

Abstract
Functional materials engineered to degrade upon triggering are in high demand due their potentially lower impact on the environment as well as their use in sensing and in medical applications. Here, stimuli‐responsive polymers are prepared by decorating a self‐immolative poly(dithiothreitol) backbone with pendant catechol units. The highly functional polymer is fashioned into stimuli‐responsive gels, formed through pH‐dependent catecholato–metal ion cross‐links. The gels degrade in response to specific environmental changes, either by addressing the pH responsive, non‐covalent, catecholato–metal complexes, or by addition of a thiol. The latter stimulus triggers end‐to‐end depolymerization of the entire self‐immolative backbone through end‐cap replacement via thiol–disufide exchanges. Gel degradation is visualized by release of a dye from the supramolecular gel as it itself is converted into smaller molecules.
Keywords: catechol, hydrogel, polymers, self-immolative, stimuli-responsive
Catechol‐functionalized self‐immolative poly(disulfides) are synthesized and fashioned into dual‐responsive gels through formation of catecholato–Al3+ cross‐links. Two distinct pathways to cargo release are engineered, namely the depolymerization in a self‐immolative fashion, or breaking catecholato–Al3+ cross‐links at low pH. Both gel degradation pathways lead to release of bound dye molecules on demand.

Introduction
Multi‐responsive gels are an attractive class of materials with applications within areas such as delivery of therapeutics, [1] actuation, [2] and sensing. [3] The ability to engineer a material to show a specific response when exposed to certain conditions such as the presence of a specific molecule or changes in the local environment eliminates the need for human intervention, as the system responds autonomously, with a pre‐programmed response. Such materials can consist of two fundamental components, a polymeric backbone structure and cross‐links responsible for gelation. For delivery of small molecules, a vanishing gel is attractive as it delivers not only cargo, but also breaks down to small molecules at the destination of the cargo. This raises demand for a degradable polymer backbone, capable of forming a gel.
Recently, highly functional materials were reported in which both the polymer backbone and the cross‐links possessed responsive chemistries. Zhang et al. utilized thermal ring opening polymerization of thioctic acid and subsequent carboxylate‐Fe3+ cross‐linking to generate a network polymer possessing two levels of self‐healing. Rapid exchange in carboxylate‐Fe3+ cross‐links was responsible for fast formation of weak bonds in broken interfaces, and a slower disulfide exchange between backbones generated new covalent bonds at interfaces. [4]
Polyamines functionalized by polyphenols such as l‐3,4‐dihydroxyphenylalanine formed doubly pH‐responsive hydrogels based on the acid/base chemistry of the amines and the pH‐dependent metal coordination of the polyphenols.[ 5 , 6 ] Lu et al. reported a multi‐responsive gel capable of actuation by virtue of a polyelectrolyte backbone, while also possessing the ability to be photocleaved and photocross‐linked through pendant coumarin groups. [7] Chen et al. presented a multi‐responsive system containing benzoxaborole ester cross‐links, responsive towards pH, ATP, fructose, and H2O2. [8]
While the above examples contain high levels of responsivity, they all leave the intact polymer backbones after triggering responses; full deconstruction of the polymer system would in many cases be advantageous. In literature, numerous examples of polymeric systems with such a property are reported. For example, disulfide‐linked cyclodextrins have been shown to exhibit responsivity toward glutathione, resulting in cleavage of polymer cross‐links. [9] Chain shattering polymers that respond to either UV or thiols and released drugs/monomeric units upon exposure to triggering conditions have been reported.[ 10 , 11 ] Zhang et al. further reported a poly(thioctic acid) material that could enter in a recycling loop complete with polymer degradation, monomer recycling, and repolymerization. [12] Recently, self‐immolative polymers (SIPs) have emerged as a unique class of stimuli‐responsive polymers with the attractive trait of controlled end‐to‐end depolymerization upon demand. [13] Reported examples include poly(benzyl carbonates) used in sensors, [14] poly(benzyl carbamates) in point‐of‐care devices, [15] poly(benzyl ethers) utilized for reversible adhesion, [16] in plastics recycling, [17] and in antibacterials, [18] poly(phthalaldehydes) in smart composites, [19] polyglyoxylates exploited in degradable packaging, [20] and poly(dithiothreithol) as vanishing plastic that depolymerizes into water‐soluble small molecules. [21]
In terms of responsive cross‐links, dynamic covalent bonds such as boronate esters, [22] imines, [23] and Diels–Alder adducts [24] have been explored, contributing to desirable material properties such as self‐healing. [25] Non‐covalent cross‐links, such as catecholato–metal complexes, or chelates of carboxylates and metal ions similarly result in formation of self‐healing gels.[ 4 , 26 , 27 ] Similar properties are also obtained through cyclodextrin host–guest interactions. [28]
