

Abstract
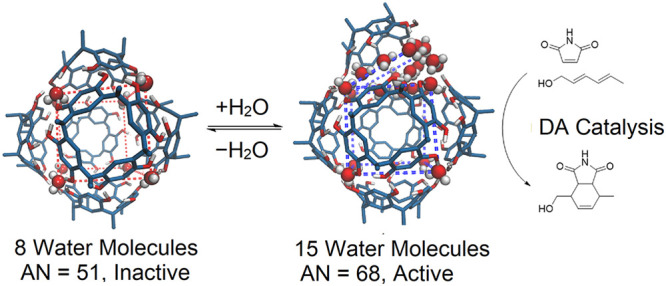
The hexameric undecyl-resorcin[4]arene capsule (C11R6) features eight discrete structural water molecules located at the vertices of its cubic suprastructure. Combining NMR spectroscopy with classical molecular dynamics (MD), we identified and characterized two distinct species of this capsule, C11R6-A and C11R6-B, respectively featuring 8 and 15 water molecules incorporated into their respective hydrogen-bonded networks. Furthermore, we found that the ratio of the C11R6-A and C11R6-B found in solution can be modulated by controlling the water content of the sample. The importance of this supramolecular modulation in C11R6 capsules is highlighted by its ability to perform acid-catalyzed transformations, which is an emergent property arising from the hydrogen bonding within the suprastructure. We show that the conversion of C11R6-A to C11R6-B enhances the catalytic rate of a model Diels–Alder cyclization by 10-fold, demonstrating the cofactor-derived control of a supramolecular catalytic process that emulates natural enzymatic systems.
Introduction
Supramolecular catalysis derives inspiration from enzymes, translating natural features into synthetic systems to attain higher levels of control in chemical processes. Approaches toward bioinspired supramolecular catalysis include the biomimicry,1−4 second coordination sphere design,5−7 and confinement of the catalytic site.8−14
Along these lines, the positioning of catalytic active sites within well-defined capsules has been demonstrated to enable the control of catalyst properties to promote selective catalytic transformations.6,7 In natural systems, enzymatic activity that enables the self-steering of catalytic processes necessary for metabolism can be modulated via allosteric modifications by physiochemical inputs. Although it is an intrinsic feature of natural systems, analogous modulation of catalyst properties in synthetic mimics are rare.15−18
It is now more than 30 years ago that the Aoyama group described the host–guest chemistry of resorcin[4]arenes in nonpolar organic solvents.19−22 As further characterization developed, the hexameric nature of these capsules was realized and its capacity for host–guest interactions were extensively characterized.23−39 Analogous to an enzyme, C11R6 exhibits catalytic function from the elevated Brønsted acidity emerging from its supramolecular structure.40 Illustrated in Figure 1, this capsule is formed in nonpolar solvents (e.g., chloroform) through the self-assembly of six facial monomers in a cubic arrangement, featuring eight water molecules (one per vertex).23 The edges of C11R6 are held together by hydrogen-bond network edges between adjacent facial monomers, with each end point capped at the vertex with a water molecule, completing the cubic structure.23
Figure 1.
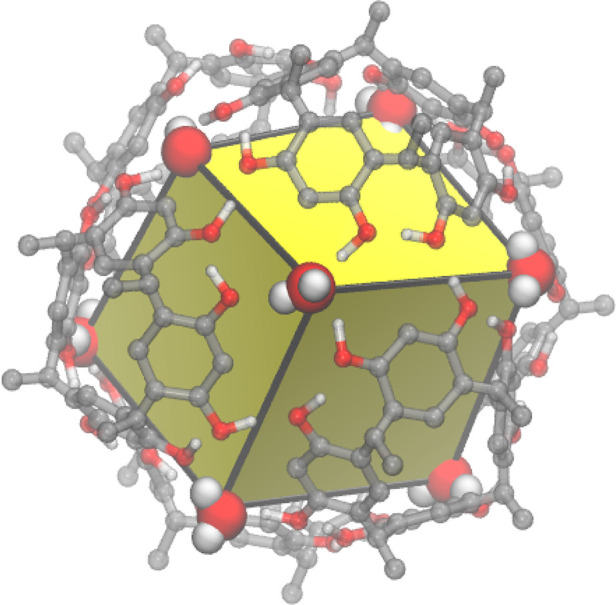
Ball-and-stick rendering of a model C11R6 showing a cubic structure with 6 resorcin[4]arenes (CPK rendering) forming the faces of the cube (yellow) held together by an hydrogen-bond network continuous along each edge (black line), and capped by eight water molecules at the vertex positions (van der Waals volume renderings). To improve clarity, pendant alkyl groups and nonhydroxy hydrogen atoms were omitted from this figure.
The hydrogen-bond network of C11R6 results in the enhanced Brønsted acidity beyond that of the individual monomer units.40 This feature has driven the application of C11R6 as a supramolecular, organic Brønsted acid catalyst for chemical transformations under mild conditions.41−44 In addition, the hydrogen-bond rich environment of the internal cavity within C11R6 has been utilized as a supramolecular organocatalyst,45−52 demonstrating a host-selective reactivity based on substrate size, and substrate–bond activation via supramolecular interactions. The use of a supplemental protic acid cocatalyst (typically HCl) extends the scope of C11R6 activity,53−64 notably for application toward facile synthesis of high-value terpene derivatives.60−64 Further reactivity has been demonstrated in host-catalyzed Diels–Alder cyclization.65
Beyond the intrinsic Brønsted acidity of C11R6, this supramolecule possesses an internal cavity (ca. 1400 Å3),23 permitting the encapsulation of transition metal catalysts66−69 or organic catalysts70−72 within its cavity. In these instances, the internal surface of the capsule serves as a second coordination-sphere to modulate or enhance catalytic function.5−7
Both the acidity and host-capacity of C11R6 are derived from its structure.23,40 Recent work by Payne and Oliver have demonstrated structural modification of C11R6 by the incorporation of alcoholic solvent molecules into the hydrogen bond network,73 complementing previous studies by Cohen74−76 and Schnatwinkel,77 which featured similar inclusion of long chain alcohols into the hydrogen-bond network. Interestingly, Katiyar has reported the association of free water to the capsule’s hydrogen-bond network,78,79 beyond the 8 molecules needed for capsule assembly.23,31,32 Studies by Merget suggest that the presence of additional water may impact the catalytic activity of the C11R6 capsule in acid-promoted cyclization of terpenes.61 Together these findings suggest that polar molecules such as water may act as cofactors able to modulate the structure and acidity of C11R6, analogous to the allosteric control of enzymes (e.g., cytochrome p450 oxidases, nitric oxide synthases, etc.). Understanding that these structural changes would provide insights into the previously observed water-dependent catalytic behavior and fine control the capsule’s catalytic activity.
