

Abstract
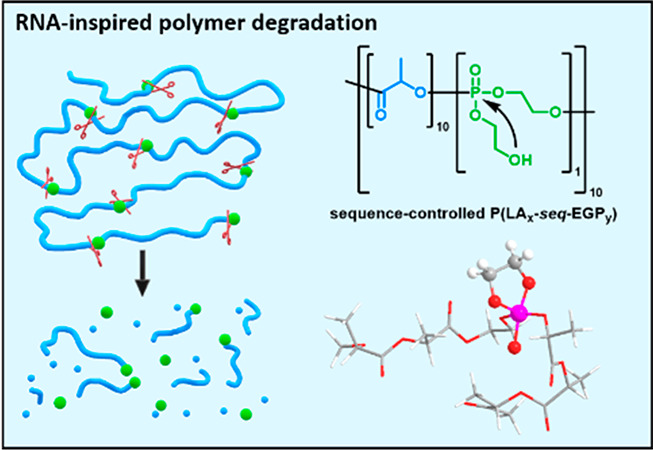
Marine plastic pollution is a worldwide challenge making advances in the field of biodegradable polymer materials necessary. Polylactide (PLA) is a promising biodegradable polymer used in various applications; however, it has a very slow seawater degradability. Herein, we present the first library of PLA derivatives with incorporated “breaking points” to vary the speed of degradation in artificial seawater from years to weeks. Inspired by the fast hydrolysis of ribonucleic acid (RNA) by intramolecular transesterification, we installed phosphoester breaking points with similar hydroxyethoxy side groups into the PLA backbone to accelerate chain scission. Sequence-controlled anionic ring-opening copolymerization of lactide and a cyclic phosphate allowed PLA to be prepared with controlled distances of the breaking points along the backbone. This general concept could be translated to other slowly degrading polymers and thereby be able to prevent additional marine pollution in the future.
Introduction
Plastics and polymers are ubiquitous and indispensable in our daily lives. However, the intensive use of plastics also has resulted in marine plastic pollution with several million tons of plastic waste entering the oceans every year.1,2 Therefore, the littering of plastics and the problem of their persistence in the environment have become a focus in research and the news.3 Biodegradable polymers such as polylactide (poly(lactic acid), PLA) are discussed as a suitable alternative to commodity plastics, as they decompose faster than commodity products.4 Especially, PLA has become a prominent material, as it has several processing and environmental benefits, such as low costs, scalability, and a biobased production from corn or potato starch.5 Notwithstanding that PLA degrades under specific conditions like via composting in weeks, several studies pointed out that PLA did not show significant signs of degradation on soil or in seawater over at least 3 years.6−10
A common strategy for biomedical applications to adjust the degradation of PLA is the random copolymerization of lactide (LA) with glycolide to poly(lactide-co-glycolide)s (PLGAs).11,12 By incorporation of the more hydrophilic glycolic acid units, the copolymer can swell and thus degrades faster. Also, oligomers of lactic acid (Mn < 550 g mol–1) were proven to be quickly attacked by enzymes and hydrolyzed. As the oligomers can dissolve, they degrade faster than solid PLA of high molar mass.13 Today, PLGA is used in medical applications and the degradation times can be adjusted by the comonomer feed ratio.14 However, the swelling of PLGA and the changed thermal properties limit its further applications.15 A similar strategy was presented by Martin et al., who copolymerized 1,3-dioxolan-4-one (DOX) with lactide to introduce hydrolysis-labile acetals, which should hydrolyze under acidic conditions and also in seawater, but only a very low mass loss (<2%) was achieved in seawater, which is probably attributed to a strong gradient or block-like copolymerization behavior of LA and DOX.16 Another strategy to induce polymer degradation is intramolecular transesterification, which had been reported for newly designed lactones carrying nucleophilic side chains that trigger main chain scission, e.g., for delivery of drugs. To date, no general approach for designing seawater-degradable PLA has been reported.6 We developed the first seawater-degradable PLA derivatives with adjustable hydrolysis times from days to years using an RNA-inspired transesterification mechanism.
To overcome the strong gradient formation during copolymerization of lactide and cyclic phosphoesters,17 we introduce the RNA-inspired breaking points into the PLA chain by sequence-controlled copolymerization of LA with a cyclic phosphate monomer, i.e., 2-(ethylene glycol vinyl ether)-1,3,2-dioxaphospholane 2-oxide (EVEP).18 Knowing the propagation rate constants, the breaking points are evenly distributed along the PLA chain by dosing of the faster polymerizing LA. The “breaking points” are protected during the polymerization by vinyl-ether groups. After mild hydrolysis of the vinyl ethers, the released 2-hydroxyethoxy side chain shall accelerate the backbone hydrolysis under seawater conditions, which is ca. pH 8.1,19 by transesterification (Figure 1).
Figure 1.
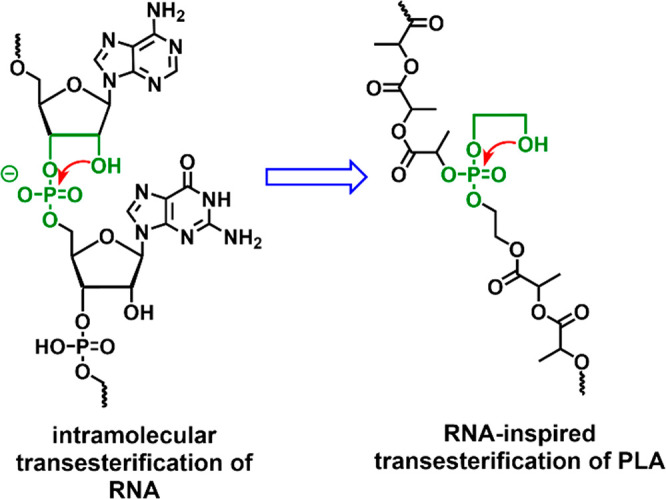
Illustration of the intramolecular transesterification of RNA transferred to PLA by using a synthetic phosphoester with a pendant hydroxyl group for intramolecular transesterification and acceleration on polymer backbone degradation.
