

Abstract
Multidrug resistance (MDR) is one of the major factors in the failure of many chemotherapy approaches. In cancer cells, MDR is mainly associated with the expression of ABC transporters such as P‐glycoprotein, MRP1 and ABCG2. Despite major efforts to develop new selective and potent inhibitors of ABC drug transporters, no ABCG2‐specific inhibitors for clinical use are yet available. Here, we report the evaluation of sixteen tetrahydroquinoline/4,5‐dihydroisoxazole derivatives as a new class of ABCG2 inhibitors. The affinity of the five best inhibitors was further investigated by the vanadate‐sensitive ATPase assay. Molecular modelling data, proposing a potential binding mode, suggest that they can inhibit the ABCG2 activity by binding on site S1, previously reported as inhibitors binding region, as well targeting site S2, a selective region for substrates, and by specifically interacting with residues Asn436, Gln398, and Leu555. Altogether, this study provided new insights into THQ/4,5‐dihydroisoxazole molecular hybrids, generating great potential for the development of novel most potent ABCG2 inhibitors.
Keywords: ABC transporter, ABCG2, Isoxazolines, Molecular dynamics simulation, Tetrahydroquinolines
Tetrahydroquinoline/4,5‐dihydroisoxazole hybrids can inhibit the ABCG2 transporter activity and increase the ATP hydrolysis in cells overexpressing ABCG2. Molecular docking and molecular dynamics simulation results suggest that molecular hybrids inhibit ABCG2 activity by binding at the inhibitor binding region, as well as the substrate binding regions.

Introduction
Multidrug resistance (MDR) is one of the major challenges to cancer treatment. [1] Cancer resistance can be intrinsic or acquired, and different mechanisms are described to trigger MDR, including the reduced drug uptake, enhancing DNA repair, failure on programmed cell death mechanisms, mutations of targeted proteins. However, the leading cause of MDR is associated with the expression of ATP‐binding cassette (ABC) proteins in cancer cells.[ 2 , 3 ] The ABC transporters are present across many different tumor types, [2] promoting the efflux of several unrelated chemotherapeutic agents.[ 2 , 4 ] The catalytic mechanism of drug transport is based on substrate binding on transmembrane domains (TMDs) followed by ATP binding and hydrolysis at nucleotide binding domains (NBDs). [5] The human genome codes 48 ABC proteins, and the three of them are undoubtedly associated with MDR, P‐glycoprotein (encoded by ABCB1), multidrug resistance‐associated protein 1 (encoded by ABCC1) and breast cancer resistance protein (encoded by ABCG2). [6]
ABCG2 transporter was discovered in 1998 by three independent laboratories, that named the protein based on the biological model used, BCRP (breast cancer resistance protein) due of its identification in breast cancer cells, [7] MXR because of the mitoxantrone resistance in cancer cells [8] and ABCP, as a result of the presence on placenta. [9] ABCG2 promotes the efflux of many substrates, including the chemotherapeutic agent topotecan, the active metabolite of irinotecan (SN‐38), mitoxantrone, methotrexate, doxorubicin and TKIs.[ 7 , 10 , 11 ] This protein is overexpressed in different types of cancer, such as breast, pancreas, kidney, liver, glioblastomas and sarcomas, triggering the MDR phenotype. [12]
One of the most promising strategies to overcome MDR mediated by ABCG2 is the development of potent inhibitors (Figure 1). The first inhibitor of ABCG2 identified was the fungal toxin fumitremorgin C (1) (FTC), obtained from A. fumigatus. Despite the selectivity toward ABCG2, FTC presents neurotoxic effects. [13] Since its discovery, several tetracyclic analogs of FTC have been synthesized aiming to increase the potency of inhibition and reduce side‐effects, such as Ko143 (2). This inhibitor shows a very high potency of inhibition; however, Ko143 is unstable in vivo and non‐specific toward ABCG2, as it also inhibits ABCB1 and ABCC1 transporters at higher concentrations.[ 14 , 15 ] Later, extensive chemical modifications of the Ko143 scaffold, as well as in vitro analyses, revealed the details of ABCG2 interactions with Ko143 and resulted in novel inhibitors such as MZ29 (3). [16] Besides the FTC and its derivatives, other natural compounds such as flavones, chalcones, and curcuminoids have been reported as ABCG2 inhibitors.[ 17 , 18 , 19 ] As part of flavonoids, the chromones (4) have been widely studied and characterized as novel potent, selective and non‐toxic ABCG2 inhibitors.[ 20 , 21 ] Despite the increased number of ABCG2 studies, no inhibitor for clinical use is yet available.[ 14 , 22 , 23 ]
Figure 1.
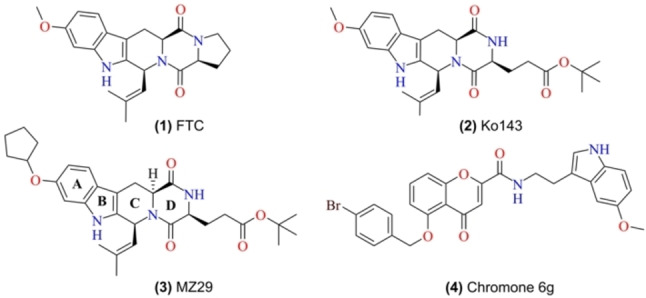
Chemical structure of reported ABCG2 inhibitors. (1) Fumitremorgin C (FTC), (2) Ko143, (3) MZ29, and (4) Chromone 6 g (MBL‐II‐141).
A critical milestone, a high‐resolution cryo‐EM structure for the ABCG2 transporter, was reached in 2017, together with the proposed mechanism of inhibition. [23] Later, the structure of ABCG2 in complex with two inhibitors revealed another ABCG2 conformation. [16] The transport and inhibition mechanism invokes ATP‐driven alternating access to the substrate/inhibitors binding sites (Figure 2). Upon the substrate binding in the Cavity 1 (Figure 2A), from the cytoplasm or the inner membrane, the nucleotide‐binding domain (NBD) approach in response to the ATP binding, inducing a conformational change towards the outward‐facing conformation of ABCG2, which can release the substrate (Figure 2B). [23]
Figure 2.
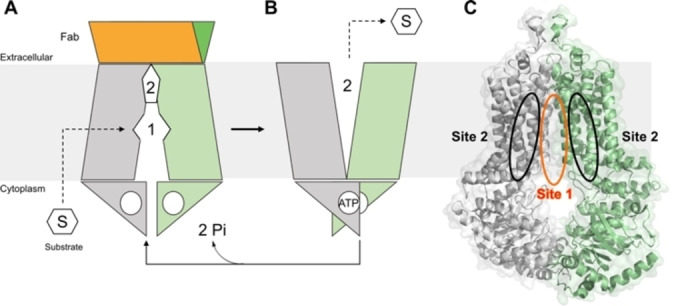
Illustrative mechanism of ABCG2 transport exchanging from the inactive state (A), where the substrate molecule can enter in the cavity 1 from the cytoplasm or inner membrane, towards the active state (B) where, upon ATP binding, the transporter opens cavity 2 releasing the substrate to outside. (C) Binding sites of ABCG2 indicated as Site 1 (S1) and Site 2 (S2), PDB ID: 6FFC.