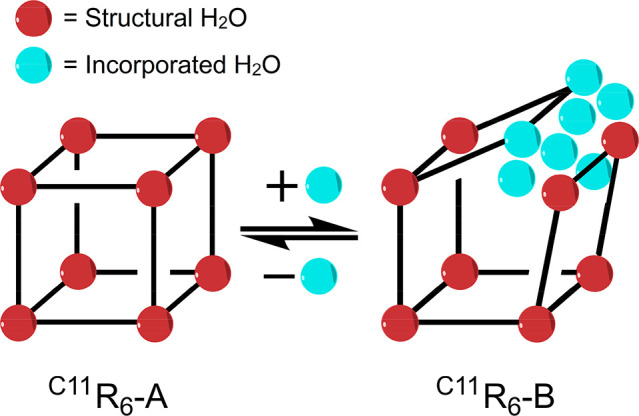
In this work, we investigate the structural changes of C11R6 capsules through classical molecular dynamics (MD) simulations, which is further supported by 1H NMR spectroscopy. Using MD, we find that C11R6 interconverts between two assemblies as summarized by Figure 2. The C11R6-A assembly features 8 water molecules at the vertex positions—in line with previous reports of C11R6 structure23—while C11R6-B has 14–15 water molecules, 6–7 of which spontaneously incorporate into a single edge of the cubic suprastructures, referred to here as “incorporated water” (Scheme 1). This computational finding is supported by NMR studies of water association, revealing a water-dependent equilibrium between the two capsule species differing significantly in their hydrogen-bond network. Differences between the C11R6-A and C11R6-B assemblies are substantiated by 31P NMR chemical shifts of an encapsulated phosphine oxide, revealing different internal acidities quantified by their Guttman–Beckett acceptor number (AN). This difference in internal acidity allows the rate modulation of C11R6 catalyzed Diels–Alder cycloaddition of maleimide and sorbic alcohol, demonstrating novel control of an abiotic homogeneous catalytic process.
Figure 2.
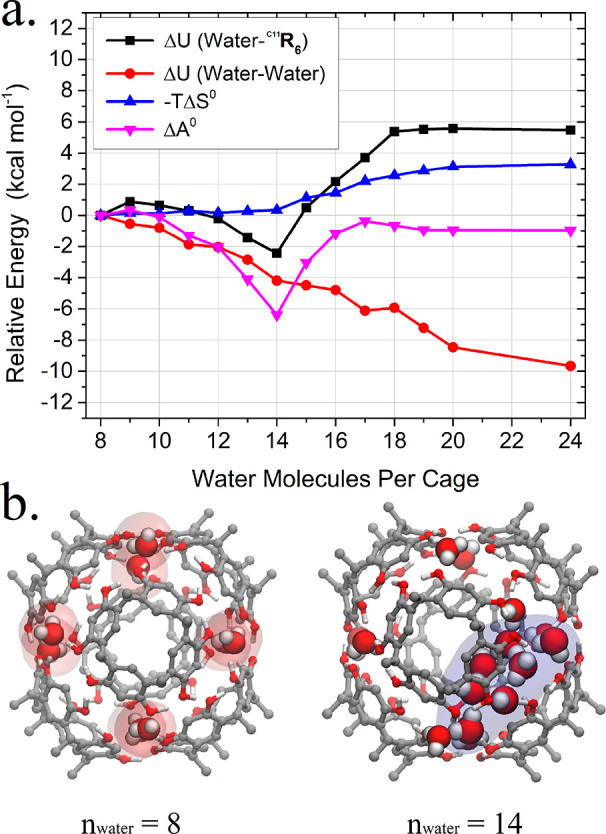
(a) Plot of the relative Helmholtz free energies (ΔA), internal energy (ΔU) of the water–water or water–C11R6 interactions, and entropy (ΔS) for the incorporation of water molecules beyond 8 determined by the GIST method.80 (b) Renderings depicting the structures of C11R6-A and C11R6-B containing 8 and 14 water molecules, respectively. Renderings feature highlights indicating structural (red) and incorporated (blue) water, which differentiate C11R6-A and C11R6-B, respectively. Note that structural water highlights for C11R6-B are omitted for clarity. Similarly, alkyl pendant groups, solvent, and nonhydroxy hydrogen atoms are omitted from both renderings for clarity. Both thermodynamic calculations (a) and model visualizations (b) were generated from 28 800 ns MD trajectories.
Scheme 1. Simplified Representation of the Water-Dependent Conversion between the Two Forms of C11R6, Highlighting the 8 Water Molecules Necessary for Capsule Formation (Red) and the 7 Additional Water Molecules (Cyan) That Effect the Transition by Association to a Capsule Edge (Black Line).
Results and Discussion
MD Simulations Reveal Distinct Species
Simulations containing explicitly solvated C11R6 with a total of 8–24 explicit water molecules were propagated as molecular dynamics trajectories for a total of 10 μs using optimized force field parameters (Figure S1). Unfortunately, simulations featuring randomly placed water molecules and undecyl-resorcin[4]arene monomers (C11R1) failed to self-assemble over several μs of MD propagation (results not reported). Therefore, we found it necessary to include the 8 structural water molecules, placed at the vertex positions of the capsule, while the remaining water molecules were positioned randomly in the periphery of the capsule.
In simulations containing 8–12 water molecules, we observe the external attachment of free water to the C11R6 in line with previous reports.78,79 Simulations containing ≥14 water molecules reveal 6 additional incorporated water molecules along a single edge of the hydrogen-bond network of the C11R6 capsule (Scheme 1), as depicted in Figure 2b. Although these incorporated water molecules are highly organized and an integral part of the hydrogen bond network (Figure S16), single water molecules still exchange rapidly with water molecules from the bulk solvent and the 8 structural waters needed to form the capsule. The mobility of the incorporated water is highlighted by the concerted migration between the hydrogen bond edges of the capsule. This migration phenomenon was qualitatively observed as a rare event in our MD simulations (Figure S15), but occur at a sub-microsecond time scale.
The incorporation of additional water into the edge of the hydrogen bond network results in a breakage of the hydrogen bond between adjacent C11R1 faces, altering the connectivity of the supramolecular system. This change in connectivity and composition distinguishes C11R6-B from the typical C11R6-A assembly. Analysis of hydrogen-bonding in our MD trajectories (Figure S2) reveal a minimum of 6 extra incorporated water molecules are required to form C11R6-B.
Energetic analysis of the MD data using GIST (Figure 2a) distinguishes between both attached water78,79 and the incorporated water we observe in C11R6-B. While GIST does not provide complete free energy differences between C11R6-A and C11R6-B, it is useful in the analysis of favorable water structures found in our MD simulations. In simulations containing 8–12 water molecules the attached water is observed. Interestingly, the GIST-determined ΔA is similar to previously reported values (ca. −2.0 kcal mol–1),78,79 and from our analysis this is driven entirely by a favorable water–water interaction (Figure 2a, ΔUwater–water). The inclusion of water along the hydrogen bond edge is optimal in the presence of 14 water molecules, where an additional favorable water-capsule interaction (Figure 2a, ΔUwater-C11R6), resulting in a very favorable association (ΔA = −6.3 kcal mol–1). While the incorporation of further water molecules within the suprastructure is possible, it incurs an increasing penalty from internal energy (Figure 2a, ΔU) and system entropy (Figure 2a, −TΔS). The specificity of C11R6-B to incorporate 6 water molecules is a “goldilocks” number, originating from the required size of the hydrogen-bond network needed to fill a capsule edge (Figure 2b), resulting in favorable internal energy (Figure 2a). These “incorporated water” molecules are more mobile than their “structural water” counterparts, and are not as strongly localized. These simulations suggest that C11R6 is found in only two forms—C11R6-A containing 8 water molecules and C11R6-B containing 14 water molecules—and the ratio between the two may depend on water content.
1H NMR Identification of C11R6-A and C11R6-B
The formation of C11R6-A and C11R6-B was investigated by 1H NMR, by measuring spectra of C11R6 solution at various concentrations of water (44.12–103.01 mM; for details see Supporting Information). Contrasting previous reports, which broadly attribute all phenolic peaks (δ = 8.5–10.0 ppm) to a singular species of C11R6,13−16 our spectra, shown in Figure 3a, reveal a changing pattern in the phenolic peaks, concomitant with the changing water content. The separation of these phenolic peaks indicates slowly exchanging environments,81 inconsistent with the 5 ns lifetimes of previously described water dynamics.79 As these peaks increase (or decrease) in a correlated fashion, we attribute these spectral features to distinct assemblies: C11R6-A (δ = 9.58 and 9.35 ppm) and C11R6-B (δ = 9.65 and 9.46 ppm). This peak assignment is further supported by inversion-relaxation measurements (Figure S21), from which identical T1 relaxation times were obtained for the phenolic peaks of either capsule indicative of a shared environment. The increased sensitivity of T1 relaxation times of the peaks belonging C11R6-B to changing water content is in line with the larger number of water molecules associate to its structure.
