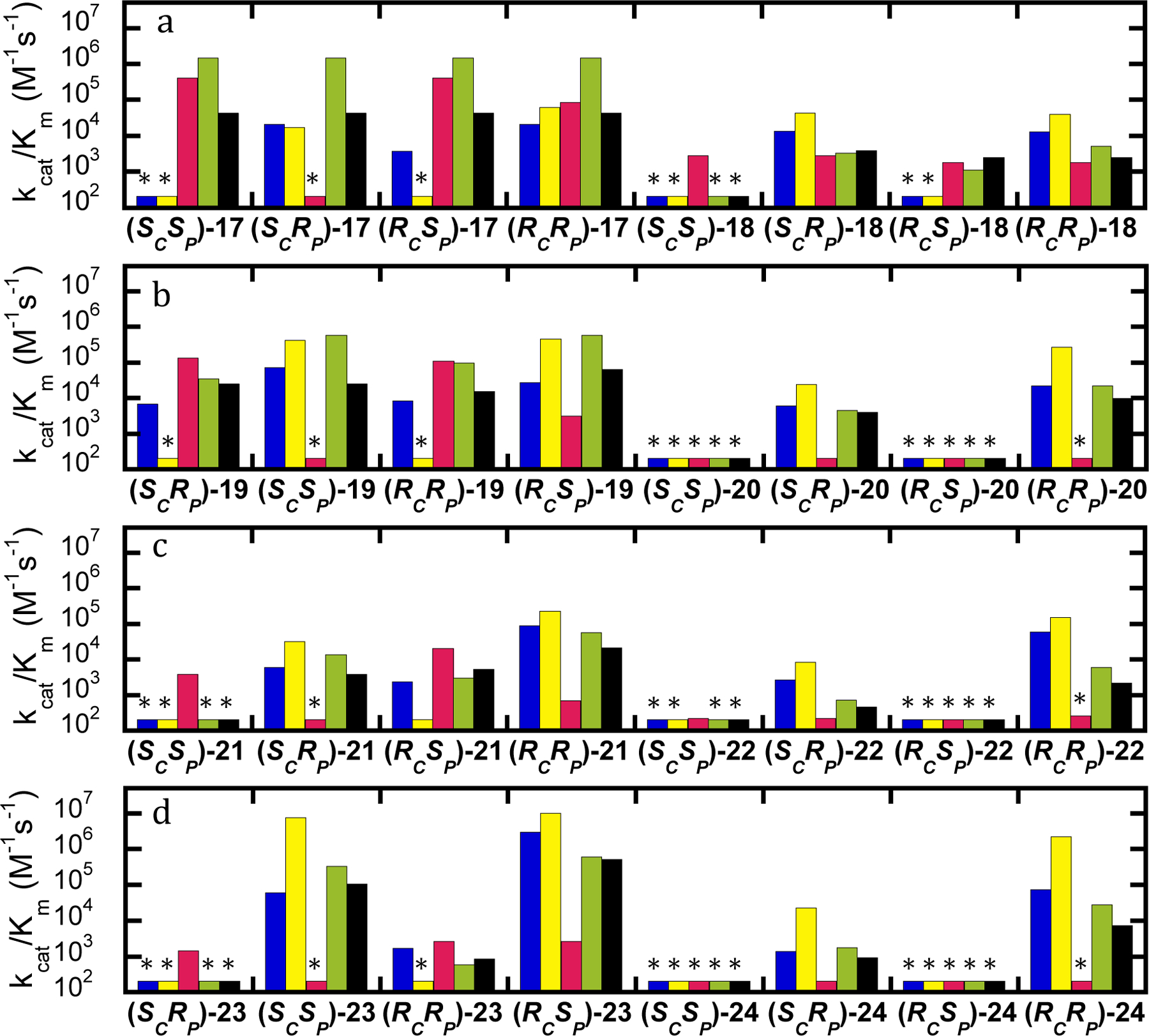
The values of kcat/Km for the hydrolysis of isomers of GD (17) and analogs (18-24). Wild-type PTE (blue), G60A (yellow), YT (red), BHR-30 (green), and BHR-72 (black). (A and C) Compounds with fluoride leaving group. (B and D) Compounds with p-nitrophenyl leaving group. Variants without quantifiable activity are marked with an asterisk and the detection limit for the assay is provided in Table 2.