Herein, our aim is to synthesize fully degradable and dual‐responsive gels addressing both the polymer backbone and cross‐linking chemistry. As for the choice of responsive backbone, we chose a SIP, given its inherent ability to be depolymerized into monomeric species in response to specific stimuli. SIP backbones are a promising class of fully degradable polymers, enabling reuse of the product at end‐of‐life. Recently, we expanded the SIP family with a new member, namely a self‐immolative disulfide backbone, poly(dithiothreitol) (pDTT), end‐capped with thiopyridine‐disulfide bonds (see structure in Scheme 1 a). [21] An attractive trait of pDTT is the ease by which it is prepared on gram scale through simple solid‐state synthesis with no need of rigorously dry and inert conditions. In addition, the backbone of pDTT has hydroxy moieties, which are apt for further functionalization, making pDTT a strong candidate for a degradable scaffold in highly functional and responsive polymer systems. A third prominent feature is its rapid depolymerization in response to external stimuli. Specifically, adding dithiothreitol (DTT) results in uncapping (i.e. initiation of depolymerization) through thiol–disulfide exchange with the activated thiopyridine disulfide bonds of the end‐cap moieties (see Scheme 1 b).
Scheme 1.

a) Functionalization of pDTT using EDC coupling of protected dihydrocaffeic acid, followed by deprotection to reveal the catecholic moiety. b) Thiol‐induced/base‐catalyzed depolymerization of catechol‐modified poly(dithiothreitol) to produce catechol‐modified cDTT via cyclization reactions upon end‐cap removal.
As for the choice of cross‐linking chemistry, we find the catechol system particularly appealing. Catechol excels with its inherently versatile chemistry involving interaction through either hydrogen bonding, π–π stacking, or cross‐linking through metal ion chelation.[ 29 , 30 ] Both catecholato–boronate [31] and catecholato–metal complexes[ 27 , 29 , 32 ] are highly responsive, and have been utilized as dynamic cross‐links in functional materials. The catecholato–metal complexation with trivalent metal ions is dependent on the redox state [33] of catechol, and solution pH. Agergaard et al. utilized both properties to synthesize detachable polymer brushes, tethered to surfaces through catecholato–metal compexes. [34] Mono‐catecholato–metal species are predominant at low pH, and tris‐catecholato–metal complexes are abundant in alkaline environments. When catechol is attached to a polymer backbone, complexation with metal ions results in cross‐links to give a pH‐responsive gel. The gels have desirable properties such as self‐healing, owing to the dynamic bond exchange nature of the catecholato–metal bond. [35] The physical properties of the catecholato–metal complex, for example, color and rupture force, depend on the metal ion used and its exact d‐electron structure.[ 6 , 36 ]
Specifically, we synthesize a dual‐responsive polymer and gel, denoted pDTT‐Cat, where the versatile and pH‐responsive metal ion cross‐linking chemistry of catechol is merged with the SIP backbone of pDTT that provides on‐demand degradation through self‐immolative depolymerization. We demonstrate how depolymerization can result in release of rhodamine 6G dye from gels, due to material degradation to small molecules. Concurrently, gel liquefaction can be achieved when exposing the material to low pH, where the catecholato–metal cross‐links are broken, resulting in disintegration of the gel, and dye release. Thus, the two mechanisms of degradation are promoted under different conditions, either at high pH in a base‐catalyzed, thiol‐induced depolymerization, or at low pH through the transition from tris‐ to mono‐catecholato–metal ion complexes, effectively reversing cross‐linking.
Results and Discussion
Scheme 1 a outlines the protocol for synthesizing catechol‐modified pDTT, pDTT‐Cat x , where the subscript designates the catechol content which could be adjusted from 0 to 100 %. First, pDTT was produced using the previously described solid‐state synthesis, applying mechanical mixing (i.e. a speedmixer for 3 min) to a mixture of monomeric DTT and a thiol–disufide activating reagent, 2,2′‐dithiodipyridine. [21]
Next, the carboxylic acid group on protected dihydrocaffeic acid (DHCA) was esterified with the alcohol groups on the pDTT backbone through a Steglich esterification. Conversion of backbone hydroxy groups was found to be quantitative when using 4‐dimethylaminopyridine (DMAP) as esterification catalyst and 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride (EDC⋅HCl) as coupling agent. Protection of catecholic hydroxy groups was essential for achieving clean reactions in fair yield. Either acetonide (Ace) or tert‐butyldimethylsilyl (TBS) protection groups were used (Figures S1 and S2), abbreviated TBS‐DHCA and Ace‐DHCA, respectively. Polymers obtained from this coupling are denoted pDTT‐TBS‐DHCA x and pDTT‐Ace‐DHCA x , respectively.
