

Abstract
Utrophin is an autosomal paralogue of dystrophin, a protein whose deficit causes Duchenne and Becker muscular dystrophies (DMD/BMD). Utrophin is naturally overexpressed at the sarcolemma of mature dystrophin‐deficient fibres in DMD and BMD patients as well as in the mdx Duchenne mouse model. Dystrophin and utrophin can co‐localise in human foetal muscle, in the dystrophin‐competent fibres from DMD/BMD carriers, and revertant fibre clusters in biopsies from DMD patients. These findings suggest that utrophin overexpression could act as a surrogate, compensating for the lack of dystrophin, and, as such, it could be used in combination with dystrophin restoration therapies. Different strategies to overexpress utrophin are currently under investigation. In recent years, many compounds have been reported to modulate utrophin expression efficiently in preclinical studies and ameliorate the dystrophic phenotype in animal models of the disease. In this manuscript, we discuss the current knowledge on utrophin protein and the different mechanisms that modulate its expression in skeletal muscle. We also include a comprehensive review of compounds proposed as utrophin regulators and, as such, potential therapeutic candidates for these muscular dystrophies.
Keywords: Becker muscular dystrophy, biglycan, Duchenne muscular dystrophy, dystrophin, ezutromid, therapy, utrophin
INTRODUCTION
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a fatal X‐linked disorder that affects approximately one in 5000 live male births worldwide. It is caused by one or more mutations in the DMD gene, which encodes dystrophin protein [1, 2, 3]. This protein provides a structural link between the skeletal muscle cytoskeleton and the extracellular matrix, and it is essential to maintain muscle integrity. DMD patients appear clinically asymptomatic at birth; however, they manifest signs of muscle weakness and walking difficulties during early childhood when they are typically diagnosed. Loss of ambulation and wheelchair dependency ensues around puberty. Thanks to improvements in palliative care, life expectancy and quality of life have improved, and many patients may survive beyond 30 years of age [3]. Becker muscular dystrophy (BMD) is a milder dystrophy form caused by in‐frame mutations in the DMD gene, leading to the expression of a shorter and partially functional dystrophin protein. Individuals with BMD share signs and symptoms with DMD patients, but they present a much later disease onset and a nearly average lifespan [4].
Although the molecular mechanisms of this disease have been extensively investigated, there is still no complete curative treatment available. The current standard of care includes corticoids, such as prednisone or deflazacort, to delay disease progression [5]. Therapies based on dystrophin replacement at the protein or gene level are challenging due to the gene's large size, the wide distribution of the skeletal muscle throughout the body, and the possibility of immune response activation. However, many of these aspects have been overcome, and several micro‐dystrophin gene therapies are currently undergoing clinical trials [6].
In recent years, regulatory agencies have conditionally approved several RNA treatments, based on read‐through (ataluren [7]) or exon‐skipping strategies (eteplirsen [8], golodirsen [9] and viltolarsen [10]). Nevertheless, these therapies are only applicable to a low percentage of DMD patients. Moreover, their delivery to the muscle is challenging [11], and their approval is controversial due to the low efficacy in dystrophin restoration and the limited clinical efficacy demonstrated so far [12].
Alternative strategies to mutation‐specific approaches have been under intense investigation in several laboratories worldwide in order to find a therapy applicable to the Becker and Duchenne community, regardless of their specific mutations. Among them, upregulation of utrophin, a structural and functional paralogue of dystrophin, is one of the most promising therapeutic strategies. Recent studies based on high‐throughput screening have identified small molecules able to induce utrophin upregulation. However, utrophin expression is subject to regulation at multiple steps throughout its synthesis and degradation pathways, which need to be studied in depth to improve pharmacological interventions.