The design of the breaking points was inspired by the intramolecular transesterification of RNA. In contrast to DNA, which is very stable against hydrolysis, RNA hydrolyzes much faster via an intramolecular transesterification.20,21 Nature is using the 2′-OH group in the ribose units of RNA, which is in the β-position of the phosphoester in the backbone and can form a cyclic intermediate during hydrolysis (Figure 1). Transferring this motif into a synthetic phosphate linkage with a pendant 2-hydroxyethoxy side chain and installing it as a breaking point in PLA should lead to multiple chain scissions in seawater, as a similar mechanism had been proven for the degradation of polyphosphoesters with a single 2-hydroxyethoxy motif at the chain end.22 After the intramolecular transesterification, shorter PLA chains are generated, which increases the number of OH end groups. As PLA degrades mainly via a backbiting mechanism under neutral and basic conditions,3 this increase of terminal OH groups also increases the overall PLA degradation rate. Further, the degradation rates were tailored by the comonomer feed ratio, i.e., the number of breaking points and additional chain ends. We introduced 3–15% of breaking points into PLA and PLLA to obtain half-life times of 3 days to half a year in seawater. From DFT calculations, the intramolecular transesterification was favored against the direct attack of water to the phosphate group, which implies the preference of the so-called RNA-inspired degradation mechanism. The RNA-inspired degradation is a general platform and might be a key strategy to control the fate of slowly degrading polymers in seawater and to prevent further marine plastic pollution.
Results and Discussion
The RNA-inspired degradation mechanism was simulated and compared to two other possible hydrolysis mechanisms of the polylactide copolymers. This was simulated at the DFT level. The first one was the single step SN2(P) nucleophilic substitution at the phosphorus atom, in which the hydroxyl attack is synchronous with the P–O bond breaking that leads directly to the break of the polymer main chain. The transition state has not been identified. An alternative mechanism, a stepwise addition–elimination (AE) process, is analogous to the SN2(P) but assumes the formation of a penta-coordinated phosphorus intermediate. The third mechanism, the “RNA-inspired” degradation due to its similarity to the cyclization presented in Figure 1, is also a stepwise mechanism, in which a cyclic intermediate is formed upon the intramolecular attack of the pendant 2-hydroxyethoxy group on the phosphorus atom. This cyclic intermediate subsequently decomposes and results in the breaking of the polymer main chain. For the above-mentioned two mechanisms, we have optimized the structures of all reactants, intermediates, and products as well as the transition states connecting them. The presence of an aqueous solution was included in the form of the continuous solvent model. We have considered the influence of different protonation states, conformations, the length of the pendant chain, the size of the model, and the site of the hydroxyl attack. Major details of the calculations are provided in the Supporting Information. For comparison, the cyclization to 2’3′-cP modeled using two ribose rings and merging the phosphate moiety has been studied computationally using the same theory level. The obtained results indicate that this reaction proceeds via a very unstable cyclic intermediate (see Figure S26), which has a Gibbs free energy of formation of 17.1 kcal mol−1. It decomposes rapidly (see data in Table S12).
Also the much less strained 3′5′ cyclization, which proceeds via the six-membered ring, was reported with a free energy of ca. 15 kcal mol−1.23 The lower energy demand for the herein presented PLA-based copolymers is probably attributed to the fact that the attacking hydroxyl group is a primary OH (not secondary as in RNA) and is placed on a flexible pendant chain. The overall results, which are illustrated in Figure 2, suggest clearly that the RNA-inspired degradation mechanism involving the cyclization (green line) is energetically favored over the addition–elimination attack (blue line). By these calculations, the installation of phosphate groups containing an ethoxyhydroxy group as a pendant chain should be able to accelerate the hydrolysis of polyester such as polylactide, which is reported in the following.
Figure 2.
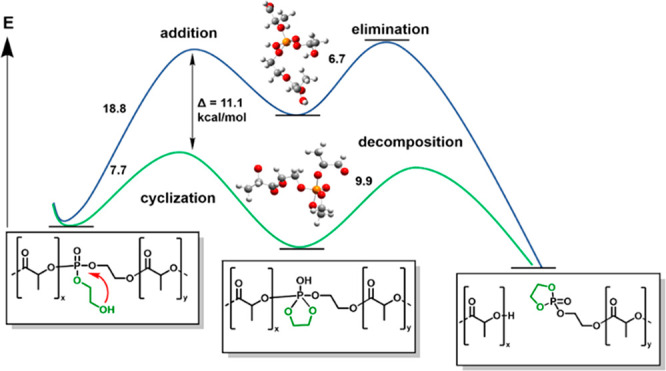
Comparison of Gibbs free energies of activation (in kcal mol−1) for the two first steps in addition–elimination (upper blue) and RNA-inspired (lower green) decomposition mechanisms resulting from the DFT calculations. The common substrate and the products of the RNA-inspired mechanism are presented schematically. Optimized structures of penta-coordinated (upper) and cyclic (lower) intermediates are illustrated with gray carbon, white hydrogen, red oxygen, and orange phosphorus atoms.
According to these theoretical considerations, we installed phosphoesters with a pendant 2-hydroxyethoxy side chain into the PLA backbone by copolymerization of LA with the RNA-inspired breaking points. As the OH group needs to be protected during the organocatalytic ring-opening polymerization to prevent branching, we prepared 2-ethylene glycol vinyl ether-1,3,2-dioxaphospholane 2-oxide (EVEP) as a comonomer, which is compatible with the polymerization conditions of LA. EVEP was synthesized by a modified protocol according to Lim et al. (cf. the Supporting Information).18 We used 1,8-diazabicyclo-[5.4.0]undec-7-en (DBU) as an organocatalyst and prepared homopolymers of PLA and PLLA with different degrees of polymerization (entries 5, 6, 9, and 10 in Table 2 for later comparison).24 To the best of our knowledge, to date, only a single paper describes the statistical copolymerization of cyclic phosphonates with LA and DBU,17 while cyclic phosphates had been studied before as comonomers for LA only once with Al(OiPr)3 as the catalyst.25 From our previous knowledge of cyclic phosphonates, a strong gradient formation was also expected during a statistical copolymerization of EVEP with LA. Using 2-(benzyloxy)ethanol as the initiator and DBU as the organocatalyst, copolymers were successfully prepared (Figure 3). GPC proved the formation of a copolymer with a moderate molar mass dispersity of Đ = 1.23; however, a slight shoulder to higher molar masses was detected, indicating the occurrence of transesterifications during the copolymerization under the chosen conditions. 1H NMR spectroscopy was used to assess the composition and the molecular weight of the polymer (Mn = 18 kg mol−1, Dlactide = 184, DPEVEP = 8; Figure S1). The degree of polymerization was determined by comparing the resonances originating from the initiator (7.37–7.28 ppm) with the CH2 protons from the ethylene glycol in the backbone and side chain of EVEP (3.83–4.52 ppm) and the resonances of the methine protons in the LA backbone (5.27–5.00 ppm). 1H-DOSY and 2D 1H,31P{H} HMBC spectra confirmed the incorporation of both monomers in the polymer during the statistical copolymerization (Figures S1–S3). All possible diad sequences (cf. Figure 3b) were successfully assigned to specific resonances in the H,31P{H} HMBC correlation spectra. From the NMR spectra, more PP diads compared to LP diads were determined (Figure S1), which indicates a strong gradient formation during the copolymerization with a polyphosphate segment. The rate constants (kp) of LA and EVEP were determined via real-time1H and 31P NMR spectroscopy taken during the statistical copolymerization (1H and 31P NMR spectra were recorded alternately to follow the conversion of LA from 1H spectra and EVEP from the 31P spectra, Figures S4 and S6).26
Table 2. Summarized Properties of the Prepared P(LA-seq-EVEP) Copolymers via Sequential Addition of Lactide before and after Deprotection with Aqueous Hydrochloric Acid.
