

Abstract
Identification of molecular alterations occurring in the adenomatous and carcinomatous components within the same tumor would greatly enhance understanding of the neoplastic progression of colorectal cancer. We examined somatic copy number alterations (SCNAs) and mRNA expression at the corresponding loci involved in the adenoma–carcinoma sequence in the isolated adenomatous and cancer glands of the same tumor in 15 cases of microsatellite‐stable “carcinoma in adenoma,” using genome‐wide SNP and global gene expression arrays. Multiple copy‐neutral loss of heterozygosity events were detected at 4q13.2, 15q15.1, and 14q24.3 in the adenomatous component and at 4q13.2, 15q15.1, and 14q24.3 in the carcinomatous component. There were significant differences in the copy number (CN) gain frequencies at 20q11.21–q13.33, 8q13.3, 8p23.1, and 8q21.2–q22.2 between the adenomatous and carcinomatous components. Finally, we found a high frequency of five genotypes involving CN gain with upregulated expression of the corresponding gene (RPS21, MIR3654, RSP20, SNORD54, or ASPH) in the carcinomatous component, whereas none of these genotypes were detected in the adenomatous component. This finding is interesting in that CN gain with upregulated gene expression may enhance gene function and play a crucial role in the progression of an adenoma into a carcinomatous lesion.
Keywords: array‐based analysis, colorectal adenoma, colorectal cancer, mRNA expression array, somatic copy number alteration
Abbreviations
- ASPH
aspartate β‐hydroxylase
- CN
copy number
- CN‐LOH
copy‐neutral loss of heterozygosity
- CRC
colorectal cancer
- LOH
loss of heterozygosity
- MSI
microsatellite instability
- SCNA
somatic copy number alteration
INTRODUCTION
There are three pathways involved in sporadic colorectal carcinogenesis: the adenoma–carcinoma sequence, serrated pathway and de novo pathway. 1 , 2 , 3 , 4 , 5 , 6 , 7 According to the adenoma–carcinoma sequence, molecular alterations essential to the development of colorectal cancer (CRC) accumulate in multiple genes that regulate cell growth and differentiation, and loss of heterozygosity (LOH) facilitates neoplastic progression (8–9). The serrated pathway is characterized by mutations in the BRAF gene and global methylation occurring in CpG islands of promoter regions. 3 , 4 In addition, specific pathological findings including a higher frequency of right‐sided colon cancers and the development of medullary and mucinous carcinomas are associated with the serrated pathway. 3 , 4 , 8 Third, the de novo pathway, proposed by the Hasegawa and Fujimori research groups in Japan, suggests that depressed lesions rapidly progress into invasive or metastatic lesions, which are characterized by TP53 mutations without KRAS mutations. 7 , 9 Among these models, the adenoma–carcinoma sequence is crucial for elucidating the molecular alterations occurring during colorectal carcinogenesis, which is the direct progression of a malignant (carcinomatous) lesion from a benign (adenomatous) lesion. 10
Somatic copy number alterations (SCNAs) are closely associated with neoplastic progression of CRC. 11 , 12 , 13 Previous studies have shown that SCNAs accumulate during the progression from adenoma to cancer, according to the adenoma–carcinoma sequence, suggesting that SCNAs are powerful driving forces in CRC. 14 , 15 However, most SCNAs are presumed not to affect the expression of the corresponding gene(s) (low or high expression based on copy number [CN] loss or gain, respectively) in CRC. Accordingly, although SCNAs may be powerful markers predicting neoplastic progression, 14 , 15 SCNAs that do not affect gene expression may not be necessary for tumor growth or acquisition of metastatic potential in CRC. On the other hand, SCNAs that do affect expression of the corresponding gene(s) might enhance neoplastic progression during colorectal carcinogenesis. 16 , 17 To detect neoplastic progression based on the adenoma–carcinoma sequence, the most representative pathway of CRC development, it is necessary to separately examine the molecular alterations occurring in the adenomatous versus carcinomatous components of the same tumor.
Our aim is to identify molecular alterations including SCNAs and global gene expression changes occurring in the adenomatous and carcinomatous components of the same tumor, obtained using a crypt isolation method, which excludes interstitial cells from the tumor tissues. In addition, we also investigated expression of the genes at the same locus as the identified SCNAs, which may be the force driving progression from an adenoma to carcinoma.
MATERIALS AND METHODS
Patients
Fifteen cases of carcinoma in/with adenoma were examined. Carcinoma in/with adenoma was diagnosed according to the modified World Health Organization 2019 criteria. 8 Briefly, low‐grade adenomas are characterized by a uniform monolayer of columnar cells with basal nuclei showing minimal atypia. In high‐grade adenomas, nuclear atypia is more frequent, with nuclear pleomorphism, nuclear enlargement and pseudostratification without stromal invasion. Carcinoma‐ in or with adenoma‐containing intramucosal cancer is defined by cytological atypia and complex architecture with cribriform groups, irregular branching, glandular anastomosis and budding of neoplastic cells into the lumen, which are considered representative of stromal invasion. Clinicopathological findings were recorded according to the general rules for management of the Japanese Colorectal Cancer Association. 18 The detailed clinicopathological findings are summarized in Table 1. This study was approved by the local ethics committee of Iwate Medical University (approval number HGH28‐26), and all patients provided informed consent.
Table 1.