Specifically, we obtained pDTT‐Cat100 via the acetonide route and pDTT‐Cat20 via the silyl ether route. We note that the yield of the acetonide synthesis of pDDT‐Cat100 is low (9 %), but we include pDTT‐Cat100 here for the sake of obtaining a clearer understanding of the polymer analysis. Figures S3, S4, and S5 show the 1H NMR spectra of pDTT, pDTT‐Ace‐DHCA100, and pDTT‐Cat100, respectively. For complete polymer composition analysis of pDTT, pDTT‐Ace‐DHCA100, pDTT‐Cat100, pDTT‐Ace‐DHCA20, pDTT‐TBS‐DHCA20, and pDTT‐Cat20, please refer to Supporting Information (Figures S3–S8).
Figure 1 a shows Size Exclusion Chromatography Multiple Angle Laser Light Scattering (SEC–MALS) traces of pDTT after successive catechol functionalization. Upon modification of pDTT with protected DHCA, the SEC trace shifts to shorter elution times as expected for larger molecules. The higher the esterification density becomes, the more unimodal the SEC trace appears, suggesting that conversion of hydroxy groups to ester functions diminishes molecular interactions in terms of hydrogen bonding. Upon protection group removal, elution volume decreases further, which we attribute to existence of strong intermolecular interactions between different polymer chains through now‐revealed catechol units. [37] Such interactions include bidentate hydrogen bonding and π–π stacking. In the case of pDTT‐Cat20, we were able to substantiate such concentration‐dependent interchain interactions between catechol units by comparing SEC traces of pDTT‐Cat20 and pDTT‐TBS‐DHCA20 at increasing concentrations. While elution times of pDTT‐Cat20 increased at higher concentrations (Figure S9), there was no such effect for pDTT‐TBS‐DHCA20 (Figure S10). Figure S11 shows the SEC traces for pDTT, pDTT‐Ace‐DHCA20, and pDTT‐Cat20, and Table S1 compiles M n, M w, and DP parameters for all studied polymers, extracted from SEC data (see Figures S9–S13 for further discussion).
Figure 1.

a) SEC traces of unmodified pDTT (blue), pDTT‐Ace‐DHCA20 (orange), pDTT‐Ace‐DHCA100 (yellow), and pDTT‐Cat100 (purple) in 0.01 m LiBr/DMF. The small‐molecule peak at ca. 24 min is ascribed to solvent impurity as confirmed by 1H NMR (Figure S5). b) SEC trace of pDTT‐Cat100 (blue) and pDTT‐Cat100 after addition of 1 equiv DTT and 0.5 equiv Et3N w.r.t. end‐caps (orange) in 0.01 m LiBr/DMF. All sample concentrations were 4 mg mL−1.
Degradability of Modified pDTT
Scheme 1 b shows the proposed reaction transforming pDTT‐Cat to small molecules on the basis of the already known depolymerization pathway of pDTT itself. [21] This ability to degrade on demand through a self‐immolative process by exposure to specific stimuli is a key property of the novel multifunctional polymer. Specifically, by cleaving off the end‐caps with a stoichiometric amount of DTT (i.e. 1 equiv per end‐cap), the unstable polymer backbone initiates depolymerization through a cascade of cyclizations. This involves a sequence of base‐catalyzed ring‐closing thiol–disulfide exchanges, to give the thermodynamically stable and esterified form of 1,2‐dithiane‐4,5‐diol cyclic product (cDTT), which is the oxidized and ring‐closed form of DTT. To promote faster thiol–disulfide exchange kinetics, a catalytic amount of base (0.5 equiv Et3N per end‐cap) was added to increase the concentration of the more reactive thiolate species.
Figure 2 a,b shows how the transformation of pDTT‐Cat100 from polymer to small molecules is reflected in 1H NMR. Upon uncapping, the peaks assigned to end‐caps all shift downfield when pyridine‐2‐thiol is released in accordance with our previous report. [21] The progress of depolymerization is indicated by decrease of the peak at 5.28 ppm, associated with C H ‐O(CO)‐ in pDTT‐Cat100, while a new peak at 4.89 ppm is ascribed to the same proton in the cyclic monomer, cDTT‐(Cat)2 (Figure S14). A peak at 5.23 ppm, assigned to CH‐O H in cDTT, originates from the DTT added to remove end‐caps (Figures S14–S16 display full spectra and assignments of depolymerization products from pDTT‐Cat100).