|
Mnb (kg mol–1) |
Đc |
|||
|---|---|---|---|---|
| polymera | p | d | p | d |
| 1 | 12.8 | 11.7 | 1.65 | 1.59 |
| 2 | 11.1 | 10.8 | 1.32 | 1.19 |
| 3 | 15.2 | 14.5 | 1.31 | 1.08 |
| 4 | 17.8 | 16.7 | 1.23 | 1.18 |
| 7 | 88.3d | 84.5d | 1.54 | 1.35 |
| 8 | 98.8d | 123.1d | 1.76 | 1.40 |
p = protected, d = deprotected.
Determined via 1H NMR spectroscopy.
Determined via GPC in THF or DMF (vs PS standards).
For the high molecular weight, the initiator signal is so small that the error is becoming big.
Figure 3.
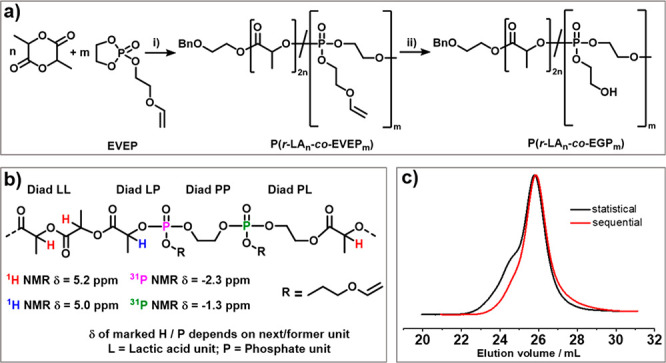
(a) Statistical copolymerization of lactide and EVEP (i) with 2-(benzyloxy)ethanol as the initiator and DBU as the organocatalyst, DCM, 2 h, 25 °C, and (ii) with 2 M HCl in dioxane, 45 min, 45 °C. (b) Different possible diad sequences for P(LAn-co-EVEPm) copolymers and the respective chemical shifts. (c) GPC elugrams of the statistical copolymerization (black) and the sequence-controlled copolymerization (red) of LA and EVEP (measured in THF, RI detection).
From the 1H NMR spectra,
the consumption of lactide
can be followed by comparing the resonances of the methine proton
in LA (at 5.10 ppm) with the polymeric resonance of the methine proton
(at 5.20 ppm) as an upfield shift occurs during the ring-opening polymerization.
Besides the methine resonances, also the methyl signals can be used
to follow the polymerization as a they are shifted downfield when
transformed from the monomeric methyl signal to the polymeric methyl
signal (Figure S4). Normalized integrals
over time taken from the methyl group have been used to plot 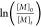 against the reaction time t to determine the apparent rate constant kp,app (Figure S5). The polymerization of EVEP
was followed by monitoring the upfield shift in the 31P
NMR spectra during the polymerization from ca. 17.3 ppm (EVEP monomer)
to ca. −1.3 ppm (polymer) (Figure S6). As the integral of the monomer resonance in the 31P
NMR spectrum is proportional to the monomer concentration ([M]t), the apparent rate constant kapp for EVEP can be determined (Figure S7). The propagation rate constant kp was calculated via
against the reaction time t to determine the apparent rate constant kp,app (Figure S5). The polymerization of EVEP
was followed by monitoring the upfield shift in the 31P
NMR spectra during the polymerization from ca. 17.3 ppm (EVEP monomer)
to ca. −1.3 ppm (polymer) (Figure S6). As the integral of the monomer resonance in the 31P
NMR spectrum is proportional to the monomer concentration ([M]t), the apparent rate constant kapp for EVEP can be determined (Figure S7). The propagation rate constant kp was calculated via 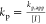 (Table 1, Table S2).
(Table 1, Table S2).
Table 1. Average Reaction Rate Constants (kp) and Reactivity Ratios (r) of the Copolymerization of Lactide with EVEP.
| monomer | kp (L mol–1 s–1) | r-parameter |
|---|---|---|
| lactide | 0.8 ± 0.3 | 20.0 ± 0.5 |
| EVEP | 1.0 × 10–2 ± 0.3 × 10–2 | 0.046 ± 0.004 |
The calculated reactivity ratios of the copolymerization of lactide with EVEP (with 2-(benzyloxy) ethanol as an initiator and DBU as the base in DCM at 25 °C, Table S1) were calculated by different models (i.e. the ideal integrated model, the Jaacks and the Beckinham−Sanoja−Lynd models) indicating r(EVEP) = 0.046 and r(LLA) = 20.0.
Since the kp of lactide was ca. 80 times faster compared to EVEP under these conditions, a strong gradient copolymer was obtained. To break up the PLA in smaller chains, the breaking points must be distributed homogeneously over the whole polymer. Statistical copolymerization cannot be used to achieve a random sequence of the phosphate comonomers along the backbone—another strategy is needed. Therefore, a setup based on the different rate constants of both monomers with the sequential addition of lactide was developed (Figure 4).
Figure 4.

Concept of the synthesis of the sequence-controlled copolymerization of EVEP and LA with multiple sequential additions of 5 equiv each of lactide after calculated time intervals tdiff,x, resulting in a P(LA-seq-EVEP) chain with evenly distributed EVEP units along the PLA chain (note: the graphical representation shows the average distances of EVEP units (green balls) in the PLA chain (blue balls) calculated from the 1H NMR kinetics.)
The polymerization of EVEP is initiated, and as it proceeds with its relatively low propagation rate, lactide is added sequentially after calculated times (depending on the kp values) to place the phosphate breaking points equally along the polymer backbone. To achieve this controlled comonomer sequence in a one-pot copolymerization, the average time needed for the incorporation of one EVEP unit was calculated (see the Supporting Information for the calculations). By adding the LA after certain periods, the sequence-controlled P(LA-seq-EVEP) copolymers with well-distributed EVEP breaking points along the chain were obtained (Figure 4).