Docking studies reported by Krapf et al., (2018) indicate the presence of at least two transporter binding sites, named as sites 1 and 2, respectively (Figure 2C).[ 24 , 25 ] Site S1 is located at the interface between the two transmembrane domains and was previously characterized as highly promiscuous (Supporting Information, Figure 1), allowing substrates and non‐substrates to bind. [26] On the other hand, site S2 is located between the transmembrane regions and contrarily to S1, it is selective for substrates in comparison with non‐substrates or inhibitors. [24]
Molecular hybrids are emerging as a new class of small molecules targeting ABC transporters, which potentially can be employed in association with chemotherapeutic agents to overcome MDR in cancer. Tetrahydroisoquinoline (THIQ), a quinoline family member can inhibit P‐glycoprotein, improving the chemotherapy efficacy on glioblastoma multiforme cell lines. [27] On the other hand, isoxazole derivatives have shown potency against MRP1, however, more studies about their specificity, toxicity, and effectivity are needed.[ 28 , 29 ] Considering the anticancer effect of our previously synthesized molecular hybrids of tetrahydroquinoline (THIQ)/4,5‐dihydroisoxazole (ISX) [30] as well as the inhibitory effect of THIQ and isoxazole against the ABC transporter family, [27] we screened sixteen THIQ/ISX hybrids, aiming to identify selective and potent ABCG2 inhibitors. The affinity of the five best inhibitors was investigated by the vanadate‐sensitive ATPase assay. In addition, the molecular mechanism of ABCG2 binding was explored using computational approaches, such as docking and molecular dynamics (MD). Our data suggest that tetrahydroquinoline/4,5‐dihydroisoxazole hybrids can inhibit the ABCG2 transporter activity binding on‐site S1 reported as inhibitors binding region, as well as S2, a selective region for substrates.
Results and Discussion
Synthesis of tetrahydroquinoline/4,5‐dihydroisoxazole hybrids
For easy access to the series of sixteen (16) tetrahydroquinoline/4,5‐dihydroisoxazole hybrids (A–D series) tested in this work, it was necessary to obtain the corresponding N‐allyl tetrahydroquinolines C‐6 substituted (R=H, CH3, OCH3, and Cl). These keys precursors were synthesized with high yields, via a “one‐pot” three‐component cationic Povarov reaction. [31] Once these tetrahydroquinoline compounds were properly purified and characterized, they reacted efficiently with a set of selected aryl aldoximes through a 1,3‐dipolar cycloaddition reaction mediated for NaClO as an inexpensive oxidant, according to our methodology previously described. [30] Tetrahydroquinoline/4,5‐dihydroisoxazole hybrids were obtained in good yields after column chromatography purification on silica gel (Table 1). The stereochemistry of all these hybrid synthesized compounds was determined by 1H NMR spectroscopy. All THQ/4,5‐dihydroisoxazole hybrids obtained are a mixture of diastereoisomers. In all cases, the properties, including NMR data for the corresponding hybrids, match with our previously reported data. [30]
Table 1.
Chemical structures and yields of new hybrids tetrahydroquinoline/4,5‐dihydroisoxazole (A–D series). The 4,5‐dihydroisoxazole heterocycle ring is highlighted in blue in all structures.
|
Structure |
Compd |
R |
Yield[a] [%] |
d.r.[b] |
|---|---|---|---|---|
|
| ||||
|
|
A1 |
H |
74 |
1 : 1 |
|
A2 |
CH3 |
64 |
1 : 0.9 |
|
|
A3 |
OCH3 |
65 |
1 : 0.9 |
|
|
A4 |
Cl |
65 |
1 : 1 |
|
|
| ||||
|
|
B1 |
H |
65 |
1 : 1 |
|
B2 |
CH3 |
70 |
1 : 0.8 |
|
|
B3 |
OCH3 |
63 |
1 : 0.7 |
|
|
B4 |
Cl |
68 |
1 : 0.7 |
|
|
| ||||
|
|
C1 |
H |
77 |
1 : 0.7 |
|
C2 |
CH3 |
70 |
1 : 0.7 |
|
|
C3 |
OCH3 |
76 |
1 : 0.8 |
|
|
C4 |
Cl |
68 |
1 : 0.9 |
|
|
| ||||
|
|
D1 |
H |
74 |
1 : 0.6 |
|
D2 |
CH3 |
64 |
1 : 0.7 |