Clinicopathological findings of the cases of colorectal adenocarcinoma in/with adenoma
| Clinicopathological findings (%) | |
|---|---|
| Total | 15 |
| Sex | |
| Male | 7 (46.7) |
| Female | 8 (53.3) |
| Age, median (range) (years) | 71 (53–83) |
| Size, median (range) (mm) | 40 (23–79) |
| Location | |
| Right | 9 (60) |
| Left | 6 (40) |
| Macroscopic type | |
| Elevated | 15 (100) |
| Depressed | 0 (0) |
| Adenoma component | |
| Histological subtype | |
| Tubular adenoma | 8 (53.3) |
| Tubulovillous adenoma | 7 (46.7) |
| Histological grade | |
| Low | 7 (46.7) |
| High | 8 (53.3) |
| Presence of KRAS mutations | 7 (46.7) |
| Carcinoma component | |
| Histological subtype | |
| Well differentiated | 12 (80) |
| Moderately differentiated | 2 (13.3) |
| Papillary | 1 (6.7) |
| Depth of cancer invasion | |
| Mucosa | 12 (80) |
| Submucosa | 0 |
Crypt isolation method
Fresh tumor and normal tissues were obtained by endoscopic resection. Normal colonic mucosa was collected from the most distal portion of the resected specimen. Adenomatous and carcinomatous components were isolated from the resected specimen separately. The adenomatous component was distinguished from the carcinomatous component based on findings observed under a dissecting microscope (SZ60; Olympus). Gland isolation from tumor and normal mucosae was performed using tumor glands obtained from both components as described previously. 19 At least 10 tumor or normal glands were obtained (tumor: range 10–25 glands, mean 15 glands; normal: range 10–28 glands, mean 18 glands). Briefly, fresh tissues were minced with a razor into small pieces and incubated at 37°C for 30 min in calcium‐ and magnesium‐free Hanks' balanced salt solution containing 30 mM EDTA. The isolated glands were immediately fixed in 70% ethanol and stored at 4°C until used for DNA extraction. The fixed glands were observed under a dissecting microscope (SZ60; Olympus). Some of the glands were routinely processed by histopathological analysis to confirm their histological nature. Contamination, such as with interstitial cells, was not evident in any of our samples. In addition, the obtained adenomatous and carcinomatous glands were confirmed using histological sections.
DNA and RNA extraction
DNA was extracted from the isolated tumors and normal glands of each patient using routine phenol–chloroform extraction. An A260/A280 ratio of 1.8 was used as the criteria for DNA, and a ratio of 2.0 was used for RNA. Total amount of yielded DNA was 500–2000 ng/μL. Total RNA was extracted from cancer and normal cells using the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions.
Analysis of microsatellite instability (MSI)
The MSI status was determined using a consensus panel of five reference microsatellite markers (BAT25, BAT26, D2S123, D3S546, and D17S250) according to a previously described method. 20 When none of the five markers was altered, the tumors were defined as microsatellite stable. When only one marker was altered, the tumors were defined as low MSI. When two or more markers were altered, the tumors were defined as high MSI.
SNP array analysis
SNP array analysis was conducted using the Cytoscan HD (Affymetrix) platform. This array contains more than 1.9 million nonpolymorphic markers and over 740 000 SNP markers with an average intragenic marker spacing of 880 bp and intergenic marker spacing of 1737 bp. These platforms consist of microarrays containing nonpolymorphic probes specific for CN variations in the coding and noncoding regions of the human genome as well as polymorphic SNP probes. All procedures were performed according to the manufacturer's instructions. We analyzed the hybridized slides containing biotin‐labeled DNA using the GeneChip Scanner 3000 7G (Affymetrix) and the Chromosome Analysis Suite Software (Affymetrix). An abnormality was defined as (a) a minimum of 50 consecutively duplicated probes, (b) a minimum of 50 consecutively deleted probes, or (c) segments of LOH larger than 3 Mb. Smaller alterations involving cancer‐associated genes were also investigated. The detailed methodology was described previously. 21 Finally, we used 25 ng of DNA for each array.
Classification of CN alterations
In the present study, SCNAs were classified into five subtypes: gain, LOH, copy‐neutral LOH (CN‐LOH), mosaic and mixed. 21 LOH was considered a cross chromosomal change resulting in loss of the entire gene and surrounding region, and gain was defined as a cross chromosomal change resulting in gain of the entire gene and surrounding region. CN‐LOH was defined as LOH without a copy number change (CN = 2). A mosaic pattern was defined as a mixture of normal and abnormal cells with SCNAs. A mixed pattern was defined as a mixture of more than two SCNA patterns within one locus, such as LOH and mosaic with LOH (LOH type), gain and LOH (gain > LOH, gain; LOH > gain, LOH), or gain and mosaic with gain (gain). The mosaic type was subclassified into mosaic gain or mosaic loss. In the present study, mosaic gain was classified into gain, while mosaic loss was assigned into LOH. Representative illustrations of the gain, mosaic gain, LOH, mosaic loss, and mixed patterns are shown in Figure S1.
Microarray analysis
RNA was extracted from isolated normal, adenomatous and carcinomatous glands, and RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent). All RNA used was confirmed to be of good quality. In addition, we used 20 ng of RNA for each array. This array measured 21 453 mRNA transcripts. Probe labeling, chip hybridization, and scanning were performed according to the manufacturer's instructions. Gene expression was determined using GeneChip Human Clariom S arrays (Affymetrix) and Transcriptome Analysis Console software (Affymetrix).
Statistical analysis
Upregulated or downregulated expression of the mRNA transcripts in tumor tissues was assessed by paired t‐test with the Benjamini–Hochberg false discovery rate procedure, compared with those of isolated normal glands. Differences between SCNAs with an upregulated or downregulated corresponding gene expression level between patterns (including gain and nongains, or LOH and non‐LOH) were assessed with a Fisher's exact test. Differences in SCNA number including gain, LOH and CN‐LOH among the groups were evaluated using the Wilcoxon matched‐pairs signed‐rank test in JMP Pro13 (SAS Institute, Cary, NC, USA). Differences in SCNAs between isolated adenomatous and carcinomatous glands were analyzed using a binomial test. Finally, differences in the genotype (e.g., CN gain/GNAS) frequency among isolated adenomatous and carcinomatous glands were determined by a binomial test. A p‐value <0.05 was accepted as significant.