We followed the chain growth after each addition by NMR spectroscopy: as in the case for the statistical copolymerization, mentioned above, we were able to distinguish the different diads in the spectra, and mainly LL diads for the PLA segment and an increasing number of LP diads for the crossover reaction, i.e., the desired breaking points, were determined. Using the kp values determined above, a theoretical sequence was calculated and plotted against the NMR integrals (Figure 5), which allows control of the monomer sequence and, thus, the average chain length of PLA segments between the phosphate breaking points.
Figure 5.
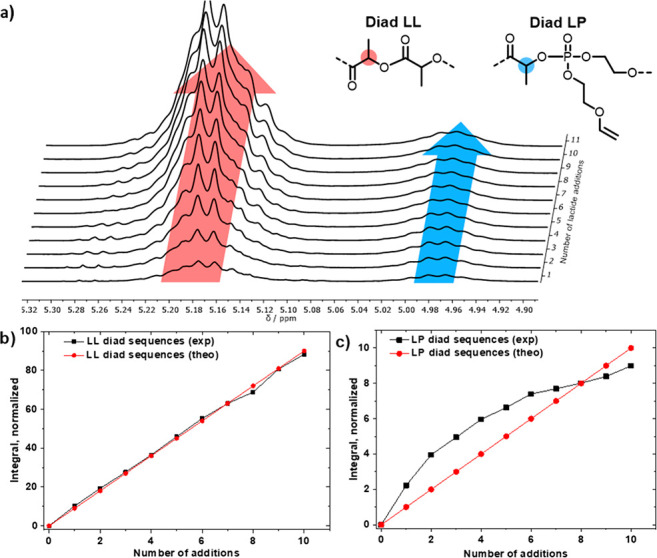
(a) Stacked 1H NMR spectra of the sequence-controlled copolymerization of EVEP and LA; zoom into the region for the LL and LP diads showing a simultaneous increase after each monomer addition step. (b, c) Plots for the experimental and theoretical integrals (b) of the methine region (5.24–5.11 ppm) for LL diad sequences and (c) of the methine region (5.02–4.93 ppm) for LP diads vs the number of sequential lactide additions. The integrals were referenced to the five aromatic initiator protons.
The evolution of both LL and LP diads follows the theoretical values, while a certain decrease of the LP diads with increasing reaction time is detected (Table S4). The lower reaction kinetics of EVEP at longer reaction times is attributed to dilution during the addition steps and the consumption of EVEP during the copolymerization. A theoretical representation of the slight increase of the segment length for PLA is shown in Figure 4 (bottom right), showing only a slight increase along one theoretical copolymer chain.
Using this strategy, we synthesized a library of copolymers using the enantiomerically pure l-lactide or the racemic r-lactide with different amounts of EVEP breaking points to control the overall half-life times of the PLA derivatives in seawater (Table 3). Only the LP diads would result in breaking points of PLA that also increase the number of OH end groups of PLA chains. In polymers 7 and 8 basically, all EVEP units resulted in an LP diad. All copolymers proved a low to moderate molar mass distribution in GPC. No “shoulders” that would indicate transesterification were detected, in contrast to the statistical copolymerization (cf. Figure 3c).
Table 3. Properties of the Prepared PLA Homopolymers and P(LA-seq-EVEP) Copolymers by Sequential Monomer Addition.
| no. | polymer | Mna (kg mol–1) | Đb | ΧEVEPa (mol %) |
 (LA)a (LA)a
|
NLLa | NLPa |
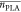 d d
|
|---|---|---|---|---|---|---|---|---|
| 1 | P(l-LA125-seq-EVEP22) | 12.8 | 1.65 | 15.5 | 125 | 112 | 12.7 | 9 |
| 2 | P(r-LA123-seq-EVEP14) | 11.1 | 1.32 | 9.0 | 123 | 116 | 7.1 | 16 |
| 3 | P(l-LA194-seq-EVEP6) | 15.2 | 1.31 | 2.9 | 193 | 190 | 4.0 | 48 |
| 4 | P(r-LA225-seq-EVEP7) | 17.8 | 1.23 | 3.2 | 225 | 221 | 4.4 | 51 |
| 5 | P(l-LA126) | 9.2 | 1.18 | 0 | 126 | |||
| 6 | P(r-LA122) | 8.9 | 1.09 | 0 | 122 | |||
| 7 | P(l-LA1024-seq-EVEP36) | 88.3c | 1.54 | 3.5 | 1024 | 988 | 36 | 28 |
| 8 | P(r-LA1180-seq-EVEP47) | 98.8c | 1.76 | 4.0 | 1180 | 1131 | 47 | 25 |
| 9 | P(l-LA1260) | 95.7c | 1.67 | 0 | 1260 | |||
| 10 | P(r-LA1400) | 106.4c | 1.84 | 0 | 1400 | |||
| 11 | P(l-LA1843-seq-EVEP54) | 150.5c | 1.75 | 2.8 | 1843 | 1789 | 54 | 34 |
| 12 | P(r-LA1490-seq-EVEP48) | 122.6c | 1.91 | 3.1 | 1490 | 1440 | 48 | 31 |
Determined via 1H NMR spectroscopy, NLL = number of LL linkages, NLP = number of LP linkages.
Determined via GPC in THF or DMF (vs PS standard).
For the high molecular weight, the initiator signal is so small that the error is becoming big.
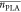 = average length of the PLA sequences.
= average length of the PLA sequences.
Representative 1H and 31P NMR spectra of the polymer after workup are shown in Figure S9. As the vinyl ether group has been reported to be stable under basic and anhydrous conditions,27 the copolymers can be stored in their protected form without any sign of degradation for at least several months.
The vinyl ether protection groups were cleaved quantitatively by treating the copolymers with diluted hydrochloric acid in dioxane, as proven by IR and NMR (Figures 8, S10, and S11). 1H NMR spectroscopy showed the disappearance of the vinyl resonances at 6.5 ppm (NMR) and the appearance of a broad OH vibration around 3300 (IR) (Table 2). Further, GPC proved a slight shift to higher elution volumes after removal of the protective group; however, the polyester remained intact under these conditions (Figure S12). The thermal properties of the copolymers were analyzed by DSC and TGA and compared to the PLA and PLLA homopolymers of a similar degree of polymerization. In TGA measurements, the PLA homopolymers started to decompose around 200 °C, with a char yield of ca. 5%. All of the phosphorus-containing copolymers decomposed at higher temperatures (220–320 °C), remarkably with increased char yield, depending on the phosphorus content, up to 15% for P(l-LA125-seq-EVEP22), as is expected for phosphorus-containing polymers (Figure S14 and Table S5).