|
|
D3 |
OCH3 |
75 |
1 : 0.7 |
|
|
D4 |
Cl |
75 |
1 : 0.7 |
|
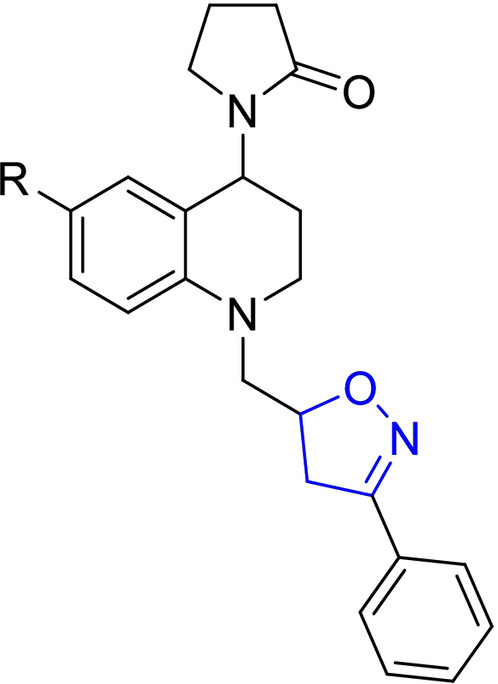
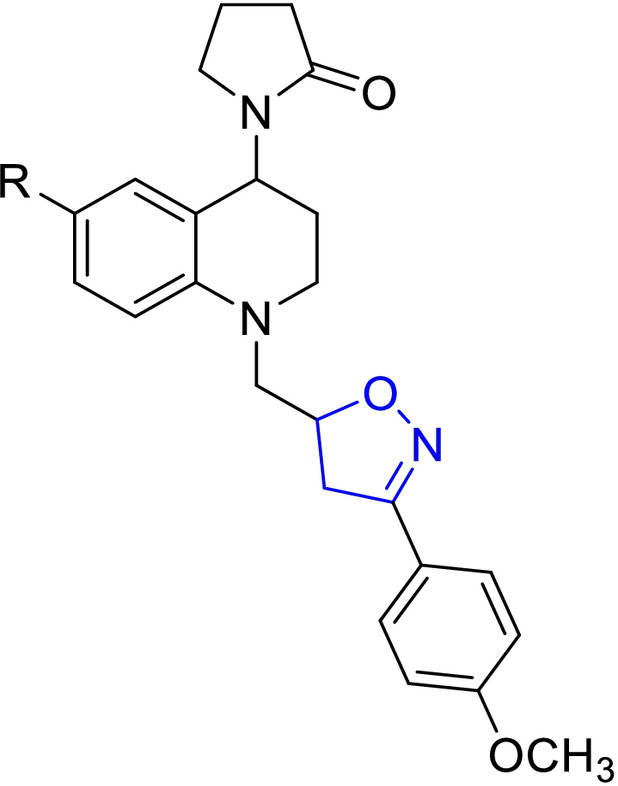
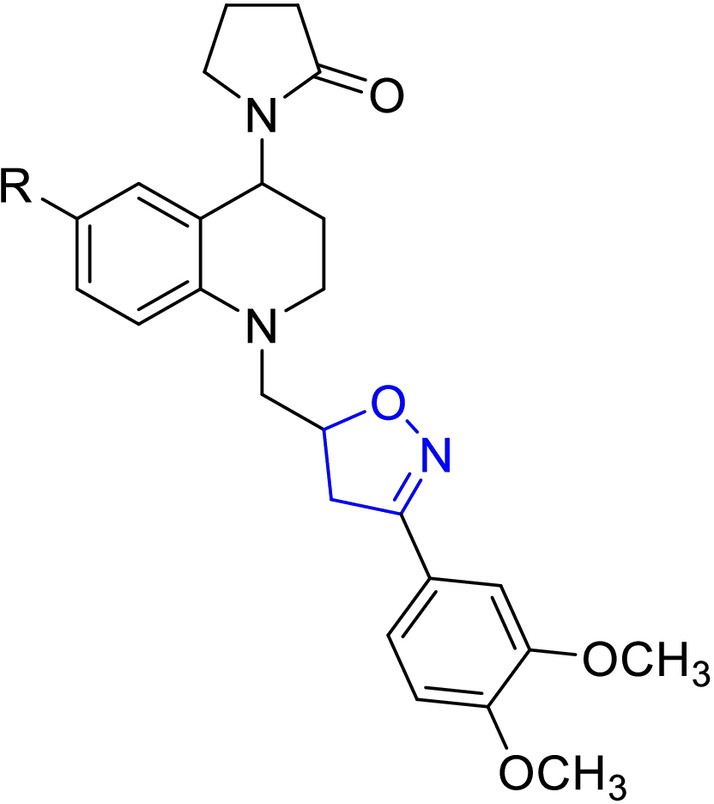
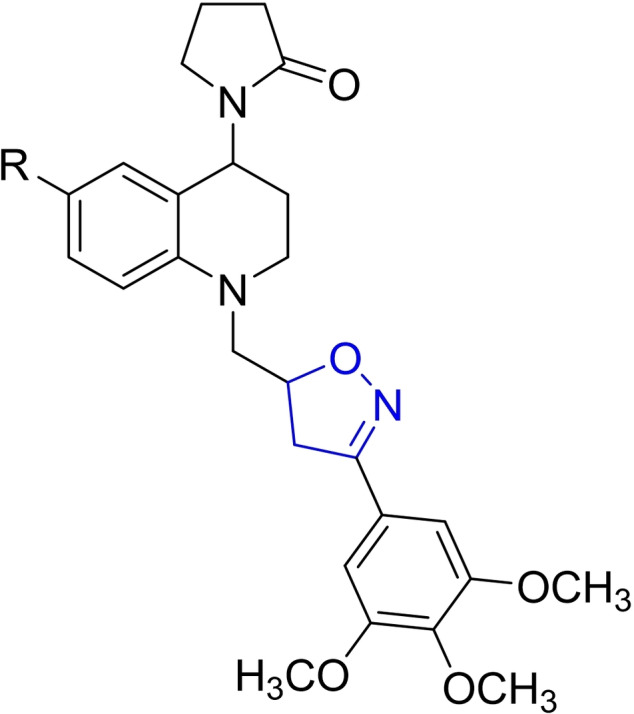
[a] Isolated yield after column chromatography. [b] Diastereomers ratio from the quantitative 1H‐NMR spectrum.
Tetrahydroquinoline/4,5‐dihydroisoxazole hybrids inhibit ABCG2 transport activity
To evaluate the capability of the tetrahydroquinoline/4,5‐dihydroisoxazole hybrids to inhibit the ABCG2 transport activity, HEK293‐ABCG2 cells stably transfected to overexpress ABCG2 transporter were used. The ABCG2 substrate used was the fluorescent chemotherapeutic mitoxantrone, and its intracellular accumulation was quantified by flow cytometry. All the sixteen molecular hybrids were assayed at 10 μM and 50 μM. As shown in Figure 3, all compounds were able to inhibit the ABCG2‐mediated mitoxantrone efflux. Five compounds (A1, C1, C4, D1 and D4), produced higher than 50 % inhibition at the 10 μM concentration and these were used to explore the molecular mechanism of ABCG2 inhibition.
Figure 3.
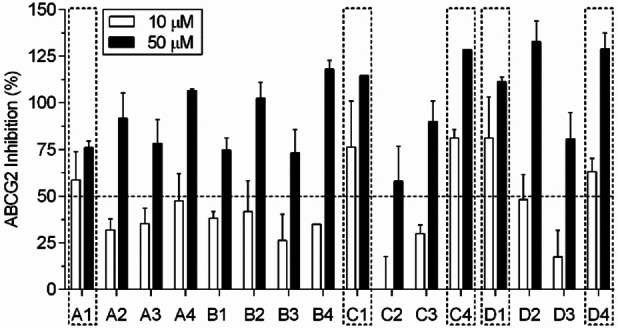
Inhibition of ABCG2‐mediated mitoxantrone efflux. HEK293‐ABCG2 stably transfected cells were concomitantly treated with mitoxantrone (10 μM) and molecular hybrids at 10 and 50 μM for 30 minutes. The mitoxantrone intracellular accumulation was quantified by flow cytometry. The percent of inhibition was determined using Ko143 at 0.5 μM as a reference inhibitor (100 % of inhibition). Values represent the mean ±SD of two independent experiments.
A preliminary SAR analysis comparing the effect on ABCG2 transport activity at 10 μM revealed some interesting features on the chemical structure of tetrahydroquinoline/4,5‐dihydroisoxazole hybrids (A–D series, Table 1). Firstly, the substituents on the C‐6 position of the tetrahydroquinoline scaffold from series A did not show relevant influence on ABCG2 inhibition, excepting the hybrid A1 (where R=H). Regarding the B series, the presence of the methoxy group on C‐4 on the phenyl moiety attached to the isoxazoline ring generates a significant decrease in ABCG2 inhibition. Contrarily, the presence of two (2) or three (3) methoxy groups increases the inhibition of ABCG2 transport activity from series C and D. Briefly, the presence of methoxy groups on C‐3 and C‐5 on the phenyl moiety attached to the isoxazoline ring seems to enhance the inhibitory effect on ABCG2 transporter activity. This is observed in compounds C1, C4, D1, and D4, which are at least 2‐fold more active than their counterparts B1, and B4 (Figure 3). Meanwhile, electron donating groups (−CH3, −OCH3) on C‐6 position of tetrahydroquinoline rings decreases the ABCG2 inhibition compared to unsubstituted and chloride derivatives. This suggests that the inductive effect of substituent group on tetrahydroquinoline has a role in ABCG2 inhibition. To confirm the role of inductive effect, additional derivatives should be synthetized and tested.
Tetrahydroquinoline/4,5‐dihydroisoxazole hybrids stimulate ATPase activity
The efflux of substrates by ABC transporters is mediated by the ATP binding and hydrolysis. [32] To get insights onto the biochemical mechanism of ABCG2 inhibition, the effect of the five most potent inhibitors (A1, C1, C4, D1, D4) was investigated on ABCG2 ATPase activity using High‐Five insect cells crude membranes overexpressing ABCG2. A range of concentrations from 0.1 to 10 μM was used to identify and compare the affinity of the five compounds on ABCG2. As shown in Figure 4, all molecular hybrids stimulated the ABCG2 ATPase activity in the respective order: C1>C4>D1>D4>A1. The EC50 of the ABCG2 ATPase stimulation varied from 0.17 to 1.64 μM, to hybrid C1 and A1, respectively.