Effects of Substitutions on Inactivation of Acetylcholinesterase.
The toxic effects of the organophosphate nerve agents are due to the inactivation of the neural enzyme acetylcholinesterase (23). The nerve agent analogs used in this investigation were tested for their ability to inactivate acetylcholine esterase (Table 1). For direct comparison to previous work, the enzyme from electric eel was used (7). GB (1) is known to be the least reactive toward AChE with a bimolecular rate constant of 1.4 × 10⁷ M−1 min-1. Inactivation by GD (17) and GF (9) is considerably more potent with bimolecular rate constants in excess of 1 × 10⁸ M−1 min−1 (5, 7). The inactivation of AChE has a strong stereochemical dependence with the observed inactivation being due primarily or exclusively to the (SP)-isomers (7).
Most of the analogs tested demonstrated the expected time-dependent inactivation of AChE, which varied with the concentration of the inactivator (Figure 6a). Replotting the apparent rate of inactivation as a function of concentration of the analog provides the bimolecular rate constant for inactivation (kinact/KI)(Figure 6b) (20). The ability of the analogs to inactivate acetylcholine esterase varied by over 8-orders of magnitude (Tables 1 and 2). Substitution of an O-methyl group in place of the methyl phosphonate moiety results in more than a 2-order of magnitude reduction in the rate of inactivation. With a bimolecular rate constant of 1 × 10⁴ M−1 min−1, (RP)-3 inactivates AChE 1400-fold more slowly than GB (SP-1). The smallest effect on the inactivation of AChE upon substitution of the P-methyl with an O-methyl group was observed with (SCRP)-19, which has a bimolecular inactivation constant of 8.6 × 10⁵ M−1 min−1, which is reduced ~200-fold compared to (SCSP)-17. Both (RP)-11 and (RCRP)-19 reduced the inactivation rate constant approximately 1000-fold compared to (SP)-9 and (RCSP)-17, respectively.