Figure 8.
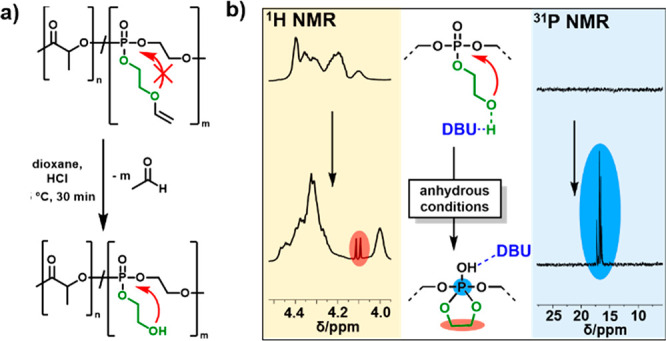
Mechanism of the RNA-inspired degradation visualized by 1H and 31P NMR spectroscopy under anhydrous conditions: (a) Deprotection of P(LA-seq-EVEP) copolymers with hydrochloric acid in dioxane yields the RNA-inspired polymer structure with pendant 2-hydroxyethoxy groups capable for transesterification. (b) Formation of the penta-coordinated phosphorus with a 5-membered cyclic intermediate monitored by 1H NMR (500 MHz, 298 K, CDCl3) (left) and 31P {H} NMR (202 MHz, 298 K, CDCl3) (right). The cyclization under anhydrous conditions was achieved by the addition to DBU to P(r-LA123-seq-EGP14).
The DSC measurements revealed that the glass transition (Tg) and melting (Tm) temperatures were influenced by the comonomer content. While the herein prepared PLA and PLLA homopolymers had a Tg of 37 °C and PLLA showed an additional melting at ca. 126 °C, our prepared copolymers showed slightly decreased values. For polymers with a low phosphorus content, less than 4%, the Tg remained relatively unchanged compared to the homopolymer PLA. For higher phosphorus contents, the Tg decreased significantly. For PLLA copolymers, the Tm remained relatively unchanged for phosphorus content up to 3%: for P(l-LA194-seq-EVEP6), the melting point was similar (ca. 128 °C), and for the deprotected version P(l-LA194-seq-EGP6), the melting started earlier at ca. 116 °C. When the comonomer content was increased, fully amorphous polymers were obtained (Figure S15 and Table S5).
The Young’s moduli of polymers 9–12 (cf. Table 3) were determined via AFM. Polymer films were produced by drop-casting the respective PLA homo- and P(LA-seq-EVEP) copolymers from a dichloromethane solution on piranha-cleaned silicon wafers. The solvent was evaporated in vacuo, and the remaining films had a smooth surface (RMS roughness of 0.6 nm) with low adhesion properties (normalized by tip radius: 0.15 N/m), as evidenced by quantitative AFM imaging (Figure S21). The elastic moduli of the polymer films were determined between 1.7 and 2.4 GPa (Figure S22), which fits the literature values and indicates that the installation of ca. 3% phosphate breaking points does not hamper the mechanical properties.28
Degradation
PLA degrades under neutral or basic conditions primarily via a backbiting mechanism.3 As seawater typically has pH values between 8.0 and 8.2,19 backbiting is supposed to be a dominant mechanism for its hydrolysis. Thus, the increase of terminal OH groups would also increase the overall degradation rate, besides a previous breakdown in shorter fragments induced by the RNA-inspired degradation units. The pendant 2-hydroxy ethoxy units in the phosphate groups will fragment the chain; the number of chain ends is multiplied by the number of breaking points, consequently accelerating the degradation. Moreover, shorter PLA oligomers are more hydrophilic or even soluble in water below a critical value (Mn < 550 g mol–1),13 which additionally increases the hydrolysis rate. Karjomaa et al. reported an increased degradation of short PLLA oligomers at 25 °C compared to higher molecular weight PLLA chains, which were only degradable in a reasonable time frame (6 months) at 58 °C.13 The shorter PLLA oligomers hydrolyzed abiotically, whereas a biotic environment (Fusarium moniliforme and Penicillium roqueforti fungi as well as Pseudomonas putida bacteria) increased the degradation even further, assumably by enzymatic chain scission of PLLA oligomers by esterases.3
To determine the potential of the herein-reported RNA-inspired degradation mechanism, polymer films were produced by drop-casting the respective P(LA-seq-EVEP) (protected) and P(LA-seq-EGP) (deprotected) copolymers from a chloroform solution on microscope coverslips. The solvent was evaporated in vacuo, and the remaining films had a smooth surface. They were immersed in buffered artificial seawater. The prepared artificial seawater was buffered using NaHCO3 to ensure a constant pH of 8.12 during the degradation, as the pH can vary through the formation of lactic acid as a hydrolysis product of PLA (which would be washed away in the open sea). The degradation was studied by a combination of techniques: first, the weight loss of the polymer films was determined gravimetrically; then, the content of released lactic acid was determined by an enzymatic assay (Figure 6). These two methods give an already detailed view on the seawater degradability of the P(LA-seq-EVEP). Additionally, GPC and NMR analyses were performed to determine the change in molar mass (Mn and Mw) as well as the molar mass dispersity Đ. Samples were taken after certain time intervals.
Figure 6.
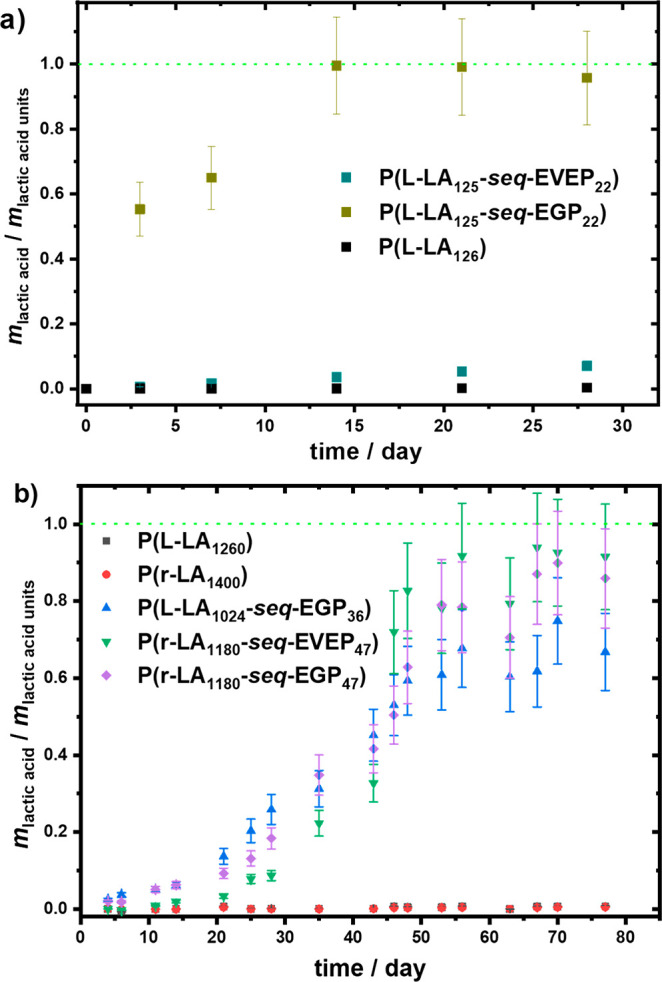
Degradation of PLA, P(LA-seq-EVEP), and P(LA-seq-EGP) copolymer films in artificial seawater (lactic acid was quantified using an enzymatic assay).