Figure 3.

Quantitative 1H NMR spectra (a) and DOSY diffusograms (b) of C11R6 (5.38 mM) observed over a range of water concentrations (44.12–103.01 mM) corresponding to 8–19 water molecules per C11R6 capsule. Water peaks and diffusion traces are highlighted in blue or accompanied by a blue circle. Peaks and diffusion traces attributed to C11R6-A (red highlight and diamond) and C11R6-B (green highlight and triangle) assemblies are annotated. Overlapping diffusion traces are shown in purple. Quantitation of water concentration was determined by integration of the water peak (blue circle) compared to a peak corresponding to both C11R6 assemblies (δ = 6.2 ppm, 24 H, 5.38 mM). Complete diffusograms are provided in the Supporting Information (Figure S4). Long recycle delays (25 s) were necessary to obtain quantitative spectra for both water (T1 = 0.7–0.9 s, data not shown) and C11R6 (T1 = 1.39 s, data not shown).
Interestingly, the relative concentrations of these species vary with water content from 44.12 mM (ca. 8 water molecules per capsule) to 103.01 mM (ca. 19 water molecules per capsule). As these differences are only apparent in the phenolic region of the NMR spectrum, we surmise that these assemblies are distinguished by the structure of their respective hydrogen-bond networks. Therefore, we putatively assigned these peaks to C11R6-A (δOH = 9.58, 9.35 ppm) and C11R6-B (δOH = 9.65, 9.46 ppm) based on the increasing concentration of water and consistent with the structures observed in MD simulations (Figure 2). The presence of incorporated water in C11R6-B is further evidenced by stronger NOE correlations between its phenolic peaks and free water (Figure S18). Deuterium exchange of the OH-groups with D2O (Figure S23) is different for the two capsules, and evidence the discontinuous hydrogen bond network in line with our MD simulations (Figure S16).
Interestingly, only two peaks of equal area are observed for the phenolic protons of either assembly, despite the asymmetry derived by incorporated water molecules in C11R6-B (Figure 2). Our MD simulations show the specific arrangement of incorporated water shift between edges of the capsule on a sub-microsecond time scale (Figure S15). The environments of the phenolic protons of C11R6-B, exchange at this rate, and as such are observed as a time-averaging signal. Exchange of water between C11R6-B and C11R6-A is relatively slow leading to distinct phenolic peaks that can be distinguished in the NMR spectra (Figure S14).81 On the basis of the relative strength of NOE correlations between the phenolic peaks and water, we assign the upfield peaks of either assembly (δ = 9.35 and 9.46 ppm) to the 24 phenolic protons adjacent to the structural water sites (Figure 1). Similarly the downfield peaks of either assembly (δ = 9.58 and 9.65 ppm), are assigned to the remaining 24 phenolic protons which participate in hydrogen bonding between and within the resorcin[4]arene monomer faces. Fortunately, the separation of the pairs (33 Hz) of the resolved C11R6-A and C11R6-B phenolic peaks constraints the rate constant for chemical exchange (kex) between the two assemblies to <155 s–1 (for a detailed discussion see, Figure S14).81
The apparent diffusion of these phenolic peaks appears faster than the other peaks (Figure 3b) due to proton exchange with water occurring within the diffusion time in the measurement (Δ = 100 ms). Fortunately, the pairing of these diffusion traces further supports the speciation of the two assemblies observed by the correlation of the peak areas (Figure 3a).
Further characterization of the capsule using 1H NMR (Figure S3), DOSY (Figures 3b and S4),31−33,78 and solution state FTIR (Figure S5)36 indicate that both assemblies are hexameric assemblies with a similar Stokes radius (16.6 Å) at [H2O] = 44 and 103 mM consistent with previous reports of C11R6 capsule structure.23−39
The single observed peak of water (Figure 3) indicates that it is in a state of fast exchange between a free state in the bulk solution and a bound state, incorporated into the C11R6 capsule (Figure S14).83 As previous reports detail, the available water is completely incorporated into the cage at low (i.e., 44 mM) water concentrations;31−33,78 therefore, the measured chemical shift (δ = 5.1 ppm) can be attributed to the structural water (Figure 1), as opposed to the free H2O water-saturated chloroform.82 As the observed chemical shift is time-averaged,81 the proportion and quantity of water associated with C11R6 (Bwat) was determined directly from 1H NMR spectra (Figure 3a).
Figure 4 shows the total number of water molecules associated with C11R6 increases linearly with the proportion of C11R6-B (θB) in the sample, with the slope showing an additional 7.27 ± 0.26 water molecules are incorporated per C11R6-B formed. Thus, combined with the 8 structural waters native toC11R6, a total of 15 water molecules are associated with C11R6-B. From our MD simulations (Figure 1) we surmise that these additional water molecules are incorporated into the hydrogen bonding network of the capsule. This number is in agreement with MD models (Figure 2) that predict a minimum of 14 water molecules for the formation of C11R6-B (Figure 2). The water-dependent conversion between C11R6-A and C11R6-B was fit using an empirical model (Figure S13) to enable estimation of the proportion of C11R6-B capsules (θB) via water content.
Figure 4.
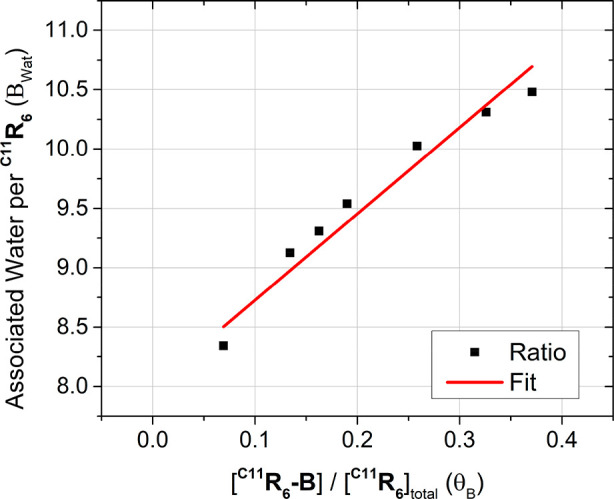
Plot of the total number of associated waters (Bwat) and proportion of C11R6-B capsules (θB) determined from 1H NMR measurements (Figure 3a). The association of an additional 7.27 ± 0.26 water molecules concomitant to conversion is determined from the slope of the linear fit (red).
31P NMR Investigation of Structure-Dependent Acidity
Many catalytic applications of C11R6 rely on the intrinsic acidity derived from its supramolecular structure.40 The 33 Hz downfield shift of the C11R6-B phenolic protons (Figure 3a) suggest an increased acidity (compared to C11R6-A),83 a feature which is further supported by their apparent diffusivities observed by DOSY (Figure 3b).
Previously the Brønsted acidity of C11R6 assemblies were measured using nitrogen bases to estimate aqueous-equivalent pKa values.40 Unfortunately, this protocol impairs the accurate determination of water content by either Karl–Fischer titration or 1H NMR integration, and could not be used to differentiate the acidity of C11R6-A and C11R6-B.