Figure 6.
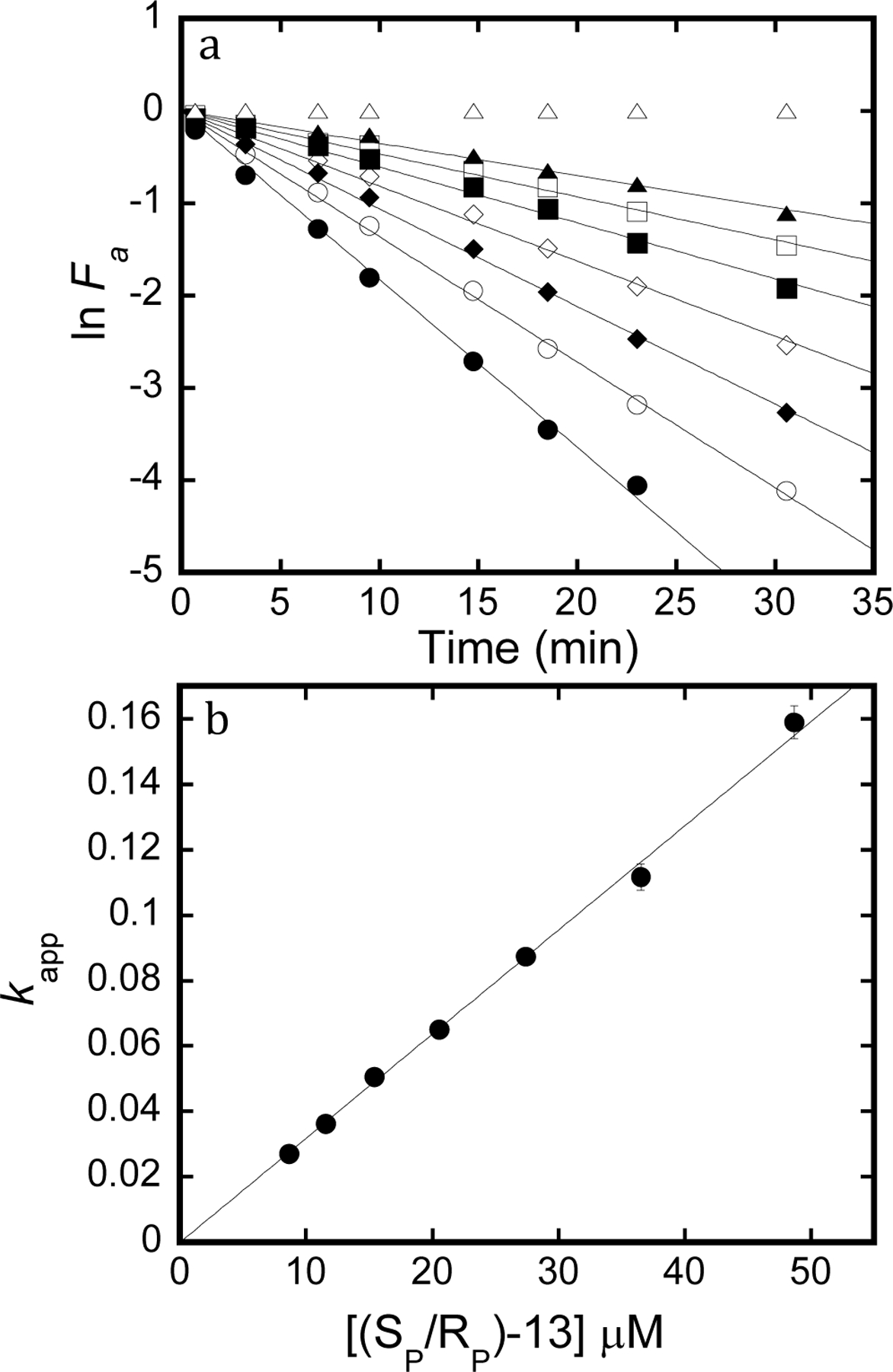
Inactivation of acetylcholine esterase. a) Natural log of fractional activity as a function of time incubated with racemic-13. Open triangles are 0 µM, filled triangles are 8.7 µM, open squares are 11.6 µM, filled squares are 15.4 µM, open diamonds are 20.5 µM, filled diamonds are 27.4 µM, open circles are 36.5 µM, and filled circles are 48.7 µM. Lines are fit to eq. 6, where slope = -kapp for inactivation. b) Replot of kapp as a function of inhibitor concentration. Line is fit to eqn. 7 where slope is the second order inactivation constant.
Substitution of the fluoride leaving group with p-nitrophenol has even larger effects with the least reduction in inactivation obtained with (SP)-5 with a rate constant of 4 × 10³ M−1 min−1, which is 3000-fold slower than the inactivation of AChE with GB ((SP)-1). (SP)-13 was similarly reduced 5000-fold compared to GF ((SP)-9). The effect was much stronger with the GD-analogs where substitution with p-nitrophenol in 21 resulted in ~106-fold reductions in the rate of inactivation for compared to (RCSP)-17 and (SCSP)-17.
Substitution of the phosphoryl oxygen for sulfur was most effective at reducing the rate of inactivation of AChE. The smallest effect was with (SP)-2, which was reduced 5000-fold compared to GB. The effects on the GF and GD analogs were stronger, with (SP)-10 inhibiting 83,000-fold less effectively than GF (SP)-9) and (SCSP)-18 and (RCSP)-18 being ~5 × 10⁴- and 3 × 10⁵-fold reduced compared to GD (SCSP)-17 and (RCSP)-17), respectively.
