

Abstract
2‐O‐Glucosylglycerol is accumulated by various bacteria and plants in response to environmental stress. It is widely applied as a bioactive moisturising ingredient in skin care products, for which it is manufactured via enzymatic glucosylation of glycerol by the sucrose phosphorylase from Leuconostoc mesenteroides. This industrial process is operated at room temperature due to the mediocre stability of the biocatalyst, often leading to microbial contamination. The highly thermostable sucrose phosphorylase from Bifidobacterium adolescentis could be a better alternative in that regard, but this enzyme is not fit for production of 2‐O‐glucosylglycerol due to its low regioselectivity and poor affinity for glycerol. In this work, the thermostable phosphorylase was engineered to alleviate these problems. Several engineering approaches were explored, ranging from site‐directed mutagenesis to conventional, binary, iterative or combinatorial randomisation of the active site, resulting in the screening of ∼3,900 variants. Variant P134Q displayed a 21‐fold increase in catalytic efficiency for glycerol, as well as a threefold improvement in regioselectivity towards the 2‐position of the substrate, while retaining its activity for several days at elevated temperatures.
Keywords: biocatalysis, glucosylglycerol, glycoside phosphorylases, protein engineering, sucrose phosphorylase
Osmolyte synthesis at elevated temperatures: 2‐O‐Glucosylglycerol is produced on industrial scale using the promiscuous transglycosylation activity of sucrose phosphorylase. We engineered the catalytic efficiency and regioselectivity of a sucrose phosphorylase that is far more thermostable than the one currently used in industry.

Introduction
Osmolytes are a structurally diverse group of highly soluble molecules that can be accumulated inside cells to counteract the effects of environmental stress. [1] Drought or high salinity can trigger rapid fluxes of cell water along the osmotic gradient, which can lead to reduction in turgor and dehydration of the cytoplasm. By importing or synthesising osmolytes to high intracellular concentrations, the osmotic pressure can effectively be counterbalanced. Furthermore, these compounds do not disturb cellular functions when amassed; hence their alternative name, compatible solutes. One of the naturally occurring osmolytes that has raised commercial interest is 2‐O‐(α‐d‐glucopyranosyl)‐glycerol (2‐GGo). Like many osmolytes, 2‐GGo has a strong stabilising effect and it can be used as an additive to protect proteins from undergoing inactivation through elevated temperature or freeze‐drying, [2] or to enhance soluble expression of aggregation‐prone proteins.[ 3 , 4 ] It also shows potential as a low‐calorie sweetener with low cariogenicity, and it was suggested to contribute to the sweet taste of sake. [5] However, 2‐GGo is primarily applied today in a range of skin care products, where it acts as a moisturizing ingredient that was demonstrated to promote skin elasticity, smoothness and thickness.[ 6 , 7 ]
2‐GGo is obtained on industrial scale through biocatalytic glucosylation of glycerol from sucrose and sold by the German company Bitop AG under the trade name Glycoin.[ 8 , 9 ] The production process is based on a side activity of the sucrose phosphorylase (SP) from Leuconostoc mesenteroides (LmSP) (Scheme 1). LmSP follows a double displacement mechanism where a carboxylic residue attacks the anomeric carbon of sucrose, forming a covalent glucosyl‐enzyme intermediate that can subsequently be attacked by an incoming acceptor molecule. That role is fulfilled by phosphate under natural conditions, leading to α‐glucose 1‐phosphate as product. However, because LmSP is rather promiscuous, it can transfer the glucosyl group of sucrose to plenty of other molecules as well, including glycerol. [10] Thanks to a careful optimisation of the reaction conditions in earlier work by Goedl et al., the reaction with glycerol can be performed with great efficiency. [8] The competing hydrolysis reaction, where water intercepts the covalent intermediate, is strongly kinetically suppressed in the presence of high concentrations of glycerol, limiting the hydrolysis of sucrose. [8] Secondary hydrolysis of the transfer product by LmSP is not observed under the optimised conditions either. Thanks to these peculiarities, transfer yields of ∼90 % can be achieved with ease.
Scheme 1.

Synthesis of glucosylglycerol (GGo) using the promiscuous transglycosylation of sucrose phosphorylases.