Therefore, we investigate the ability of structure-dependent acidity to modulate the interaction strength with tri-n-butyl phosphine oxide (Bu3PO) as guest through 31P NMR (Figure 5).84,85 The encapsulation of Bu3PO was readily confirmed by 1H NMR, showing the development of broad upfield peaks (δ = −2.0–0.5 ppm), typically observed for encapsulated guests.24−39 The binding of Bu3PO within the capsule was further evidenced by 1H DOSY measurements (Figure S12), with similar diffusion for the C11R6 host and upfield peaks (log D = −9.0, see Figure 3b).
Figure 5.
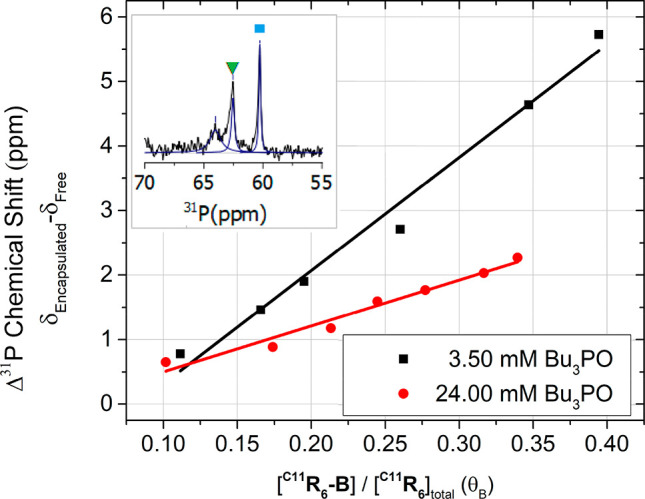
Chemical shift difference between free and encapsulated Bu3PO observed by 31P NMR at two concentrations, 3.50 mM (black) and 24.00 mM (red) in the presence of C11R6 (5.38 mM). Spectra were obtained at water contents spanning 43.76–110.19 mM (3.50 mM Bu3PO) and 43.05–86.53 mM (24.00 mM Bu3PO), which were subsequently converted to the proportion of C11R6-B (θB) by an empirical model (Figure S13). Inset, a 31P NMR spectrum showing peaks corresponding to encapsulated (▼, green) and free (■, blue) Bu3PO.
A downfield chemical shift in 31P NMR is expected when a Bu3PO forms a hydrogen-bond adduct with another species, such as when encapsulated within C11R6 and the degree of this shift is proportionate to the acidity of the hydrogen-bond donor.84,85 Three peaks (31Pδ ≈ 55.0–65.0 ppm) were consistently observed in the 31P NMR spectra of the encapsulated Bu3PO (Figures S9 and S10). The upfield peak (31Pδ ≈ 55.0–64.0 ppm) was assigned to the free Bu3PO by observed correlations to the protons of the free species by 1H–31P HMBC (Figure S11). A low intensity peak (31Pδ ≈ 64.0–65.0 ppm) was observed in all spectra, with a low intensity that waned with increasing water content. This spectral feature is particularly evident at a minimal water concentration (44.18 mM water, Figure S8), where the majority of the Bu3PO (3.50 mM) was observed to be encapsulated. Unfortunately, two-dimensional techniques (e.g., HMBC) could not provide sufficient cross-peaks with which to identify the originating species by other means. With additional water this minor peak broadens and diverges compared to the major peaks, and we infer that exchange between this minor species and the observed major peak is unlikely based on the diverging chemical shift. On the basis of the low intensity of the 31P signal, we surmise that this spectral feature does not correspond to the free or encapsulated Bu3PO, and its identity is unlikely to interfere with measurements of the C11R6 capsule’s internal acidity. The remaining peak was attributed to the C11R6-associated Bu3PO (31Pδ ≈ 60.0–64 ppm) based on its apparent intensity (Figures S9 and S10). All three peaks were observed to move in a concerted fashion with changes in water content, which we ascribe to changes in bulk dielectric of the solvent medium.86
The free and encapsulated Bu3PO afford distinct peaks in slow exchange (Figure 5, inset). Similar to observations made with 1H NMR (Figure 3), differentiation between phosphine oxide encapsulated within C11R6-A and C11R6-B was not observed by 31P due to the similarities of the magnetic environments experienced by the phosphorus nuclei. Due to this similarity, the shift of the observable peak corresponds to the time weighted average of the Bu3PO encapsulated within C11R6-A and C11R6-B (see Figure S14a for an example of the exchange of indistinguishable nuclei).81 Further complications arise as a phosphine oxide guest within C11R6-A or C11R6-B may exchange hydrogen bonding partners within the capsule at a time scale faster than NMR measurement,81 resulting in a single observable peak with a shift that is the time weighted average of the hydrogen bonding states (see Figure S14b for a detailed example of the exchange of a rapid process). The result of these exchange processes is a single observable peak corresponding to Bu3PO encapsulated by C11R6-A or C11R6-B, in all states of hydrogen bonding (see Figure S14 for a detailed explanation).81
Despite these limits in observation, the strength of the interaction between C11R6 and Bu3PO can be correlated to the downfield chemical shift of the single observable peak (31Pδ = 64.0–60.0 ppm). The strength of the interaction between Bu3PO and C11R6 can be determined by modulating the Brønsted acidity through changing the content of the sample (i.e., varying the water content of the sample) as shown in Figure 5. Two sets of experiments were performed where the C11R6-A/C11R6-B ratio was modulated through controlling water content (44.18–110.19 mM and 43.05–86.53 mM, respectively) in the presence of either a low (3.50 mM) or high (24.00 mM) concentration of Bu3PO. While the high concentration is analogous to catalytic conditions, at lower concentrations the Bu3PO probe selectively associates to the stronger interacting (i.e., more acidic) assembly. From these contrasting measurements we determine that the environment of C11R6-B is more acidic than C11R6-A, which may enhance its catalytic activity. We rationalize the increased acidity of C11R6-B by the increased availability of protons within the capsule from the weakly bound incorporated water molecules (Scheme 1).
Similar to 4 of the structural water molecules of C11R6-A,40 the 7 incorporated water molecules found in C11R6-B are capable hydrogen-bond donors, and may also act as acids stabilized by the edge hydrogen-bond network (Figure S16).
The Guttman–Becket acceptor number (AN) is a measure of Lewis acidity that quantifies the differences in acidity between the two capsules, and allows comparison of acid catalysts in solvent media.85 On the basis of 31P NMR spectra obtained at a minimal water concentration ([H2O] = 44.18 mM, Figure S8), we have estimated the Lewis acidity of C11R6-A (AN = 51), similar to B(OMe)3 (AN = 51).85 By extrapolating the chemical shift difference observed with Bu3PO (3.5 mM, Figure 5), we estimate the Lewis acidity of C11R6-B assemblies (AN = 68 ± 1), similar to TiCl4 (AN = 70).87
Structural Modulation of the C11R6-Catalyzed Diels–Alder Cycloaddition
We investigated the catalytic activity of the two C11R6 assemblies in the Diels–Alder cycloaddition of maleimide and sorbic alcohol to produce 4-(hydroxymethyl)-7-methyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (Figure 6, inset). The Diels–Alder reaction was explicitly chosen as a probe for the structure-dependent catalytic activity of C11R6 as it proceeds without the generation of water or acid as a byproduct. Specifically, catalysis was performed at different water concentrations ([H2O] = 8.76–25.95 mM) enabling modulation of the C11R6-B proportion (θB = 0.12–0.44) within the mixture. The dependency of catalytic activity on the proportion of C11R6-B was revealed, with the result depicted in Figure 6.
Figure 6.