Utrophin vs dystrophin: structure, distribution and function
Dystrophin is a 427 kDa protein encoded by the DMD gene, the largest human gene, localised on the X chromosome. Utrophin, known initially as ‘dystrophin‐related protein’, is a 395 kDa autosomal paralogue of dystrophin encoded by the UTRN gene localised in the human chromosome 6q24 [13]. While four full‐length dystrophin isoforms driven by different promoters have been described, only two full‐length utrophin isoforms have been identified to date, utrophin A and B (Figure 1). These isoforms are transcribed from two different promoters, A and B. The two mRNAs vary at their 5’ ends, resulting in two identical functional proteins with slightly different N‐terminal domains and different expression patterns. While utrophin A is expressed in a variety of structures, including neuromuscular junctions, choroid plexus, pia mater and renal glomerulus [14], utrophin B remains restricted to the endothelial cells [15]. Interestingly, five novel 5’ utrophin isoforms (A’, B’, C, D and F) have been recently identified in human adult and embryonic tissues, but they remain to be fully characterised [16]. Both the DMD and the UTRN gene also encode for shorter dystrophin and utrophin transcripts. Shorter dystrophin isoforms, including Dp260, Dp140, Dp116 and Dp71, have been identified in different non‐muscle tissues such as the brain [17] and retina [18] (Figure 1A,C). Utrophin's internal promoters produce shorter transcripts such as Up71, Up140 and G‐utrophin, which are expressed in many tissues with functions not fully understood [19] (Figure 1B, C).
FIGURE 1.
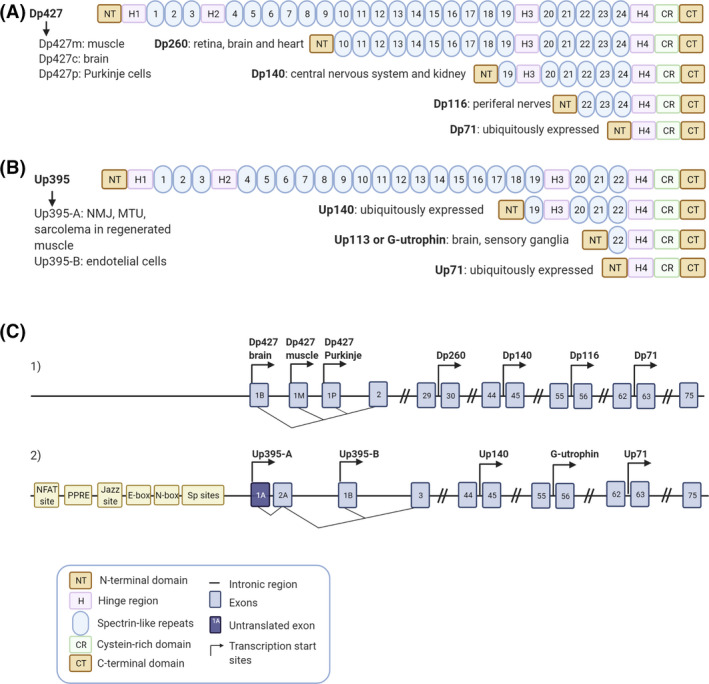
Dystrophin and utrophin isoforms. Schematic representation of the full length and truncated dystrophin (A) and utrophin (B) protein isoforms including their most representative expression in tissues. (C1 and C2) Blue boxes show the specific exons, the black line represents the intronic regions and the transcription start sites of the different promoters are indicated by arrows within the dystrophin (C1) and the utrophin (C2) gene. (C1) Full‐length dystrophin expression is driven by three promoters Dp427 brain, muscle and Purkinje and the smaller isoforms are produced from four internal promoters, Dp260, Dp140, Dp116 and Dp71. (C2) Full‐length utrophin expression is driven by two promoters Up395‐A and Up395‐B and the smaller isoforms are produced from three internal promoters Up140, G‐utrophin and Up71. Different elements of the utrophin A promoter are also specified in the panel. Created with BioRender.com
In the adult skeletal muscle, dystrophin is an essential structural protein that links the extracellular matrix to the actin cytoskeleton through assembly to the dystrophin–glycoprotein complex (DGC) (Figure 2A). This large multi‐protein complex is critical for maintaining the fibre's structural integrity, the stability of the neuromuscular synapse, and the muscle fibre's strength and flexibility while protecting the membrane from contraction‐induced damage [20]. In DMD patients, loss of dystrophin leads to destabilisation and deterioration of the whole complex. Mutations in genes encoding different components of the DGC result in a variety of muscular dystrophies, which highlights the importance of this complex [21].