In contrast to the PLA and PLLA homopolymers, a significant change in the GPC elugrams was observed for the investigated copolymers P(LA-seq-EGP) after immersion of the films in artificial seawater for 28 days (Figure S18). The lactic acid assay proved the formation of lactic acid during the time, with kinetics depending on the amount of EGP units installed in the polymer chain (Figure 6a and Figure S20). GPC showed monomodal but broadened elugrams shifted to higher elution volumes (i.e., lower Mn), observed after incubation of the film of P(r-LA123-seq-EGP14) in seawater (Figure S18a). The broadened elugram can be explained by slow chain scission, in the beginning, followed by the backbiting mechanism of the PLA oligomers (the oligomers have an average length of 16 units, which makes them faster to degrade), which increases as more end groups are released. A bimodal and significantly broadened elugram was observed for the copolymer P(l-LA194-seq-EGP6) after degradation. This can be explained by the formation of different lengths of polymer chains upon chain scission by hydrolysis of phosphoester breaking points. The PLA fragments (average length of 47 units) produced by hydrolysis (following pathway 1 in Figure 7) will undergo slower further degradation, as the secondary OH-group at the chain end of PLA is still connected to the phosphate unit. A second ring closing is less favored, since the phosphate OH-group is deprotonated and negatively charged under the degradation conditions (Figure 7). Following pathway 2 in Figure 7, free OH-end groups in PLA are generated, which will undergo hydrolysis by backbiting. With the increased number of chain ends on the one hand and the increased hydrophilicity on the other hand, this leads to an accelerated degradation process.29 A second intramolecular attack of the pendant OH-group at the P-atom will lead to the final cleavage of the phosphate unit and PLA oligomers (ultimately lactic acid (Figure 7 bottom)). The degradation values from polymers 1–4 after 28 days of degradation are summarized in Table 4 (Table S6).
Figure 7.
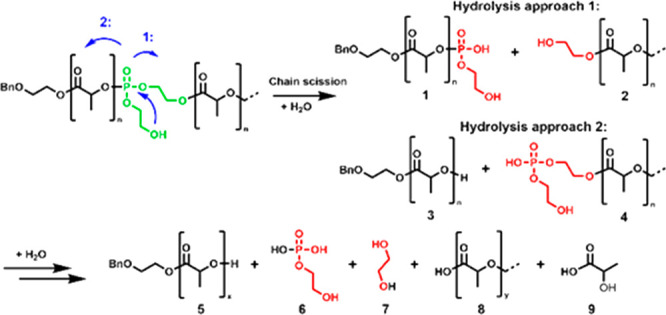
Degradation patterns of P(LA123-seq-EGP14) copolymers: two possible ways of hydrolysis in the first step leading to different intermediates. Subsequent hydrolysis forms ethylene glycol, 2-hydroxyethylphosphate, and the corresponding lactic acid oligomers (proven by 1H NMR, Figure S19).
Table 4. Summarized Experimental Results from the Lactic Acid Enzymatic Assay with Estimated Degradation Times to Full Biomineralization.
| polymer | degradation ratio (%)a | tdeg,100% (weeks)b |
|---|---|---|
| P(l-LA125-seq-EVEP22) | 7.0 ± 1.1 | 58 |
| P(l-LA125-seq-EGP22) | 95.7 ± 14.4 | 2 |
| P(r-LA123-seq-EGP14) | 16.4 ± 2.5 | 20 |
| P(l-LA194-seq-EGP6) | 25.2 ± 3.8 | 11 |
| P(r-LA225-seq-EGP7) | 1.1 ± 0.2 | 166 |
After 28 days of degradation in artificial seawater, determined by an enzymatic lactic acid assay. Degradation ratio: m(lactic acid)/m(lactic acid units).
Degradation was extrapolated linearly to obtain estimated full degradation times.
It is important to prove further a complete degradation to lactic acid under these conditions, which we detected by a commercial enzyme assay for l-lactic acid; in the case of a racemic mixture, the value was multiplied by a factor of 2. For the P(l-LA125-seq-EGP22) with the highest content of RNA-inspired breaking points (of 15%), the copolymer was fully hydrolyzed to lactic acid after 2 weeks (Figure 6a). PLA homopolymers did not show any significant degradation after incubation in artificial seawater for 28 days. Only trace amounts of lactic acid were detected by the enzyme assay and also only a slight shift of the molar mass distribution in the GPC elugram was detected, indicating a much slower degradation process compared to the copolymers (Figure S18). The protected copolymer, which carries the vinyl ether protective group in the phosphoester units, proved a much slower degradation compared to the deprotected version under the same conditions (Figure 6a). However, it is only significant for the fast degrading polymer 1 P(l-LA125-seq-EGP22). The vinyl ether protective group is stable under basic and anhydrous conditions; it can be slowly hydrolyzed under aqueous conditions to release the pendant OH groups. Therefore, for the slower degrading polymer 8, the kinetics of the vinyl ether hydrolysis seemed to be as fast as the backbone degradation. Therefore, only a slight delay for the degradation was determined (Figure 6b). However, they showed a significant enhancement compared to the PLLA homopolymer. Interestingly, PLLA copolymers proved a faster degradation compared to racemic PLA (Figure S20 and Table S7), which might be explained by the fact that the OH chain ends, as well as the phosphoester breaking points, act as defects during the crystallization and are expelled to the surface of crystallites, similar to other phosphoesters in polyethylene-like materials.30
The polymers 7 and 8 show a high amount of LP diads; therefore, the PLA segments are short, while the overall amount of phosphorus units is lover than 4%. With these polymers, we conducted a longer degradation study proving almost full degradation after 70 days (Figure 6). The PLA homopolymers 9 and 10 did not show any degradation at all in this time period.