Figure 2.
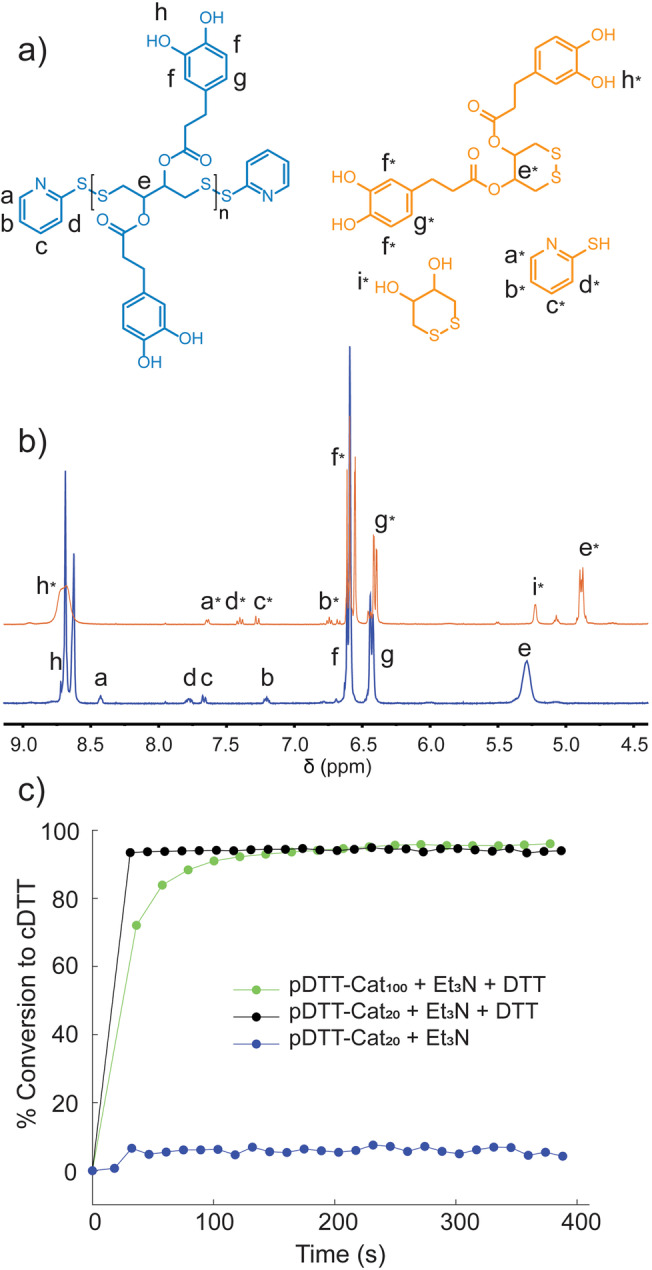
a) Chemical structure of depolymerization products from pDTT‐Cat100. b) 1H NMR of pDTT‐Cat100 (blue) and pDTT‐Cat100 depolymerized in presence of 1 equiv DTT and 0.5 equiv Et3N with respect to polymer end‐caps (orange) in DMSO‐d 6. c) Conversion of polymers, pDTT‐Cat100 (green), and pDTT‐Cat20 (black) to cyclic monomer in response to adding 1 equiv DTT and 0.5 equiv Et3N, along with the response of adding only 0.5 equiv Et3N to pDTT‐Cat20 (blue), measured by 1H NMR spectroscopy (lines to guide the eye). In all measurements the polymer concentration was 1.6 mm.
Figure 1 b shows corresponding SEC data where a change occurs from large hydrodynamic radius with elution times of 16–21 min to small molecule fragments with elution time of 24 min upon uncapping and depolymerization. This signifies that the self‐immolative property of the pDTT backbone is preserved after modification with catechol. Similarly, degradation of pDTT‐Cat20 was characterized by 1H NMR (Figures S17 and S18) and SEC (Figure S19). Evident from the SEC data, the products obtained from degradation of pDTT‐Cat20 result in several peaks, corresponding to cDTT (27 min) and cDTT‐(Cat)1 with one catechol unit (25 min) along with a peak ascribed to an unknown solvent impurity (24 min). Figure S19 also shows the SEC trace of degradation products from pDTT‐Cat100, which results in a single peak assigned to cDTT‐(Cat)2 (23 min).