RESULTS
Microsatellite status of the adenomatous and carcinomatous components of the same tumor
All isolated components were classified as microsatellite stable according to previously reported criteria. 20
SCNAs in the adenomatous and carcinomatous components of the same tumor
In the isolated adenomatous component, the median number of total chromosomal aberrations per patient was 40 (range 16–233), with a median of 15 gains (range 2–145), 19 LOH events (range 10–88), and 2 copy‐neutral LOH events (range 0–65). On the other hand, the median number of total chromosomal aberrations per patient in the carcinomatous component was 115 (range 28–659), with a median of 67 gains (range 5–551), 29 LOH events (range 9–108), and 3 copy‐neutral LOH events (range 0–65). There were significant differences in the total number of gains between the adenomatous and carcinomatous components (p = 0.0125). However, the total numbers of LOH and CN‐LOH events were common between the two components. The results are illustrated in Figure 1.
Figure 1.
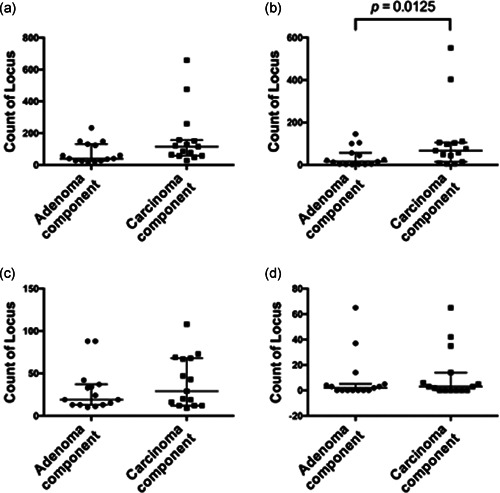
Total number of somatic copy number alterations (SCNAs), including gain, loss of heterozygosity (LOH) and copy‐neutral (CN)‐LOH events, in the isolated adenomatous and carcinomatous components of the same tumor
We examined differences in SCNAs between the two isolated components. Regions harboring SCNAs detected in more than 30% of cases were selected for comparison between the two components. CN gain events detected in more than 30% of cases were located at 14q32.33, 7q34, and 7q36.2 in the adenomatous component, whereas those in the carcinomatous components were found (in decreasing order of frequency) at 14q32.33, 20q11.21–q13.33, 8q11.1–q24.3, 7q11.21–q36.3, 7p22.3–p11.1, 8p23.3–p11.1, 20p12.1–p11.1, 13q11–q14.12, 13q14.3–q34, and 19q13.2–q13.32. LOH events detected in more than 30% of cases were found at 4q13.2, 15q15.1, 14q24.3, 6q22.31, 10q26.13, 19q13.42, 2q12.3, 17p13.3‐p12, 18q12.1–q23, and 11q11 in the carcinomatous components, compared with 4q13.2, 15q15.1, 14q24.3, 2q12.3, 10q26.13, 11q11, 19q13.42, 6q22.31, 7q34, 3q25.33, 17p13.1, 17p11.2, and 17q12 in the adenomatous component. No CN‐LOH events were detected in more than 30% of cases in either component. These results are shown in Table S1, and ideograms showing the SCNAs in the adenomatous versus carcinomatous components are presented in Figure S2. Significant differences in the frequency of the CN gains between the adenomatous and carcinomatous components were found at 20q11.21–q13.33, 8p23.1, 8q13.3, and 8q21.2–q22.2 (higher frequency in the carcinomatous than adenomatous component) (Table S2). No significant differences in the frequency of the LOH or CN‐LOH events were observed between the adenomatous and carcinomatous components.
Individual differences in SCNAs of each case between the adenomatous and carcinomatous components within the same tumor are depicted in Figure S3. Several SCNA changes in each case were frequently found in carcinomatous components compared with adenomatous components.
mRNA expression profiling in the adenomatous and carcinomatous components of the same tumor
We performed global mRNA expression profiling in the 15 isolated adenomatous and carcinomatous components and compared the results with expression in the isolated normal gland samples. The criteria for differential expression were a fold‐change in expression <−1.5 or >1.5 and p < 0.05 (Benjamini–Hochberg false discovery rate). First, we compared the expression levels between the adenomatous and normal glands and identified 36 differentially expressed mRNAs (36 upregulated and 0 downregulated in the adenomatous component) (Table 2). Second, we compared the expression levels between the carcinomatous and normal glands and found 73 differentially expressed mRNAs (71 upregulated and 2 downregulated in the carcinomatous component) (Tables 3 and 4). Of these differentially expressed mRNAs, 31 were common to both the isolated adenomatous and carcinomatous components (Figure 2), whereas five mRNAs were found in the adenomatous component only and 42 in the carcinomatous component only (Figure 2).
Table 2.
