

Abstract
Despite much progress in improving graft outcome during cardiac transplantation, chronic allograft vasculopathy (CAV) remains an impediment to long‐term graft survival. MicroRNAs (miRNAs) emerged as regulators of the immune response. Here, we aimed to examine the miRNA network involved in CAV. miRNA profiling of heart samples obtained from a murine model of CAV and from cardiac‐transplanted patients with CAV demonstrated that miR‐21 was most significantly expressed and was primarily localized to macrophages. Interestingly, macrophage depletion with clodronate did not significantly prolong allograft survival in mice, while conditional deletion of miR‐21 in macrophages or the use of a specific miR‐21 antagomir resulted in indefinite cardiac allograft survival and abrogated CAV. The immunophenotype, secretome, ability to phagocytose, migration, and antigen presentation of macrophages were unaffected by miR‐21 targeting, while macrophage metabolism was reprogrammed, with a shift toward oxidative phosphorylation in naïve macrophages and with an inhibition of glycolysis in pro‐inflammatory macrophages. The aforementioned effects resulted in an increase in M2‐like macrophages, which could be reverted by the addition of L‐arginine. RNA‐seq analysis confirmed alterations in arginase‐associated pathways associated with miR‐21 antagonism. In conclusion, miR‐21 is overexpressed in murine and human CAV, and its targeting delays CAV onset by reprogramming macrophages metabolism.
Keywords: basic (laboratory) research / science, heart (allograft) function / dysfunction, heart transplantation / cardiology, immunobiology, macrophage / monocyte biology: activation, molecular biology: micro RNA, rejection: vascular, translational research / science
Short abstract
The authors show that, after heart transplantation, microRNA‐21 is overexpressed in chronic allograft vasculopathy (CAV), that pharmacological and genetic modulation of microRNA‐21 mitigated CAV, and that microRNA‐21 antagonism abrogated CAV by reprogramming macrophage metabolism.
Abbreviations
- Arg1
Arginase 1
- ATG
thymoglobulin
- bm12
C57BL/6 J, B6.C‐H2bm12
- BMDM
bone marrow‐derived macrophages
- CAV
chronic allograft vasculopathy
- CPM
counts per million
- Csa
cyclosporine
- CVA
cerebrovascular accident
- ddPCR
digital droplet PCR
- ECAR
extracellular acidification rate
- ELISA
enzyme‐linked immunosorbent assay
- FACS
flow cytometric analysis
- FCCP
carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone
- LysMcre
B6.129P2‐Lyz2tm1(cre)Ifo/J
- miRNAs
MicroRNAs
- MMF
mycophenolate
- nc miRNA
negative control miRNA antagomir
- OCR
oxygen consumption rate
- PBS
phosphate‐buffered saline
- qRT‐PCR
real‐time quantitative reverse transcription polymerase chain reaction
- SEM
standard error mean
1. INTRODUCTION
Cardiac transplantation is the most effective therapy for prolonging survival in patients with end‐stage heart failure, and it also provides better survival and quality of life as compared with conventional medical or pharmacological treatment. 1 , 2 However, despite much improvement in patient selection, heart preservation, immunosuppression, and cytomegalovirus prophylaxis, the late outcomes of allograft survival have only slightly improved. 1 , 3 , 4 , 5 Chronic allograft vasculopathy (CAV) occurs in 50% of cardiac transplant recipients by 5 years after transplantation 6 , 7 and is characterized by neointimal hyperplasia, vascular inflammation, and occlusion; is a major feature of chronic rejection; and continues to limit long‐term survival following cardiac transplantation. 1 , 8 This type of rejection occurs in the presence of T cell‐related immunosuppression confirming the role of other immune cells like macrophages. Indeed, the degree of macrophages infiltration correlates with CAV severity. 9 , 10 , 11 While the etiology of CAV varies from immune to nonimmune events, 12 , 13 no specific treatments are available. MicroRNAs (miRNAs) are short noncoding single‐stranded RNAs, many of which have been highly conserved throughout evolution. miRNAs regulate gene expression post‐transcriptionally by targeting mRNA for degradation or translational repression 14 , 15 , 16 and have been implicated in regulation of the immune system and of transplant outcomes. Particularly, miR‐21 has been identified as modulator of the alloimmune response in kidney transplant and fibrosis. 14 , 15 , 17 , 18 , 19 , 20 , 21 , 22 , 23 Interestingly, the miRNA pathway involved in CAV has not yet been discovered. 19 In this study, we used an unbiased approach, in which we profiled the miRNome of samples obtained from a murine model heterotopic vascularized cardiac transplantation in which a minor HLA mismatching between donor/recipient immune repertoire evokes a CAV by 8 weeks after transplantation thus resembling the human CAV. 13 , 24 We have also performed miRNomic in samples obtained from patients with CAV to delineate the exact miRNA profiles of human CAV. Furthermore, we evaluated the effect of targeting the miRNAs identified with specific miRNA antagomirs in order to determine the effect on CAV.
2. MATERIALS AND METHODS
A detailed description of the methods used in this study is provided in the Data S1.
2.1. Patients
Heart biopsies were obtained from the right side of the interventricular septum of cardiac transplant recipients. Samples were formalin‐fixed, paraffin‐embedded, sectioned, and histologically graded by a cardiac pathologist, as per the criteria of the 2005 International Society for Heart and Lung Transplantation. 25 Characteristics of patients and a description of the immunosuppressive regimens 26 are listed in Table 1 and in Table S1.
TABLE 1.
Patient characteristics (n = 10)
| Year of transplantation | 2005–2014 | |
|---|---|---|
| Sex (M/F) | 7/3 | |
| Age, years | 39.7 ± 13.75 | |
| End‐stage heart failure etiology, n (%) | Ischemic cardiomyopathy | 3 (30%) |
| Dilated cardiomyopathy | 4 (40%) | |
| Post‐myocarditic cardiomyopathy | 1 (10%) | |
| Retransplant | 1 (10%) | |
| Postpartum cardiomyopathy | 1 (10%) | |
| Immunosuppressive regimen a | Induction ATG | |
| Maintenance | ||
| Csa + MMF + prednisone | ||
| Csa + azathioprine + prednisone | ||
| Tacrolimus + MMF + prednison | ||
|
Acute graft rejection episodes b , n (%) |
8 (80%) | |
| Mortality, n (%) | Squamous cell carcinoma, 1 (10%) | |
| Donor | Sex (M/F) | 6/4 |
| Age, year | 41.4 ± 15.67 | |
| Ischemia, min | 164.2 ± 72.51 | |
Abbreviations: ATG, thymoglobulin; Csa, cyclosporine; CVA, cerebrovascular accident; MMF, mycophenolate.
Data are expressed as mean ± SD.
Immunosuppressive regimen was subsequently tuned according to patient conditions.
bRequiring intravenous steroid treatment.
Human studies were approved by the appropriate institutional review board.
2.2. Mice
C57BL/6 J, B6.C‐H2bm12 (bm12), and B6.129P2‐Lyz2tm1(cre)Ifo/J (LysMcre, stock number 004781) mice were obtained from the Jackson Laboratory. miR‐21 fl/fl mice were kindly provided by Eric Olson (UT Southwestern). miR‐21 fl/flLysMCre C57BL/6 mice were generated as follows: miR‐21 fl/fl mice were bred with LysMCre mice in order to obtain miR‐21 fl/flLysMCre C57BL/6 mice, in which miR‐21 is flanked by loxP sites and is selectively removed by Cre recombinase expression in LysM‐expressing cells, allowing for specific deletion of miR‐21 in macrophages. All mice were used and cared for in accordance with institutional guidelines, and animal protocols were approved by the Boston Children's Hospital Institutional Animal Care and Use Committee.
2.3. Cardiac transplantation
Vascularized cardiac allografts were transplanted intra‐abdominally using microsurgical techniques as described by Corry et al. 27 Rejection was determined as complete cessation of cardiac contractility and was confirmed by direct visualization.
2.4. miR‐21 targeting in vivo
Adult (8–12 weeks) C57BL/6 J mice were transplanted with sex‐matched bm12 hearts and received miR‐21‐specific antagomir or nonspecific negative control oligonucleotide (Regulus and Exiqon) (80 mg/kg) in phosphate‐buffered saline (PBS) by intravenous injection. The miR‐21 antagomir is a high‐affinity oligonucleotide complementary to the active site of miR‐21, with a modified backbone. 15 , 22 Mice were injected twice per week at Weeks 1 and 2 and once per week at Weeks 3 and 4 after cardiac surgery.