To prove that the degradation followed the RNA-inspired mechanism via a five-membered intermediate ring as predicted by the DFT calculations, 1H and 31P NMR studies were performed. The deprotected copolymer P(r-LA123-seq-EVEP14) was dissolved in anhydrous CDCl3, and an excess of DBU was added to the solution. Under anhydrous conditions, DBU was able to activate the pendant OH group and the five-membered cyclic intermediate was proven in 1H NMR by the doublet at 4.1 ppm22 and the distinctive 31P NMR resonance for strained 5-membered phospholanes at ca. 17 ppm (Figure 8b and Figure S13).
Taking the possible degradation products in seawater into account, one of the intermediate products (3 in Figure 7) after the first main chain cleavage can undergo backbiting degradation of the PLA chain, while the second intermediate (1, in Figure 7) undergoes a second hydrolysis step to release 2-hydroxyethylphosphate (6 in Figure 7), after which backbiting will occur. The integral of the signal in the methine region of LP diad sequences (5.05–4.89 ppm) reduced about 49% after 28 days in seawater for P(r-LA123-seq-EGP14). From this context, it can be concluded that 49% of the LP diads underwent hydrolytic cleavage via hydrolysis approach 2 (Figure 7, Figure S17). Degradation products via hydrolysis approach 1 (1, in Figure 7) cannot be differentiated from intact LP diads in the polymer backbone by 1H NMR spectroscopy. With a quantitative degradation to lactic acid and the presence of 2-hydroxyethylphosphate, the two-step RNA-inspired degradation pathway is very likely (Figure S19).
For both PLA and PLLA copolymers, by varying the ratio between LA and EVEP in the copolymers, the degradation rates in seawater were effectively adjusted (Figure S20). Using the RNA-inspired degradation strategy, we were able to adjust the degradation rate of PLA/PLLA in artificial seawater from several days over weeks and months up to two years, which might be utilized as a general approach to other polyesters as well.
Conclusion
We were able to transfer the motif of intramolecular transesterification of RNA to synthetic PLA and accelerate its degradation in seawater to adjustable half-life times from weeks to years. The thermal and mechanical properties remained unchanged at least up to a comonomer content of 3 mol %. Initial DFT calculations indicated that the RNA-inspired degradation mechanism would be energetically preferred to random hydrolysis also in artificial polyesters. To achieve a tailored degradation rate of PLA, we prepared sequence-controlled copolymers of lactide with a cyclic phosphoester monomer (EVEP) capable of intramolecular transesterification in seawater. As the propagation rate constant of lactide (0.8 ± 0.3 L mol–1 s–1) was ca. 80 times faster than that of EVEP (1.0 × 10–2 ± 0.3 × 10–2 L mol–1 s–1), an equal distribution of EVEP breaking points was achieved along the PLA chain by continuous addition of lactide to a running polymerization of EVEP. After the release of the pendant 2-hydroxy ethoxy groups at the phosphoester units, polymer films were drop-cast and immersed into artificial seawater. Depending on the amount of phosphoester breaking points, degradation half-lives in seawater ranging from 2 weeks to 2 years were achieved with complete degradation to lactic acid.
We believe that the RNA-inspired degradation pathway is a general strategy to accelerate polymer degradation and will be applicable for other synthetic polymers as well. Equipped with additional orthogonal chemistry at the pendant phosphoester group, this concept can be extended to highly controlled polymer stability in natural environments such as seawater or soil.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c07508.
Detailed synthetic protocols, polymerization kinetics, and degradation studies as well as all characterization data (NMR and IR spectra, thermogravimetry, differential scanning calorimetry data, gel permeation chromatography, etc.) and additional calculations (PDF)
The authors thank the Deutsche Forschungsgemeinschaft (WU 750/6-2) for funding.
The authors declare no competing financial interest.
Supplementary Material
References
- Chitrakar P.; Baawain M. S.; Sana A.; Al-Mamun A. Current Status of Marine Pollution and Mitigation Strategies in Arid Region: A Detailed Review. Ocean Sci. J. 2019, 54 (3), 317–348. 10.1007/s12601-019-0027-5. [DOI] [Google Scholar]
- Suzuki M.; Tachibana Y.; Kasuya K.-i. Biodegradability of poly(3-hydroxyalkanoate) and poly(ε-caprolactone) via biological carbon cycles in marine environments. Polym. J. 2021, 53 (1), 47–66. 10.1038/s41428-020-00396-5. [DOI] [Google Scholar]
- Haider T. P.; Völker C.; Kramm J.; Landfester K.; Wurm F. R. Plastics of the Future? The Impact of Biodegradable Polymers on the Environment and on Society. Angew. Chem., Int. Ed. 2019, 58 (1), 50–62. 10.1002/anie.201805766. [DOI] [PubMed] [Google Scholar]
- Drumright R. E.; Gruber P. R.; Henton D. E. Polylactic Acid Technology. Adv. Mater. 2000, 12 (23), 1841–1846. . [DOI] [Google Scholar]
- Karamanlioglu M.; Preziosi R.; Robson G. D. Abiotic and biotic environmental degradation of the Bioplastic polymer poly(lactic acid): A review. Polym. Degrad. Stab. 2017, 137, 122–130. 10.1016/j.polymdegradstab.2017.01.009. [DOI] [Google Scholar]
- Wang G.-X.; Huang D.; Ji J.-H.; Völker C.; Wurm F. R. Seawater-Degradable Polymers—Fighting the Marine Plastic Pollution. Advanced Science 2021, 8 (1), 2001121. 10.1002/advs.202001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri A. R.; Laforsch C.; Greiner A.; Agarwal S. Fate of So-Called Biodegradable Polymers in Seawater and Freshwater. Global Challenges 2017, 1 (4), 1700048. 10.1002/gch2.201700048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Duigou A.; Bourmaud A.; Davies P.; Baley C. Long term immersion in natural seawater of Flax/PLA biocomposite. Ocean Eng. 2014, 90, 140–148. 10.1016/j.oceaneng.2014.07.021. [DOI] [Google Scholar]
- Greene J.Biodegradable Bottle Development and Testing: Evaluation of the Manufacturing Process and Performance Properties for Polyhydroxyalkanoate (PHA) Materials. California Department of Resources Recycling and Recovery, 2012.