There are two major downsides to the use of LmSP as catalyst for the large‐scale synthesis of 2‐GGo. The first is its mediocre thermostability causing rapid inactivation at even modestly elevated temperatures. The process thus has to be operated at room temperature, which in turn increases the risk of microbial contamination substantially. Second, a large excess of glycerol must be added to the reaction in order to achieve satisfactory reaction rates, implying a low affinity of the enzyme for the substrate. As a result, much of the glycerol is left unconverted. Very recently, an alternative two‐enzyme process was suggested where the native phosphorolytic reaction of LmSP was coupled to the activity of a natural glucosylglycerol phosphorylase (GGoP), which can generate the osmolyte very efficiently but requires α‐glucose 1‐phosphate as donor substrate instead of sucrose. [11] This coupled system was claimed to reach far higher productivities than the current industrial process even at low glycerol concentrations, but the low thermostability of LmSP was identified as an important hurdle of this reaction set‐up as well. [12] Hence, there is a clear and economically relevant need for an SP that is better suited for the industrial production of 2‐O‐glucosylglycerol.
An attractive alternative SP would be the one from Bifidobacterium adolescentis (BaSP), which can catalyse the same phosphorolysis or transglycosylation reactions as LmSP, but with a far higher thermostability. The half‐life time of wild‐type BaSP at 60 °C is 12 h and successful efforts have been made to increase that number even further by means of immobilisation and mutagenesis.[ 13 , 14 , 15 ] Despite this obvious advantage, BaSP is not currently applied for industrial production of 2‐GGo. This work describes the development of a useful BaSP variant for the synthesis of 2‐GGo at elevated temperatures.
Results and Discussion
Comparison of sucrose phosphorylases
To evaluate whether BaSP could be a suitable substitute for LmSP for the production of 2‐GGo from sucrose and glycerol, the kinetic parameters in this transglycosylation reaction were compared for both enzymes. For LmSP, the enzyme used in industry today, the turnover number and K M for glycerol were 2.4±0.1 s−1 and 0.97±0.09 M, respectively. However, no characteristic Michaelis‐Menten profile could be obtained for BaSP because its dependency on substrate concentration was almost linear up to 5 M. Nevertheless, the estimated K M of 10.2±3.2 M and turnover number of 1.5±0.1 s−1 indicate a 16‐fold lower catalytic efficiency compared to LmSP. Furthermore, BaSP suffered from a lower regioselectivity that results in a product mixture containing 65 % of 2‐GGo and 35 % of its isomer 1‐O‐(α‐d‐glucopyranosyl)‐glycerol (1‐GGo). In contrast, LmSP generates ∼88 % of the desired product. [16] Even though both enzymes are capable of catalysing the target reaction, BaSP clearly is not fit to compete with its less thermostable homologue in terms of catalytic performance.
Rational engineering inspired by natural phosphorylases
Although BaSP and LmSP belong to the same subfamily of glycoside hydrolases (GH13_18), [10] they only share 35 % sequence identity. Some of the differences between the two enzymes are undoubtedly responsible for their variation in activity, substrate affinity, regioselectivity and thermostability. A thorough comparison of their amino acid sequences could thus reveal which positions of BaSP should be mutated in order to introduce some of the favourable properties of LmSP. Furthermore, glucosylglycerol phosphorylase (GGoP) from the same subfamily can also be included in the analysis to pinpoint residues responsible for the correct binding of glycerol. Indeed, the GGoP from Marinobacter adhaerens (∼37 % sequence identity to LmSP and BaSP) is able to generate 2‐GGo from α‐glucose 1‐phosphate and glycerol with perfect regioselectivity and much higher activity (39±2 s−1) as well as affinity for glycerol (7±1 mM). [11] Unfortunately, GGoP is not viable for the industrial production of 2‐GGo because of its low thermostability and, most importantly, because it cannot use the cheap substrate sucrose as glucosyl donor. Nevertheless, its sequence may contain valuable information about the determinants for stimulating binding of glycerol in the preferred productive orientation in the active site.