2.5. miR‐21 targeting in vitro
Bone marrow‐derived macrophages (BMDMs) were treated in vitro with 100 nM of miR‐21 antagomir (Exiqon) for 24 h according to the manufacturer's instructions. Cells and supernatant were collected for analysis.
2.6. Statistics
Data are expressed as mean ± standard error mean (SEM) from at least three independent experiments. Kaplan–Meier curves were used for analysis of survival followed by Log‐rank (Mantel–Cox) test. Statistical analysis, other than for RNA‐seq results, was performed using Student's t test with Welch's correction (if applicable). For multiple comparisons, one‐way ANOVA followed by Tukey multiple comparisons analysis between the group of interest and all other groups was used. Volcano plot and differential expression analysis of miRNAs were generated using the edgeR Bioconductor package in R. Differences between the experimental groups were considered statistically significant (*) or highly significant (**) (***), when the p value was <.05, <.01, or <.001, respectively. Graphs were generated using GraphPad Prism software version 6.0 (GraphPad).
2.7. Study approval
Human and animal studies were approved by the appropriate institutional review board(s) of Niguarda Hospital (Milan, Italy) (80–032013) and Boston Children's Hospital, Harvard Medical School (Boston, MA) (16–04‐3127R). Written informed consent was received from participants prior to inclusion in the study.
3. RESULTS
3.1. miR‐21 is upregulated in murine heart allografts during CAV
To determine miRNAs potentially involved in CAV, we first profiled the miRNome in the well‐characterized minor MHC Class II mismatch murine model of cardiac transplantation (Figure 1A,B). In this model, bm12 hearts are transplanted into C57BL/6 recipients (bm12 into C57BL/6), and cardiac allografts typically survive for approximately 50–70 days without immunosuppression. 28 , 29 Bm12, cardiac allografts are not acutely rejected by C57BL/6 mice but show instead minimal cellular infiltration starting from 1 week after the transplant procedure. 30 By Day 40 after the transplant, allografts develop features of chronic rejection with vasculopathy, interstitial inflammation, and myocardial fibrosis, with a scattered distribution of CD4 or CD8 T cells infiltrating the graft and with perivascular clustering of macrophages resembling the features of human CAV. 3 , 31 , 32 , 33 To identify dysregulated miRNAs expressed in murine transplanted hearts and involved in CAV, we chose an early time‐point when preclinical signs began to appear (4 weeks) and a later time‐point (8 weeks) when the phenotype becomes more severe and was concomitant with the high infiltration of macrophages rather than of T cells or of B cells (Figure S1A–J). Following miRNA microarray analysis, we identified 192 miRNAs that were upregulated or downregulated in murine transplanted heart allografts with CAV compared with syngeneic transplants (bm12 into bm12), which were used as controls (Figure 1A,B). Those with a p value <.05 and mean >6 were considered upregulated and those with a p value <.05 and a mean <6 were considered downregulated. Among these miRNAs, miR‐21 was both the most highly expressed and the most robustly upregulated in allogeneic transplanted hearts as compared with syngeneic transplants, displaying a 20‐fold increase at 8 weeks (Figure 1B). A common set of 17 miRNAs was found to be upregulated at both 4 and 8 weeks post‐transplant (Figure 1C,D). miR‐21 upregulation was confirmed by real‐time quantitative reverse transcription polymerase chain reaction (PCR) (qRT‐PCR) and digital droplet PCR (ddPCR) (Figure 1E,F), thus validating our array profiling data. In situ hybridization demonstrated that the miR‐21 hybridization signal was greatly enhanced in allogeneic transplanted hearts as compared with syngeneic transplanted hearts (Figure 1G,H1–H4,I1–I4). Noteworthy, our data confirmed that miR‐21 was the most highly upregulated miRNA in murine transplanted heart allografts with CAV as compared with controls.
FIGURE 1.
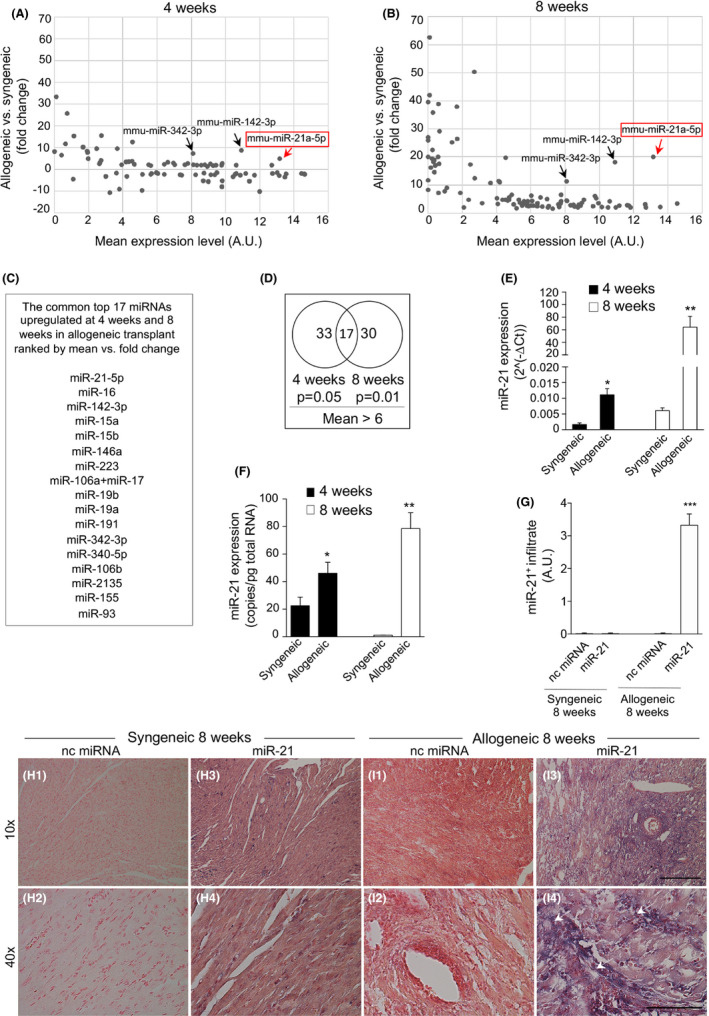
miR‐21 is upregulated in murine heart allografts during chronic allograft vasculopathy (CAV). (A,B) Differentially expressed miRNAs detected using miRNA microarray analysis at 4 weeks (A) following murine cardiac allogeneic transplant compared with syngeneic transplant and at 8 weeks (B) following cardiac allogeneic transplant compared with syngeneic transplant. A common set of 17 miRNAs was upregulated at both 4 and 8 weeks post‐allogeneic transplant; the list is shown in (C), ranked by mean versus fold change. (D) Venn diagram showing the set of upregulated miRNAs at 4 and 8 weeks after murine cardiac allogeneic transplant compared with syngeneic transplant. For statistical analysis, R was used to detect miRNAs with different expression levels between study groups (n = 4 samples per group). The resulting p values <.05 were considered statistically significant. (E,F) miR‐21 expression by qRT‐PCR (E) and ddPCR (F) in cardiac grafts after syngeneic and allogeneic transplant at 4 and 8 weeks post‐transplant; statistical significance was determined using Student's t test with Welch's correction (*p < .05; **p < .01; at least n = 3 per group). (G) Quantification of miR‐21 in (H1–H4, I1–I4) is shown (at least n = 3 samples per group). (H1–H4, I1–I4) Representative images of in situ hybridization (ISH) of murine cardiac sections from syngeneic (H1–H4) and allogeneic (I1–I4) transplant for miR‐21 at 8 weeks post‐transplant (n = 3 per group). Staining is shown in purple, and white arrows indicate the presence of miR‐21. Original magnification: ×10 in (H1, H3, I1, I3), scale bar 300 µm; ×40 in (H2, H4, I2, I4), scale bar 100 µm. ddPCR, digital droplet polymerase chain reaction; miRNAs, micro‐RNA; nc miRNA, negative control miRNA antagomir; qRT‐PCR, real‐time quantitative reverse transcription polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.2. miR‐21 is upregulated in human heart allografts during CAV
We next obtained human transplanted heart samples from cardiac‐transplanted patients, with and without features of CAV (Table 1 and Table S1). miRNome profiling of human heart biopsies revealed 666 miRNAs of interest after filtering out nondetectable miRNAs (miRNAs having an expression level of >1 counts per million [CPM] in at least two samples were considered detectable). miR‐21 again showed the highest expression and most robust upregulation in heart samples obtained from cardiac‐transplanted patients with CAV compared with samples obtained from patients without CAV (Figure 2A). In addition to miR‐21, we found 16 other miRNAs potentially important for cardiac‐transplanted patients that were upregulated in CAV (Figure 2B) but to a lesser degree than miR‐21. miR‐21 upregulation in CAV subjects was confirmed by qRT‐PCR and ddPCR (Figure 2C,D). In situ hybridization showed some upregulation of miR‐21 within heart samples of cardiac‐transplanted patients with CAV as compared with samples from patients without CAV (Figure 2E1–E4,F1–F4,G). The presence of a robust miR‐21 upregulation in our samples was particularly associated with the presence of heart allograft‐infiltrating macrophages Mac‐2+ cells as shown in Figure 2H1–H2 and I. Altogether, our data demonstrated that miR‐21 was the highly upregulated miRNA in heart allograft samples from cardiac‐transplanted patients with CAV as compared to those without CAV.