FIGURE 2.
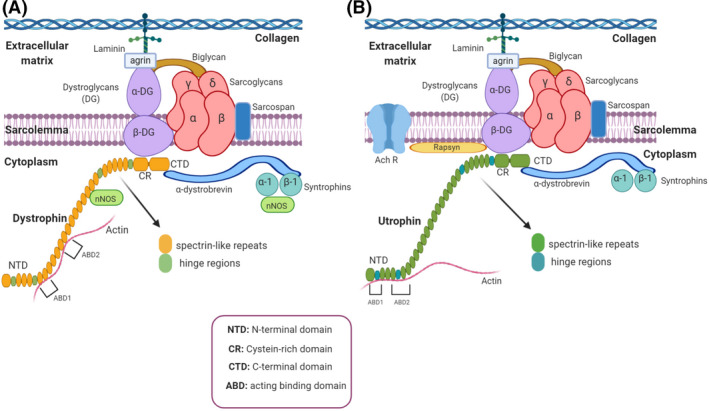
Schematic representation of dystrophin and utrophin glycoprotein complexes (DGC/UGC). (A) Dystrophin glycoprotein complex (DGC) and (B) utrophin glycoprotein complex (UGC) consist of dystrophin (or utrophin), syntrophins, dystrobrevins, sarcoglycans, sarcospan and dystroglycans distributed in cytoplasmic, transmembrane and extracellular protein complex. The cytoplasmic part includes α1 and β1 syntrophin isoforms and α‐dystrobrevin; transmembrane part includes the sarcoglycan (α, β, γ, δ) and sarcospan complex. Dystroglycan complex consists in the extracellular component, α‐dystroglycan (α‐DG) which binds to agrin and laminin in the extracellular matrix and the transmembrane isoform β‐dystroglycan (β‐DG). Biglycan is another extracellular matrix component of the DGC/UGC that binds to α‐dystroglycan and α‐ and γ‐sarcoglycan [22]. Finally, β‐DG binds to dystrophin or utrophin, completing the link between the actin‐based cytoskeleton and the extracellular matrix [23]. Furthermore, utrophin is associated with large acetylcholine receptors (AChR) clusters at the crests of post‐junctional folds in neuromuscular junctions (NMJs) [24]. Notice that the main differences between dystrophin and utrophin are their lateral interactions with actin and the impossibility of the UGC to recruit nNOS. Created with BioRender.com
Dystrophin has four main domains: an N‐terminal actin‐binding domain (NTD), a central spectrin‐like repeat region, a cysteine‐rich domain (CR), which binds the DGC, and a C‐terminal domain (CTD) (Figure 2A). Utrophin shares those domains with dystrophin, but there are some structural and mechanical differences. Both proteins differ in their lateral interactions with actin [20] (Figure 2B), utrophin containing fewer spectrin‐like repeats, and sharing only a 35% homology in the central domain with dystrophin. Moreover, a significant difference in the mechanical behaviour between spectrin repeats has been recently demonstrated [25]. Crucially, they also differ in their capacity to recruit neuronal nitric oxide synthase (nNOS), which cannot be recruited by utrophin [26]. nNOS, a signalling protein associated with the DGC that produces nitric oxide (NO), is considerably reduced in dystrophic muscle fibres, leading to functional ischaemia due to decreased contraction‐induced vasodilation.
While dystrophin is predominantly expressed in muscle and to a lesser extent in the brain, utrophin is widely expressed in several non‐skeletal muscle tissues such as lung, kidney and liver [27]. During foetal muscle development and at early gestational stages, utrophin is present at the sarcolemma of muscle fibres. After birth, utrophin is progressively silenced by the Ets‐2 repressor factor and replaced by dystrophin in adult myofibres. Thereafter, utrophin disappears from the membrane, and its expression is confined to the neuromuscular and myotendinous junctions, where it participates in post‐synaptic membrane maintenance and acetylcholine receptor clustering [28, 29]. However, there is an increase in utrophin expression and redistribution of this protein to the sarcolemma in the dystrophic muscle, in mature dystrophin‐deficient fibres, regenerating fibres and dystrophin‐competent revertant fibres found both in DMD and BMD patients, as well as in mdx mice [30, 31, 32].