A sequence alignment of BaSP, LmSP and GGoP was generated and scrutinised to identify hotspots for mutagenesis. The search was focused on first‐shell residues that are in direct contact with the substrate, as well as second‐shell residues that may influence substrate binding indirectly (Figure S1). The alignment shows that all residues involved in binding of the glucosyl moiety in subsite −1 (such as Asp50, His289 and Arg399, numbering according to BaSP) are conserved among the three enzymes. Contrarily, the residues that are known to be essential for binding the fructosyl moiety of sucrose in SP (such as Tyr132 and Asp342) [17] are unsurprisingly replaced by a different amino acid in GGoP. However, in the context of this study, the most relevant residues are those that are identical in the two phosphorylases with high selectivity towards 2‐GGo (i. e. LmSP and GGoP), but are different in the enzyme with lower regioselectivity (i. e. BaSP). Seven positions in the first or second shell fit that criterion (Table 1, Figure 1). Three of them (i. e. Pro134, Arg135 and Leu343) are part of two highly dynamic loops that constitute an essential part of the glucosyl acceptor site of BaSP. [18] Positions 135 and 343 are especially important for binding of the phosphate group when SP catalyses its native phosphorolytic reaction, whereas position 134 has a moderate and indirect influence on acceptor substrate recognition. [17] Given that SP does not need to bind phosphate in order to perform the relevant transglycosylation reaction, these residues can safely be mutated despite their central position in the active site. The four other residues of interest are Ser155, Val194, Ile231 and Asn397, which are further away from the binding pocket and their exact role has not yet been elucidated.
Table 1.
Performance of BaSP variants inspired by other natural phosphorylases.[a]
|
Variant |
Regioselectivity[b] |
Relative yield [%][c] |
|---|---|---|
|
BaSP |
1.9±0.2 |
7±1 |
|
P134R |
5.3±0.4 |
100±5 |
|
R135 K |
1.9±0.2 |
30±3 |
|
S155T |
1.9±0.3 |
9±2 |
|
V194F |
n.a.[d] |
n.a.[d] |
|
I231P |
2.1±0.3 |
6±1 |
|
L343I |
2.0±0.2 |
13±4 |
|
N397E |
2.1±0.2 |
7±2 |
[a] After 24 h incubation of 5 % (v/v) heat‐purified lysate with 0.35 M sucrose and 2 M glycerol. [b] Ratio of 2‐GGo over 1‐GGo. [c] Yield for 2‐GGo relative to the yield obtained using variant P134R, which was 64 % with respect to sucrose. [d] No activity detected.
Figure 1.
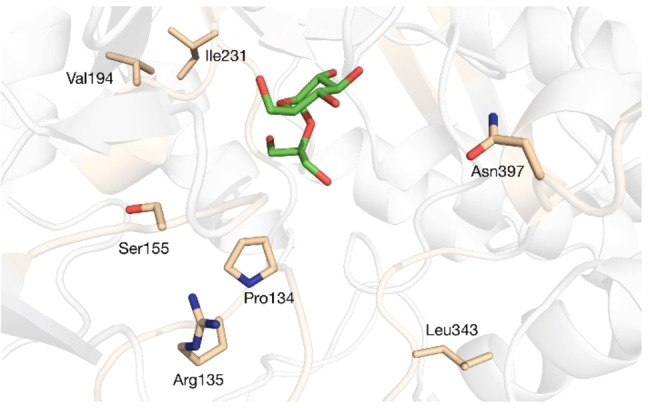
Docking of 2‐GGo in the active site of BaSP (PDB code 2GDV). Shown are the residues selected for mutagenesis based on a sequence comparison of BaSP to LmSP and GGoP.
The seven residues of interest in BaSP were substituted by their counterparts present at the corresponding positions in LmSP and GGoP. These substitutions were introduced in a variant of BaSP that was previously engineered to improve its thermostability even further without any observed impact on its catalytic behaviour. [14] The variants were expressed, partially purified by heat treatment, and used to synthesise glucosylglycerol from sucrose and glycerol. Mutant V194F seemingly experienced a total loss of transglycosylation activity (Table 1). Of the other enzymes, one demonstrated a significant change in both regioselectivity and activity. Indeed, the product ratio between 2‐GGo and 1‐GGo increased from 1.9 in BaSP to 5.3 in variant P134R, while the yield of 2‐GGo under the screening conditions increased 14‐fold (Table 1).
To verify whether better properties may be obtained by the cooperative action between multiple mutations, [19] a binary combinatorial library was designed where only two codons were allowed at each of the seven target positions: one that represents the wild‐type amino acid in BaSP, and another that represents the amino acid found in LmSP and GGoP. Approximately 400 clones were screened in microtiter plate format, which should ensure sufficient coverage of this binary library with a theoretical size of 128 variants. Unfortunately, none of these enzymes displayed a higher regioselectivity than variant P134R. Furthermore, the best results were obtained when mutation P134R was present, which thus seems to be the determining factor. Oddly, two of those hits carried the V194F mutation that was earlier demonstrated to eliminate transglycosylation activity entirely, suggesting that its destructive nature can be compensated by other substitutions.