FIGURE 2.
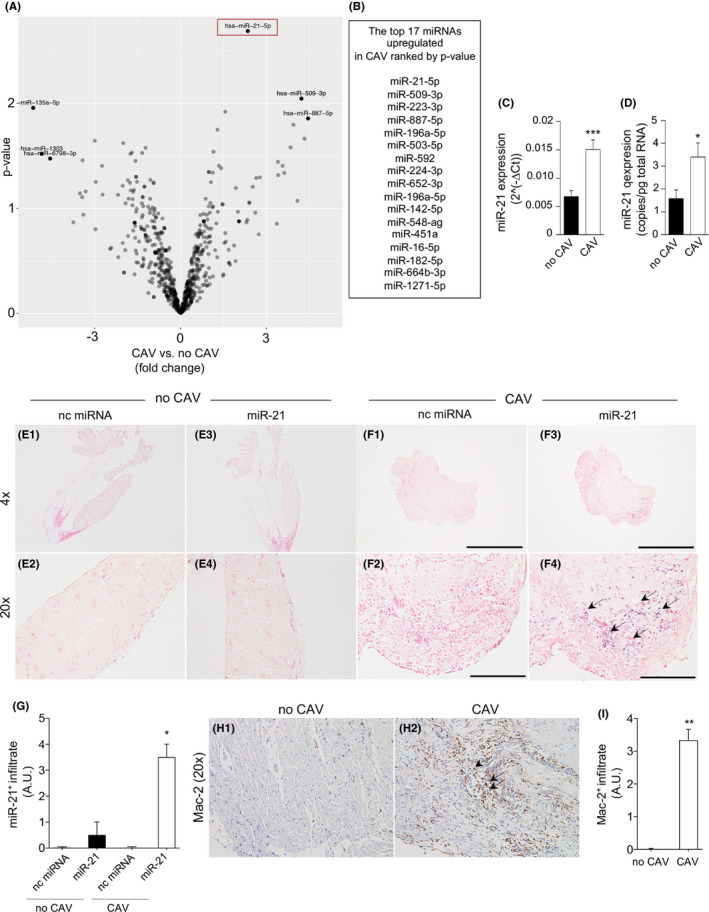
miR‐21 is upregulated in human heart allografts during chronic allograft vasculopathy (CAV). (A) Volcano plot showing significantly upregulated and downregulated miRNAs in cardiac transplants of CAV patients relative to cardiac transplants of patients without CAV (log10 p value on the y‐axis and log2 fold change chronic allograft vasculopathy (CAV) versus no CAV on the x‐axis) (n = 3 samples per group). (B) The top 17 miRNAs upregulated in CAV ranked by p value; R was used to draw volcano plots and for comparison of miRNA expression analysis. (C,D) miR‐21 expression by qRT‐PCR (C) and ddPCR (D) in human cardiac graft biopsies from cardiac‐transplanted patients with CAV or without CAV (at least n = 6 per group), and statistical significance was determined using Student's t test with Welch's correction (*p < .05; **p < .01; ***p < .001). (E1–E4, F1–F4) Representative examples of in situ hybridization (ISH) of cardiac sections for miR‐21 from human cardiac transplants of patients with CAV or without CAV (n = 3 per group). Original magnification: ×10 in (E1, E3, F1, F3), scale bar 300 µm; ×40 in (E2, E4, F2, F4), scale bar 100 µm. (G) Quantification of miR‐21 in (E1–E4, F1–F4) is shown (at least n = 3 samples per group); staining is shown in purple, and black arrows indicate the presence of miR‐21. (H1, H2) Representative examples of immunohistochemistry of cardiac sections for Mac‐2 from human cardiac transplants of patients with CAV or without CAV (n = 3 per group). Staining is shown in brown, and black arrows indicate the presence of Mac‐2. (I) Quantification of Mac‐2 in (H1, H2) is shown (at least n = 3 samples per group). CAV, cardiac allograft vasculopathy; ddPCR, digital droplet polymerase chain reaction; miRNAs, micro‐RNA; nc miRNA, negative control miRNA antagomir; qRT‐PCR, real‐time quantitative reverse transcription polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.3. miR‐21‐expressing macrophages play a role in CAV onset
In order to understand which cells were responsible for miR‐21 upregulation during CAV, we injected Cy3‐labeled anti‐miR‐21 oligonucleotide into C57BL/6 mice transplanted with bm12 hearts. Mice were injected after 8 weeks post‐transplantation on Day 55, and heart allografts were examined after 24 h on Day 56, allowing for examination of miR‐21 localization within heart allografts. Interestingly, miR‐21 was detected in graft‐infiltrating macrophages (Mac‐2+), but not in B (B220+) or T cells (CD3+) (Figure 3A1–A6,B), while myocytes appeared completely negative for miR‐21 (data not shown). We further quantified the percentage of miR‐21+ macrophages within the total population of macrophages and miR‐21+ macrophages appeared to be approximately the 40% of the total macrophages (Figure 3C). qRT‐PCR analysis of splenocytes obtained from naïve C57BL/6 mice confirmed that in F4/80+ cells, the expression of miR‐21 was significantly higher in those cells as compared with CD11c+, CD3+ and CD19+ cells (Figure S1K). To further determine whether miR‐21 upregulation in macrophages is relevant during CAV onset, we used a murine model of CAV (C57BL/6 mice transplanted with bm12 hearts), in which we first treated heart‐transplanted mice with clodronate in order to deplete the circulating macrophage pool. Clodronate, which has been extensively used previously to deplete macrophages, 34 , 35 was injected every 5 days beginning at Day 1 following heart transplantation; another group of mice were left untreated and used as controls (Figure 3D). The depletion of macrophages from peripheral blood was confirmed by flow cytometry as revealed by a decrease in CD115+ monocytes cells (Figure 3E,F) and in heart‐allograft by a reduced infiltration of macrophages as shown by the scarce immunohistochemical staining of Mac‐2 (Figure 3G1–G3) and by a decrease in F4/80 expression as revealed by qRT‐PCR (Figure 3H). A nonsignificant delay in CAV onset was observed in clodronate‐treated mice as compared with controls (Figure 3D). Interestingly, macrophage depletion following clodronate treatment was shown to correlate miR‐21 expression in heart‐allografts as assessed by qRT‐PCR (Figure 3I) and by in situ hybridization (Figure 3J1–J6,K). Due to the fact that clodronate removes all macrophages regardless of miR‐21 expression, we then decided to conditionally delete miR‐21 in macrophages by using the miR‐21 fl/flLysMCre C57BL/6 mouse. In this mouse, the miR‐21 is flanked by loxP sites and is selectively removed by Cre recombinase expression in LysM‐expressing cells (under the control of the LysM promoter, present in macrophages and some myeloid cells), such that miR‐21 deletion occurs specifically in macrophages. miR‐21 fl/flLysMCre C57BL/6 mice receiving bm12 hearts showed a prolonged and indefinite cardiac allograft survival as compared to control cardiac‐transplanted littermate mice (Figure 3D). miR‐21 fl/flLysMCre cardiac‐transplanted mice displayed a reduction in miR‐21 expression in heart allografts as analyzed by qRT‐PCR (Figure 3I) and in situ hybridization (Figure 3J7–J8,K) as compared with controls (Figure 3J3–J4,K). In order to understand if the observed beneficial effect on allograft survival was associated with any altered survival of the macrophages, we quantified the percentage of apoptotic macrophages within the graft by using caspase 3 staining and we confirmed the absence of any differences (Figure S2A). Lastly, as an aberrant proliferation of activated vascular smooth muscle cells (VSMCs) has been associated to CAV, we thus sought to investigate if any effect by miR‐21 targeting was evident on VSMCs. Importantly, VSMCs within the cardiac allograft from miR‐21 LysMCre mice kept their quiescent no‐proliferating status, as revealed by a negative PCNA staining in the smooth muscle cells of the tonaca media (Figure S2B,C). Altogether, these data suggest that specific depletion of miR‐21+ macrophages rather than complete macrophage depletion might have a beneficial effect over prolongation of heart allograft survival in a murine model of CAV.