To investigate the possibility of inter‐chain disulfide exchange and macrocycle formation, we studied degradation of unmodified pDTT in a more concentrated solution (40 mm vs. 1.6 mm in standard degradation experiments) to promote possible inter‐chain reactions. By adding 0.25 equiv DTT w.r.t. end‐caps, only every other chain is expected to depolymerize if no inter‐chain depolymerization occurs. SEC traces showed formation of only small molecules (Figure S20) which indicates that some inter‐chain depolymerization may take place, however without resulting in larger macrocyclic structures. Most likely, this observation can be attributed to a situation where the smallest (and thus fastest diffusing) depolymerizing thiolate ends attack new chains, thus inducing depolymerization.
Figure 2 c displays kinetic traces of the degradation processes of pDTT‐Cat100 and pDTT‐Cat20, respectively. Depolymerization was taken as the percentage conversion to cyclic monomer species against time, determined by 1H NMR analysis (Figures S16 and S17). As seen, both polymers exhibit complete degradation in DMSO‐d 6 within a few minutes of adding DTT and Et3N. In fact, degradation of pDTT‐Cat20 occurs essentially within the 30 s needed to record the first NMR spectrum. Depolymerization of pDTT‐Cat100 is slower than that of DTT‐Cat20, which we ascribe to the higher content of bulky catechol substituents, slowing the ring‐closing reaction. In addition to steric effects, the relatively low pK a of the catechol hydroxy groups may reduce the efficiency of the base catalysis. [38] Nevertheless, the response of pDTT‐Cat100 to chemical stimuli (i.e. DTT/Et3N) is rapid, with full depolymerization accomplished within 3 min. Evident from Figure 2 c, adding Et3N alone does not induce depolymerization or end‐cap removal on the timescale of these degradation experiments.
The facile depolymerization of pDTT‐Cat x is crucial for the intended degradation of gels cross‐linked by catecholato–metal ion complexes, as steric constraints imposed by the cross‐links is expected to slow down the disulfide cyclization reaction further, thus reducing depolymerization rate. In this study, we did not attempt to isolate and recycle the monomeric species from the depolymerization, but we have previously reported the recovery and repolymerization of cDTT obtained from depolymerization of pristine pDTT. [21]
Hydrogel Formation
Figure 3 establishes a key feature of polymers decorated with catechol pendant groups, which is the ability to form gel networks through formation of catecholato–metal cross‐links. In this study, hydrogels were formed and loaded with dye (for visualization) by first dissolving pDTT‐Cat20 in a solution of rhodamine 6G in MeOH, and adding a solution of Al3+ ions to produce the mono‐catecholato–Al3+ species (Figure 3 a). To induce cross‐linking, pH was raised by addition of aqueous NaOH, resulting in immediate hydrogelation with the dye trapped in the gel (Figure 3 b). Formation of catecholato–Al3+ species is pH‐dependent, forming predominantly mono‐, bis‐, and tris‐complexes at acidic, intermediate, and alkaline pH respectively.[ 6 , 39 ]
Figure 3.

a) pDTT‐Cat20, Al3+, and rhodamine 6G in solution and b) after inducing hydrogel formation by addition of NaOH to raise pH (>10). c) Mechanism of gel breakdown, induced either by depolymerization through decapping by addition of 10 equiv DTT and 5 equiv Et3N w.r.t. end caps (top), or by cleavage of Al3+–catecholato cross‐links at low pH, by addition of 1 m HCl in MeOH (bottom). d) Resulting liquefaction of the hydrogel and concomitant release of rhodamine 6G into solution after 100 min (depolymerization) and 17 min (catecholato–metal cross‐link cleavage).