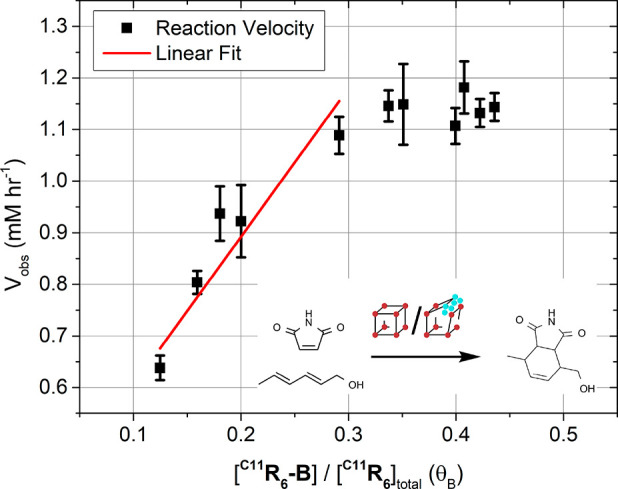
Initial reaction velocities, Vobs, for the Diels–Alder cycloaddition of sorbic alcohol (6.00 mM), and maleimide (6.00 mM), catalyzed by C11R6 (1.34 mM, 22 mol %) in CDCl3 under ambient conditions (25 °C). Reaction velocity was measured by the depletion of maleimide (δ = 6.72 ppm) referenced to a TMS standard by 1H NMR. Reaction water content spanned 11.77–34.87 mM (8.76–25.95 molecules per capsule) as determined by Karl–Fischer titration. The water content was used to estimate C11R6-B conversion by an empirical model (eq S1). Error bars correspond to the standard error in the linear fitting of the initial reaction velocity (Figure S6). A linear fit (red) reveals the turnover frequency of C11R6-A (0.24 ± 0.06 h–1) and C11R6-B (2.16 ± 0.29 h–1).
The initial reaction rates reveal that increases in water content afforded a doubling of the observed reaction rate (0.65–1.15 h–1), an effect not observed in the absence of C11R6 (Figure S7). As the ratio of C11R6-A and C11R6-B could not be directly observed by NMR, they were computed from the measured water content in conjunction with our empirical model (eq S1). The observed reaction velocity increases linearly (θB = 0.1–0.3) with the formation of C11R6-B until it plateaus (θB = 0.3–0.5), where another process becomes rate limiting. We propose that this rate limitation is due to the slow isomerization of sorbyl alcohol from its inactive s-trans isomer to the active s-cis isomer (Figure S17). From this limitation we surmise that C11R6 acts primarily as an acid-catalyst for the activation of maleimide. A linear fit of the reaction rate to the proportion of C11R6-B (θB) between 0.1–0.3 decomposes the overall reaction rate to the activity of either C11R6-A or C11R6-B assemblies. From this linear fit we find the more acidic C11R6-B (2.16 ± 0.29 h–1) is significantly more active than C11R6-A (0.24 ± 0.06 h–1). As the computed rate of C11R6-A catalyzed cycloadditions is close to the uncatalyzed reaction (0.21 ± 0.01 h–1, Figure S7) we surmise that C11R6-B is the sole active catalytic species. This result highlights the similarities between biological and supramolecular catalytic systems, where subtle changes in the arrangement of (supra)molecular features yield significant changes in catalytic output under mild conditions.
Conclusion
On the basis of NMR spectroscopy and computational data we demonstrate that the self-assembled hexameric undecyl-resorcin[4]arene capsule C11R6 can be switched between two distinct species—C11R6-A and C11R6-B—respectively featuring 8 and 15 water molecules within their hydrogen-bond networks. The internal environments of the two assemblies were probed by the binding of Bu3PO, revealing substantial shifts in the 31P NMR peak of this guest through changing the C11R6-A/C11R6-B ratio by the addition of water to the sample. These NMR experiments suggest a stronger acidity of C11R6-B assemblies that translate into differences in catalytic activity. The catalytic activity of these two assemblies were investigated in a Diels–Alder cycloaddition reaction, revealing that C11R6-B exhibits greater catalytic output by an order of magnitude. This study demonstrates the ability of water to effect structural changes in C11R6 capsules by modulating the structure-derived catalytic properties of the supramolecular assembly. We envisage that the present work will enable subsequent study of other small-molecules as structural effectors of C11R6 (and related supramolecular structures) with the goal of gated and self-steering catalytic applications.
Acknowledgments
We thank our colleagues Dr. Anne Jans, Eline Meijer, and Dr. Andreas Ehlers for their assistance and advice for this study. We thank the European Research Council (ERC Adv. Grant 339786-5354 NAT_CAT) and the sustainable chemistry research program from the University of Amsterdam for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c04924.
Computational simulation parameters, experimental conditions, spectral data for all measurements (PDF)
Coordinates and connectivity of a representative structure for C11R6-A (PDB)
Coordinates and connectivity of a representative structure for C11R6-B (PDB)
Coordinates, charge and connectivity of undecyl-resorcin[4]arene monomer subunit used in MD simulations (monomer.mol2); Force field parameters used for MD simulation provided in Amber format (sim.frcmod) (ZIP)
The authors declare no competing financial interest.
Supplementary Material
References
- Rebilly J.-N.; Colasson B.; Bistri O.; Over D.; Reinaud O. Biomimetic cavity-based metal complexes. Chem. Soc. Rev. 2015, 44 (2), 467–489. 10.1039/C4CS00211C. [DOI] [PubMed] [Google Scholar]
- Breslow R.; Dong S. D. Biomimetic Reactions Catalyzed by Cyclodextrins and Their Derivatives. Chem. Rev. 1998, 98 (5), 1997–2012. 10.1021/cr970011j. [DOI] [PubMed] [Google Scholar]
- Marchetti L.; Levine M. Biomimetic Catalysis. ACS Catal. 2011, 1 (9), 1090–1118. 10.1021/cs200171u. [DOI] [Google Scholar]
- Tu Y.; Peng F.; Adawy A.; Men Y.; Abdelmohsen L. K. E. A.; Wilson D. A. Mimicking the Cell: Bio-Inspired Functions of Supramolecular Assemblies. Chem. Rev. 2016, 116 (4), 2023–2078. 10.1021/acs.chemrev.5b00344. [DOI] [PubMed] [Google Scholar]
- Elsby M. R.; Baker R. T. Strategies and mechanisms of metal–ligand cooperativity in first-row transition metal complex catalysts. Chem. Soc. Rev. 2020, 49, 8933–8987. 10.1039/D0CS00509F. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Wang H.-B.; Ji L.-N.; Mao Z.-W. Insights into metalloenzyme microenvironments: biomimetic metal complexes with a functional second coordination sphere. Chem. Soc. Rev. 2013, 42 (21), 8360. 10.1039/c3cs60162e. [DOI] [PubMed] [Google Scholar]
- Trouve J.; Gramage-Doria R. Beyond hydrogen bonding: recent trends of outer sphere interactions in transition metal catalysis. Chem. Soc. Rev. 2021, 50, 3565–3584. 10.1039/D0CS01339K. [DOI] [PubMed] [Google Scholar]
- Nurttila S. S.; Linnebank P. R.; Krachko T.; Reek J. N. H. Supramolecular Approaches to Control Activity and Selectivity in Hydroformylation Catalysis. ACS Catal. 2018, 8, 3469–3488. 10.1021/acscatal.8b00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeuwissen J.; Reek J. N. H. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. 10.1038/nchem.744. [DOI] [PubMed] [Google Scholar]