Combining the substitutions further reduced the rate of inactivation of AChE, though not in an additive manner. Thiophosphates are less soluble than their phosphate or phosphonate counterparts. Combination of the p-nitrophenol and thiophosphate moieties resulted in substantially reduced rates of inactivation with no inactivation detected at the maximal solubility for (SP)-6, (SP)-8, (SP)-14, (SCSP)-22, (RCSP)-22, (SCSP)-24 or (RCSP)-24. The only sulfur containing p-nitrophenyl analog which had measurable inactivation of AChE was (SP)-16, which showed an inactivation rate constant of 4.4 × 10² M−1 min−1, which is 6 × 10⁵-fold reduced compared to GF, (SP)-9. Combination of the O-methyl and sulfur substitutions decreased the inactivation rate constant to between 1 × 10² M−1 min−1 and 3 × 10³ M−1 min-1. For the GB analog, (SP)-4, the inactivation rate is reduced 80,000-fold compared to GB, (SP)-1, and 16-fold compared to the sulfur substitution alone for (SP)-2. There was little additional effect on the addition of the p-nitrophenyl group with the GF analog (SP)-12, which is reduced 85,000-fold compared to GF, (SP)-9. The GD analogs, (SCSP)-20 and (RCSP)-20, reduced the inactivation rate 2- to 3-fold compared to the sulfur analog (18). The combination of O-methyl and the p-nitrophenol leaving group further reduced the rate of inactivation by the GB and GF analogs, compared to each substitution separately with inactivation constants of 1 × 10³ M−1 min−1 for (RP)-7 and 8 × 10³ M−1 min−1 for (RP)-15. Surprisingly, inclusion of the O-methyl substituent in the GD analog 23 increases the rate of inactivation compared to the p-nitrophenyl analog alone, though with an inhibition constants < 1 × 10³ M−1 min−1 they are more than 6 × 10⁵ reduced compared to GD (17). The relative values of kinact/Ki for compounds 1–24 are graphically provided in Figure S6.
Discussion
The use of analogs in enzyme evolution is often required to allow for high throughput screening typically using a chromogenic substrate analog (24). Selection of appropriate analogs is a major concern in any enzyme evolution project because ultimately the activity being selected for is not the activity desired, and as activity improves the selection for the analog over the actual target becomes larger. These problems are enhanced in the case of evolving enzymes for the decontamination of the G-type nerve agents because of the stereochemistry of the phosphorus center and the extreme toxicity of the G-agents. For an analog to be useful in developing enzymes for the decontamination of the G-agents it must take into account the proper stereochemistry of the phosphorus center, faithfully report on relative activity variants, and be sufficiently reduced in toxicity to be used in large-scale experiments.
The toxicity of analogs has typically been ignored in the literature with the general assumption of reduced toxicity based on comparison to the less toxic insecticides which contain O-methyl or O-ethyl constituents rather than P-methyl and generally utilize an aromatic alcohol leaving group (16). Here it is demonstrated that for the G-agent analogs, the replacement of the P-methyl with an O-methyl substituent reduces the rate of inactivation of AChE by at least 2-orders of magnitude, while substitution of the fluoride leaving group for p-nitrophenol reduces the rate of inactivation by at least 3-orders of magnitude. In a novel approach, the replacement of the phosphoryl oxygen with sulfur shows at least a 5,000-fold reduced rate of inactivation with greater than 105-fold reductions observed in some cases. While the toxicity of any compound will depend on multiple factors including species tested, route of administration, and metabolism, the dramatic reduction in ability to inactivate AChE indicates that the goal of reducing the toxicity of the analogs to levels acceptable for academic labs has been met (7, 25–27).
None of the analogs tested proved to be a perfect reporter of activity with all variants, but it has been previously demonstrated that using sets of analogs rather than a single analog provides a more robust evolution strategy (1). Here the individual substitutions of the constituents of the phosphorus center proved more reliable than the combination of substitutions. The use of the p-nitrophenyl analogs provides a reasonable means of high throughput screening, especially against the SP-isomers of GD where the activity is too low to be measured with other analogs. As activity is improved, the ability to screen against the O-methyl and thiophosphonate analogs provides an opportunity to evaluate the activity against both the authentic leaving group and to evaluate the correct stereochemistry. In particular the novel concept of utilizing the thiophosphates and thiophosphonates provides for a dramatic reduction in AChE inactivation while still allowing the evaluation of the authentic leaving group and stereochemistry.
Conclusions
The O-methyl analogs seem particularly good at predicting the activity against the authentic G-agents. The addition of the O-methyl substitutent sufficiently reduces toxicity while the combination of the stereocenter with the authentic leaving group provides a unique opportunity to evaluate the activity of variants. The use of this analog accurately predicted the substantial increases in activity for the YT-variant against all of the G-agents, and allowed the identification of BHR-30 as being enhanced, especially against GD. Based on other analogs this variant would likely not have been selected, but 19 correctly predicted substantial improvements with the SP-isomers. Testing with authentic agent now confirms that with a kcat/Km of 1.4 × 10⁶ M−1 s−1 and no significant stereoselectivity BHR-30 is ~4-fold more efficient than other variants reported in the literature (6, 12, 28).