Utrophin overexpression in Duchenne muscular dystrophy
Utrophin is naturally increased at the sarcolemma of skeletal muscle samples in DMD and BMD patients compared to healthy individuals [31, 32, 33] by a repair process that also occurs in animal models of the disease, proposed as a compensatory mechanism to mitigate the lack of dystrophin. Moreover, preclinical studies indicate an inverse correlation between utrophin expression and disease severity in DMD, suggesting that utrophin could play a role as a dystrophin surrogate. However, while some human studies report a positive effect of this utrophin expression on disease severity, delaying its progression [34], others do not find any correlation [35].
The most widely used animal model for DMD research is the mdx mouse, carrying a nonsense point mutation (C‐to‐T transition) in exon 23 of the Dmd gene, which completely abolishes dystrophin expression. Despite being dystrophin‐deficient, mdx mice have mild clinical symptoms and a long lifespan, in contrast to DMD patients [36]. Utrophin levels are increased at the sarcolemma of regenerating myofibres in the adult mdx skeletal muscle [37, 38], but this increase may also occur independently of regeneration [39]. Moreover, experimental data suggest that upregulation of utrophin may compensate for dystrophin deficiency. The potential compensatory role of utrophin has been assessed by generating double knockout mice for both dystrophin and utrophin genes (dko). These mice display a much more severe pathology compared to mdx mutants, as well as multiple systemic degenerative changes, in addition to earlier muscle degeneration [40]. On the other hand, the Fiona mouse, a dystrophin‐deficient mdx transgenic mouse that overexpresses utrophin, shows a correction of the dystrophic phenotype [38, 41].
Over the years, preclinical studies have demonstrated that transgenic overexpression and pharmacological modulation of utrophin prevent skeletal muscle pathology in mdx mice. These studies reveal that a 2‐fold increase in sarcolemmal utrophin completely rescues the mechanical function and effectively normalises classical markers of DMD‐related muscle damage [42, 43]. However, even a 1.5 fold increase may be beneficial for mdx mice, given that utrophin localises at the sarcolemma of dystrophic fibres [38]. Utrophin levels also influence mitochondrial pathology that contributes to oxidative stress and propagates muscle damage in DMD. While utrophin deficiency aggravates the pathology, utrophin over‐expression in the dystrophic muscle supports mitochondrial function in mouse models [44]. Interestingly, another study focused on the role of utrophin replacing dystrophin in the male reproductive system discovered that full‐length dystrophin deficiency disturbed the balance between proliferation and apoptosis of germ cells during spermatogenesis. In this case, there is also a utrophin upregulation and relocation as a compensatory response to dystrophin deficiency [45]. Taken together, data in animal models suggest that utrophin can functionally compensate for the lack of dystrophin.
Utrophin overexpression in patients is a promising therapeutic strategy for treating muscle dystrophies, since it targets the primary cause of the disease and would apply to all DMD and BMD patients regardless of their genetic mutation. Several approaches may be used to modulate utrophin levels including direct mechanisms, such as gene or protein replacement, or indirect ones, such as transcriptional upregulation of the utrophin promoter, post‐transcriptional regulation and protein/mRNA stabilisation (see Table 1).
TABLE 1.