Triggered Hydrogel Degradation
Figure 3 c,d demonstrates the stimuli‐triggered response of gels made from pDTT‐Cat20, rhodamine 6G dye, and Al3+, after addition of DTT/Et3N in MeOH or 1 m HCl in MeOH, either of which results in complete gel liquefaction. Importantly, this confirms successful introduction of dual stimuli‐responsiveness into the gels through the self‐immolative backbone and the catecholato–metal cross‐links. Notably, the time scale for these two types of gel liquefaction is different, in that cleavage of cross‐links is complete within 17 min, while depolymerization takes at least 100 min (Figure S21 and Movie S1 in Supporting Information). As such, the two functionalities represent two orthogonal pathways to hydrogel degradation, working in different time regimes. While catecholato–metal cross‐links addressed by lowering pH respond relatively fast, the depolymerization triggered by addition of DTT is, at least, five times slower. We note that the response time is minutes to a few hours, and thus the translation of a molecular trigger to a macroscopic response can be considered as rapid. Compared to previously reported degradable gels based on breaking disulfides, this depolymerization reaction results in a swift degradation, with the option for even faster responsivity through the acid‐labile cross‐links.[ 9 , 10 , 40 ]
Figure 4 shows the absorbance in UV/Vis caused by release of rhodamine 6G from the gels as function of time using the two stimuli (see Figures S22–S24 for raw UV spectra). The important revelation of this experiment is that the macroscopic response (increase in absorbance from the released dye) to a molecular signal is significant, no matter which of the stimuli‐responsive moieties is addressed. Thus, the pDTT‐Cat20 gel can, potentially, work as drug carrier with the self‐immolative backbone or the metal–catecholato cross‐links as release functions. Note that the release is, by and large, completed within one hour, independent of the selected stimuli‐responsive moiety. This similarity of the results is not particularly surprising, as the release of a given molecule from a hydrogel breaking up consists of additional steps than the responsiveness actions themselves, for example, diffusion of small molecules and changes in polymer chain conformation. Thus, different regimes of mass transport would need to be considered if a thorough evaluation of the kinetics was the goal. In addition, the experimental conditions were not exactly the same in the experiments presented in Figures 3 and 4. While the gels depicted in Figure 3 were left unperturbed for gel degradation until pictures were captured, the corresponding solutions above the gels for the small molecule release measurements (Figure 4) were homogenized immediately prior to withdrawing each sample (see Supporting Information). Nonetheless, the experiments work nicely as proof‐of‐concept illustrations of the dual‐responsiveness of the gels.
Figure 4.

Dye release from pDTT‐Cat20 gels in different environments measured as absorbance at 529 nm; 1 m HCl in MeOH (blue), 10 equiv DTT and 5 equiv Et3N (with respect to end‐caps) in MeOH (yellow), MeOH without stimuli (orange). Dashed lines to guide the eye.
Finally, regeneration of gels from the acidic solution resulting from cleaving catecholato–Al3+ cross‐links was attempted by raising pH once again. However, addition of aqueous NaOH resulted in formation of smaller, individual gel particles rather than a single, large gel. We ascribe this to the significant dilution of the polymer chains when attempting direct gel reforming (starting concentration of ≈110 mg mL−1 compared to a final concentration of ≈25 mg mL−1). Formation of smaller gel particles may find use in for example, drug formulation within the gels.
Conclusion
We have presented a responsive polymer consisting of a self‐immolative backbone decorated with catechol units. Through catecholato–Al3+ cross‐links, the polymers form dual‐responsive hydrogels in a pH‐responsive fashion. Gel degradation and concomitant release of trapped cargo are triggered either by lowering pH and cleaving the cross‐links, or by translation of a molecular signal, dithiothreitol, to a macroscopic response by virtue of depolymerization of the self‐immolative backbone. As such, the two functionalities represent two orthogonal pathways to hydrogel degradation, working in different time regimes. Both processes result in vanishing gels and release of small molecule cargo. Thus, molecular signals are amplified to a macroscopic response, with the option of leaving only small molecules behind. Controlling material response in different environments is useful for storing and releasing small molecules, which we illustrated by release of rhodamine 6G. This work on highly functionalized, degradable polymers expands the scope of self‐immolative polymers, and presents a material capable of releasing small molecules on demand, with the option of concomitant transformation from a polymeric structure into small molecules. Thus, we show that functionalization of self‐immolative polymer backbones results in versatile and stimuli‐responsive polymeric materials.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Funding from the Independent Research Fund Denmark (grant no. 9041‐00096B) and Aarhus University is gratefully acknowledged. Affiliation with the Center for Integrated Materials Research (iMAT) at Aarhus University is gratefully acknowledged. Affiliation with the Smart Polymer Materials and Nano‐Composites (SPOMAN) Open Science Initiative is gratefully acknowledged.
A. H. Agergaard, A. Sommerfeldt, S. U. Pedersen, H. Birkedal, K. Daasbjerg, Angew. Chem. Int. Ed. 2021, 60, 21543.
Contributor Information
Prof. Henrik Birkedal, Email: hbirkedal@chem.au.dk.
Prof. Kim Daasbjerg, Email: kdaa@chem.au.dk.