Somatic copy number alterations and expression of the corresponding gene in the adenomatous component compared with normal glands
| Official symbol | Location | Upregulated case (n = 15) (%) | Pattern of copy number alteration (n = 15) (%) | p‐value | |
|---|---|---|---|---|---|
| Gain | Nongain | ||||
| CEACAM5 | 19q13.2 | 8 (53.3) | 4 (26.7) | 11 (73.3) | 0.5692 |
| MIR3654 | 7q33 | 8 (53.3) | 4 (26.7) | 11 (73.3) | 1.0000 |
| GNAS | 20q13.32 | 9 (60) | 3 (20) | 12 (80) | 0.5253 |
| RPS21 | 20q13.33 | 8 (53.3) | 3 (20) | 12 (80) | 1.0000 |
| RPL13A | 19q13.33 | 7 (46.7) | 3 (20) | 12 (80) | 1.0000 |
| SNORD32A | 19q13.33 | 7 (46.7) | 3 (20) | 12 (80) | 1.0000 |
| SNORD33 | 19q13.33 | 7 (46.7) | 3 (20) | 12 (80) | 1.0000 |
| SNORD34 | 19q13.33 | 7 (46.7) | 3 (20) | 12 (80) | 1.0000 |
| SNORD35A | 19q13.33 | 7 (46.7) | 3 (20) | 12 (80) | 1.0000 |
| RPS28 | 19p13.2 | 8 (53.3) | 2 (13.3) | 13 (86.7) | 0.2000 |
| CAST | 5q15 | 9 (60) | 1 (6.7) | 14 (93.3) | 0.4000 |
| SNORD95 | 5q35.3 | 9 (60) | 1 (6.7) | 14 (93.3) | 0.4000 |
| SNORD96A | 5q35.3 | 9 (60) | 1 (6.7) | 14 (93.3) | 0.4000 |
| SLC12A2 | 5q23.3 | 7 (46.7) | 1 (6.7) | 14 (93.3) | 1.0000 |
| NAP1L1 | 12q21.2 | 5 (33.3) | 1 (6.7) | 14 (93.3) | 1.0000 |
| PIGR | 1q32.1 | 11 (73.3) | 0 (0) | 15 (100) | 1.0000 |
| HSPA8 | 11q24.1 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| RPL3 | 22q13.1 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| SNORD14C | 11q24.1 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| SNORD14D | 11q24.1 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| SNORD43 | 22q13.1 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| SNORD83B | 22q13.1 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| EEF1A1 | 6q13 | 9 (60) | 0 (0) | 15 (100) | 1.0000 |
| RACK1 | 5q35.3 | 9 (60) | 0 (0) | 15 (100) | 1.0000 |
| H4C3 | 6p22.2 | 9 (60) | 0 (0) | 15 (100) | 1.0000 |
| RPS24 | 10q22.3 | 9 (60) | 0 (0) | 15 (100) | 1.0000 |
| EEF1G | 11q12.3 | 8 (53.3) | 0 (0) | 15 (100) | 1.0000 |
| ITM2C | 2q37.1 | 8 (53.3) | 0 (0) | 15 (100) | 1.0000 |
| MTRNR2L9 | 6q11.1 | 7 (46.7) | 0 (0) | 15 (100) | 1.0000 |
| RPS3 | 11q13.4 | 7 (46.7) | 0 (0) | 15 (100) | 1.0000 |
| SNORD15A | 11q13.4 | 7 (46.7) | 0 (0) | 15 (100) | 1.0000 |
| TMEM123 | 11q22.2 | 7 (46.7) | 0 (0) | 15 (100) | 1.0000 |
| TPM1 | 15q22.2 | 7 (46.7) | 0 (0) | 15 (100) | 1.0000 |
| EEF1A1P5 | 9q34.13 | 6 (40) | 0 (0) | 15 (100) | 1.0000 |
| EPCAM | 2p21 | 6 (40) | 0 (0) | 15 (100) | 1.0000 |
| CBWD5 | 9q21.11 | 5 (33.3) | 0 (0) | 15 (100) | 1.0000 |
Table 3.
Somatic copy number alterations and expression of the corresponding gene in the carcinomatous component compared with normal glands
| Official symbol | Location | Upregulated case (n = 15) (%) | Pattern of copy number alteration (n = 15) (%) | p‐value | |
|---|---|---|---|---|---|
| Gain | Nongain | ||||
| GNAS | 20q13.32 | 12 (80) | 8 (53.3) | 7 (46.7) | 0.0769 |
| RPS21 | 20q13.33 | 11 (73.3) | 8 (53.3) | 7 (46.7) | 0.0256 |
| MIR3654 | 7q33 | 9 (60) | 7 (46.7) | 8 (53.3) | 0.0070 |
| ASPH | 8q12.3 | 7 (46.7) | 6 (40) | 9 (60) | 0.0406 |
| RPS20 | 8q12.1 | 6 (40) | 6 (40) | 9 (60) | 0.0110 |
| SNORD54 | 8q12.1 | 6 (40) | 6 (40) | 9 (60) | 0.0110 |
| CEACAM5 | 19q13.2 | 10 (66.7) | 4 (26.7) | 11 (73.3) | 0.2308 |
| FBL | 19q13.2 | 9 (60) | 4 (26.7) | 11 (73.3) | 1.0000 |
| OLFM4 | 13q14.3 | 9 (60) | 4 (26.7) | 11 (73.3) | 1.0000 |
| RPL13A | 19q13.33 | 11 (73.3) | 3 (20) | 12 (80) | 0.5165 |
| SNORD32A | 19q13.33 | 11 (73.3) | 3 (20) | 12 (80) | 0.5165 |
| SNORD33 | 19q13.33 | 11 (73.3) | 3 (20) | 12 (80) | 0.5165 |
| SNORD34 | 19q13.33 | 11 (73.3) | 3 (20) | 12 (80) | 0.5165 |
| SNORD35A | 19q13.33 | 11 (73.3) | 3 (20) | 12 (80) | 0.5165 |
| NAP1L1 | 12q21.2 | 9 (60) | 3 (20) | 12 (80) | 0.5253 |
| RPL38 | 17q25.1 | 9 (60) | 3 (20) | 12 (80) | 0.2286 |
| RPL41 | 12q13.2 | 9 (60) | 3 (20) | 12 (80) | 0.5253 |