Figure 4.

Effect of THQ/4,5‐dihydroisoxazole at increasing concentrations (0.01 to 10 μM) on basal vanadate‐sensitive ATPase assay. Values are represented by the mean ±SD of three independent experiments performed in duplicate.
Tetrahydroquinoline/4,5‐dihydroisoxazole hybrids proposed binding mode on ABCG2
To explore potential binding modes for tetrahydroquinoline/4,5‐dihydroisoxazole derivatives and tetrahydroquinoline/isoxazoline scaffolds, we performed molecular docking calculations withing the site 1 in ABCG2 cryo‐EM structure followed by molecular dynamics (MD) simulations. Docking poses were selected by visual inspection based on their common interactions with important residues such as Asn436, Thr435, and Arg482 and docking scores (Supporting information – Table 1). Additionally, co‐crystalized ligand MZ29 was re‐docked (RMSD 2.1 Å, Supporting Information – Figure 2).
MD simulations suggests that the derivative A1 can stably occupy the binding site located between the two transmembrane domains. This site was initially described by Krapf et al., (2018) as S1 [24] (Figure 5A,B) and residues such as Thr435–Phe439 are included within this site. Molecular hybrid A1 was docked and simulated between the two transmembrane domains, where docking results suggest two possible orientations of hybrids A1 within the S1 (Supporting Information – Figure 3A,B). Furthermore, molecular dynamics simulation of hybrid A1 in these two orientations inside S1 reveals a strong hydrogen bonding interaction with residue Thr435 along with the simulation. Moreover, hydrophobic interactions with residues Phe439 and Leu555 were also observed as well as stable ligand position within the proposed binding pocket (Supporting Information – Figure 4A–D).
Figure 5.
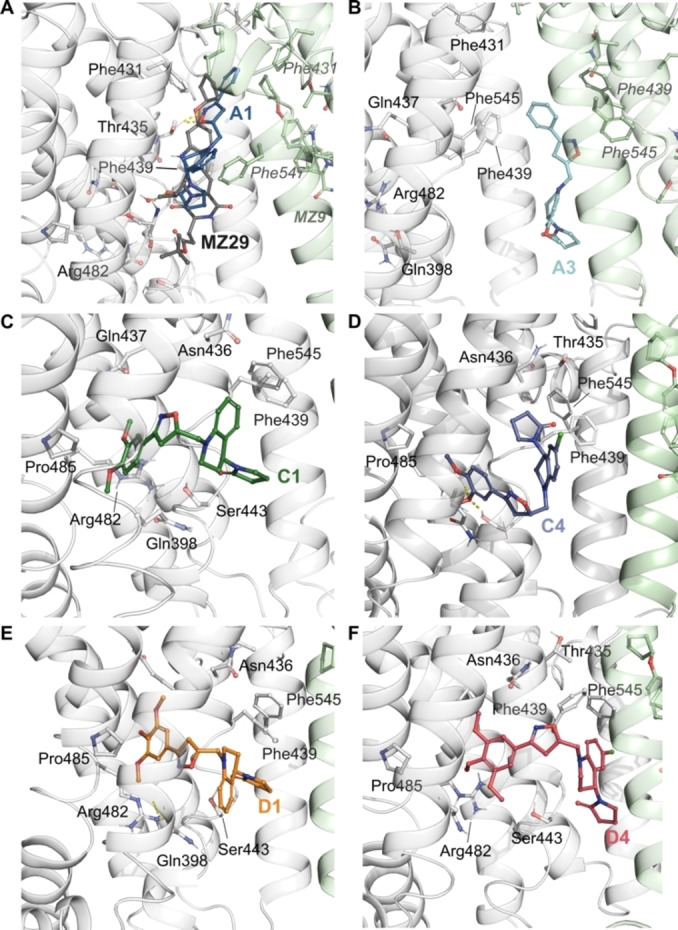
Snapshots of the poses following molecular dynamics simulations of THQ/4,5‐dihydroisoxazole hybrids. Co‐crystallized ligand, MZ29 (A) and molecular hybrids A1 (B), C1 (C), C4 (D), D1 (E) and D4 (F). ABCG2’s residues are colored according to the atom type of the interacting amino‐acid residues (protein's carbon, light grey (chain A) and pale green (chain B); oxygen, red; nitrogen, blue). The protein‐ligand interactions are represented by dash lines as follow: hydrogen bond interactions are colored in yellow, and π‐π interactions are colored in blue.
On the other hand, the derivatives C1, C4, D1, and D4 stably occupied the binding S2 (Figure 5C–F), located between the transmembrane helix 1 and 4 (TH1 and TH4) (see Supporting Information Figure 1, for a general structure of ABCG2 transporter). The binding S2 has the residues Arg482 and Pro485, which are shown to strongly affect substrate selectivity. [26] Interestingly, the binding of non‐substrates into S2, partially responsible by the substrate recognition, is limited. Site S1 was shown to be highly promiscuous, binding substrates and non‐substrates. [26] Furthermore, molecular hybrids with two different scaffolds were synthesized. Tetrahydroquinoline and isoxazoline cores were docked and simulated inside the two transmembrane domains, following the same docking protocol previously described. Throughout the simulation, tetrahydroquinoline and isooxazoline cores kept stable within the proposed binding site (Supporting Information – Figure 5D and E). Regarding protein‐ligand interactions, THQ core upholds similar hydrogen bond interaction with Thr435 like molecular hybrid A1. On the other hand, hydrophobic interactions with residues Phe439 and Phe545 are present in THQ and ISX cores in a similar fashion with our molecular hybrids (Supporting Information – Figure 5A–C).
Since our synthesized hybrids were obtained as a mixture of diastereoisomers (see synthesis section), all docking calculations included all possible isomers, trying to explore and explain a possible stereoselectivity against ABCG2 transporter inhibition. However, it was not possible to discriminate between the diastereoisomers based only on the docking score values. The docking poses were then selected based on their interactions with relevant amino‐acids, such as Gln398, Asn436, Thr435, Asn436, Phe439, Phe545 and Leu555. We also took into consideration poses without the clashes with nearby residues. We then proceeded to simulate the diastereoisomers that better suited our docking criteria.
ABCG2’s simulation with MZ29 shows stable hydrogen bond interactions between its indole ring and Asn436 (100 % of analyzed trajectories) and between its carboxyl group Gln398 (48 %) (Figure 5A, Supporting Information – Figure 6A,B). Comparatively, simulations with hybrid A1 and A3 shows a hydrogen bond interaction between Thr435 and oxygen of isoxazoline scaffold, while Asn436 forms a water‐mediated hydrogen bond (22 %) with the oxygen of pyrrolidone group (Figure 6A–D).
Figure 6.