Mechanisms of action of potential drugs that could modulate utrophin expression
| Direct mechanisms | Protein replacement | TAT‐μUtrn [46, 47] |
|---|---|---|
| Gene therapy | μUtrn [48] | |
| Indirect mechanisms | Acting at utrophin A promoter level |
Artificial zinc finger transcription factors (ZF‐ATFs): Jazz [49], Bagly [50], Utroup [51], JZif1 [52]. Aryl hydrocarbon receptors (AhR) antagonists [53]: Ezutromid or SMTC0011 [54] and SMT022357[42] Other small molecules: Nabumetone [55], Heregulin [56, 57], Okadaic acid [58], Adiponectin [59, 60] |
| Oxidative phenotype promoters |
Via peroxisome proliferator‐activated receptor (PPAR) agonists: Via AMPK activators: AICAR Metformin [61] Adiponectin Obestatin [62] Quercetin Resveratrol [63] |
|
| nNOS activation |
L‐arginine L‐citrulline |
|
| mRNA stabilisation at 5’ UTR and 3’UTR level |
eEF1A2/IRES‐mediated translation: Betaxolol [64, 65], Pravastatina and 6α‐methylprednisolone‐21 sodium succinate (PDN) [66] via microRNA targeting: Let‐7c, miR‐150, miR‐196b, miR‐296‐5p, miR‐133b AntimiR 206 via p38 MAPK/KSRP: Heparin [67], Heparin/AICAR [68], Heparin/GALGT2 [69] Celecoxib[70] Anisomycin [71] Trichostatin A |
|
| Protein stabilisation |
GalNAc2 [75] |
DIRECT UTROPHIN REPLACEMENT
Protein replacement
Direct protein replacement using recombinant full‐length or truncated utrophin is an attractive potential method to increase utrophin levels in vivo directly.
Systemic administration of a recombinant ‘micro‐utrophin’ (μUtrn) protein combined with the cell‐penetrating TAT protein (TAT‐μUtrn), the transduction domain of the HIV‐1, can functionally form a μUtrophin‐glycoprotein complex at the sarcolemma. This therapeutic strategy is able to mitigate the dystrophic phenotype of mdx mice, improving contractile strength [35]. TAT‐μUtrn also ameliorates the phenotype of dystrophin/utrophin double‐knockout (dko) mice, increasing skeletal muscle strength and improving activity and life span compared to placebo [36].
Although this therapeutic strategy looks promising, posology and administration limit its use; it would be necessary to give frequent, high‐dose injections that could eventually trigger a harmful immune response. Nevertheless, this approach might be combined with other therapies to increase utrophin expression.
Gene therapy
Developing gene therapy treatments for Duchene muscular dystrophy is challenging for three main reasons: first, both full‐length DMD or UTRN genes, and even their cDNAs, are too large and need to be engineered into truncated (‘mini’ or ‘micro’) constructs in order to be packaged into adeno‐associated virus (AAV), which are currently the most commonly used delivery vectors [80]; the second limitation is the possibility of inducing a cellular immune response to the new dystrophin generated and/or against the AAV vector [81]; the third and major challenge is the difficulty to achieve body‐wide transduction into human muscle fibres. Nevertheless, micro‐dystrophin (µDys) gene therapy using AAV vectors has recently been carefully optimised, leading to promising data in murine and canine DMD models [82, 83, 84] and phase I/II clinical trials in DMD patients that are currently ongoing.[85]
A similar pathway has been followed in the development of utrophin gene therapy alternatives. Several preclinical studies using ‘micro‐utrophin’ (µUtrn) gene delivery have been reported in the last years; studies conducted using AAV‐µUtrn in mdx mice reported restoration of the DGC, prevention of myofibre degeneration, normalisation of serum CK levels and improvement of muscle function [86]. Moreover, additional studies in double knockout (dko) mice and canine X‐linked muscular dystrophy dogs have shown that µUtrn improves their severe pathological dystrophic phenotype [87]. Modulation of utrophin expression could potentially treat many disease manifestations since AAV‐μUtrn transgene administration functionally replaces dystrophin in the heart and ameliorates the skeletal and cardiac muscle phenotype in the D2/mdx mouse model [88]. In addition, the ex vivo UTRN gene correction of mouse dystrophic iPS cells by µUtrn gene transfection and subsequent transplantation into dystrophic dko mice has also demonstrated DGC restoration and improvement of contractile strength [89].