| CD63 | 12q13.2 | 7 (46.7) | 3 (20) | 12 (80) | 0.5692 |
| MGST1 | 12p12.3 | 7 (46.7) | 3 (20) | 12 (80) | 1.0000 |
| RPL18 | 19q13.33 | 7 (46.7) | 3 (20) | 12 (80) | 0.5692 |
| MIR21 | 17q23.1 | 5 (33.3) | 3 (20) | 12 (80) | 0.2418 |
| VMP1 | 17q23.1 | 5 (33.3) | 3 (20) | 12 (80) | 0.2418 |
| PIGR | 1q32.1 | 12 (80) | 2 (13.3) | 13 (86.7) | 1.0000 |
| RPL23 | 17q12 | 11 (73.3) | 2 (13.3) | 13 (86.7) | 1.0000 |
| SNORA21 | 17q12 | 11 (73.3) | 2 (13.3) | 13 (86.7) | 1.0000 |
| CAST | 5q15 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| RPS8 | 1p34.1 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| SNORD38A | 1p34.1 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| SNORD38B | 1p34.1 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| SNORD46 | 1p34.1 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| SNORD55 | 1p34.1 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| SNORD95 | 5q35.3 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| SNORD96A | 5q35.3 | 10 (66.7) | 2 (13.3) | 13 (86.7) | 0.5238 |
| HNRNPM | 19p13.2 | 9 (60) | 2 (13.3) | 13 (86.7) | 0.4857 |
| RPS28 | 19p13.2 | 9 (60) | 2 (13.3) | 13 (86.7) | 1.0000 |
| H4C14 | 1q21.2 | 8 (53.3) | 2 (13.3) | 13 (86.7) | 1.0000 |
| H4C15 | 1q21.2 | 8 (53.3) | 2 (13.3) | 13 (86.7) | 1.0000 |
| RPS6 | 9p22.1 | 8 (53.3) | 2 (13.3) | 13 (86.7) | 0.4667 |
| SLC12A2 | 5q23.3 | 8 (53.3) | 2 (13.3) | 13 (86.7) | 0.2000 |
| CDH1 | 16q22.1 | 7 (46.7) | 2 (13.3) | 13 (86.7) | 0.2000 |
| RPL3 | 22q13.1 | 12 (80) | 1 (6.7) | 14 (93.3) | 1.0000 |
| SNORD43 | 22q13.1 | 12 (80) | 1 (6.7) | 14 (93.3) | 1.0000 |
| SNORD83B | 22q13.1 | 12 (80) | 1 (6.7) | 14 (93.3) | 1.0000 |
| APP | 21q21.3 | 10 (66.7) | 1 (6.7) | 14 (93.3) | 1.0000 |
| CSTB | 21q22.3 | 10 (66.7) | 1 (6.7) | 14 (93.3) | 1.0000 |
| H4C3 | 6p22.2 | 10 (66.7) | 1 (6.7) | 14 (93.3) | 1.0000 |
| ITM2C | 2q37.1 | 10 (66.7) | 1 (6.7) | 14 (93.3) | 1.0000 |
| PARK7 | 1p36.23 | 10 (66.7) | 1 (6.7) | 14 (93.3) | 1.0000 |
| SPTBN1 | 2p16.2 | 9 (60) | 1 (6.7) | 14 (93.3) | 1.0000 |
| TMSB10 | 2p11.2 | 9 (60) | 1 (6.7) | 14 (93.3) | 1.0000 |
| EPCAM | 2p21 | 8 (53.3) | 1 (6.7) | 14 (93.3) | 1.0000 |
| ITGB1 | 10p11.22 | 8 (53.3) | 1 (6.7) | 14 (93.3) | 1.0000 |
| PTMA | 2q37.1 | 6 (40) | 1 (6.7) | 14 (93.3) | 0.4000 |
| UQCR10 | 22q12.2 | 6 (40) | 1 (6.7) | 14 (93.3) | 0.4000 |
| RPS29 | 14q21.3 | 5 (33.3) | 1 (6.7) | 14 (93.3) | 0.3333 |
| HSPA8 | 11q24.1 | 12 (80) | 0 (0) | 15 (100) | 1.0000 |
| SNORD14C | 11q24.1 | 12 (80) | 0 (0) | 15 (100) | 1.0000 |
| SNORD14D | 11q24.1 | 12 (80) | 0 (0) | 15 (100) | 1.0000 |
| RPS3 | 11q13.4 | 11 (73.3) | 0 (0) | 15 (100) | 1.0000 |
| SNORD15A | 11q13.4 | 11 (73.3) | 0 (0) | 15 (100) | 1.0000 |
| RACK1 | 5q35.3 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| RPS12 | 6q23.2 | 10 (66.7) | 0 (0) | 15 (100) | 1.0000 |
| EEF1G | 11q12.3 | 9 (60) | 0 (0) | 15 (100) | 1.0000 |
| EEF1A1 | 6q13 | 8 (53.3) | 0 (0) | 15 (100) | 1.0000 |
| EIF4G2 | 11p15.4 | 8 (53.3) | 0 (0) | 15 (100) | 1.0000 |
| RPS7 | 2p25.3 | 8 (53.3) | 0 (0) | 15 (100) | 1.0000 |
| SNORD97 | 11p15.4 | 8 (53.3) | 0 (0) | 15 (100) | 1.0000 |
| SELENOW | 19q13.33 | 7 (46.7) | 0 (0) | 15 (100) | 1.0000 |
| LAPTM4A | 2p24.1 | 6 (40) | 0 (0) | 15 (100) | 1.0000 |
| TMEM123 | 11q22.2 | 6 (40) | 0 (0) | 15 (100) | 1.0000 |
| RPL32P29 | 14q21.3 | 5 (33.3) | 0 (0) | 15 (100) | 1.0000 |
Table 4.
Somatic copy number alterations and expression of the corresponding gene in the carcinomatous component compared with normal glands
| Pattern of copy number alteration (n = 15) (%) | |||||
|---|---|---|---|---|---|
| Official symbol | Location | Downregulated case (n = 15) (%) | LOH | Non‐LOH | p‐value |
| CCDC102B | 18q22.1‐q22.2 | 6 (40) | 5 (33.3) | 10 (66.7) | 0.3287 |
| PTPRM | 18p11.23 | 5 (33.3) | 3 (20) | 12 (80) | 0.5055 |
Abbreviation: LOH, loss of heterozygosity.
Figure 2.