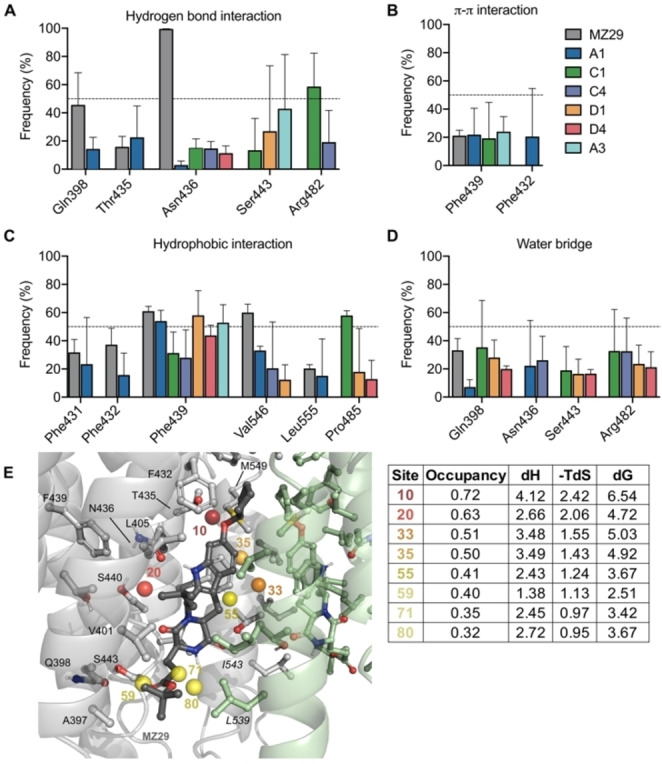
Frequency of hydrogen bonding interactions (A); π‐π interactions (B); hydrophobic interactions (C); and water mediated hydrogen bonding interactions (D) observed along the molecular dynamics simulation (three independent runs of 500 ns per system). Each bar represents the standard error of three independent simulations. (E) Superimposed ABCG2 sidechains, MZ29 and conserved water molecules are presented colored spheres, according to their WaterMap based free energy values (ΔG), also described in the adjacent table. The table contains the thermodynamic parameters for the solvation of the seven hydration sites, within the compound pocket. Green spheres represent regions where stable water molecules could be placed and therefore less likely to contributed to free energy gain upon ligand binding; alternatively, red and orange spheres represent regions where ligand occupancy could contribute to binding affinity by enthalpic energy gain. Occupancy is calculated from the number of water‐oxygen atoms found occupying a given hydration site during the 5 ns of molecular dynamics simulation, enthalpic energy (ΔH) and free energy value (ΔG) are given in kcal/mol.
In terms of hydrophobic interactions, the ring A of MZ29 has contacts with Phe439 (60 % of the simulated time, Figure 6C), while the O‐cyclopentyl substituent occupies a hydrophobic pocket composed by the Phe431 and Val546 (Figure 6C). This hydrophobic pocket also houses hydration sites with high free‐energy values (Figure 6E). These sites, when occupied by hydrophobic moieties can contribute to the overall binding energy of the compound by preventing water molecules to be trapped in unfavorable high energy positions (Figure 6E and respective table).
Simulations also suggest that derivatives A1 and A3 would share a similar hydrophobic interaction profile with MZ29. Specifically, the pyrrolidone group and tetrahydroquinoline moieties in hybrids A1 and A3 can occupy a pocket formed by the Phe439 and Phe432 (Figure 5A and 6E), where aromatic interactions between Phe439 and inhibitors or substrates have been widely reported. [33] Although both hybrids A1 and A3 can occupy the same binding pocket, hybrid A3 does not make interactions with relevant residues, such as Gln398 and Thr435. Regarding the phenyl group of the isoxazoline scaffold, occupies another hydrophobic sub pocket composed by Phe431 and Leu555 (Figure 6C,E), remaining a similar orientation to the original MZ29 ligand (Figure 5A). The hydrophobic interaction between A1’s phenyl ring with Leu555 could play an important role in the inhibition of drug transport given its relevance on ABCG2’s regulation. [34] ABCG2 transmembrane domain contains two apparent cavities, separated by a hydrophobic di‐leucine seal (Leu554 and Leu555). This seal separates the central/larger cavity, which is essential for substrate recognition, from the upper cavity, which is most related to the control of drug release. [34] This sub pocket composed of Leu555 and Phe431 could also be relevant to other inhibitors, such as Ko143, fumitremorgin C and sunitinib.[ 16 , 35 , 36 ] Consequently, we believe the hydrophobic interaction with Leu555, shared by MZ29 and hybrid A1, could be relevant to transport inhibition, given its critical role for translocating substrate towards the upper cavity. [34]
Differently from derivatives MZ29, A1 and A3, the ligands C1, C4, D1, and D4 share similar binding modes, located in S2 (Figure 5C–F). These ligands are stabilized by hydrogen bond interactions between the Arg482 with its methoxy substituents (Figure 5A,D) and intermittent hydrophobic interactions with Pro485 (Figure 5C). Arg482 and Pro485 are located at TH3 (Supporting Information, Figure 4). Further evidence supports Arg482’s role in translating the conformational changes induced by ATP binding in the NBD to the transmembrane domains.[ 35 , 37 , 38 , 39 ] The relevance of this large conformational change can be highlighted by mutants, such as Arg482Thr and Arg482Lys, that show an increased ATP hydrolysis rate, [40] as a compensatory mechanism, and differential affinity for substrates.[ 38 , 39 ]
Additionally, Pro485 confers this helix the structural kink that generates a dynamic binding cavity. Accordingly, Pro485Ala mutant is reported to affect the transport activity for some substrates such as prazosin and SN‐38, without influencing mitoxantrone and Hoechst 33342 transport.[ 35 , 41 ] In our simulations, Pro485 was shown to have similar flexibility, as exemplified by similar RMSF values, where downstream residues were prone to larger conformational changes (Supporting Information, Figure 6C). We hypothesize that differences on the transport are due to changes in conformation and flexibility of the binding cavity, as seem be our compounds that can transit between sub‐pockets. However, the precise structural bases of Pro485 and its contribution to drug binding it is still uncertain.
Given the highly hydrophobic nature of the S1/S2 sites and the relevance of hydrophobic elements in the recognition of inhibitors, we do not disregard that changes in the hydrophobicity profile of the inhibitors might play a role in their inhibitory capacity. This was previously reported by the work of Egido et al., (2015), [42] where non‐amphiphilic scaffolds (i. e. a hydrophobic moiety flanked by two lightly polar parts) were more likely to interact with ABCG2 than other distributions. When comparing the A series against the others (B–D), we observe an increase in this non‐amphiphilic character, which can partially explain the increase in potency.
Interestingly, ABC inhibitors, such as trans stilbenes, can increase the ATPase activity without being transported.[ 43 , 44 ] Alternatively, some inhibitory compounds that have a strong effect on ATP hydrolysis, at the same time compete with other substrates, e. g. Hoechst 33342, by rapidly diffusing into the cell (also called “fast‐diffusers”).[ 45 , 46 ] In this sense, we hypothesize that hybrids C1, C4, D1, and D4 can compete with ABCG2 substrates without being transported. The foregoing due to its effect on the increase of the vanadate‐sensitive ATPase activity (Figure 4), as well as its suggested binding site by docking and MD simulations results (Figure 5). However, more studies about their inhibition type are needed.
Conclusion
The presence of methoxy groups on the phenyl moiety appear to be positive to tetrahydroquinoline/4,5‐dihydroisoxazole hybrids bearing H or Cl on the conjugated aromatic ring. Molecular modelling suggests that hybrid A1 has a similar interaction profile as MZ29 inhibitor, preferentially binding in the site S1, where it shows stable interactions with residues Asn436, Gln398, and Leu555. Particularly, Leu555 is known to play an important role in substrates recognition and its translocation towards the upper cavity.