Supplementary Material
Funding
This work was supported in part by grants from the National Institutes of Health (GM116894 and GM139428) and the Robert A. Welch Foundation (A-840).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Additional NMR spectra and tables.
Accession Codes
PTE, UniProt entry: P0A434
The authors declare no competing financial interest.
References
- [1].Bigley AN, Desormeaux E, Xiang DF, Bae SY, Harvey SP, and Raushel FM (2019) Overcoming the Challenges of Enzyme Evolution To Adapt Phosphotriesterase for V-Agent Decontamination, Biochemistry 58, 2039–2053. [DOI] [PubMed] [Google Scholar]
- [2].Sun Z, Wikmark Y, Backvall JE, and Reetz MT (2016) New Concepts for Increasing the Efficiency in Directed Evolution of Stereoselective Enzymes, Chemistry 22, 5046–5054. [DOI] [PubMed] [Google Scholar]
- [3].Choi S, Han S, Lee H, Chun YJ, and Kim D (2013) Evaluation of Luminescent P450 Analysis for Directed Evolution of Human CYP4A11, Biomol. Ther. (Seoul) 21, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sugrue E, Scott C, and Jackson CJ (2017) Constrained evolution of a bispecific enzyme: lessons for biocatalyst design, Org. Biomol. Chem 15, 937–946. [DOI] [PubMed] [Google Scholar]
- [5].Gupta RD, Goldsmith M, Ashani Y, Simo Y, Mullokandov G, Bar H, Ben-David M, Leader H, Margalit R, Silman I, Sussman JL, and Tawfik DS (2011) Directed evolution of hydrolases for prevention of G-type nerve agent intoxication, Nat. Chem. Biol 7, 120–125. [DOI] [PubMed] [Google Scholar]
- [6].Tsai PC, Fox N, Bigley AN, Harvey SP, Barondeau DP, and Raushel FM (2012) Enzymes for the homeland defense: optimizing phosphotriesterase for the hydrolysis of organophosphate nerve agents, Biochemistry 51, 6463–6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Benschop HP, and De Jong LPA (1988) Nerve agent stereoisomers: analysis, isolation and toxicology, Acc. Chem. Res 21, 368–374. [Google Scholar]
- [8].Koplovitz I, Gresham VC, Dochterman LW, Kaminskis A, and Stewart JR (1992) Evaluation of the toxicity, pathology, and treatment of cyclohexylmethylphosphonofluoridate (CMPF) poisoning in rhesus monkeys, Arch. Toxicol 66, 622–628. [DOI] [PubMed] [Google Scholar]
- [9].Tsai PC, Bigley A, Li Y, Ghanem E, Cadieux CL, Kasten SA, Reeves TE, Cerasoli DM, and Raushel FM (2010) Stereoselective hydrolysis of organophosphate nerve agents by the bacterial phosphotriesterase, Biochemistry 49, 7978–7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang P, Liu EJ, Tsao C, Kasten SA, Boeri MV, Dao TL, DeBus SJ, Cadieux CL, Baker CA, Otto TC, Cerasoli DM, Chen Y, Jain P, Sun F, Li W, Hung HC, Yuan Z, Ma J, Bigley AN, Raushel FM, and Jiang S (2019) Nanoscavenger provides long-term prophylactic protection against nerve agents in rodents, Sci. Trans.l Med 11. [DOI] [PubMed] [Google Scholar]
- [11].Cherny I, Greisen P Jr., Ashani Y, Khare SD, Oberdorfer G, Leader H, Baker D, and Tawfik DS (2013) Engineering V-type nerve agents detoxifying enzymes using computationally focused libraries, ACS Chem. Biol 8, 2394–2403. [DOI] [PubMed] [Google Scholar]
- [12].Goldsmith M, Eckstein S, Ashani Y, Greisen P Jr., Leader H, Sussman JL, Aggarwal N, Ovchinnikov S, Tawfik DS, Baker D, Thiermann H, and Worek F (2016) Catalytic efficiencies of directly evolved phosphotriesterase variants with structurally different organophosphorus compounds in vitro, Arch. Toxicol 90, 2711–2724. [DOI] [PubMed] [Google Scholar]
- [13].Watkins LM, Mahoney HJ, McCulloch JK, and Raushel FM (1997) Augmented hydrolysis of diisopropyl fluorophosphate in engineered mutants of phosphotriesterase, J. Biol. Chem 272, 25596–25601. [DOI] [PubMed] [Google Scholar]