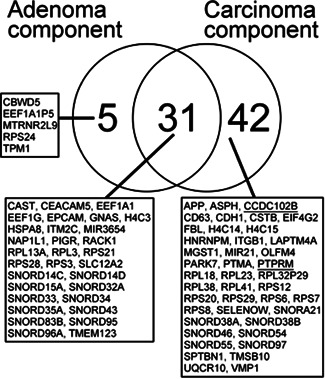
Global transcriptome analysis of the isolated adenomatous and carcinomatous glands. The right circle of the Venn diagram shows the abnormally expressed genes in the adenomatous glands compared with the normal glands, and the left circle shows the abnormally expressed genes in the carcinomatous glands compared with the normal glands. The central overlapping circle indicates the abnormally expressed genes common to both the adenomatous and carcinomatous glands
Integrated genome and transcriptome analyses
To examine whether there was a correlation of SCNA status with its corresponding gene expression, we utilized a statistical approach. First, each mRNA and its corresponding chromosomal location (e.g., RPS21 located at 20q13.33) was explored in our two groups (isolated adenomatous and carcinomatous glands). Second, we examined the association of the corresponding chromosomal location with the type of SCNA, including gain and nongain, or LOH or non‐LOH, in isolated adenomatous and carcinomatous glands (e.g., SCNA at 20q13.33 was a CN gain). In addition, we determined whether the SCNA pattern correlated with the change in the expression level of its corresponding gene (upregulated or downregulated) using a Fisher's exact test (Tables 2, 3, 4). As a result, integrated analysis of the SCNA and mRNA expression data revealed that none of the SCNAs involved a correspondingly differentially expressed gene in the adenomatous component (Table 2). However, in the carcinomatous component, we observed five SCNAs (gains) with upregulated expression of the corresponding gene. Next, In the carcinomatous component, five of the 73 genes with significant differential expression compared with the normal gland (RPS21 (ribosomal protein 21), MIR3654, RPS20, SNORD54 (small nucleolar RNA, C/D Box 54), and ASPH (aspartate beta‐hydroxylase) showed concordance with the corresponding SCNA (Table 3). Finally, using copy number status and gene expression levels, we showed that CN gain was correlated with upregulation of its corresponding gene (Figure 3).
Figure 3.
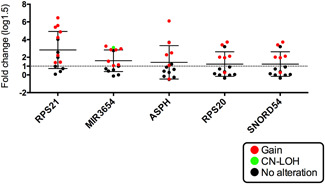
Expression levels of pooled mRNA in array‐based tests of isolated adenomatous and cancerous glands. Different patterns of somatic copy number alterations (SCNAs) are marked by different colors connecting copy number levels and gene expression levels. Red, gain; Blue, loss of heterozygosity (LOH), green, copy‐neutral LOH and black, no alteration
We defined these CN gains with upregulated expression of the corresponding gene (RPS21, MIR3654, RPS20, SNORD54, and ASPH) as a specific genotype (e.g., 20q13.33/RSP21). There were statistical differences in the frequency of each of these five genotypes between the adenomatous and carcinomatous components of the same tumor (p < 0.01). A representative case is shown in Figure S4.
DISCUSSION
Previous studies have shown that SCNAs accumulate during the progression of CRC. 11 , 12 , 13 According to Vogelstein's hypothesis, which is characterized by multi‐step carcinogenesis, genetic loss such as LOH in tumor suppressor genes is an essential genetic event in the neoplastic progression of CRC. 10 In the present study, however, we found that CN gain, involving elevated CNs of particular genes, plays a major role in colorectal carcinogenesis. CN gain at 14q32–33 was identified as a common chromosomal alteration between the adenomatous and carcinomatous components, suggesting that this alteration contributes to early colorectal tumorigenesis. The 14q32–33 region includes the gene AKT1, encoding AKT serine/threonine kinase 1, which regulates cell growth and proliferation and may be responsible for some human cancers, 22 , 23 although not often CRC. High expression of AKT1 was not found in either the adenomatous or carcinomatous component, suggesting that CN gain at 14q32–33 does not play a functional role in the progression from adenoma to carcinoma.
In the present study, there were significant differences in the frequency of CN gain at 20q11.21–q13.33, 8q13.3, 8p23.1, and 8q21.2–q22.2 between the adenomatous and carcinomatous components. However, none of the genes corresponding to these gains showed altered expression. This finding suggests that although CN gains at 20q11.21–q13.33, 8q13.3, 8p23.1, or 8q21.2–q22.2 may be an epiphenomenon in the progression from adenoma to carcinoma, as suggested previously, 24 , 25 CN gain in the carcinomatous component may play a role in colorectal carcinogenesis. SCNAs may become candidate molecular markers predicting the risk of progression from adenoma to carcinoma. 24 , 25 , 26
In the present study, there were no differences in total SCNAs between adenoma and carcinoma gland samples, which contradicts findings in previous studies. 1 , 4 , 5 In addition, the frequency of LOH or CN‐LOH of the APC gene located at 5q22.2 may be low, compared with previous studies. 4 , 5 This finding might be novel or it might be a consequence of the technical issues, but this could not be resolved in the present study. We performed crypt isolation in adenoma and carcinoma glands, and conducted genome‐wide analyses on all material. To verify the results, further study is required.
Recently, we found that a specific genotype defined by SCNAs with altered expression of the corresponding gene plays an important role in CRC progression. 25 In the present study, we showed that five genotypes involving CN gain with upregulated expression of the corresponding gene (RPS21, MIR3654, RSP20, SNORD54, and ASPH) are closely associated with progression from the adenomatous to carcinomatous component of the same tumor. This finding is interesting in that CN gain associated with upregulated gene expression may enhance gene function. Such genotypes play a leading role in the development of carcinoma from adenoma.
The present results demonstrated that five genotypes are closely associated with early carcinogenesis in the colon/rectum. Of these affected genes, RPS family members have been reported in not only breast and prostate cancers 26 , 27 but also CRC, in which alterations in RPS genes result in ribosomopathies. 28 Ribosomal proteins, the essential components of the ribosome, are a family of RNA‐binding proteins that play a prime role in ribosome biogenesis and protein translation. 27 , 29 Broderick et al. reported a single germline truncating mutation in RPS20 in familial CRC type X. 28 , 30 Although the mechanisms by which mutations in RPS20, RPS21, or other genes encoding the RPS genes result in phenotypic diseases such as CRC remain largely speculative, upregulation of RPS20 or RPS21 expression caused by CN gain may help elucidate the role of ribosomal proteins in the progression of CRC. 27 , 28 , 29 , 30 In addition, elucidation of the role of the SCNAs associated with RPS20 or RPS21 in early colorectal carcinogenesis is of interest, as such alterations may be a powerful driving force in neoplastic progression.