References
- 1. Nguyen Q. V., Huynh D. P., Park J. H., Lee D. S., Eur. Polym. J. 2015, 72, 602–619. [Google Scholar]
- 2. Shi Q., Liu H., Tang D. D., Li Y. H., Li X. J., Xu F., NPG Asia Mater. 2019, 11, 64. [Google Scholar]
- 3. Zhang D., Ren B., Zhang Y., Xu L., Huang Q., He Y., Li X., Wu J., Yang J., Chen Q., Chang Y., Zheng J., J. Mater. Chem. B 2020, 8, 3171–3191; [DOI] [PubMed] [Google Scholar]; Xia S., Zhang Q., Song S. X., Duan L. J., Gao G. H., Chem. Mater. 2019, 31, 9522–9531. [Google Scholar]
- 4. Zhang Q., Shi C. Y., Qu D. H., Long Y. T., Feringa B. L., Tian H., Sci. Adv. 2018, 4, eaat8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andersen A., Krogsgaard M., Birkedal H., Biomacromolecules 2018, 19, 1402–1409; [DOI] [PubMed] [Google Scholar]; Krogsgaard M., Behrens M. A., Pedersen J. S., Birkedal H., Biomacromolecules 2013, 14, 297–301. [DOI] [PubMed] [Google Scholar]
- 6. Krogsgaard M., Hansen M. R., Birkedal H., J. Mater. Chem. B 2014, 2, 8292–8297. [DOI] [PubMed] [Google Scholar]
- 7. Lu D., Zhu M., Wu S., Lian Q., Wang W., Adlam D., Hoyland J. A., Saunders B. R., Adv. Funct. Mater. 2020, 30, 1909359. [Google Scholar]
- 8. Chen Y., Wang W., Wu D., Zeng H., Hall D. G., Narain R., ACS Appl. Mater. Interfaces 2019, 11, 44742–44750. [DOI] [PubMed] [Google Scholar]
- 9. Trotta F., Caldera F., Dianzani C., Argenziano M., Barrera G., Cavalli R., ChemPlusChem 2016, 81, 439–443. [DOI] [PubMed] [Google Scholar]
- 10. Cai K., Yen J., Yin Q., Liu Y., Song Z., Lezmi S., Zhang Y., Yang X., Helferich W. G., Cheng J., Biomater. Sci. 2015, 3, 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y., Yin Q., Yin L., Ma L., Tang L., Cheng J., Angew. Chem. Int. Ed. 2013, 52, 6435–6439; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6563–6567. [Google Scholar]
- 12. Zhang Q., Deng Y. X., Shi C. Y., Feringa B. L., Tian H., Qu D. H., Matter 2021, 4, 1352–1364. [Google Scholar]
- 13. Xiao Y., Tan X., Li Z., Zhang K., J. Mater. Chem. B 2020, 8, 6697–6709; [DOI] [PubMed] [Google Scholar]; Sirianni Q. E. A., Rabiee Kenaree A., Gillies E. R., Macromolecules 2019, 52, 262–270; [Google Scholar]; Esser-Kahn A. P., Sottos N. R., White S. R., Moore J. S., J. Am. Chem. Soc. 2010, 132, 10266–10268. [DOI] [PubMed] [Google Scholar]
- 14. Gnaim S., Shabat D., J. Am. Chem. Soc. 2017, 139, 10002–10008. [DOI] [PubMed] [Google Scholar]
- 15. Lewis G. G., Robbins J. S., Phillips S. T., Chem. Commun. 2014, 50, 5352–5354; [DOI] [PubMed] [Google Scholar]; Lewis G. G., Robbins J. S., Phillips S. T., Macromolecules 2013, 46, 5177–5183. [Google Scholar]
- 16. Kim H., Mohapatra H., Phillips S. T., Angew. Chem. Int. Ed. 2015, 54, 13063–13067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13255–13259. [Google Scholar]
- 17. Baker M. S., Kim H., Olah M. G., Lewis G. G., Phillips S. T., Green Chem. 2015, 17, 4541–4545. [Google Scholar]
- 18. Ergene C., Palermo E. F., J. Mater. Chem. B 2018, 6, 7217–7229. [DOI] [PubMed] [Google Scholar]
- 19. Lloyd E. M., Lopez Hernandez H., Feinberg E. C., Yourdkhani M., Zen E. K., Mejia E. B., Sottos N. R., Moore J. S., White S. R., Chem. Mater. 2019, 31, 398–406. [Google Scholar]
- 20. Fan B., Salazar R., Gillies E. R., Macromol. Rapid Commun. 2018, 39, 1800173. [DOI] [PubMed] [Google Scholar]
- 21. Pal S., Sommerfeldt A., Davidsen M. B., Hinge M., Pedersen S. U., Daasbjerg K., Macromolecules 2020, 53, 4685–4691. [Google Scholar]
- 22. Bapat A. P., Sumerlin B. S., Sutti A., Mater. Horiz. 2020, 7, 694–714; [Google Scholar]; Guo R., Su Q., Zhang J., Dong A., Lin C., Zhang J., Biomacromolecules 2017, 18, 1356–1364. [DOI] [PubMed] [Google Scholar]