- Gaeta C.; La Manna P.; De Rosa M.; Soriente A.; Talotta C.; Neri P. Supramolecular Catalysis with Self-Assembled Capsules and Cages: What Happens in Confined Spaces. ChemCatChem 2021, 13 (7), 1638–1658. 10.1002/cctc.202001570. [DOI] [Google Scholar]
- Grommet A. B.; Feller M.; Klajn R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 2020, 15, 256–271. 10.1038/s41565-020-0652-2. [DOI] [PubMed] [Google Scholar]
- Leenders S. H. A. M.; Gramage-Doria R.; de Bruin B.; Reek J. N. H. Transition metal catalysis in confined spaces. Chem. Soc. Rev. 2015, 44, 433–448. 10.1039/C4CS00192C. [DOI] [PubMed] [Google Scholar]
- Morimoto M.; Bierschenk S. M.; Xia K. T.; Bergman R. G.; Raymond K. N.; Toste F. D. Advances in supramolecular host-mediated reactivity. Nature Catalysis 2020, 3, 969–984. 10.1038/s41929-020-00528-3. [DOI] [Google Scholar]
- Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem., Int. Ed. 2009, 48 (19), 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- Rieth S.; Hermann K.; Wang B.-Y.; Badjic J. D. Controlling the dynamics of molecular encapsulation and gating. Chem. Soc. Rev. 2011, 40, 1609–1622. 10.1039/C005254J. [DOI] [PubMed] [Google Scholar]
- Blanco-Gómez A.; Cortón P.; Barravecchia L.; Neira I.; Pazos E.; Peinador C.; García M. Controlled binding of organic guests by stimuli-responsive macrocycles. Chem. Soc. Rev. 2020, 49, 3834–3862. 10.1039/D0CS00109K. [DOI] [PubMed] [Google Scholar]
- Hermann K.; Ruan Y.; Hardin A. M.; Hadad C. M.; Badjić J. D. Gated molecular baskets. Chem. Soc. Rev. 2015, 44, 500–514. 10.1039/C4CS00140K. [DOI] [PubMed] [Google Scholar]
- Díaz-Moscoso A.; Ballester P. Light-responsive molecular containers. Chem. Commun. 2017, 53, 4635–4652. 10.1039/C7CC01568B. [DOI] [PubMed] [Google Scholar]
- Aoyama Y.; Tanaka Y.; Sugahara S. Molecular recognition. 5. Molecular recognition of sugars via hydrogen-bonding interaction with a synthetic polyhydroxy macrocycle. J. Am. Chem. Soc. 1989, 111 (14), 5397–5404. 10.1021/ja00196a052. [DOI] [Google Scholar]
- Tanaka Y.; Ubukata Y.; Aoyama Y. Selective Sugar Binding with a Synthetic Polyhydroxy Macrocycle. A Remarkable Selectivity for Fructose over Glucose. Chem. Lett. 1989, 18 (11), 1905–1908. 10.1246/cl.1989.1905. [DOI] [Google Scholar]
- Tanaka Y.; Kato Y.; Aoyama Y. Molecular recognition. Two-point hydrogen-bonding interaction: a remarkable chain-length selectivity in the binding of dicarboxylic acids with resorcinol-aldehyde cyclotetramer as a multidentate host. J. Am. Chem. Soc. 1990, 112 (7), 2807–2808. 10.1021/ja00163a056. [DOI] [Google Scholar]
- Kikuchi Y.; Tanaka Y.; Sutarto S.; Kobayashi K.; Toi H.; Aoyama Y. Highly cooperative binding of alkyl glucopyranosides to the resorcinol cyclic tetramer due to intracomplex guest-guest hydrogen-bonding: solvophobicity/solvophilicity control by an alkyl group of the geometry, stoichiometry, stereoselectivity, and cooperativity. J. Am. Chem. Soc. 1992, 114 (26), 10302–10306. 10.1021/ja00052a029. [DOI] [Google Scholar]
- MacGillivray L. R.; Atwood J. L. A chiral spherical molecular assembly held together by 60 hydrogen bonds. Nature 1997, 389 (6650), 469–472. 10.1038/38985. [DOI] [Google Scholar]
- MacGillivray L. R.; Atwood J. L. Rational Design of Multicomponent Calix[4]arenes and Control of Their Alignment in the Solid State. J. Am. Chem. Soc. 1997, 119 (29), 6931–6932. 10.1021/ja970758r. [DOI] [Google Scholar]
- Cohen Y.; Avram L.; Frish L. Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights. Angew. Chem., Int. Ed. 2005, 44 (4), 520–554. 10.1002/anie.200300637. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y. Spontaneous Formation of Hexameric Resorcinarene Capsule in Chloroform Solution as Detected by Diffusion NMR. J. Am. Chem. Soc. 2002, 124 (51), 15148–15149. 10.1021/ja0272686. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y. Effect of a Cationic Guest on the Characteristics of the Molecular Capsule of Resorcinarene: A Diffusion NMR Study. Org. Lett. 2003, 5 (7), 1099–1102. 10.1021/ol034156q. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y. Discrimination of Guests Encapsulation in Large Hexameric Molecular Capsules in Solution: Pyrogallol[4]arene versus Resorcin[4]arene Capsules. J. Am. Chem. Soc. 2003, 125 (52), 16180–16181. 10.1021/ja0377394. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y. Hexameric Capsules of Lipophilic Pyrogallolarene and Resorcinarene in Solutions as Probed by Diffusion NMR: One Hydroxyl Makes the Difference. Org. Lett. 2003, 5 (18), 3329–3332. 10.1021/ol035211y. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y. Self-Recognition, Structure, Stability, and Guest Affinity of Pyrogallol[4]arene and Resorcin[4]arene Capsules in Solution. J. Am. Chem. Soc. 2004, 126 (37), 11556–11563. 10.1021/ja047698r. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y.; Rebek J. Jr. Recent advances in hydrogen-bonded hexameric encapsulation complexes. Chem. Commun. 2011, 47 (19), 5368–5375. 10.1039/C1CC10150A. [DOI] [PubMed] [Google Scholar]
- Avram L.; Cohen Y. Diffusion NMR of molecular cages and capsules. Chem. Soc. Rev. 2015, 44, 586–602. 10.1039/C4CS00197D. [DOI] [PubMed] [Google Scholar]
- Evan-Salem T.; Baruch I.; Avram L.; Cohen Y.; Palmer L. C.; Rebek J. Resorcinarenes are hexameric capsules in solution. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (33), 12296–12300. 10.1073/pnas.0604757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivanyuk A.; Friese J. C.; Döring S.; Rebek J. Jr. Solvent-Stabilized Molecular Capsules. J. Org. Chem. 2003, 68 (17), 6489–6496. 10.1021/jo034791+. [DOI] [PubMed] [Google Scholar]
- Shivanyuk A.; Rebek J. Reversible encapsulation by self-assembling resorcinarene subunits. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (14), 7662–7665. 10.1073/pnas.141226898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling C. J. M.; Fundin L. J.; Williams N. H. Trimorphism in solid resorcinarenes. Chem. Commun. 2007, 1748–1750. 10.1039/b700469a. [DOI] [PubMed] [Google Scholar]
- Palmer L. C.; Shivanyuk A.; Yamanaka M.; Rebek J. Jr. Resorcinarene assemblies as synthetic receptors. Chem. Commun. 2005, (7), 857. 10.1039/b414252g. [DOI] [PubMed] [Google Scholar]