Apart from all these promising results, delivering µUtrn instead of µDys has a potential advantage: a lower risk to elicit an immune response since utrophin is naturally expressed at low levels in DMD patients. A recent study performed in the German shorthaired pointer deletional‐null canine model (GSHPMD), reported a strong systemic cell‐mediated immune response against µDys but not to µUtrn. This supports the use of non‐immunogenic utrophin‐based gene therapy approach for DMD [90]. Moreover, overexpression of utrophin rather than dystrophin could prevent the use of expensive and potentially toxic adjuvant immunosuppressive drug therapies [86].
INDIRECT MECHANISMS FOR UTROPHIN OVEREXPRESSION
Transcriptional upregulation
The utrophin A promoter contains several regulatory motifs that could activate utrophin overexpression (Figure 3). The E‐box and N‐box motifs are essential for myogenic differentiation, and synaptic expression of utrophin A [91]. The E‐box motif is a binding site to myogenic factors like MyoD, myogenin, Myf5 and MRF4 [91]. In contrast, the N‐box motif is targeted by the ETS‐related transcription factor complex GA‐binding protein (GABP) α/β, which is activated by nerve‐derived and transcription factors. Besides, Sp binding sites targeted by Sp1 and Sp3 zinc finger transcription factors may establish a cooperative interaction with GABP to stimulate the utrophin promoter [92] (Figure 3). Moreover, it has been recently shown that utrophin A promoter contains a PPRE site targeted by the peroxisome‐proliferator‐activated receptor beta/delta (PPAR‐β/δ). This PPRE site can also be stimulated through 5′ adenosine monophosphate‐activated protein kinase (AMPK) and sirtuin 1 (SIRT‐1) signalling pathways that activate the peroxisome proliferator‐activated receptor‐gamma coactivator 1α (PGC‐1α) which, in turn, activates either PPARβ/δ or GABPα/β.
FIGURE 3.
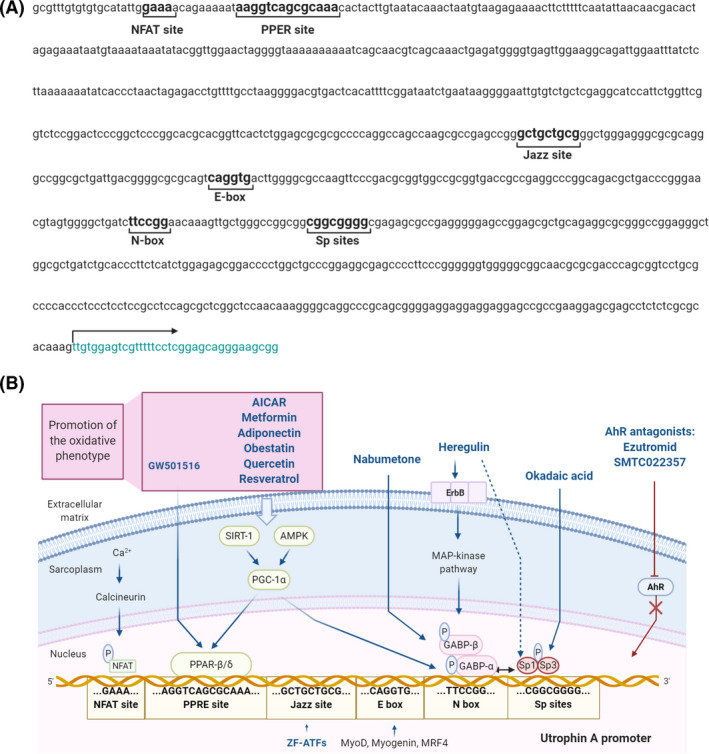
Utrophin A promoter transcriptional activation pathways. (A) Nucleotide sequence of the human UTRN gene promoter including the transcriptional regulatory elements: NFAT binding site, PPRE site, Jazz binding site, E‐box site, N‐box site and Sp binding sites. The arrow indicates the transcription starting site. (B) Representation of the utrophin A promoter regulatory binding sites, their transcriptional upregulation mechanisms and the compounds involved in utrophin upregulation through these signalling pathways (in blue). The compounds that act through promotion of the slow and oxidative phenotype are included in the purple box. Created with BioRender.com
The calcineurin‐nuclear factor of activated T‐cells (NFAT) calcium‐dependent signalling cascade is another pathway that positively regulates utrophin expression in the skeletal muscle. In this pathway, calcineurin dephosphorylates NFAT, enabling its entry into the nucleus and subsequent activation of the utrophin A promoter [93] (Figure 3).