Randomisation of the glucosyl acceptor site
The comparison between enzymes with high and low regioselectivity for 2‐GGo exposed the positive effect of the mutation P134R. In pursuit of further improvements, the hotspot position 134 was fully randomised to determine whether other amino acids besides arginine could be more beneficial. One could argue that arginine is probably the optimal residue at the corresponding position in the active site of GGoP, which has been optimised for binding 2‐GGo by natural evolution, but the same is not necessarily true in the active site of SPs. Indeed, the ability of LmSP to synthesise 2‐GGo with high regioselectivity and affinity with an active site that is actually finetuned for processing sucrose is probably merely coincidental, given that this side activity is not relevant in vivo. Hence, more suitable residues may exist. One microtiter plate of 96 clones was screened for library P134X, using a relatively low concentration of glycerol (100 mM) to provide a competitive advantage to variants that exhibit a higher affinity for the substrate, in accordance with the first law of directed evolution (“you get what you screen for”). [20] Three hits were found, expressed at larger scale, and evaluated in more detail (Table 2). Although none of them turned out to be significantly more selective than P134R, markedly higher yields could be obtained in some cases. In particular, variant P134Q offered yields that were 33‐fold or 1.75‐fold higher than that of BaSP or variant P134R, respectively.
Table 2.
Performance of selected BaSP variants.[a]
|
Variant |
Regioselectivity[b] |
Relative yield [%][c] |
|---|---|---|
|
BaSP |
1.9±0.2 |
3±0.2 |
|
P134R |
5.3±0.4 |
57±3 |
|
P134C |
4.3±0.2 |
65±2 |
|
P134G |
3.0±0.4 |
90±5 |
|
P134Q |
6.0±0.4 |
100±4 |
|
R135P |
6.1±0.5 |
13±2 |
|
L341W |
5.6±0.5 |
80±4 |
|
P134Q+R135P |
4.6±0.3 |
57±4 |
|
P134Q+L341W |
4.6±0.5 |
8±1 |
|
R135P+L341W |
5.3±0.4 |
5±0.2 |
|
P134Q+R135P+L341W |
5.3±0.3 |
<1 |
[a] After 24 h incubation of 0.5 mg/mL purified enzyme with 0.35 M sucrose and 2 M glycerol. [b] Ratio of 2‐GGo over 1‐GGo. [c] Yield for 2‐GGo relative to the yield obtained using variant P134Q, which was 95 % with respect to sucrose.
Seven additional positions in the two dynamic acceptor site loops of BaSP were evaluated next. Libraries Y132X, R135X, L341X, D342X, L343X, Y344X and Q345X were already constructed in earlier work, where BaSP was engineered for the efficient production of the rare sugar kojibiose. [21] Their re‐screening for variants with an increased regioselectivity towards 2‐GGo revealed two new hits, i. e. R135P and L341 W (Table 2). Although their selectivity is comparable to that of P134Q, they yield fewer products. All combinations of these three favourable mutations were made, but no further improvements could be observed (Table 2).
Because the mutational effects of P134R, R135P and L341W on selectivity were not additive, the route of screening additional single‐site saturation libraries was abandoned. Instead, we opted for an iterative approach where the same residues were again randomised, but this time using the best mutant P134Q as starting point. [22] Doing so, the synergy between P134Q and the second mutation could immediately be taken into consideration. Seven new libraries were constructed (P134Q+Y132X, R135X, L341X, D342X, L343X, Y344X or Q345X) and 96 clones were screened for each. Additionally, three large libraries were constructed where two positions were randomised simultaneously. The underlying idea was to sample sequence space for synergistic combinations of mutations that would not be retained as hits individually, but that could greatly improve the enzyme‘s properties when they occur together. The target residue combinations were chosen rationally, based on the proximity of these residues to glycerol and to one another in the three‐dimensional structure of BaSP.[ 18 , 23 ] In two libraries, hotspot residue Pro134 was randomised together with either Arg135 or Tyr344. In the third, Tyr344 and Gln345 were simultaneously saturated. Each library represented a high degree of structural diversity with 400 different possible variants. Approximately 800 clones were screened in each case. Despite this effort, none of the variants displayed a convincingly higher regioselectivity or product yield when compared to variant P134Q.