- Teixeira S.; Eblagon K. M.; Miranda F.; Pereira M. F. R.; Figueiredo J. L. Towards Controlled Degradation of Poly(lactic) Acid in Technical Applications. C 2021, 7 (2), 42. 10.3390/c7020042. [DOI] [Google Scholar]
- Samadi K.; Francisco M.; Hegde S.; Diaz C. A.; Trabold T. A.; Dell E. M.; Lewis C. L. Mechanical, rheological and anaerobic biodegradation behavior of a Poly(lactic acid) blend containing a Poly(lactic acid)-co-poly(glycolic acid) copolymer. Polym. Degrad. Stab. 2019, 170, 109018. 10.1016/j.polymdegradstab.2019.109018. [DOI] [Google Scholar]
- Jem K. J.; Tan B. The development and challenges of poly (lactic acid) and poly (glycolic acid). Advanced Industrial and Engineering Polymer Research 2020, 3 (2), 60–70. 10.1016/j.aiepr.2020.01.002. [DOI] [Google Scholar]
- Karjomaa S.; Suortti T.; Lempiäinen R.; Selin J. F.; Itävaara M. Microbial degradation of poly-(l-lactic acid) oligomers. Polym. Degrad. Stab. 1998, 59 (1), 333–336. 10.1016/S0141-3910(97)00204-8. [DOI] [Google Scholar]
- Xu Y.; Kim C.-S.; Saylor D. M.; Koo D. Polymer degradation and drug delivery in PLGA-based drug–polymer applications: A review of experiments and theories. J. Biomed. Mater. Res., Part B 2017, 105 (6), 1692–1716. 10.1002/jbm.b.33648. [DOI] [PubMed] [Google Scholar]
- Kranz H.; Ubrich N.; Maincent P.; Bodmeier R. Physicomechanical Properties of Biodegradable Poly(D,L-lactide) and Poly(D,L-lactide-co-glycolide) Films in the Dry and Wet States. J. Pharm. Sci. 2000, 89 (12), 1558–1566. . [DOI] [PubMed] [Google Scholar]
- Martin R. T.; Camargo L. P.; Miller S. A. Marine-degradable polylactic acid. Green Chem. 2014, 16 (4), 1768–1773. 10.1039/C3GC42604A. [DOI] [Google Scholar]
- Beament J.; Wolf T.; Markwart J. C.; Wurm F. R.; Jones M. D.; Buchard A. Copolymerization of Cyclic Phosphonate and Lactide: Synthetic Strategies toward Control of Amphiphilic Microstructure. Macromolecules 2019, 52 (3), 1220–1226. 10.1021/acs.macromol.8b02385. [DOI] [Google Scholar]
- Lim Y. H.; Heo G. S.; Rezenom Y. H.; Pollack S.; Raymond J. E.; Elsabahy M.; Wooley K. L. Development of a Vinyl Ether-Functionalized Polyphosphoester as a Template for Multiple Postpolymerization Conjugation Chemistries and Study of Core Degradable Polymeric Nanoparticles. Macromolecules 2014, 47 (14), 4634–4644. 10.1021/ma402480a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doney S. C.; Fabry V. J.; Feely R. A.; Kleypas J. A. Ocean Acidification: The Other CO2 Problem. Annual Review of Marine Science 2009, 1 (1), 169–192. 10.1146/annurev.marine.010908.163834. [DOI] [PubMed] [Google Scholar]
- Perreault D. M.; Anslyn E. V. Unifying the Current Data on the Mechanism of Cleavage–Transesterification of RNA. Angew. Chem., Int. Ed. Engl. 1997, 36 (5), 432–450. 10.1002/anie.199704321. [DOI] [Google Scholar]
- Li Y.; Breaker R. R. Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2‘-Hydroxyl Group. J. Am. Chem. Soc. 1999, 121 (23), 5364–5372. 10.1021/ja990592p. [DOI] [Google Scholar]
- Bauer K. N.; Liu L.; Wagner M.; Andrienko D.; Wurm F. R. Mechanistic study on the hydrolytic degradation of polyphosphates. Eur. Polym. J. 2018, 108, 286–294. 10.1016/j.eurpolymj.2018.08.058. [DOI] [Google Scholar]
- Grigorenko B.; Polyakov I.; Nemukhin A. Mechanisms of ATP to cAMP Conversion Catalyzed by the Mammalian Adenylyl Cyclase: A Role of Magnesium Coordination Shells and Proton Wires. J. Phys. Chem. B 2020, 124 (3), 451–460. 10.1021/acs.jpcb.9b07349. [DOI] [PubMed] [Google Scholar]
- Lohmeijer B. G. G.; Pratt R. C.; Leibfarth F.; Logan J. W.; Long D. A.; Dove A. P.; Nederberg F.; Choi J.; Wade C.; Waymouth R. M.; Hedrick J. L. Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters. Macromolecules 2006, 39 (25), 8574–8583. 10.1021/ma0619381. [DOI] [Google Scholar]
- Wen J.; Kim G. J. A.; Leong K. W. Poly(d,llactide–co-ethyl ethylene phosphate)s as new drug carriers. J. Controlled Release 2003, 92 (1), 39–48. 10.1016/S0168-3659(03)00294-3. [DOI] [PubMed] [Google Scholar]
- Becker G.; Marquetant T. A.; Wagner M.; Wurm F. R. Multifunctional Poly(phosphoester)s for Reversible Diels–Alder Postmodification To Tune the LCST in Water. Macromolecules 2017, 50 (20), 7852–7862. 10.1021/acs.macromol.7b01716. [DOI] [Google Scholar]
- Mangold C.; Dingels C.; Obermeier B.; Frey H.; Wurm F. PEG-based Multifunctional Polyethers with Highly Reactive Vinyl-Ether Side Chains for Click-Type Functionalization. Macromolecules 2011, 44 (16), 6326–6334. 10.1021/ma200898n. [DOI] [Google Scholar]
- Farah S.; Anderson D. G.; Langer R. Physical and mechanical properties of PLA, and their functions in widespread applications — A comprehensive review. Adv. Drug Delivery Rev. 2016, 107, 367–392. 10.1016/j.addr.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Codari F.; Lazzari S.; Soos M.; Storti G.; Morbidelli M.; Moscatelli D. Kinetics of the hydrolytic degradation of poly(lactic acid). Polym. Degrad. Stab. 2012, 97 (11), 2460–2466. 10.1016/j.polymdegradstab.2012.06.026. [DOI] [Google Scholar]
- Haider T.; Suraeva O.; O’Duill M. L.; Mars J.; Mezger M.; Lieberwirth I.; Wurm F. R. Controlling the crystal structure of precisely spaced polyethylene-like polyphosphoesters. Polym. Chem. 2020, 11 (20), 3404–3415. 10.1039/D0PY00272K. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.