In sharp contrast, the hybrids C1, C4, D1 and D4 would preferably target S2. Relevant residues associated with stimulation of ATP hydrolysis and ABCG2 transport activity, such as Arg482 and Pro485 are present in this site. We herein propose that the interaction between hybrids C1, C4, D1, and D4 and Arg482 could be related to a strong effect observed on the ATP hydrolysis.[ 35 , 38 , 39 ] Thus, more studies are required to explore the molecular mechanism of inhibition promoted the molecular hybrids.
In conclusion, docking and molecular dynamics simulations provided new insights into THQ/4,5‐dihydroisoxazole molecular hybrids, generating great potential for the development of novel potent ABCG2 inhibitors.
Experimental Section
Synthesis of tetrahydroquinoline/4,5‐dihydroisoxazole hybrids
Tetrahydroquinoline/4,5‐dihydroisoxazole molecular hybrids (A–D series) were easily and efficiently prepared as previously reported. [30] For this reaction, a two‐step synthetic pathway was considered. Initially, the N‐allyl tetrahydroquinoline derivatives were synthesized via a three‐component cationic Povarov reaction mediated by p‐toluenesulfonic acid (PTSA). Once these tetrahydroquinoline precursors are prepared, the corresponding THQ/4,5‐dihydroisoxazole hybrids were obtained, through the 1,3‐dipolar cycloaddition reaction mediated for NaOCl. All hybrid derivatives obtained were purified and characterized by spectroscopy IR and NMR mono‐ and bi‐dimensional experiments and a Mass Spectrometric technique.
ABCG2 transport assay
The effect of test compounds on the transport function was determines using HEK293 stably transfected to overexpress ABCG2 (HEK293‐ABCG2 cells). Cells were seeded (1×105 cells/well) onto 24 well culture plates. After 48 h at 37 °C under 5 % CO2, cells were treated with compounds (10 and 50 μM) and mitoxantrone (10 μM) for 30 min at 37 °C and 5 % CO2. After treatment, culture media was removed and cells were washed with PBS, trypsinized and resuspended in 300 μL of ice‐cold PBS. The intracellular fluorescence of mitoxantrone was monitored by flow cytometry (FACS Calibur ‐ Becton Dickinson) using the FL4 channel. At least 10,000 events were collected for each sample. The maximal fluorescence, considered as 100 % of inhibition, was determined by the difference between the mean fluorescence of HEK‐ABCG2 cells treated with the reference inhibitor Ko143 (0.5 μM) Ko143 and cells without inhibitor, only with the substrate mitoxantrone. The percent of inhibition was determined as previously described. [47]
Vanadate‐sensitive ABCG2 ATPase activity
The ATPase assay was carried out as previously described. [48] High‐Five insect cell total membranes overexpressing ABCG2 were used at a concentration of 5 μg protein (final volume of 100 μL). The membranes were incubated in assay buffer (50 mM Tris‐HCl, pH 6.8, 150 mM N‐methyl‐D‐glucamine (NMDG)‐Cl, 5 mM sodium azide, 1 mM EGTA, 1 mM ouabain, 2 mM DTT, and 10 mM MgCl2) in the presence or absence of sodium orthovanadate (0.3 mM). The protein‐buffer was treated with the compounds at increasing concentrations, as indicated in the graphs, and incubated for 20 minutes at 37 °C in the presence of ATP (5 mM). After incubation, the reaction was stopped with the addition of 5 % SDS (100 μL). For color development, 400 μL of Pi solution (deionized water, sulfuric acid 36.2 N, ammonium molybdate and antimony potassium tartrate), deionized water (500 μL) and 1 % ascorbic acid (200 μL) was added. The absorbance was measured after 10 min at 880 nm using the spectrophotometer Evolution 201 (Thermo Scientific).
Protein preparation and molecular docking
The system preparation and docking calculations were performed using the Schrödinger Drug Discovery suite for molecular modeling (version 2019.4). The ABCG2 atomic structure (PDB ID: 6FFC, resolution 3.56 Å [16] ) was obtained from the Protein Data Bank (PDB, www.rcsb.org). The structure was chosen since it is one of the cryo‐EM structures with high‐resolution reported. ABCG2 structure was prepared with the Protein preparation wizard [49] to fix protonation states of amino acids residues, adding hydrogens and also fixing missing side‐chain atoms. Missing loops between Asp301‐Leu328 and Gly354‐Tyr369 were generated and optimized using Prime. [50] During the preparation of this manuscript, the work from Orlando et al., 2020, provided novel substrate bound structures. In the novel structures, the originally described S1/S2 pockets are fused into a single larger cavity. This is mainly due to conformational changes in the TMD (represented by the interaction between Phe439 of different subunits) induced by the close proximity between the NBDs. We, however, proceeded with the chosen structure, due to the similarity between the co‐determined ligand and our series of compounds in terms of compound size and further studies with those novel structures are planned for follow‐up publications.
All tetrahydroquinoline/4,5‐dihydroisoxazole hybrids as well as the co‐crystallized ligand (tert‐butyl‐3‐((3S,6S,12aS)‐9‐(cyclopentyloxy)‐6‐iso‐butyl‐1,4‐dioxo1,2,3,4,6,7,12,12a‐octahydropyrazino[1′,2′:1,6]pyrido[3,4‐b]indol‐3‐yl)propanoate, herein referred as MZ29) were drawn using maestro and prepared using LigPrep [51] to generate the three‐dimensional conformation, adjust protonation state to physiological pH (7.4), and calculate the partial atomic charges, with the force field OPLS3e. [52]
Docking studies with the prepared ligands were performed using Glide[ 53 , 54 ] (Glide V7.7), with the flexible modality of Induced‐fit docking with extra precision (XP), followed by a side‐chain minimization step using Prime. [55] Ligands were docked within a grid around 12 Å from the centroid of the co‐crystallized ligand (MZ29) generating 20 poses per ligand, this grid size is large enough to encompass both sites 1 and 2. The refinement of protein‐ligand complexes was then performed using Prime with standard options. In this step, all sidechains within 5 Å of each docked ligand pose were minimized. Docking poses were selected by visual inspection based on their common interactions with relevant residues such as Phe439, Phe545 and Thr435, as well as their docking score.
Molecular dynamics simulation
Selected docking poses were further validated by molecular dynamics simulation, where ligand stability within the proposed pocket and its interactions were evaluated. MD simulations were carried out using Desmond [56] engine with the OPLS3e force‐field,[ 57 , 58 ] which leads to improved performance in predicting protein‐ligand binding affinities. Protein was embedded within a DMPC lipid bilayer taken from the OPM database, [59] using the System Builder, where the membrane positioning was controlled by the orientation of the alpha‐helices based on the transmembrane sequences.
The protein‐membrane system was placed in a cubic box with 15 Å from the box edges to any atom of the protein, using PBC conditions, and, filled with TIP3P [60] water. Then, all systems were equilibrated by short simulations under the NPT ensemble for 5 ns implementing the Berendsen thermostat and barostat methods. A constant temperature of 310 K and 1 atm of pressure throughout the simulation using the Nose‐Hoover thermostat algorithm and Martyna‐Tobias‐Klein Barostat algorithm, respectively. After minimization and relaxing steps, we proceeded with the production step of at least 500 ns. All MD simulations were performed at least three independent runs with randomly generated seeds. Trajectories and interaction data are available on Zenodo repository (under the code: 10.5281/zenodo.3746123).