Utrophin upregulation by stimulating utrophin A promoter activity is a promising pharmacological approach that has been extensively investigated using different strategies. One laboratory's proposal was the engineered artificial zinc finger transcription factors (ZF‐ATFs) called ‘Jazz’, capable of binding the utrophin A promoter both in humans and mice. Systemic delivery of ZF‐ATFs with AAVs can induce a significant rescue of muscle function in dystrophic mdx mice through utrophin upregulation [94]. Indeed, several ‘Jazz’ factors have shown remarkable efficacy in ameliorating the pathological phenotype of mdx mice and improving the morphology and plasticity of neuromuscular junctions [52]. Among them, ‘JZif1’, the most recently upgraded version, was developed using the backbone of the well‐characterised Zif268/EGR1 human transcription factor to minimise immunogenicity and facilitate its clinical application.
Thousands of candidates from drug libraries have been tested by high‐throughput screening (HTS) assays in order to find small molecules acting at the utrophin A promotor level. In these cell‐based assays, a reporter gene (usually luciferase) is linked to the utrophin promoter [55, 95]. Small molecules offer several advantages, such as improved delivery and bioavailability compared to gene therapy or protein replacement and the possibility of testing compounds already approved for clinical use. Indeed, drug repurposing to other indications may accelerate their transfer to the clinic and improve their chances of success. In different studies, both repurposing and newly synthesised compounds have shown promising results at preclinical level, with dose‐dependent activation of the utrophin promoter such as nabumetone, heregulin and okadaic acid. Some of them, like ezutromid, have already reached clinical trials.
Nabumetone is a long‐acting nonsteroidal anti‐inflammatory drug, specifically a COX‐1/COX‐2 inhibitor that shows a preference for COX‐2 inhibition in vitro. It is used for pain and inflammation management in osteoarthritis and rheumatoid arthritis, and it is an example of pharmacological repurposing for DMD. HTS assays in C2C12 muscle cells demonstrated that nabumetone could activate utrophin A promoter and upregulate endogenous utrophin at mRNA and protein level [55].
Heregulin is a small nerve‐derived grow factor capable of transactivating utrophin A promoter via the N‐box motif. Utrophin transcription induced by heregulin‐mediated activation of GABPα/β occurs through the extracellular‐signal related kinase (ERK) signalling pathway via the interaction of heregulin with the ErbB tyrosine kinase receptor [56, 57]. Intraperitoneal injections of a small heregulin peptide in mdx mice resulted in upregulation of utrophin, together with a marked functional improvement of the muscle pathology [96].
Recently, it has been shown that okadaic acid, a selective inhibitor of PP1 and PP2A phosphatases, can induce utrophin A promoter activation during myogenesis through Sp1 phosphorylation. There is evidence that okadaic acid increases utrophin A mRNA levels increased by around two‐fold in C2C12 myoblasts, but not in myotubes [58].
Ezutromid (SMTC1100) was the first orally bioavailable utrophin regulator that showed increased UTRN transcription. It was identified following a HTS strategy with a luciferase reporter‐linked assay in murine H2K cells. Later, in vitro assays in human myoblasts demonstrated an increase in utrophin expression at mRNA and protein levels after ezutromid treatment, and further in vivo assays demonstrated that once‐a‐day daily‐dosing of ezutromid in mdx mice increased utrophin levels, as well as muscle strength and resistance to exercise.