- Atwood J. L.; Barbour L. J.; Jerga A. Organization of the interior of molecular capsules by hydrogen bonding. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (8), 4837–4841. 10.1073/pnas.082659799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Tiefenbacher K. Hexameric Resorcinarene Capsule is a Brønsted Acid: Investigation and Application to Synthesis and Catalysis. J. Am. Chem. Soc. 2013, 135 (43), 16213–16219. 10.1021/ja4080375. [DOI] [PubMed] [Google Scholar]
- Gaeta C.; Talotta C.; De Rosa M.; La Manna P.; Soriente A.; Neri P. The Hexameric Resorcinarene Capsule at Work: Supramolecular Catalysis in Confined Spaces. Chem. - Eur. J. 2019, 25, 4899–4913. 10.1002/chem.201805206. [DOI] [PubMed] [Google Scholar]
- Gambaro S.; Talotta C.; Sala P. D.; Soriente A.; De Rosa M.; Gaeta C.; Neri P. Kinetic and Thermodynamic Modulation of Dynamic Imine Libraries Driven by the Hexameric Resorcinarene Capsule. J. Am. Chem. Soc. 2020, 142 (35), 14914–14923. 10.1021/jacs.0c04705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaro S.; La Manna P.; De Rosa M.; Soriente A.; Talotta C.; Gaeta C.; Neri P. The Hexameric Resorcinarene Capsule as a Brønsted Acid Catalyst for the Synthesis of Bis(heteroaryl)methanes in a Nanoconfined Space. Front. Chem. 2019, 7, 687. 10.3389/fchem.2019.00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Sorella G.; Sperni L.; Strukul G.; Scarso A. Supramolecular Activation of Hydrogen Peroxide in the Selective Sulfoxidation of Thioethers by a Self-Assembled Hexameric Capsule. Adv. Synth. Catal. 2016, 358, 3443–3449. 10.1002/adsc.201600430. [DOI] [Google Scholar]
- La Sorella G.; Sperni L.; Strukul G.; Scarso A. Supramolecular Encapsulation of Neutral Diazoacetate Esters and Catalyzed 1,3-Dipolar Cycloaddition Reaction by a Self-Assembled Hexameric Capsule. ChemCatChem 2015, 7 (2), 291–296. 10.1002/cctc.201402631. [DOI] [Google Scholar]
- Giust S.; La Sorella G.; Sperni L.; Fabris F.; Strukul G.; Scarso A. Supramolecular Catalysis in the Synthesis of Substituted 1 H-Tetrazoles from Isonitriles by a Self-Assembled Hexameric Capsule. Asian J. Org. Chem. 2015, 4 (3), 217–220. 10.1002/ajoc.201402229. [DOI] [Google Scholar]
- Gambaro S.; De Rosa M.; Soriente A.; Talotta C.; Floresta G.; Rescifina A.; Gaeta C.; Neri P. A hexameric resorcinarene capsule as a hydrogen bonding catalyst in the conjugate addition of pyrroles and indoles to nitroalkenes. Org. Chem. Front. 2019, 6, 2339–2347. 10.1039/C9QO00224C. [DOI] [Google Scholar]
- La Manna P.; De Rosa M.; Talotta C.; Rescifina A.; Floresta G.; Soriente A.; Gaeta C.; Neri P. Synergic Interplay Between Halogen Bonding and Hydrogen Bonding in the Activation of a Neutral Substrate in a Nanoconfined Space. Angew. Chem., Int. Ed. 2020, 59 (2), 811–818. 10.1002/anie.201909865. [DOI] [PubMed] [Google Scholar]
- Giust S.; La Sorella G.; Sperni L.; Fabris F.; Strukul G.; Scarso A. Supramolecular Catalysis in the Synthesis of Substituted 1 H-Tetrazoles from Isonitriles by a Self-Assembled Hexameric Capsule. Asian J. Org. Chem. 2015, 4 (3), 217–220. 10.1002/ajoc.201402229. [DOI] [Google Scholar]
- Köster J. M.; Häussinger D.; Tiefenbacher K. Activation of Primary and Secondary Benzylic and Tertiary Alkyl (sp3)C-F Bonds Inside a Self-Assembled Molecular Container. Front. Chem. 2019, 6, 639. 10.3389/fchem.2018.00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manna P.; De Rosa M.; Talotta C.; Gaeta C.; Soriente A.; Floresta G.; Rescifina A.; Neri P. The hexameric resorcinarene capsule as an artificial enzyme: ruling the regio and stereochemistry of a 1,3-dipolar cycloaddition between nitrones and unsaturated aldehydes. Org. Chem. Front. 2018, 5, 827–837. 10.1039/C7QO00942A. [DOI] [Google Scholar]
- La Manna P.; Talotta C.; Floresta G.; De Rosa M.; Soriente A.; Rescifina A.; Gaeta C.; Neri P. Mild Friedel–Crafts Reactions inside a Hexameric Resorcinarene Capsule: C–Cl Bond Activation through Hydrogen Bonding to Bridging Water Molecules. Angew. Chem., Int. Ed. 2018, 57, 5423–5428. 10.1002/anie.201801642. [DOI] [PubMed] [Google Scholar]
- Catti L.; Zhang Q.; Tiefenbacher K. Advantages of Catalysis in Self-Assembled Molecular Capsules. Chem. - Eur. J. 2016, 22, 9060–9066. 10.1002/chem.201600726. [DOI] [PubMed] [Google Scholar]
- Köster J. M.; Tiefenbacher K. Elucidating the Importance of Hydrochloric Acid as a Cocatalyst for Resorcinarene-Capsule-Catalyzed Reactions. ChemCatChem 2018, 10, 2941–2944. 10.1002/cctc.201800326. [DOI] [Google Scholar]
- La Sorella G.; Sperni L.; Ballester P.; Strukul G.; Scarso A. Hydration of aromatic alkynes catalyzed by a self-assembled hexameric organic capsule. Catal. Sci. Technol. 2016, 6, 6031–6036. 10.1039/C6CY00307A. [DOI] [Google Scholar]
- Catti L.; Zhang Q.; Tiefenbacher K. Self-Assembled Supramolecular Structures as Catalysts for Reactions Involving Cationic Transition States. Synthesis 2016, 48, 313–328. 10.1055/s-0035-1560362. [DOI] [Google Scholar]
- Zhang Q.; Catti L.; Kaila V. R. I.; Tiefenbacher K. To catalyze or not to catalyze: elucidation of the subtle differences between the hexameric capsules of pyrogallolarene and resorcinarene. J. Am. Chem. Sci. 2017, 8 (2), 1653–1657. 10.1039/C6SC04565K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S.; Zhang Q.; Major D. T. Thermodynamic and Kinetic Control Determine the Sesquiterpene Reaction Pathways Inside Nanocapsules. ACS Catal. 2020, 10 (12), 6843–6853. 10.1021/acscatal.0c00278. [DOI] [Google Scholar]
- Zhang Q.; Catti L.; Pleiss J.; Tiefenbacher K. Terpene Cyclizations inside a Supramolecular Catalyst: Leaving-Group-Controlled Product Selectivity and Mechanistic Studies. J. Am. Chem. Soc. 2017, 139 (33), 11482–11492. 10.1021/jacs.7b04480. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Rebek J. Jr.; Yu Y. Cyclizations catalyzed inside a hexameric resorcinarene capsule. Chem. Commun. 2019, 55, 3573–3577. 10.1039/C9CC01611B. [DOI] [PubMed] [Google Scholar]