Protein‐ligand interactions and protein conformational changes were analyzed using the Simulation Interaction Diagram (SID) tool. The stability of the MD simulations was monitored by looking specifically on the root mean square deviation (RMSD) (Supporting Information Figure 7, represents of RMSD variation along the simulation), root mean square fluctuation (RMSF, Supporting Information Figure 8) of the ligand and protein along the simulation time. Variation in the RMSD values stabilized after 100 ns for all the ligands (Supporting Information, Figure 7), which is related to the migration of the compound from the docked towards the final binding site. Meanwhile, RMSF values for the transmembrane helices did not show significant difference between chains, suggesting again an equilibrated system (Supporting Information, Figure 8).
For principal component analyses, the backbone of each frame was extracted and aligned using trj_selection_dl.py and trj_align.py scripts from Schrodinger. Individual simulations from all the runs were merged using trj_merge.py into a final trajectory and CMS file which was further used for the generation of the principal components. Principal components of protein C‐alpha atoms were calculated using trj_essential_dynamics.py script. Most of the large moments were captured in the first two components with first component having 24.3 % and second component having 10.3 % of the total motion. Despite the relatively long simulation time, only small changes were observed in the transmembrane domains, in comparison to the NBD (Supporting Information, Figure 9).
WaterMap calculation
WaterMap calculations [61] were performed using the same initial apo structure protein system treated for docking. The system was solvated in TIP3P water box extending at least 10 Å beyond the original MZ29 binding pocket and amino acids being this cutoff were restrained. A single 5 ns molecular dynamics simulation was performed, and the waters molecules trajectories were then clustered into distinct hydration sites. Enthalpy values of the hydration sites were obtained by averaging over the non‐bonded interaction for each water molecule in the cluster. Entropy and enthalpy values for each hydration site were calculated using inhomogeneous solvation theory.
Statistical analyses and figures
Structural images were generated using PyMOL 2.4.0 [62] and graphs were plotted using GraphPad Prism version 8.1 for windows, GraphPad Software, (San Diego, California USA, www.graphpad.com).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors want to thank the CSC – IT Center for Science, Finland, for the generous computational resources. L.C.V. was supported by Colciencias Scholarship fellowships 647 Doctorados nacionales and EDUFI fellowship from Finnish National Agency for Education for a research stay in Finland. This project was supported by: CNPq (grant number 400953/2016‐1), Fundação Araucária (Code 006 – 09/2016), Vicerrectoría de investigación y Extensión of Universidad Industrial de Santander, Colombia and COLCIENCIAS project code 110265842934. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. D.H.K. and S.V.A. were supported by the Intramural Research Program of the National Institutes of Health, the National Cancer Institute, Center for Cancer Research. D.H.K. received a scholarship from CAPES PrInt UFPR program.
L. C. Vesga, T. Kronenberger, A. K. Tonduru, D. H. Kita, I. F. Zattoni, C. C. Bernal, A. R. R. Bohórquez, S. C. Mendez-Sánchez, S. V. Ambudkar, G. Valdameri, A. Poso, ChemMedChem 2021, 16, 2686.
References
- 1. Robey R. W., Pluchino K. M., Hall M. D., Fojo A. T., Bates S. E., Gottesman M. M., Nat. Rev. Cancer 2018, 18, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gottesman M. M., Fojo T., Bates S. E., Nat. Rev. Cancer. 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- 3. Sarkadi B., Homolya L., Szakács G., Váradi A., Physiol. Rev. 2006, 86, 1179–1236. [DOI] [PubMed] [Google Scholar]
- 4. Fletcher J. I., Haber M., Henderson M. J., Norris M. D., Nat. Rev. Cancer. 2010, 10, 147–156. [DOI] [PubMed] [Google Scholar]
- 5. Xiong J., Feng J., Yuan D., Zhou J., Miao W., Sci. Rep. 2015, 5, 16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Locher K. P., Nat. Struct. Mol. Biol. 2016, 23, 487–493. [DOI] [PubMed] [Google Scholar]
- 7. Doyle L. A., Yang W., Abruzzo L. V., Krogmann T., Gao Y., Rishi A. K., Ross D. D., PNAS 1998, 95, 15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyake K., Mickley L., Litman T., Zhan Z., Robey R., Cristensen B., Brangi M., Greenberger L., Dean M., Fojo T., Bates S. E., Cancer Res. 1999, 59, 8–13. [PubMed] [Google Scholar]
- 9. Allikmets R., Schriml L. M., Hutchinson A., Romano-Spica V., Dean M., Cancer Res. 1998, 58, 5337–5339. [PubMed] [Google Scholar]
- 10. Mao Q., Unadkat J. D., AAPS J. 2005, 7, E118–E133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boumendjel A., Macalou S., Ahmed-Belkacem A., Blanc M., Di Pietro A., Bioorg. Med. Chem. 2007, 15, 2892–2897. [DOI] [PubMed] [Google Scholar]
- 12. Basseville A., Hall M. D., Chau C. H., Robey R. W., Gottesman M., Figg W. D., Bates S. E., in ABC Transporters – 40 Years On (Ed.: George A. M.), Springer International Publishing, Cham, 2016, pp. 195–226. [Google Scholar]
- 13. Rabindran S. K., He H., Singh M., Brown E., Collins K. I., Annable T., Greenberger L. M., Cancer Res. 1998, 58, 5850–5858. [PubMed] [Google Scholar]
- 14. Weidner L. D., Zoghbi S. S., Lu S., Shukla S., Ambudkar S. V., Pike V. W., Mulder J., Gottesman M. M., Innis R. B., Hall M. D., J. Pharmacol. Exp. Ther. 2015, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu K., Zhu J., Huang Y., Li C., Lu J., Sachar M., Li S., Ma X., Drug Metab. Pharmacokinet. 2017, 32, 193–200. [DOI] [PubMed] [Google Scholar]
- 16. Jackson S. M., Manolaridis I., Kowal J., Zechner M., Taylor N. M. I., Bause M., Bauer S., Bartholomaeus R., Bernhardt G., Koenig B., Buschauer A., Stahlberg H., Altmann K., Locher K. P., Nat. Struct. Mol. Biol. 2018, 25, 333–340. [DOI] [PubMed] [Google Scholar]
- 17. Juvale K., Stefan K., Wiese M., Eur. J. Med. Chem. 2013, 67, 115–126. [DOI] [PubMed] [Google Scholar]
- 18. Gallus J., Juvale K., Wiese M., Biochim. Biophys. Acta Biomembr. 2014, 1838, 2929–2938. [DOI] [PubMed] [Google Scholar]
- 19. Chearwae W., Shukla S., Limtrakul P., Ambudkar S. V., Mol. Cancer Ther. 2006, 5, 1995–2006. [DOI] [PubMed] [Google Scholar]
- 20. Valdameri G., Genoux-Bastide E., Peres B., Gauthier C., Guitton J., Terreux R., Winnischofer S. M. B., Rocha M. E. M., Boumendjel A., Di Pietro A., J. Med. Chem. 2012, 55, 966–970. [DOI] [PubMed] [Google Scholar]