FIGURE 3.
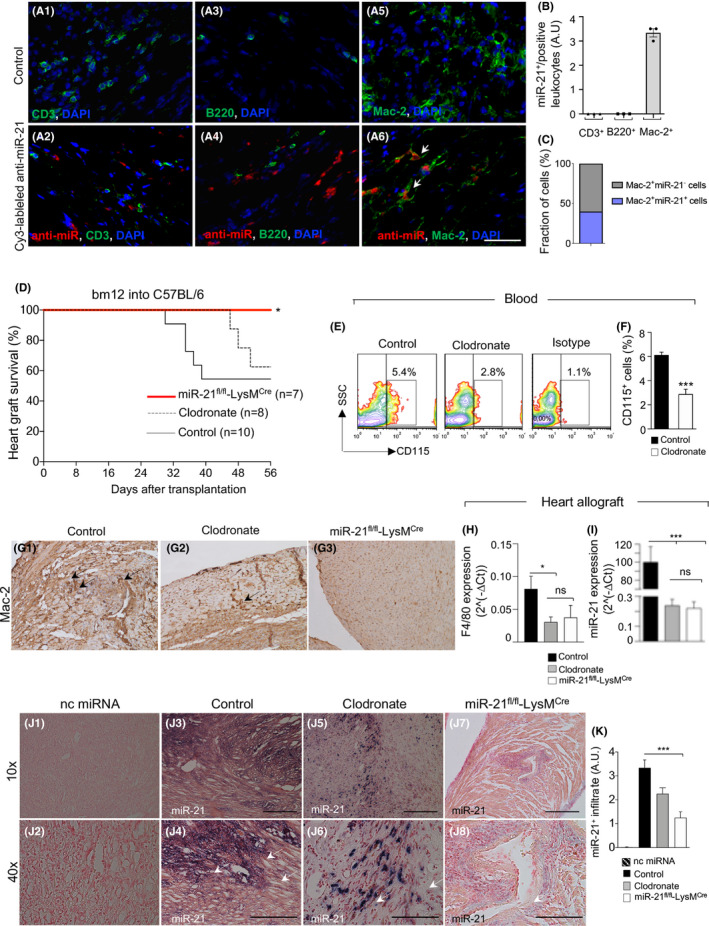
miR‐21‐expressing macrophages play a role in chronic allograft vasculopathy (CAV) onset. (A1–A6) Representative immunofluorescence images of the graft (hearts transplanted from bm12 into B6) in control mice (A1–A3–A5) and in mice treated with in vivo injection of Cy3‐labeled anti‐miR‐21 oligonucleotide (A2–A4–A6) demonstrate that in mouse heart allografts, miR‐21 colocalizes with macrophages (Mac‐2+, shown in A6) but not with B (B220+, A4) or T cells (CD3+, A2); blue: nucleus; green: Mac‐2+/B220+/CD3+; red: Cy3‐labeled anti‐miR‐21 oligonucleotide. Original magnification: ×63 in (A1–A6), scale bar 40 µm. (B) Quantification of CD3+/miR‐21+ cells, of B220+/miR‐21+ cells, and of Mac‐2+/miR‐21+ cells confirmed the exclusive expression/presence of miR‐21 by macrophages; data are representative of n = 3 sections analysis per cell type and one‐way ANOVA with Tukey's multiple comparison test was used for determining statistical significance; ns; not significant; *p < .05; **p < .01; ***p < .001. (C) Fraction of Mac‐2+ cells that are positive for miR‐21. (D) Effect of macrophage depletion in vivo on allograft survival. Selective depletion of macrophages by treatment of cardiac‐transplanted recipient mice with clodronate liposomes. Treatment promotes long‐term allograft survival in 65% of treated mice; p = ns by log‐rank (Mantel–Cox) test (n = 10 in CTRL vs. n = 8 in clodronate‐treated). Conditional deletion of miR‐21 in macrophages using miR‐21 fl/flLysMCre transplanted with cardiac bm12 allografts resulted in long‐term allograft survival in 100% of recipient mice (*p < .05 by log‐rank [Mantel–Cox] test, miR‐21 fl/flLysMCre [n =7] vs. control [n =10]). (E,F) Efficiency of CD115+ cell depletion in blood samples after clodronate treatment, as assessed by FACS (E, representative samples shown); statistical significance in F was performed by Student's t test with Welch's correction (***p < .001). (G1–G3) Mac‐2 immunohistochemical staining of heart allograft sections retrieved from C57BL/6 mice after allogeneic transplant (CTRL), in transplanted C57BL/6 mice treated with clodronate, and in miR‐21 fl/flLysMcre transplant recipient mice. (H) Decreased F4/80 and miR‐21 expression (I), analyzed by qRT‐PCR and (J1–J8,K) in situ hybridization, in hearts of C57BL/6 mice after allogeneic transplant, in transplanted C57BL/6 mice treated with clodronate, and in miR‐21 fl/flLysMcre transplant recipient mice; quantification of miR‐21 in the graft from different transplanted groups is shown in (K). For group analysis comparison in H and I, one‐way ANOVA with Tukey's multiple comparison test was used (at least n = 3 per group); ns; not significant; *p < .05; **p < .01; ***p < .001. Original magnification: ×20 in (G1–G3); ×10 in (J1, J3, J5, J7), scale bar 300 µm; ×40 in (J2, J4, J6, J8), scale bar 100 µm. ddPCR, digital droplet polymerase chain reaction; FACS, flow cytometric analysis; miRNAs, micro‐RNA; nc miRNA, negative control miRNA antagomir; qRT‐PCR, real‐time quantitative reverse transcription polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.4. miR‐21 antagonism abrogates CAV
In order to establish a proof of concept for the potential role and effect of miR‐21 targeting in CAV, we tested in vivo the efficacy of a miR‐21 specific antagomir. The miR‐21‐specific antagomir is a nontoxic oligonucleotide complementary to miR‐21, with a chemically modified backbone as described in Section 2. While untreated C57BL/6 mice transplanted with bm12 hearts rejected heart allografts between 4 and 8 weeks post‐transplant (Figure 4A), mice treated with miR‐21 antagomir displayed long‐term heart allograft survival (>95 days) in 100% of recipients (Figure 4A). To further characterize the effect of in vivo miR‐21 targeting, we analyzed the infiltrate of cardiac transplants isolated from miR‐21 antagomir‐treated and in control mice. Pathological examination of allografts obtained at 8 weeks post‐transplantation (Figure 4B–J) revealed that miR‐21 antagomir‐treated mice showed reduced mononuclear cell infiltration, particularly with respect to Mac‐2+ macrophages (Figure 4E4,F4,G), in the graft infiltrate as compared to controls (Figure 4E3,F3,G). Reduced vasculopathy in the cardiac perivascular space was evident by hematoxylin and eosin staining respectively in grafted heart tissue obtained from miR‐21 antagomir‐treated mice (Figure 4E2,F2) as compared with controls (Figure 4E1,F1). Significant reduction in the number of CD3+ cells was observed when comparing miR‐21 antagomir‐treated (Figure 4E6,F6,H) to control mice (Figure 4E5,F5,H). Quantitative analysis confirmed a reduction in degree of cell infiltration (Figure 4B), vasculopathy (Figure 4C), and luminal stenosis (Figure 4D) in miR‐21 antagomir‐treated mice as compared with controls. Moreover, we observed a reduction in the degree of fibrosis as evaluated with Masson's trichrome staining in miR‐21 antagomir‐treated mice (Figure 4I5,I6,J) compared with controls (Figure 4I2,I3,J). We then evaluated the immune profile of miR‐21 antagomir‐treated mice by flow cytometry and gene expression analysis by qRT‐PCR (Figure S3). In miR‐21 antagomir‐treated mice, immunophenotyping the percentage of macrophages in the allograft and in the spleen of miR‐21 antagomir‐treated mice and controls was similar (Figure S3A,K). Moreover, no differences were detected in the M1 phenotype markers CD80, CD86 (Figure S3B,C,L,M) or in the M2 phenotype marker CD206 (Figure S3D,N) in the allograft and in the spleen of miR‐21‐specific antagomir‐treated as compared to controls. We found high levels of Arg1 mRNA and of Nos2 mRNA in the spleen, but not in the allograft of miR‐21 antagomir treated mice as compared with controls (Figure S3E,F,O,P). Interestingly, in miR‐21 antagomir‐treated mice, T cells expressed higher levels of GATA‐3 in both allograft (Figure S3G) and spleen (Figure S3Q) as compared with controls. Moreover, in miR‐21 antagomir‐treated mice, T cells obtained from allograft (Figure S3H,I), but not spleen (Figure S3R,S), expressed higher levels of T‐bet and RORc, with no differences in the overall percentage of CD4+ T cells (Figure S2J,T). To further assess whether the treatment with miR‐21 antagomir have any effect on T cells, we evaluated the percentage of IFN‐γ+CD4+ and IFN‐γ+CD8+ T cells generated during an anti‐CD3/28 stimulation of murine splenocytes in the presence/absence of miR‐21 antagomir. Our data showed no significant difference in the percentage of the different T cell subsets during miR‐21 targeting (Figure S4A–D). On the contrary, there was a slight significant difference when comparing CD4 and CD8 proliferating T cells exposed or not to miR‐21 antagomir (Figure S4E,F). miR‐21 antagonism promotes allograft survival thereby reducing cell infiltration, vasculopathy and fibrosis.