- Merget S.; Catti L.; Piccini G.; Tiefenbacher K. Requirements for Terpene Cyclizations inside the Supramolecular Resorcinarene Capsule: Bound Water and Its Protonation Determine the Catalytic Activity. J. Am. Chem. Soc. 2020, 142 (9), 4400–4410. 10.1021/jacs.9b13239. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Rinkel J.; Goldfuss B.; Dickschat J. S.; Tiefenbacher K. Sesquiterpene cyclizations catalysed inside the resorcinarene capsule and application in the short synthesis of isolongifolene and isolongifolenone. Nat. Catal. 2018, 1 (8), 609–615. 10.1038/s41929-018-0115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntrivanis L. D.; Némethová I.; Schmid D.; Levi S.; Prescimone A.; Bissegger F.; Major D.; Tiefenbacher K. Four-Step Access to the Sesquiterpene Natural Product Presilphiperfolan-1β-ol and Unnatural Derivatives via Supramolecular Catalysis. J. Am. Chem. Soc. 2020, 142, 5894–5900. 10.1021/jacs.0c01464. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Tiefenbacher K. Sesquiterpene Cyclizations inside the Hexameric Resorcinarene Capsule: Total Synthesis of δ-Selinene and Mechanistic Studies. Angew. Chem., Int. Ed. 2019, 58 (36), 12688–12695. 10.1002/anie.201906753. [DOI] [PubMed] [Google Scholar]
- Shimizu S.; Usui A.; Sugai M.; Suematsu Y.; Shirakawa S.; Ichikawa H. Hexameric Capsule of a Resorcinarene Bearing Fluorous Feet as a Self-Assembled Nanoreactor: A Diels–Alder Reaction in a Fluorous Biphasic System. Eur. J. Org. Chem. 2013, 22, 4734–4737. 10.1002/ejoc.201300652. [DOI] [Google Scholar]
- Jans A. C. H.; Caumes X.; Reek J. N. H. Gold Catalysis in (Supra)Molecular Cages to Control Reactivity and Selectivity. ChemCatChem 2018, 10, 1–12. 10.1002/cctc.201801399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavarzan A.; Reek J. N. H.; Trentin F.; Scarso A.; Strukul G. Substrate selectivity in the alkyne hydration mediated by NHC–Au(i) controlled by encapsulation of the catalyst within a hydrogen bonded hexameric host. Catal. Sci. Technol. 2013, 3, 2898–2901. 10.1039/c3cy00300k. [DOI] [Google Scholar]
- Jans A. C. H.; Gómez-Suárez A.; Nolan S. P.; Reek J. N. H. A Switchable Gold Catalyst by Encapsulation in a Self-Assembled Cage. Chem. - Eur. J. 2016, 22 (42), 14836–14839. 10.1002/chem.201603162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongkind L. J.; Rahimi M.; Poole D.; Ton S. J.; Fogg D. E.; Reek J. N. H. Protection of Ruthenium Olefin Metathesis Catalysts by Encapsulation in a Self-assembled Resorcinarene Capsule. ChemCatChem 2020, 12, 4019–4023. 10.1002/cctc.202000111. [DOI] [Google Scholar]
- Zhang Q.; Catti L.; Tiefenbacher K. Catalysis inside the Hexameric Resorcinarene Capsule. Acc. Chem. Res. 2018, 51, 2107–2114. 10.1021/acs.accounts.8b00320. [DOI] [PubMed] [Google Scholar]
- Bräuer T. M.; Zhang Q.; Tiefenbacher K. Iminium Catalysis inside a Self-Assembled Supramolecular Capsule: Modulation of Enantiomeric Excess. Angew. Chem., Int. Ed. 2016, 55, 7698–7701. 10.1002/anie.201602382. [DOI] [PubMed] [Google Scholar]
- Bräuer T. M.; Zhang Q.; Tiefenbacher K. Iminium Catalysis inside a Self-Assembled Supramolecular Capsule: Scope and Mechanistic Studies. J. Am. Chem. Soc. 2017, 139 (48), 17500–17507. 10.1021/jacs.7b08976. [DOI] [PubMed] [Google Scholar]
- Payne R. M.; Oliver C. L. A propanol-seamed C-methylcalix[4]resorcinarene hexamer accessible via solution crystallization, liquid-assisted grinding and vapour sorption. CrystEngComm 2018, 20 (14), 1919–1922. 10.1039/C8CE00294K. [DOI] [Google Scholar]
- Avram L.; Cohen Y. The Role of Water Molecules in a Resorcinarene Capsule As Probed by NMR Diffusion Measurements. Org. Lett. 2002, 4 (24), 4365–4368. 10.1021/ol0271077. [DOI] [PubMed] [Google Scholar]
- Slovak S.; Avram L.; Cohen Y. Encapsulated or Not Encapsulated? Mapping Alcohol Sites in Hexameric Capsules of Resorcin[4]arenes in Solution by Diffusion NMR Spectroscopy. Angew. Chem., Int. Ed. 2010, 49 (2), 428–431. 10.1002/anie.200904952. [DOI] [PubMed] [Google Scholar]
- Slovak S.; Cohen Y. The Effect of Alcohol Structures on the Interaction Mode with the Hexameric Capsule of Resorcin[4]arene. Chem. - Eur. J. 2012, 18, 8515–8520. 10.1002/chem.201102809. [DOI] [PubMed] [Google Scholar]
- Schnatwinkel B.; Stoll I.; Mix A.; Rekharsky M. V.; Borovkov V. V.; Inoue Y.; Mattay J. Monomeric, dimeric and hexameric resorcin[4]arene assemblies with alcohols in apolar solvents. Chem. Commun. 2008, (33), 3873–3875. 10.1039/b803890b. [DOI] [PubMed] [Google Scholar]
- Katiyar A.; Freire Sovierzoski J. C.; Calio P. B.; Vartia A. A.; Thompson W. H. Water plays a diverse role in a hydrogen-bonded, hexameric supramolecular assembly. Chem. Commun. 2019, 55 (46), 6591–6594. 10.1039/C9CC03151K. [DOI] [PubMed] [Google Scholar]
- Katiyar A.; Freire Sovierzoski J. C.; Calio P. B.; Vartia A. A.; Thompson W. H. Water plays a dynamical role in a hydrogen-bonded, hexameric supramolecular assembly. Phys. Chem. Chem. Phys. 2020, 22, 6167–6175. 10.1039/C9CP06874K. [DOI] [PubMed] [Google Scholar]
- Ramsey S.; Nguyen C.; Salomon-Ferrer R.; Walker R. C.; Gilson M. K.; Kurtzman T. Solvation thermodynamic mapping of molecular surfaces in AmberTools: GIST. J. Comput. Chem. 2016, 37 (21), 2029–2037. 10.1002/jcc.24417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant R. G. The NMR time scale. J. Chem. Educ. 1983, 60 (11), 933–935. 10.1021/ed060p933. [DOI] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- Ouellette R. J. the Chemical Shift of the Hydroxyl Proton of Phenols in Dimethyl Sulfoxide. Can. J. Chem. 1965, 43, 707–709. 10.1139/v65-091. [DOI] [Google Scholar]
- Beckett M. A.; Strickland G. C.; Holland J. R.; Sukumar Varma K. A convenient n.m.r. method for the measurement of Lewis acidity at boron centres: correlation of reaction rates of Lewis acid initiated epoxide polymerizations with Lewis acidity. Polymer 1996, 37 (20), 4629–4631. 10.1016/0032-3861(96)00323-0. [DOI] [Google Scholar]
- Mayer U.; Gutmann V.; Gerger W. The acceptor number: A quantitative empirical parameter for the electrophilic properties of solvents. Monatsh. Chem. 1975, 106 (6), 1235–1257. 10.1007/BF00913599. [DOI] [Google Scholar]
- Oka K.; Shibue T.; Sugimura N.; Watabe Y.; Winther-Jensen B.; Nishide H. Long-lived water clusters in hydrophobic solvents investigated by standard NMR techniques. Sci. Rep. 2019, 9 (1), 1–6. 10.1038/s41598-018-36787-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubczyk M.; Adamczyk-Woźniak A.; Sporzyński A.. Acceptor number of organoboron molecules quantitative determination of Lewis acidity. In Molecular Receptors.; East Publisher House, 2011. pp 53–68. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.