- 23. Xu Y., Li Y., Chen Q., Fu L., Tao L., Wei Y., Int. J. Mol. Sci. 2018, 19, 2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei Z., Yang J. H., Du X. J., Xu F., Zrinyi M., Osada Y., Li F., Chen Y. M., Macromol. Rapid Commun. 2013, 34, 1464–1470. [DOI] [PubMed] [Google Scholar]
- 25. An Q., Wessely I. D., Matt Y., Hassan Z., Bräse S., Tsotsalas M., Polym. Chem. 2019, 10, 672–678; [Google Scholar]; Perera M. M., Ayres N., Polym. Chem. 2020, 11, 1410–1423. [Google Scholar]
- 26. Lai J. C., Li L., Wang D. P., Zhang M. H., Mo S. R., Wang X., Zeng K. Y., Li C. H., Jiang Q., You X. Z., Zuo J. L., Nat. Commun. 2018, 9, 2725; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lei Y. F., Huang W. Y., Huang Q. P., Zhang A. Q., New J. Chem. 2019, 43, 261–268. [Google Scholar]
- 27. Holten-Andersen N., Harrington M. J., Birkedal H., Lee B. P., Messersmith P. B., Lee K. Y., Waite J. H., Proc. Natl. Acad. Sci. USA 2011, 108, 2651–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu G., Yuan Q., Hollett G., Zhao W., Kang Y., Wu J., Polym. Chem. 2018, 9, 3436–3449. [Google Scholar]
- 29. Andersen A., Chen Y., Birkedal H., Biomimetics 2019, 4, 30–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saiz-Poseu J., Mancebo-Aracil J., Nador F., Busque F., Ruiz-Molina D., Angew. Chem. Int. Ed. 2019, 58, 696–714; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 706–725; [Google Scholar]; Zhou J., Lin Z., Ju Y., Rahim M. A., Richardson J. J., Caruso F., Acc. Chem. Res. 2020, 53, 1269–1278; [DOI] [PubMed] [Google Scholar]; Yang J., Cohen Stuart M. A., Kamperman M., Chem. Soc. Rev. 2014, 43, 8271–8298. [DOI] [PubMed] [Google Scholar]
- 31. Chen Y., Tan Z., Wang W., Peng Y. Y., Narain R., Biomacromolecules 2019, 20, 1028–1035; [DOI] [PubMed] [Google Scholar]; Narkar A. R., Barker B., Clisch M., Jiang J., Lee B. P., Chem. Mater. 2016, 28, 5432–5439; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kan Y. J., Danner E. W., Israelachvili J. N., Chen Y. F., Waite J. H., PloS One 2014, 9, e108869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krogsgaard M., Nue V., Birkedal H., Chem. Eur. J. 2016, 22, 844–857; [DOI] [PubMed] [Google Scholar]; Krogsgaard M., Andersen A., Birkedal H., Chem. Commun. 2014, 50, 13278–13281. [DOI] [PubMed] [Google Scholar]
- 33. Lee H., Scherer N. F., Messersmith P. B., Proc. Natl. Acad. Sci. USA 2006, 103, 12999–13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Agergaard A. H., Pedersen S. U., Birkedal H., Daasbjerg K., Polym. Chem. 2020, 11, 5572–5577. [Google Scholar]
- 35. Kord Forooshani P., Lee B. P., J. Polym. Sci. Part A 2017, 55, 9–33; [DOI] [PMC free article] [PubMed] [Google Scholar]; Moulay S., Polym. Rev. 2014, 54, 436–513. [Google Scholar]
- 36. Li Y., Wen J., Qin M., Cao Y., Ma H., Wang W., ACS Biomater. Sci. Eng. 2017, 3, 979–989; [DOI] [PubMed] [Google Scholar]; Xu Z., Sci. Rep. 2013, 3, 1–7. [Google Scholar]
- 37. Tiu B. D. B., Delparastan P., Ney M. R., Gerst M., Messersmith P. B., Angew. Chem. Int. Ed. 2020, 59, 16616–16624; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 16759–16767. [Google Scholar]
- 38. Romero R., Salgado P. R., Soto C., Contreras D., Melin V., Front. Chem. 2018, 6, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Menyo M. S., Hawker C. J., Waite J. H., Soft Matter 2013, 9, 10314–10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tran V. T., Mredha M. T. I., Na J. Y., Seon J.-K., Cui J., Jeon I., Chem. Eng. J. 2020, 394, 124941–124951. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information