Characterisation of variant P134Q
The most favourable variant from the extensive mutagenesis study, in terms of regioselectivity and product yield after 24 h, turned out to be variant P134Q. Hence, this enzyme was characterized in more detail, including its kinetic parameters on glycerol (Table 3). The affinity for this acceptor substrate was found to have improved more than 5‐fold compared to that of the starting enzyme, which demonstrates that our strategy of screening variants at low glycerol concentrations was successful. Moreover, the P134Q variant offers a higher turnover number than both BaSP and LmSP.
Table 3.
Apparent kinetic parameters for glycerol as acceptor.[a]
|
Enzyme |
K M [M] |
vmax [U mg−1] |
k cat [s−1] |
k cat/K M [M−1s−1] |
|---|---|---|---|---|
|
LmSP[b] |
0.97±0.09 |
2.6±0.1 |
2.4±0.1 |
2.5 |
|
BaSP[c] |
10.2±3.2 |
1.7±0.2 |
1.5±0.1 |
0.15 |
|
BaSP P134Q[c] |
1.85±0.25 |
6.4±0.4 |
6.0±0.4 |
3.2 |
[a] With 0.35 M sucrose as donor at pH 7 and [b] 30 °C or [c] 52 °C.
To elucidate possible reasons for the drastic increase in affinity for glycerol, 2‐GGo was docked in the crystal structure of BaSP and in a model of the P134Q variant (Figure 2). Molecule B from the BaSP structure with PDB code 2GDV was used as template, because this subunit captures the state of the acceptor site as it binds the incoming glucosyl acceptor substrate. [18] In the mutant model, the introduced residue Gln134 recognizes one of the hydroxyl groups of glycerol through a hydrogen bond and also appears to shift the position of His234, allowing that residue to establish a hydrogen bond as well. As both hydrogen bonds have a predicted binding energy of about 15 kJ/mol, these two new interactions could well explain the lower KM of the mutant enzyme.
Figure 2.

Docking of 2‐GGo in the active site of (a) BaSP and (b) a model of variant P134Q.
Interestingly, the high turnover number of variant P134Q compensates for its lower affinity for glycerol in comparison to LmSP. The mutant enzyme shows a higher initial specific activity than LmSP at any glycerol concentration over 100 mM. Furthermore, hydrolysis of the sucrose is strongly suppressed in the presence of 2 M glycerol in both enzymes. As a result, high conversion of sucrose (>95 %) could be achieved within 24 h under relevant process conditions (0.35 M sucrose, 2 M glycerol, 0.15 mg mL−1 enzyme), with a transfer yield to glucosylglycerol of 92 % with respect to sucrose (Figure S2). The regioselectivity for 2‐GGo was 83±3%. Taken together, these results closely match those obtained using LmSP in earlier work.[ 8 , 16 ] Additionally, the mutation does not affect the superior thermostability of BaSP. The P134Q variant was shown to retain all of its activity after 72 h incubation at 52 °C, whereas LmSP loses half of its activity after just 10 minutes at that temperature (Figure 3). At 60 °C, BaSP P134Q retains over half of its activity for 48 h (Figure S3). Replacing LmSP by the novel enzyme would thus increase the robustness of the biocatalyst considerably, and allow operating the process at a higher temperature, thereby avoiding microbial contamination and lowering the viscosity of the reaction mixture.
Figure 3.
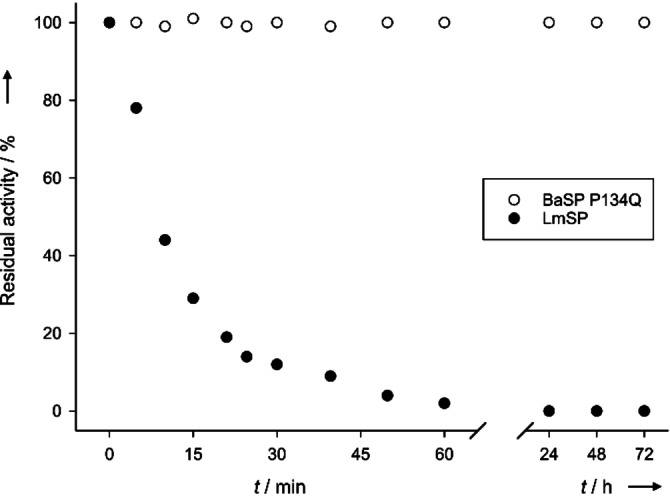
Kinetic stability of BaSP P134Q and LmSP. The enzymes were incubated at 52 °C and samples were taken regularly, after which their residual activity was compared to that of untreated enzyme (CV<10 %).