FIGURE 4.
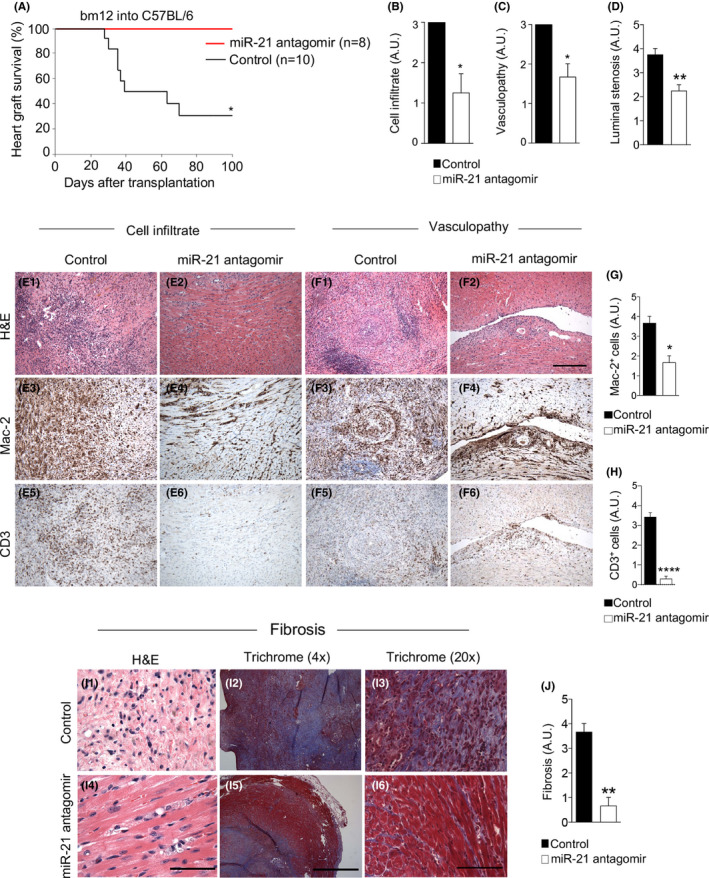
miR‐21 antagonism abrogates chronic allograft vasculopathy (CAV). (A) miR‐21 antagomir treatment, in the bm12 into C57BL/6 cardiac transplantation model, prevented cardiac transplant rejection in 100% of recipients; statistical significance was performed by log‐rank (Mantel–Cox) test; (*p < .05 miR‐21 antagomir [n =8] vs. control [n =10]). (B–H) Representative histological analysis of cardiac transplants at 8 weeks after transplantation revealed fewer numbers of macrophages, Mac‐2+, in the graft infiltrate and in the perivascular space in mice treated with miR‐21 antagomir (E4, F4, and G) compared with control mice (E3, F3), whereas significant differences were observed in CD3+ cells between treated (E6, F6, and H) and control mice (E5, F5, and H). Hematoxylin‐eosin staining showed less mononuclear cell infiltrate (E2, F2 in treated Vs E1, F1 in CTRL, shown in violet) and vasculopathy (F2 in treated Vs F1 in CTRL) and with Masson's trichrome, fewer collagen fibers (I5, I6 in treated Vs I2, I3 in CTRL, shown in blue staining) in miR‐21 antagomir‐treated mice as compared with control. (J) Quantification of fibrosis in CTRL versus anti‐mi‐R21 treated mice. Original magnification: ×10 in (E1–E6, F1–F6), scale bar 300 µm; ×40 in (I1, I4), scale bar 100 µm. Four times in (I2, I5), scale bar 1 mm; ×20 in (I3, I6), scale bar 200 µm. Quantification of (B) cell graft infiltrate and (C) coronary vasculopathy score and (D) luminal stenosis (at least n = 3 samples per group) confirmed the protection conferred by miR‐21 antagomir treatment; statistical significance was determined by Mann–Whitney test (*p < .05). Data are representative of at least n = 3 samples per group; statistical significance was determined by Student's t test with Welch's correction (if applicable); (*p < .05). miRNAs, micro‐RNA; qRT‐PCR, real‐time quantitative reverse transcription polymerase chain reaction; FACS, flow cytometric analysis; Arg1, Arginase 1 [Color figure can be viewed at wileyonlinelibrary.com]
3.5. miR‐21 antagonism reshapes macrophages profiles in vitro
We next expanded our investigation to further understand the effect of miR‐21 antagomir on the macrophage immune profile by using a BMDM‐based in vitro assay. Uncommitted BMDMs M0 were polarized into M1, using IFN‐γ, or to M2, using IL‐4, IL‐10 and IL‐13 (Figure S5A). M1 or M2 differentiation was confirmed by the presence of M1‐specific markers (CD80+ and CD86+) or M2‐specific markers (CD206+) (Figure S5B). We then challenged BMDMs with the miR‐21 antagomir, which reduced miR‐21 expression in M0, M1, and M2 macrophages (Figure 5A–C). miR‐21 antagonism modified the macrophage immune profile as shown by an increase in Arg1 gene expression, while the levels of NOS2 remained unaltered (Figure 5A–C). No differences were observed in expression of CD80, CD86, or CD206 in M0, M1, and M2 macrophages following challenge with the miR‐21 antagomir (Figure 5D–F). Next, we analyzed the secretome profile of macrophages following treatment with the miR‐21 antagomir, which did not reveal any significant differences in levels of IL‐1β and IL‐6 (Figure 5G–I). A slight increase in TNF‐α secretion was observed in M2 macrophages following miR‐21 antagonism (Figure 5I). Levels of TGF‐β, MIG, and IL‐12 decreased in M0 macrophages (Figure 5G) but not in M1 and M2 macrophages (Figure 5H,I). Finally, the use of miR‐21 antagomir was found to exert no significant effects on macrophage phagocytosis, migratory ability, or antigen‐presenting properties (Figure 5J–M). We also assessed the effect of miR‐21 targeting on M0‐, M1‐, and M2‐derived macrophages; indeed, the percentage of apoptotic macrophages was not affected by miR‐21 antagomir, while, a slight reduction in the percentage of necrosis was observed in M2‐derived macrophages (Figure S4G–K). Taken together, our results indicate that miR‐21 antagonism only slightly modulates the immune profile of in vitro generated macrophages without affecting their main functional properties (phagocytosis, migration, and antigen presentation).
FIGURE 5.