- [14].Chen-Goodspeed M, Sogorb MA, Wu F, and Raushel FM (2001) Enhancement, relaxation, and reversal of the stereoselectivity for phosphotriesterase by rational evolution of active site residues, Biochemistry 40, 1332–1339. [DOI] [PubMed] [Google Scholar]
- [15].Hill CM, Li WS, Thoden JB, Holden HM, and Raushel FM (2003) Enhanced degradation of chemical warfare agents through molecular engineering of the phosphotriesterase active site, Journal of the American Chemical Society 125, 8990–8991. [DOI] [PubMed] [Google Scholar]
- [16].The World Health Organization (1986) ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION, Geneva. (ISBN 92 4 154263 2) [Google Scholar]
- [17].Bigley AN, Narindoshvili T, Xiang DF, and Raushel FM (2018) Multiple Reaction Products from the Hydrolysis of Chiral and Prochiral Organophosphate Substrates by the Phosphotriesterase from Sphingobium sp. TCM1, Biochemistry 57, 1842–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hong SB, and Raushel FM (1999) Stereochemical constraints on the substrate specificity of phosphotriesterase, Biochemistry 38, 1159–1165. [DOI] [PubMed] [Google Scholar]
- [19].Bigley AN, Mabanglo MF, Harvey SP, and Raushel FM (2015) Variants of Phosphotriesterase for the Enhanced Detoxification of the Chemical Warfare Agent VR, Biochemistry 54, 5502–5512. [DOI] [PubMed] [Google Scholar]
- [20].Silverman RB (2000) The Organic Chemistry of Enzyme-Catalyzed Reactions, Academic Press, San Diego. [Google Scholar]
- [21].Bigley AN, Xu C, Henderson TJ, Harvey SP, and Raushel FM (2013) Enzymatic neutralization of the chemical warfare agent VX: evolution of phosphotriesterase for phosphorothiolate hydrolysis, Journal of the American Chemical Society 135, 10426–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen-Goodspeed M, Sogorb MA, Wu F, Hong SB, and Raushel FM (2001) Structural determinants of the substrate and stereochemical specificity of phosphotriesterase, Biochemistry 40, 1325–1331. [DOI] [PubMed] [Google Scholar]
- [23].Maxwell DM, Brecht KM, Koplovitz I, and Sweeney RE (2006) Acetylcholinesterase inhibition: does it explain the toxicity of organophosphorus compounds?, Arch. Toxicol 80, 756–760. [DOI] [PubMed] [Google Scholar]
- [24].Markel U, Essani KD, Besirlioglu V, Schiffels J, Streit WR, and Schwaneberg U (2020) Advances in ultrahigh-throughput screening for directed enzyme evolution, Chem Soc Rev 49, 233–262. [DOI] [PubMed] [Google Scholar]
- [25].Ellison CA, Tian Y, Knaak JB, Kostyniak PJ, and Olson JR (2012) Human hepatic cytochrome P450-specific metabolism of the organophosphorus pesticides methyl parathion and diazinon, Drug Metab Dispos 40, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].El-Sebae AH, Soliman SA, Ahmed NS, and Curley A (1981) Biochemical interaction of six OP delayed neurotoxicants with several neurotargets, J. Environ. Sci. Health B 16, 465–474. [DOI] [PubMed] [Google Scholar]
- [27].Forsyth CS, and Chambers JE (1989) Activation and degradation of the phosphorothionate insecticides parathion and EPN by rat brain, Biochemical Pharmacology 38, 1597–1603. [DOI] [PubMed] [Google Scholar]
- [28].Khersonsky O, Lipsh R, Avizemer Z, Ashani Y, Goldsmith M, Leader H, Dym O, Rogotner S, Trudeau DL, Prilusky J, Amengual-Rigo P, Guallar V, Tawfik DS, and Fleishman SJ (2018) Automated Design of Efficient and Functionally Diverse Enzyme Repertoires, Mol. Cell 72, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.