Conclusion
In this work, several strategies are described for the engineering of the thermostable sucrose phosphorylase from B. adolescentis for improved performance during the synthesis of 2‐GGo. The best variant (P134Q) showed a 5‐fold increase in affinity for glycerol as well as a 3‐fold improvement in regioselectivity towards the 2‐position of the acceptor. Moreover, its transglycosylation activity at the substrate concentration used in industry is more than 2‐fold higher than that of LmSP, the enzyme currently used in the process. The new variant was uncovered by comparing the sequences of related enzymes and then randomising the most promising hotspot. Other engineering endeavours unearthed a number of additional hits, but none could achieve better results than BaSP P134Q. Our results yet again illustrate that natural enzyme sequences are a rich source of information for engineers to draw inspiration from, as well as the power of enzyme engineering for improving biocatalysts for industrial applications.
Experimental Section
Mutagenesis: All mutations were introduced in a constitutive expression plasmid containing the gene of a thermostable BaSP variant (pCXP34 h_BaSP_NFLI; BaSP Q331E/R393 N/D445P/D446T/Q460E/E485H).[ 14 , 24 ] For the individual mutations and the iterative and combinatorial libraries, DNA fragments containing the desired mutations were first amplified using the primers in Table S1 and Q5 polymerase (standard protocol; New England Biolabs). Next, these fragments were used as megaprimers in a whole‐plasmid PCR to obtain mutated plasmids. The PCR mixtures contained 0.05 U/μL PfuUltra high‐fidelity DNA polymerase (Agilent), 0.2 mM deoxynucleoside triphosphate mix, 50 ng template and 20 ng megaprimer in a total volume of 50 μL. The program started with an initial denaturation step of 3 min at 95 °C, then 20 cycles of 30 s at 95 °C, annealing for 1 min at 65 °C and extension for 2 min/kb at 72 °C, followed by one final extension of 10 min at 72 °C. Template DNA was digested by 20 U DpnI (New England Biolabs) for 1 h at 37 °C. For libraries P134Q+L343X, P134Q+Y344X and P134X+Y344X, the mutagenic megaprimer fragments were ordered from Integrated DNA Technologies instead (Table S2). Library P134Q+D342X was created using the QuickChange Site‐Directed Mutagenesis kit (Agilent) and the primers in Table S1. The single‐site saturation libraries were available in‐house and their construction was described elsewhere. [21] The binary combinatorial library was created via gene synthesis (SGI‐DNA).
High‐performance anion exchange chromatography: Reaction samples were monitored by high‐performance anion exchange chromatography (HPAEC; Dionex ICS‐6000, Thermo Scientific) with a CarboPac PA20 pH‐stable column and pulsed amperometric detection. A 10‐μL sample was analysed at a constant flow rate of 0.5 mL/min at 30 °C. The eluent composition was 15 mM NaOH for 10 min (isocratic).
Screening of libraries: Individual colonies were picked from solid LB medium containing ampicillin (100 μg/mL) using an automated colony picker (QPix2, Genetix) and inoculated into sterile 96‐well flat‐bottomed microtiter plates (Nunc) containing 175 μL LB (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) supplemented with 100 μg/mL ampicillin. The plates were grown at 37 °C and 250 rpm (50 mm shaking amplitude) for 16 h, replicated into new plates with fresh medium and grown for another 16 h. Cells were harvested by centrifugation (5000 g, 20 min), the supernatant was discarded and the cell pellets were frozen at −20 °C for at least 2 h. After thawing, cells were lysed by incubation for 1 h at 37 °C after addition of 100 μL lysis buffer (1 mg/mL lysozyme, 0.1 mM phenylmethylsulfonyl fluoride, 50 mM Na2SO4, 4 mM MgSO4, 1 mM ethylenediaminetetraacetic acid and 50 mM 3‐morpholinopropane‐1‐sulfonic acid (MOPS) buffer, pH 7). The plates were centrifuged again. Each screening reaction contained 80 μL cell‐free lysate, 200 mM sucrose and 100 mM glycerol in a total volume of 200 μL. Reactions were incubated at 37 °C for 18 h, inactivated by addition of 200 μL 0.01 M NaOH and centrifuged for 30 min. Supernatants (15 μL) were transferred to deep‐well plates and diluted with ultra‐pure water to a final dilution of 400x. These samples were analysed by HPAEC. For the single‐site saturation libraries, hits were defined as variants that show a regioselectivity ratio (2‐GGo over 1‐GGo)≥3. From each plate of subsequent libraries, the clone that showed the highest yield of 2‐GGo was retained. Cell lysates from those top clones were evaluated again in comparison to lysate containing BaSP P134Q, in terms of yield and regioselectivity ratio.