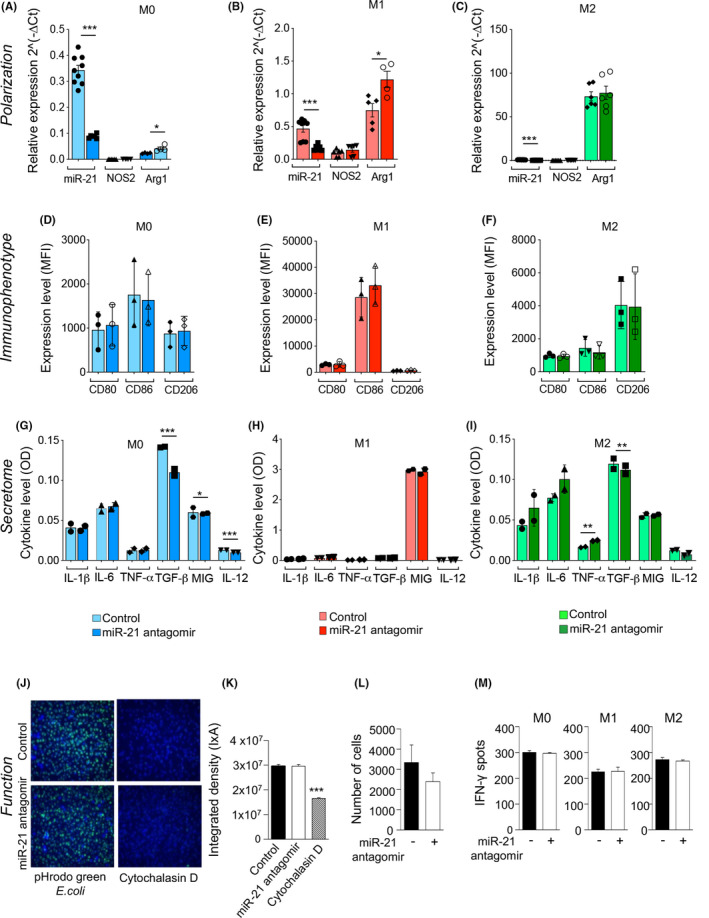
miR‐21 antagonism reshapes macrophages profiles in vitro. (A–C) In vitro challenge with miR‐21 antagomir skewed bone marrow‐derived macrophages (BMDMs) to express Arg1. M0, M1, and M2 macrophages were analyzed by (A–C) qRT‐PCR for miR‐21 and Arg1 (U6snRNA and HPRT were used as housekeeping genes), (D–F) by FACS analysis for CD80, CD86, CD206 markers gated on CD11b+F4/80+ cells (at least n = 3 samples per group) and (G–I) by ELISA for cytokine expression levels. (J–M) miR‐21 antagomir has no effect on phagocytosis (J–K), migration (L), or antigen‐presenting cell capacity analyzed with Elispot (M) of macrophages. Data are expressed as mean ± standard error of the mean (SEM). Statistical significance was performed by Student's t test with Welch's correction (if applicable) (*p < .05; **p < .01; ***p < .001). ELISA, enzyme‐linked immunosorbent assay; FACS, flow cytometric analysis; miRNAs, micro‐RNA; qRT‐PCR, real‐time quantitative reverse transcription polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
3.6. miR‐21 antagonism reprograms macrophage metabolism
We then performed RNA‐seq profiling of BMDMs after in vitro challenge with the miR‐21 antagomir. Our data revealed that 230 genes were altered by miR‐21 antagonism (Figure 6A and S6A), with 116 upregulated genes and 114 downregulated genes (Figure S6B,C). As shown in Figure 6B, among those genes that were upregulated, many are known to be involved in determining or accompanying the M2 phenotype (Figure 6B and S6B,D), or correlated with anti‐inflammatory pathways, while others are involved in fatty acid oxidation and primary metabolic pathways (Figure 6B and S6B). Consistent with these data, within the downregulated genes were found those linked with M1 signature and IFN‐γ and other inflammatory pathways (Figure 6C and S6C). Finally, using ingenuity pathway analysis in an attempt to connect those upregulated/downregulated genes following miR‐21 antagonism revealed a rewiring of the transcriptional regulatory network of macrophages to adopt an anti‐inflammatory phenotype consistent with the acquisition of some anti‐inflammatory signatures (Figure S7). We next more extensively characterized the macrophage metabolic profile upon challenge with a miR‐21 antagomir by evaluating cellular metabolic function (Figure 6D–Q). Our data demonstrated an increase in oxygen consumption rate (OCR), an indicator of an optimal mitochondrial function, in M0 macrophages following treatment with the miR‐21 antagomir (Figure 6D,E,H). Conversely, a reduction in OCR was observed in M1 macrophages after challenge with the miR‐21 antagomir as compared with baseline conditions (Figure 6D,E,I), while in M2 macrophages, OCR was unchanged (Figure 6D,E,J). Given the changes in Arginase expression, we previously observed with miR‐21 antagonism, we evaluated the effect of reconstituting l‐arginine during challenge with the miR‐21 antagomir. l‐Arginine added during treatment with the miR‐21 antagomir significantly increased OCR in M1 macrophages (Figure 6F,I), decreased OCR in M2 macrophages (Figure 6F,J), and had no effect on M0 macrophages (Figure 6F,H). Interestingly, the use of the miR‐21 antagomir robustly reduced the extracellular acidification rate (ECAR), an indicator of glycolysis, in M1 macrophages, while L‐Arginine was shown to abrogate this effect (Figure 6K,L,P). In M0 macrophages, the use of the miR‐21 antagomir enhanced ECAR, while L‐Arginine was poorly effective (Figure 6K–O). Finally, in M2 macrophages a marked increase in ECAR was evident with miR‐21 antagomir treatment and it was slightly decreased by the addition of L‐arginine (Figure 6K–N,Q). Interestingly, a decrease in glycolysis, maximum glycolytic capacity and glycolytic reserve were evident in M1 macrophages in the presence of the miR‐21 antagomir, while l‐arginine abrogated its effect (Figure S8B,E,H). An increase in glycolysis, maximum glycolytic capacity and glycolytic reserve was observed in M0 and M2 macrophages in the presence of the miR‐21 antagomir (Figure S8A,C,D,F,G,I), and the addition of l‐arginine following miR‐21 antagomir treatment contributed to these effects (Figure S8A,C,D,F,G,I). Collectively, these data suggest that miR‐21 antagonism modulates the metabolic profile of in vitro generated macrophages, with altered wide modifications of the catabolic metabolism in pro‐inflammatory M1 macrophages in an arginine‐dependent manner.
FIGURE 6.
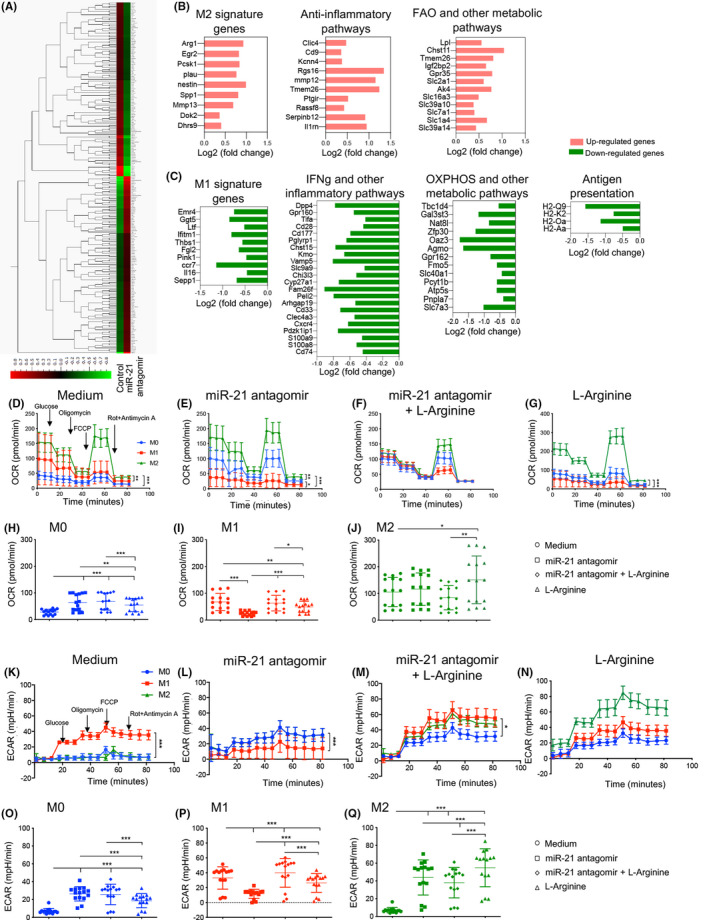
miR‐21 antagonism reprograms macrophage metabolism. (A) Heat map generated using Qlucore Omics Explorer representing the gene profile of bone marrow derived‐macrophages (BMDM) in vitro challenged with miR‐21 antagomir as compared with control (n = 3 samples per group). The complete dataset of identified and quantified genes was subjected to statistical analysis determined with Cuffdiff (v2.2.1) (p < .05). (B,C) Significantly and differentially expressed genes were further analyzed linking the upregulated genes (B) and downregulated genes (C) (determined by p < .05) with the major related pathways. Control (n = 3) and miR‐21 antagomir‐treated (n = 3) bone marrow‐derived macrophages (BMDMs) were analyzed. (D–Q) BMDM‐M0 macrophages were treated with IFN‐γ, or with IL‐4, IL‐10, IL‐13 or were left untreated; the polarization process was subsequently followed by treatment with miR‐21 antagomir or a combination of miR‐21 antagomir + l‐arginine. (D–J) Oxygen consumption rate (OCR) values were determined in real time using a Seahorse XF96 Analyzer in M0‐, M1‐, and M2‐derived macrophages, as described in Section 2, in which cells were subjected to oligomycin allowing inhibition of ATP synthase, then to the uncoupler FCCP, allowing for maximal respiration and OCR production. Treatment with rotenone/antimycin A was then performed, which blocks mitochondrial Complexes I and III and contributes to nonmitochondrial OCR. (K–Q) The extracellular acidification rate (ECAR) response of M0, M1, and M2 macrophages was obtained using a Seahorse XF96 Analyzer upon glucose injection, followed by treatment with oligomycin and rotenone/antimycin A. All measurements were performed at baseline conditions (medium) or after addition of miR‐21 antagomir or after challenge with a combination of miR‐21 antagomir + l‐arginine or with l‐arginine. Data are representative of at least n = 4 wells per condition; statistical analysis was performed using one‐way ANOVA with Bonferroni correction. BMDM, bone marrow‐derived macrophages; ECAR, extracellular acidification rate; FCCP, carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone; OCR, oxygen consumption rate [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