- 21. do R. A. Pires A., Lecerf-Schmidt F., Guragossian N., Pazinato J., Gozzi G. J., Winter E., Valdameri G., Veale A., Boumendjel A., Di Pietro A., Pérès B., Eur. J. Med. Chem. 2016, 122, 291–301. [DOI] [PubMed] [Google Scholar]
- 22. Robey R. W., Polgar O., Deeken J., To K. W., Bates S. E., Cancer Metastasis Rev. 2007, 1, 39–57. [DOI] [PubMed] [Google Scholar]
- 23. Taylor N. M. I., Manolaridis I., Jackson S. M., Kowal J., Stahlberg H., Locher K. P., Nature 2017, 546, 504–509. [DOI] [PubMed] [Google Scholar]
- 24. Krapf M. K., Gallus J., Namasivayam V., Wiese M., J. Med. Chem. 2018, 61, 7952–7976. [DOI] [PubMed] [Google Scholar]
- 25. Ferreira R. J., Bonito C. A., Cordeiro M. N. D. S., Ferreira M. J. U., Dos Santos D. J. V. A., Sci. Rep. 2017, 7, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lászloá L., Sarkadi B., Hegedüs T., PLoS One 2016, 11, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salaroglio I. C., Gazzano E., Kopecka J., Id K. C., Costamagna C., Fruttero R., Guglielmo S., Riganti C., Molecules. 2018, 23, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Norman B. H., Lander P. A., Gruber J. M., Kroin J. S., Cohen J. D., Jungheim L. N., Starling J. J., Law K. L., Self T. D., Tabas L. B., Williams D. C., Paul D. C., Dantzig A. H., Bioorg. Med. Chem. Lett. 2005, 15, 5526–5530. [DOI] [PubMed] [Google Scholar]
- 29. Jaramillo A. C., Al Saig F., Cloos J., Jansen G., Peters G. J., Cancer Drug Resist. 2018, 1, 6–29. [Google Scholar]
- 30. Bernal C. C., Vesga L. C., Mendez-Sánchez S. C., Romero Bohórquez A. R., Med. Chem. Res. 2020, 29, 675–689. [Google Scholar]
- 31. Rodríguez Y. A., Gutiérrez M., Ramírez D., Alzate-Morales J., Bernal C. C., Güiza F. M., Romero Bohórquez A. R., Chem. Biol. Drug Des. 2016, 88, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Livnat-Levanon N., Gilson A. I., Ben-Tal N., Lewinson O., Sci. Rep. 2016, 6, 21696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Orlando B. J., Liao M., Nat. Commun. 2020, 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khunweeraphong N., Szöllősi D., Stockner T., Kuchler K., Nat. Commun. 2019, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox M. H., Kapoor P., Briggs D. A., Kerr I. D., Biochem. J. 2018, 475, 1553–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manolaridis I., Jackson S. M., Taylor N. M. I., Kowal J., Stahlberg H., Locher K. P., Nature. 2018, 563, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pozza A., Perez-Victoria J. M., Sardo A., Ahmed-Belkacem A., Di Pietro A., Cell. Mol. Life Sci. 2006, 63, 1912–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ejendal K. F. K., Diop N. K., Schweiger L. C., Hrycyna C. A., Protein Sci. 2006, 15, 1597–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robey R. W., Honjo Y., Morisaki K., Nadjem T. A., Runge S., Risbood M., Poruchybsky M. S., Bates S. E., Br. J. Cancer. 2003, 89, 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Honjo Y., Hrycyna C. A., Yan Q. W., Medina-Pérez W. Y., Robey R. W., van de Laar A., Litman T., Dean M., Bates S. E., Cancer Res. 2001, 61, 6635–6639. [PubMed] [Google Scholar]
- 41. Ni Z., Bikadi Z., Shuster D. L., Zhao C., Rosenberg M. F., Mao Q., Biochemistry. 2011, 50, 8057–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Egido E., Müller R., Li-Blatter X., Merino G., Seelig A., Mol. Pharmaceutics. 2015, 12, 4026–4037. [DOI] [PubMed] [Google Scholar]
- 43. Kraege S., Stefan K., Juvale K., Ross T., Willmes T., Wiese M., Eur. J. Med. Chem. 2016, 117, 212–229. [DOI] [PubMed] [Google Scholar]
- 44. Valdameri G., Pereira Rangel L., Spatafora C., Guitton J., Gauthier C., Arnaud O., Ferreira-Pereira A., Falson P., Winnischofer S. M. B., Rocha M. E. M., Tringali C., Di Pietro A., ACS Chem. Biol. 2012, 7, 322–330. [DOI] [PubMed] [Google Scholar]
- 45. Litman T., Druley T. E., Stein W. D., Bates S. E., Cell. Mol. Life Sci. 2001, 58, 931–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Antoni F., Bause M., Scholler M., Bauer S., Stark S. A., Jackson S. M., Manolaridis I., Locher K. P., König B., Buschauer A., Bernhardt G., Eur. J. Med. Chem. 2020, 191, 112133. [DOI] [PubMed] [Google Scholar]
- 47. Valdameri G., Gauthier C., Terreux R., Kachadourian R., Day B. J., Winnischofer S. M. B., Rocha M. E. M., Frachet V., Ronot X., Di Pietro A., Boumendjel A., J. Med. Chem. 2012, 55, 3193–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ambudkar S. V., Methods Enzymol. 1998, 292, 504–514. [DOI] [PubMed] [Google Scholar]
- 49. Adzhigirey M., Day T., Sherman W., Madhavi Sastry G., Annabhimoju R., J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 50. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J. F., Honig B., Shaw D. E., Friesner R. A., Proteins 2004, 367, 351–367. [DOI] [PubMed] [Google Scholar]
- 51. Shelley J. C., Cholleti A., Frye L. L., Greenwood J. R., Timlin M. R., Uchimaya M., J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [DOI] [PubMed] [Google Scholar]
- 52. Roos K., Wu C., Damm W., Reboul M., Stevenson J. M., Lu C., Dahlgren M. K., Mondal S., Chen W., Wang L., Abel R., Friesner R. A., Harder E. D., J. Chem. Theory Comput. 2019, 15, 1863–1874. [DOI] [PubMed] [Google Scholar]
- 53. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 54. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 55. Sherman W., Day T., Jacobson M. P., Friesner R. A., Farid R., J. Med. Chem. 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- 56. Bowers K. J., Bowers K. J., Chow E., Xu H., Dror R. O., Eastwood M. P., Gregersen B. A., Klepeis J. L., Kolossvary I., Moraes M. A., Sacerdoti F. D., Salmon J. K., Shan Y., Shaw D. E., IN SC'06: PROCEEDINGS OF THE 2006 ACM/IEEE CONFERENCE ON SUPERCOMPUTING 2006. [Google Scholar]
- 57. Roos K., Wu C., Damm W., Reboul M., Stevenson J. M., Lu C., Dahlgren M. K., Mondal S., Chen W., Wang L., Abel R., Friesner R. A., Harder E. D., J. Chem. Theory Comput. 2019, 15, acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- 58. Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J. Y., Wang L., Lupyan D., Dahlgren M. K., Knight J. L., Kaus J. W., Cerutti D. S., Krilov G., Jorgensen W. L., Abel R., Friesner R. A., J. Chem. Theory Comput. 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- 59. Lomize M. A., Lomize A. L., Pogozheva I. D., Mosberg H. I., Bioinformatics. 2006, 22, 623–625. [DOI] [PubMed] [Google Scholar]
- 60. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- 61. Abel R., Young T., Farid R., Berne B. J., Friesner R. A., J. Am. Chem. Soc. 2008, 130, 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schrödinger, LLC, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information