Enzyme characterisation: The thermostable BaSP variant that was used as engineering template, [14] the novel BaSP variants designed in this study, and LmSP were constitutively expressed by inoculating 2 % of an overnight culture of E. coli CGSC 8974 with the pCXP34 h expression plasmid in 250 mL LB medium containing 100 μg/mL ampicillin in a 1‐liter shake flask. The cultures were incubated at 37 °C and 200 rpm for 8 h, centrifuged and the resulting cell pellets were frozen at −20 °C for at least 2 h. To obtain heat‐purified cell extracts, pellets were thawed and resuspended in 8 mL lysis buffer (1 mg/mL lysozyme, 0.1 mM phenylmethylsulfonyl fluoride, 50 mM MOPS, pH 7), incubated on ice for 30 min and sonicated 3 times for 3 min (Branson sonifier 250, level 3, 50 % duty cycle). Cell debris was removed by centrifugation (20,000 g for 1 h) and the extracts were incubated at 60 °C for 1 h, followed by another hour of centrifugation. Alternatively, lysates could be further purified by nickel‐nitrilotriacetic acid chromatography by using a different lysis buffer (10 mM imidazole, 300 mM NaCl, 1 mg/mL lysozyme, 0.1 mM phenylmethylsulfonyl fluoride, 50 mM phosphate buffer, pH 7.4), processing the lysates as described by the supplier (Thermo Scientific) and subsequently exchanging the buffer to MOPS buffer (pH 7) using 30‐kDa Amicon Ultra centrifugal filters (Merck).
The selectivity and relative yield of BaSP variants was determined by incubating the enzymes (5 % (v/v) for heat‐purified lysates, 0.1–1.0 mg/mL for purified enzymes) with a substrate solution for 24 h at 52 °C in 50 mM MOPS buffer, pH 7, in a total reaction volume of 1 mL. Substrate concentrations were 350 mM sucrose and 2 M glycerol.
Kinetic parameters for glycerol were determined by performing reactions using 0.5 mg/mL purified enzyme with 350 mM sucrose and various concentrations of glycerol at 30 °C (LmSP) or at 52 °C (BaSP) in 50 mM MOPS buffer, pH 7.
All samples were analysed by HPAEC after inactivation using 0.01 M NaOH. Activities and kinetic parameters were determined in triplicate and are shown as the mean ± standard deviation.
Computational analyses: The sequences of BaSP, LmSP and GGoP were aligned by ClustalO using default parameters. [25] Binding of 2‐GGo in BaSP was simulated by ligand docking in YASARA, using the implemented AutoDock VINA module with default parameters, except the numbers of runs which was increased to 100. The most accurate pose was selected on the basis of known interactions in the −1 subsite of sucrose phosphorylase. [10]
Availability of data and materials: Data obtained in this study are available in the Zenodo repository, https://doi.org/10.5281/zenodo.4775448. The plasmid for expression of BaSP P134Q has been deposited at BCCM/GeneCorner plasmid collection with accession number LMBP 12705.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors wish to thank Research Foundation ‐ Flanders (FWO‐Vlaanderen; postdoctoral fellowship J.F.; grant n 12ZD821N) and the European Union's Horizon 2020 research and innovation programme (CARBAFIN, grant n° 761030) for financial support.