A recent study has shown that downregulation of miR‐3654 plays an important role in prostate cancer progression. 31 In the present study, we found that miR‐3654 was involved in the progression of CRC and hypothesized that the CN gain of 7q33, with an upregulation of miR‐3654, might be responsible for the transformation from an adenomatous lesion to a carcinomatous lesion. However, such a mechanism has not been described in CRC. Further studies are required to investigate the molecular mechanisms regarding miR‐3654 expression in colorectal carcinogenesis.
A previous study suggested that dysregulation of SNORD54, which is a noncoding RNA, is a new biomarker in chronic lymphocytic leukemia. 32 Indeed, the relevance of noncoding RNAs in human disease has increased remarkably over the past few years. 33 miRNAs represent the most extensively investigated category of noncoding RNAs in cancer, since genetic and epigenetic defects causing miRNA deregulation and contributing to tumorigenesis have been identified. 33 SNORD54 is an epigenetic molecular category that may be associated with early development of colorectal carcinogenesis. The combination of CN gain and upregulated expression of the corresponding gene suggests the possibility of increasing epigenetic processes occurring during neoplastic progression.
Finally, aspartate β‐hydroxylase (ASPH) is a highly conserved dioxygenase overexpressed in multiple malignancies, including pancreatic cancer. 34 ASPH appears to be involved in regulating the proliferation, invasion and metastasis of pancreatic cancer cells via multiple signaling pathways, suggesting its role as a tumor biomarker and therapeutic target in many types of cancers, including CRC. 34 , 35 In the present study, CN gain with high expression of ASPH was found in isolated carcinomatous glands. Therefore, we suggest that CN gain/ASPH upregulation may be a potential mechanism that accelerates the action of ASPH in CRC.
There are some limitations to this study. First, the sample size was small. However, it is difficult to evaluate molecular alterations separately in the isolated adenomatous and carcinomatous components of a tumor. This is the first study to examine SCNAs and expression of the involved gene(s) separately in the isolated adenomatous and carcinomatous components of the same tumor. We believe that the molecular alterations responsible for neoplastic progression occurring in adenomatous and carcinomatous glands were accurately evaluated in the present study. Second, we could not examine SCNAs and global gene expression in a second cohort for validation of the present results, which was also due to the difficulty in separating the adenomatous and carcinomatous glands from the same tumor and in obtaining glands from multiple cases. Nevertheless, we believe that our study provides a novel approach to evaluating the molecular mechanisms necessary for the neoplastic progression from an adenoma to carcinoma.
In conclusion, we examined SCNAs and global mRNA expression in isolated adenomatous and carcinomatous glands of the same tumor. Although no SCNAs were associated with altered expression of the corresponding gene(s) in the adenomatous component, CN gain with upregulated expression of the corresponding gene may enhance the progression of CRC. Finally, we suggest that such a genotype is important for understanding the molecular mechanisms responsible for accelerating neoplastic progression from an adenoma to a carcinomatous lesion.
CONFLICT OF INTERESTS
None declared.
AUTHOR CONTRIBUTIONS
Tamotsu Sugai, who is first author and corresponding author, contributed to the preparation of the manuscript, including all aspects of the data collection and analysis. Mitsumasa Osakabe performed all data collection and statistical analyses. Yoshihito Tanaka and Makoto Eizuka performed the collection of the tumors and normal glands. Naoki Yanagawa supported the interpretation of pathological findings. Takayuki Matsumoto provided clinical support during the preparation of the manuscript. Wataru Habano and Hiromu Suzuki assisted with the molecular analyses.
ETHICS STATEMENT
Informed consent was obtained from each patient according to institutional guidelines, and the research protocols were approved by the ethics committee of Iwate Medical University Hospital (reference number: HG2018‐528).
CONSENT FOR PUBLICATION
We guarantee that (a) the work is original, (b) the work has not been and will not be published in whole, or in part, in any other journal, and (c) all of the authors have agreed to the contents of the manuscript in its submitted form.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We gratefully acknowledge the technical assistance of Ms. E. Sugawara and Mrs. C. Ishikawa. We also thank the members of the Department of Molecular Diagnostic Pathology, Iwate Medical University for their support.
Sugai T, Osakabe M, Habano W, Tanaka Y, Eizuka M, Sugimoto R, et al. A genome‐wide analysis of the molecular alterations occurring in the adenomatous and carcinomatous components of the same tumor based on the adenoma–carcinoma sequence. Pathology International. 2021;71:582–593. 10.1111/pin.13129
DATA AVAILABILITY STATEMENT
The data that support the findings of our study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–7. [DOI] [PubMed] [Google Scholar]
- 3. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–100. [DOI] [PubMed] [Google Scholar]
- 4. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10:13–27. 10.2353/jmoldx.2008.070082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bedenne L, Faivre J, Boutron MC, Piard F, Cauvin JM, Hillon P. Adenoma–carcinoma sequence or “de novo” carcinogenesis? A study of adenomatous remnants in a population‐based series of large bowel cancers. Cancer. 1992;69:883–8. [DOI] [PubMed] [Google Scholar]
- 7. Hasegawa H, Ueda M, Furukawa K, Watanabe M, Teramoto T, Mukai M, et al. p53 gene mutations in early colorectal carcinoma. De novo vs. adenoma‐carcinoma sequence. Int J Cancer. 1995;64:47–51. 10.1002/ijc.2910640110 [DOI] [PubMed] [Google Scholar]
- 8. Hamilton SR, Sekine S. Conventional colorectal adenoma. In: WHO Classification of Tumor Editorial Board, editor. WHO classification of tumours of the digestive system. Lyon: International Agency for Research on Cancer; 2019. p. 170–3.