Cardiac allograft vasculopathy (CAV) is a hallmark of cardiac chronic rejection and constitutes the primary limitation in long‐term survival of allografts. 36 , 37 It is therefore critical to develop novel strategies to achieve stable graft acceptance and to reduce the incidence of CAV. 38 , 39 , 40 In order to discover pathways that may be involved in CAV onset and identify new therapeutic targets, we first performed miRNomic profiling of murine and human transplanted hearts, which demonstrated that miR‐21 is the most highly expressed miRNA in transplanted hearts with allograft vasculopathy. Most, if not all, of the miR‐21 expressed within the transplanted heart was found to be localized into the cytoplasm of cardiac‐infiltrating inflammatory cells, which are predominantly macrophages. Several studies have identified miR‐21 as a key controller of immune regulation, 41 , 42 and so we sought to delineate the role of miR‐21‐expressing macrophages in the onset of CAV. We first depleted circulating macrophages with clodronate and then conditionally deleted miR‐21 in macrophages. Only conditional deletion of miR‐21 in macrophages, but not complete macrophage depletion, was successful in prolonging cardiac allograft survival. Indefinite graft survival was also observed using a miR‐21‐specific antagomir. While clodronate‐mediated macrophage depletion is nonspecific and macrophages are depleted independently of their activation state and function, specific genetic deletion of miR‐21 as well as the use of a miR‐21‐specific antagomir specifically targets miR‐21‐expressing macrophages, thus eventually involved in chronic alloimmune processes. This might be explained by the fact that total macrophage deletion may also target and eliminate regulatory macrophages, while miR‐21 genetic or pharmacologic targeting on macrophages, may have instead targeted pro‐inflammatory macrophages only, thus contributing to the observed beneficial effect on allograft survival. We then showed that the use of a miR‐21 antagomir reshapes macrophage phenotype, as demonstrated by phenotypic and RNA‐seq profiling as shown by the increased expression of Arginase‐1 activity in addition to a number of other genes, which indicate that these macrophages may exhibit increased anti‐inflammatory properties. 43 , 44 , 45 , 46 Interestingly our in vitro studies confirm a wide reshaping of macrophages metabolism when miR‐21 is antagonized, with an increase of macrophages glycolytic activity following miR‐21 antagonism. The M1 macrophage bioenergetic profile was completely altered by the use of a miR‐21 antagomir, with downregulation of OXPHOS activity and glycolytic capacity. Interestingly, the addition of l‐arginine during challenge with the miR‐21 antagomir reinstated the M1 metabolic profile to baseline status, thus confirming that the miR‐21 antagomir effect is dependent upon the l‐arginine metabolic pathway. M0 macrophages adopted a more M2‐like bioenergetic profile following challenge with a miR‐21 antagomir, as demonstrated by an upregulation of their related OXPHOS and glycometabolic activities, with no effects upon the addition of l‐arginine. Altogether, miR‐21 antagonism skews M0 macrophages such that they adopt an M2‐like bioenergetic profile, as well as abrogates M1‐related metabolic activity, thus favoring the establishment of an anti‐inflammatory environment. Indeed, our data demonstrated a shift in the macrophages metabolic profile upon miR‐21 targeting, which appeared to be in line with our recent paper in which conditional deletion of miR‐21 in macrophages reprogrammed macrophages metabolism and created an anti‐tumor immunity. 47 , 48 We recognized that the downstream mechanism underlying miR21‐macrophages particularly in CAV may not be completely elucidated by our study. We suggest that miR‐21genetic or pharmacologic deletion on macrophages modified the macrophage metabolism. An inhibition of glycolysis in pro‐inflammatory M1 macrophages may dampen macrophages inflammatory response. Indeed, immune metabolic reprogramming and particularly reprogramming of aerobic glycolysis plays a crucial role during macrophage‐mediated inflammatory response. Aberrant aerobic glycolysis was reported to be a main mediator of systemic inflammation and thus it may trigger an overall inflammatory reaction leading to organ damage and allograft rejection. 49 Furthermore, the role of miR‐21 in macrophages during LPS‐induced sepsis and in controlling glycolysis in myeloid cells has been recently observed thus confirming a relevant metabolic/inflammatory role for miR‐21. 50 , 51 Interestingly, in a different setting, miR‐21 has been described as an oncomir and its genetic targeting led to the development of smaller tumors as compared with WT. 47 The aforementioned anti‐tumor effect was ascribed to a reshaping of tumor‐associated macrophages, with a rewiring of their regulatory activity toward a pro‐inflammatory angiostatic phenotype. 47 This was described in a different setting, in which tumor microenvironment may play a role. Interestingly, a miR‐21 antagomir‐based phase II clinical trial is planned in patients with Alport syndrome. 22 , 52 In summary, our study indicates that miR‐21 plays a role in the onset of CAV and that miR‐21 antagonism has the potential to abrogate the onset of CAV by reprogramming macrophages metabolism.
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
AUTHOR CONTRIBUTIONS
V.U. and M.B.N. designed the study, performed experiments, analyzed data, and wrote the paper; F.D., L.K., A.V., M.U., C.R., E.C.L., A.M., and B.N.C. performed experiments and analyzed data. B.E., J.Y., A.S., G.A.V., G.V.Z., and R.A. coordinated research. E.A., C.L., and A.J.S. assisted with sample collection and data analysis. A.C. and E.R. performed experiments and assisted with data interpretation. L.P., M.V., M.S., E.A., and M.F. collected samples and clinical data. G.B. and D.C. performed immunostaining, in situ hybridization, and histology analysis. M.G.P. performed experiments. P.F. conceived the study, designed research, and wrote and edited the paper.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank the Fondazione Romeo and Enrica Invernizzi for the enormous support. We thank Mollie Jurewicz for editing. We are thankful to Eric Olson (UT Southwestern) for providing miR‐21fl/fl mice. We thank Edoardo Bonacina (Niguarda Hospital) for collection and analysis of human cardiac biopsies. We would also like to thank Maya Fedeli (San Raffaele Scientific Institute) for helpful discussions. P. F. and F. D are supported by the Italian Ministry of Health grant RF‐2016‐02362512. R. A. is supported by K24 AI116925. V. U. is supported by Fondazione Diabete Ricerca (FO.DI.RI) Società Italiana di Diabetologia (SID) fellowship.
Vera Usuelli and Moufida Ben Nasr contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the main text and in the supporting information of this article.