J. Franceus, Z. Ubiparip, K. Beerens, T. Desmet, ChemBioChem 2021, 22, 2777.
References
- 1. Kempf B., Bremer E., Arch. Microbiol. 1998, 170, 319–330. [DOI] [PubMed] [Google Scholar]
- 2. Sawangwan T., Goedl C., Nidetzky B., Biotechnol. J. 2010, 5, 187–191. [DOI] [PubMed] [Google Scholar]
- 3. Lentzen G., Schwarz T., Appl. Microbiol. Biotechnol. 2006, 72, 623–634. [DOI] [PubMed] [Google Scholar]
- 4. Rabbani G., Choi I., Int. J. Biol. Macromol. 2018, 109, 483–491. [DOI] [PubMed] [Google Scholar]
- 5. Takenaka F., Uchiyama H., Biosci. Biotechnol. Biochem. 2000, 64, 1821–1826. [DOI] [PubMed] [Google Scholar]
- 6. Thiem J., Scheel O., Schneider G., Eur. Pat. Appl. EP 1997, 770378, 16. [Google Scholar]
- 7. Harada N., Zhao J., Kurihara H., Nakagata N., Okajima K., Biosci. Biotechnol. Biochem. 2010, 74, 759–765. [DOI] [PubMed] [Google Scholar]
- 8. Goedl C., Sawangwan T., Mueller M., Schwarz A., Nidetzky B., Angew. Chem. Int. Ed. 2008, 47, 10086; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 10240–10089. [Google Scholar]
- 9. Bolivar J. M., Luley-Goedl C., Leitner E., Sawangwan T., Nidetzky B., J. Biotechnol. 2017, 257, 131–138. [DOI] [PubMed] [Google Scholar]
- 10. Franceus J., Desmet T., Int. J. Mol. Sci. 2020, 21, 2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franceus J., Decuyper L., D'hooghe M., Desmet T., Appl. Microbiol. Biotechnol. 2018, 102, 3183–3191. [DOI] [PubMed] [Google Scholar]
- 12. Zhang T., Yang J., Tian C., Ren C., Chen P., Men Y., Sun Y., J. Agric. Food Chem. 2020, 68, 15249–15256. [DOI] [PubMed] [Google Scholar]
- 13. Cerdobbel A., Desmet T., De Winter K., Maertens J., Soetaert W., J. Biotechnol. 2010, 150, 125–130. [DOI] [PubMed] [Google Scholar]
- 14. Cerdobbel A., De Winter K., Aerts D., Kuipers R., Joosten H. J., Soetaert W., Desmet T., Protein Eng. Des. Sel. 2011, 24, 829–834. [DOI] [PubMed] [Google Scholar]
- 15. De Winter K., Cerdobbel A., Soetaert W., Desmet T., Process Biochem. 2011, 46, 1074–1078. [Google Scholar]
- 16. Schwaiger K. N., Puschmann M. C., Striedner G., Nidetzky B., Microb. Cell Fact. 2021, 20, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verhaeghe T., Diricks M., Aerts D., Soetaert W., Desmet T., J. Mol. Catal. B 2013, 96, 81–88. [Google Scholar]
- 18. Mirza O., Skov L. K., Sprogøe D., van den Broek L. a. M., Beldman G., Kastrup J. S., Gajhede M., J. Biol. Chem. 2006, 281, 35576–35584. [DOI] [PubMed] [Google Scholar]
- 19. Franceus J., Verhaeghe T., Desmet T., J. Ind. Microbiol. Biotechnol. 2017, 44, 687–695. [DOI] [PubMed] [Google Scholar]
- 20. Arnold F. H., Angew. Chem. Int. Ed. 2019, 58, 14420–14426; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14558–14426. [Google Scholar]
- 21. Verhaeghe T., De Winter K., Berland M., De Vreese R., D'hooghe M., Offmann B., Desmet T., Chem. Commun. 2016, 52, 3687–3689. [DOI] [PubMed] [Google Scholar]
- 22. Reetz M. T., Carballeira J. D., Nat. Protoc. 2007, 2, 891–903. [DOI] [PubMed] [Google Scholar]
- 23. Sprogoe D., van den Broek L. A. M., Mirza O., Kastrup J. S., Voragen A. G. J., Gajhede M., Skov L. K., Biochemistry 2004, 43, 1156–1162. [DOI] [PubMed] [Google Scholar]
- 24. Aerts D., Verhaeghe T., De Mey M., Desmet T., Soetaert W., Eng. Life Sci. 2011, 11, 10–19. [Google Scholar]
- 25. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G., Mol. Syst. Biol. 2011, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information