- 9. Fujimori T, Satonaka K, Yamamura‐Idei Y, Nagasako K, Maeda S. Non‐involvement of ras mutations in flat colorectal adenomas and carcinomas. Int J Cancer. 1994;57:51–5. 10.1002/ijc.2910570110 [DOI] [PubMed] [Google Scholar]
- 10. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal tumor development. N Eng J Med. 1988;319:525–32. 10.1056/NEJM198809013190901 [DOI] [PubMed] [Google Scholar]
- 11. Wang H, Liang L, Fang JY, Xu J. Somatic gene copy number alterations in colorectal cancer: new quest for cancer drivers and biomarkers. Oncogene. 2016;35:2011–9. 10.1038/onc.2015.304 [DOI] [PubMed] [Google Scholar]
- 12. Redon R, Ishikawa S, Fitch KR, Feuk L., Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. 10.1038/nature05329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–64. 10.1038/nrg2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sugai T, Eizuka M, Habano W, Fujita Y, Sato A, Sugimoto R, et al. Comprehensive molecular analysis based on somatic copy number alterations in intramucosal colorectal neoplasias and early invasive colorectal cancers. Oncotarget. 2018;9:22895–906. 10.18632/oncotarget.25112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eizuka M, Sugai T, Habano W, Uesugi N, Takahashi Y, Kawasaki K, et al. Molecular alterations in colorectal adenomas and intramucosal adenocarcinomas defined by high‐density single‐nucleotide polymorphism arrays. J Gastroenterol. 2017;52:1158–68. 10.1007/s00535-017-1317-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006 Feb 15;66:2129–37. 10.1158/0008-5472.CAN-05-2569 [DOI] [PubMed] [Google Scholar]
- 17. Berg M, Agesen TH, Thiis‐Evensen E, INFAC‐study G, Merok MA, Teixeira MR, et al. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol Cancer. 2010;9:100. 10.1186/1476-4598-9-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Japanese Society for Cancer of the Colon and Rectum . Japanese classification of colorectal carcinoma. 2nd English ed. Tokyo: Kanehara Co.; 2009. p. 30–63. [Google Scholar]
- 19. Habano W, Sugai T, Nakamura S, Yoshida T. A novel method for gene analysis of colorectal carcinomas using a crypt isolation technique. Lab Invest. 1996;74:933–40. [PubMed] [Google Scholar]
- 20. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 21. Tsuyukubo T, Ishida K, Osakabe M, Shiomi E, Kato R, Takata R, et al. Comprehensive analysis of somatic copy number alterations in clear cell renal cell carcinoma. Mol Carcinog. 2020;59:412–24. 10.1002/mc.23164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korn WM, Yasutake T, Kuo WL, Warren RS, Collins C, Tomita M, et al. Chromosome arm 20q gains and other genomic alterations in colorectal cancer metastatic to liver, as analyzed by comparative genomic hybridization and fluorescence in situ hybridization. Genes Chromosomes Cancer. 1999;25:82–90. [DOI] [PubMed] [Google Scholar]
- 23. Diep CB, Parada LA, Teixeira MR, Eknaes M, Nesland JM, Johansson B, et al. Genetic profiling of colorectal cancer liver metastases by combined comparative genomic hybridization and G‐banding analysis. Genes Chromosomes Cancer. 2003;36:189–97. 10.1002/gcc.10162 [DOI] [PubMed] [Google Scholar]
- 24. Hermsen M, Postma C, Baak J, Weiss M, Rapallo A, Sciutto A, et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology. 2002;123:1109–19. 10.1053/gast.2002.36051 [DOI] [PubMed] [Google Scholar]
- 25. Sugai T, Osakabe M, Sugimoto R, Eizuka M, Tanaka Y, Yanagawa N, et al. A genome‐wide study of the relationship between chromosomal abnormalities and gene expression in colorectal tumors. Genes Chromosomes Cancer. 2021;60:250–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang Z, Mou Q, Pan Z, Zhang Q, Gao G, Cao Y, et al. Identification of candidate diagnostic and prognostic biomarkers for human prostate cancer: RPL22L1 and RPS21. Med Oncol. 2019;36:56. 10.1007/s12032-019-1283-z [DOI] [PubMed] [Google Scholar]
- 27. Pelletier J, Thomas G, Volarević S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18:51–63. 10.1038/nrc.2017.104 [DOI] [PubMed] [Google Scholar]
- 28. Kessel R, Vlachos A, Lipton JM. Ribosomopathy association with colorectal cancer. Gastroenterology. 2015;148:258. 10.1053/j.gastro.2014.08.046 [DOI] [PubMed] [Google Scholar]
- 29. Nieminen TT, O'Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology. 2014;147:595–8. 10.1053/j.gastro.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Broderick P, Dobbins SE, Chubb D, Kinnersley B, Dunlop MG, Tomlinson I, et al. Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients – a systematic review. Gastroenterology. 2017;152:75–7. 10.1053/j.gastro.2016.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saravanan S, Islam VI, Thirugnanasambantham K, Sekar D. In silico identification of human miR 3654 and its targets revealed its involvement in prostate cancer progression. Microrna. 2016;5:140–5. 10.2174/2211536605666160610094230 [DOI] [PubMed] [Google Scholar]
- 32. Ronchetti D, Mosca L, Cutrona G, Tuana G, Gentile M, Fabris S, et al. Small nucleolar RNAs as new biomarkers in chronic lymphocytic leukemia. BMC Med Genomics. 2013;6:27. 10.1186/1755-8794-6-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nallar SC, Kalvakolanu DV. Regulation of snoRNAs in cancer: close encounters with interferon. J Interferon Cytokine Res. 2013;33:189–98. 10.1089/jir.2012.0106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benelli R, Costa D, Mastracci L, Grillo F, Olsen MJ, Barboro P, et al. A promising target to limit the local invasiveness of colorectal cancer. Cancers (Basel). 2020;12:971. 10.3390/cancers12040971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dong X, Lin Q, Aihara A, Li Y, Huang CK, Chung W, et al. Aspartate β‐hydroxylase expression promotes a malignant pancreatic cellular phenotype. Oncotarget. 2015;6:1231–48. 10.18632/oncotarget.2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of our study are available from the corresponding author upon reasonable request.