REFERENCES
- 1. Davis MK, Hunt SA. State of the art: cardiac transplantation. Trends Cardiovasc Med. 2014;24(8):341‐349. [DOI] [PubMed] [Google Scholar]
- 2. Johnson MR, Aaronson KD, Canter CE, et al. Heart retransplantation. Am J Transplant. 2007;7(9):2075‐2081. [DOI] [PubMed] [Google Scholar]
- 3. Vergani A, Tezza S, D’Addio F, et al. Long‐term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation. 2013;127(4):463‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D’Addio F, Ueno T, Clarkson M, et al. CD160Ig fusion protein targets a novel costimulatory pathway and prolongs allograft survival. PLoS One. 2013;8(4):e60391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D’Addio F, Boenisch O, Magee CN, et al. Prolonged, low‐dose anti‐thymocyte globulin, combined with CTLA4‐Ig, promotes engraftment in a stringent transplant model. PLoS One. 2013;8(1):e53797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hosenpud JD, Shipley GD, Wagner CR. Cardiac allograft vasculopathy: current concepts, recent developments, and future directions. J Heart Lung Transplant. 1992;11(1 Pt 1):9‐23. [PubMed] [Google Scholar]
- 7. Stehlik J, Edwards LB, Kucheryavaya AY, et al. The registry of the international society for heart and lung transplantation: 29th official adult heart transplant report–2012. J Heart Lung Transplant. 2012;31(10):1052‐1064. [DOI] [PubMed] [Google Scholar]
- 8. Sarraj B, Ye J, Akl AI, et al. Impaired selectin‐dependent leukocyte recruitment induces T‐cell exhaustion and prevents chronic allograft vasculopathy and rejection. Proc Natl Acad Sci U S A. 2014;111(33):12145‐12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu G, Friggeri A, Yang Y, et al. miR‐21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207(8):1589‐1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mannon RB. Macrophages: contributors to allograft dysfunction, repair, or innocent bystanders? Curr Opin Organ Transplant. 2012;17(1):20‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toki D, Zhang W, Hor KLM, et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant. 2014;14(9):2126‐2136. [DOI] [PubMed] [Google Scholar]
- 12. Seki A, Fishbein MC. Predicting the development of cardiac allograft vasculopathy. Cardiovasc Pathol. 2014;23(5):253‐260. [DOI] [PubMed] [Google Scholar]
- 13. Edwards LA, Nowocin AK, Jafari NV, et al. Chronic rejection of cardiac allografts is associated with increased lymphatic flow and cellular trafficking. Circulation. 2018;137(5):488‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lodish HF, Zhou B, Liu G, Chen CZ. Micromanagement of the immune system by microRNAs. Nat Rev Immunol. 2008;8(2):120‐130. [DOI] [PubMed] [Google Scholar]
- 15. Chau BN, Xin C, Hartner J, et al. MicroRNA‐21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4(121):121ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ben Nasr M, Tezza S, D’Addio F, et al. PD‐L1 genetic overexpression or pharmacological restoration in hematopoietic stem and progenitor cells reverses autoimmune diabetes. Sci Transl Med. 2017;9(416). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thum T, Gross C, Fiedler J, et al. MicroRNA‐21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980‐984. [DOI] [PubMed] [Google Scholar]
- 18. Harris A, Krams SM, Martinez OM. MicroRNAs as immune regulators: implications for transplantation. Am J Transplant. 2010;10(4):713‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shan J, Feng LI, Luo L, et al. MicroRNAs: potential biomarker in organ transplantation. Transpl Immunol. 2011;24(4):210‐215. [DOI] [PubMed] [Google Scholar]
- 20. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203‐222. [DOI] [PubMed] [Google Scholar]
- 21. Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci U S A. 2010;107(32):14339‐14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gomez IG, MacKenna DA, Johnson BG, et al. Anti‐microRNA‐21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015;125(1):141‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilflingseder J, Regele H, Perco P, et al. miRNA profiling discriminates types of rejection and injury in human renal allografts. Transplantation. 2013;95(6):835‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hasegawa T, Visovatti SH, Hyman MC, Hayasaki T, Pinsky DJ. Heterotopic vascularized murine cardiac transplantation to study graft arteriopathy. Nat Protoc. 2007;2(3):471‐480. [DOI] [PubMed] [Google Scholar]
- 25. Stewart S, Fishbein MC, Snell GI, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26(12):1229‐1242. [DOI] [PubMed] [Google Scholar]
- 26. Bruschi G, Colombo T, Oliva F, et al. Orthotopic heart transplantation with donors greater than or equal to 60 years of age: a single‐center experience. Eur J Cardiothorac Surg. 2011;40(1):e55‐e61. [DOI] [PubMed] [Google Scholar]
- 27. Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H‐2D, H‐2 K, and non‐H‐2 antigens in rejection. Transplantation. 1973;16(4):343‐350. [DOI] [PubMed] [Google Scholar]
- 28. Sayegh MH, Wu Z, Hancock WW, et al. Allograft rejection in a new allospecific CD4 + TCR transgenic mouse. Am J Transplant. 2003;3(4):381‐389. [DOI] [PubMed] [Google Scholar]
- 29. Yang J, Popoola J, Khandwala S, et al. Critical role of donor tissue expression of programmed death ligand‐1 in regulating cardiac allograft rejection and vasculopathy. Circulation. 2008;117(5):660‐669. [DOI] [PubMed] [Google Scholar]
- 30. Schenk S, Kish DD, He C, et al. Alloreactive T cell responses and acute rejection of single class II MHC‐disparate heart allografts are under strict regulation by CD4 + CD25 + T cells. J Immunol. 2005;174(6):3741‐3748. [DOI] [PubMed] [Google Scholar]
- 31. Salomon RN, Hughes CC, Schoen FJ, Payne DD, Pober JS, Libby P. Human coronary transplantation‐associated arteriosclerosis. Evidence for a chronic immune reaction to activated graft endothelial cells. Am J Pathol. 1991;138(4):791‐798. [PMC free article] [PubMed] [Google Scholar]
- 32. Fischbein MP, Yun J, Laks H, et al. Role of CD8 + lymphocytes in chronic rejection of transplanted hearts. J Thorac Cardiovasc Surg. 2002;123(4):803‐809. [DOI] [PubMed] [Google Scholar]
- 33. Kashizuka H, Sho M, Nomi T, et al. Role of the ICOS‐B7h costimulatory pathway in the pathophysiology of chronic allograft rejection. Transplantation. 2005;79(9):1045‐1050. [DOI] [PubMed] [Google Scholar]
- 34. Falkenham A, de Antueno R, Rosin N, et al. Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis. Am J Pathol. 2015;185(4):927‐942. [DOI] [PubMed] [Google Scholar]
- 35. Danenberg HD, Fishbein I, Gao J, et al. Macrophage depletion by clodronate‐containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106(5):599‐605. [DOI] [PubMed] [Google Scholar]
- 36. von Rossum A, Laher I, Choy JC. Immune‐mediated vascular injury and dysfunction in transplant arteriosclerosis. Front Immunol. 2014;5:684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hunt SA, Haddad F. The changing face of heart transplantation. J Am Coll Cardiol. 2008;52(8):587‐598. [DOI] [PubMed] [Google Scholar]
- 38. Fiorina P, Jurewicz M, Tanaka K, et al. Characterization of donor dendritic cells and enhancement of dendritic cell efflux with CC‐chemokine ligand 21: a novel strategy to prolong islet allograft survival. Diabetes. 2007;56(4):912‐920. [DOI] [PubMed] [Google Scholar]
- 39. Vergani A, Fotino C, D'Addio F, et al. Effect of the purinergic inhibitor oxidized ATP in a model of islet allograft rejection. Diabetes. 2013;62(5):1665‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. D’Addio F, Vergani A, Potena L, et al. P2X7R mutation disrupts the NLRP3‐mediated Th program and predicts poor cardiac allograft outcomes. J Clin Invest. 2018;128(8):3490‐3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krichevsky AM, Gabriely G. miR‐21: a small multi‐faceted RNA. J Cell Mol Med. 2009;13(1):39‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fiorina P, Lattuada G, Ponari O, Silvestrini C, DallAglio P. Impaired nocturnal melatonin excretion and changes of immunological status in ischaemic stroke patients. Lancet. 1996;347(9002):692‐693. [DOI] [PubMed] [Google Scholar]
- 43. Corna G, Campana L, Pignatti E, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica. 2010;95(11):1814‐1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Das A, Yang C‐S, Arifuzzaman S, et al. High‐resolution mapping and dynamics of the transcriptome, transcription factors, and transcription co‐factor networks in classically and alternatively activated macrophages. Front Immunol. 2018;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jablonski KA, Amici SA, Webb LM, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS One. 2015;10(12):e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Das A, Sinha M, Datta S, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. 2015;185(10):2596‐2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sahraei M, Chaube B, Liu Y, et al. Suppressing miR‐21 activity in tumor‐associated macrophages promotes an antitumor immune response. J Clin Invest. 2019;129(12):5518‐5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sheedy FJ, Palsson‐McDermott E, Hennessy EJ, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR‐21. Nat Immunol. 2010;11(2):141‐147. [DOI] [PubMed] [Google Scholar]
- 49. Soto‐Heredero G, de Las G, Heras MM, Gabande‐Rodriguez E, Oller J, Mittelbrunn M. Glycolysis ‐ a key player in the inflammatory response. FEBS J. 2020;287(16):3350‐3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van den Bossche J, Baardman J, Otto N, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17(3):684‐696. [DOI] [PubMed] [Google Scholar]
- 51. Paulo Henrique Melo ARPAaCHS . microRNA‐21 controls inflammatory and metabolic program of macrophage and neutrophils during sepsis. J Immunol. 2019;202. [Google Scholar]
- 52. Kölling M, Kaucsar T, Schauerte C, et al. Therapeutic miR‐21 silencing ameliorates diabetic kidney disease in mice. Mol Ther. 2017;25(1):165‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that supports the findings of this study are available in the main text and in the supporting information of this article.