After these initial results, ezutromid was developed by Summit Therapeutics as a potential treatment for DMD and BMD. A Phase 1 placebo‐controlled randomised clinical trial in healthy male volunteers and a Phase 1b placebo‐controlled, randomised, double‐blind study in boys with DMD showed that it was safe and well‐tolerated. However, a Phase 2 clinical study (NCT02858362) failed to achieve both the primary (changes in leg muscle magnetic resonance parameters) and secondary endpoints (increased utrophin levels and decreased muscle damage). Based on these results, Summit Therapeutics abandoned the development program of ezutromid [97, 98]. Recent studies have elucidated the ezutromid mechanism of action as an aryl hydrocarbon receptor (AhR) antagonist [53, 99]. Similarly, other molecules that ameliorate mdx pathology like SMT022357 [53] or resveratrol [100] have also shown activity as AhR antagonists [100]. While the pathway between AhR antagonism and utrophin upregulation remains unknown, it seems to involve the stabilisation of active peroxisome proliferator‐activated receptor gamma coactivator (PGC1α) [101]. Indeed, moderately elevated levels of PGC1α ameliorate the dystrophic phenotype of mdx mice at the biochemical, histological and functional levels [102].
SMT022357, is a second‐generation compound structurally related to ezutromid, sharing the same mechanism of action but with improved physicochemical properties and a more robust metabolic profile. SMT022357 administration has been associated with an increase in utrophin expression in skeletal, respiratory and cardiac muscles and prevention of the dystrophic pathology in mdx mice [42].
Oxidative phenotype promotion
An alternative therapeutic strategy to increase utrophin expression in the skeletal muscle focuses on the upregulation of the slow oxidative myogenic program. Promotion of the slow oxidative phenotype has been achieved by different transcriptional and post‐transcriptional pathways showing utrophin overexpression (Figure 3). This strategy has demonstrated attenuation of the dystrophic pathology in mdx animals [103].
One mechanism reported is PPAR‐β/δ stimulation using the synthetic agonist GW501516. This molecule has also been found to stimulate utrophin A promoter in C2C12 muscle cells and improve sarcolemmal integrity in mdx mice, conferring protection against eccentric contraction‐induced damage to muscle [104].
Chronic activation of AMPK also promotes the slow oxidative phenotype. Treatment of mdx mice with 5‐aminoimidazole‐4‐carboxamide‐1‐β‐D‐ribofuranoside (AICAR) and other AMPK/PGC‐1α activators significantly enhanced utrophin expression and have proved to be beneficial for the dystrophic phenotype and rescue muscle function [103].
One of the best known pharmacologically AMPK activators is metformin, a widely prescribed oral antidiabetic drug that has reached clinical trials for DMD in combination with the NOS modulators L‐arginine and L‐citrulline. Metformin increases skeletal muscle utrophin content via AMPK activation and parallel or reciprocal increments in PGC‐1α and PPAR‐δ expression [61]. Skeletal muscle nNOS activation is also AMPK dependent [105]. However, the partial response to metformin treatment in mdx muscles combined with the reduced quantity of NO in some studies supports the notion of combined therapy for DMD patients [61, 106]. In combination with L‐arginine, metformin showed evident amelioration of muscular metabolism in the first proof‐of‐concept pilot study (NCT02516085) carried out in DMD patients. Results from another study, a randomised, double‐blind placebo‐controlled clinical trial with 47 ambulant DMD patients, combining L‐citrulline (an L‐arginine precursor) and metformin (NCT01995032), showed a clinically relevant but not statistically significant reduction in motor function decline in a specific subgroup of patients with no apparent side effects. Therefore, additional clinical trials are needed to validate this approach [107]. Interestingly, NOS‐based therapy by itself has also proved to increase utrophin expression. In this context, L‐arginine administration in mdx mice resulted in a nearly 2‐fold increase in utrophin in skeletal muscle, heart and brain, accompanied by an improvement of the dystrophic phenotype [108]. This study demonstrates that NOS expression has beneficial effects on skeletal muscle metabolism both in vitro and in vivo.
The hormone adiponectin protects the skeletal muscle against inflammation and injury via the AMPK‐SIRT1‐PGC‐1α signalling pathway. Treatment of myotubes from DMD patients with adiponectin leads to downregulation of the nuclear factor kappa B (NF‐κB) and inflammatory genes, together with an upregulation of utrophin [59]. Transgenic upregulation of adiponectin has demonstrated significant beneficial properties in dystrophic mdx muscles [109]. Recently, an orally administrable active adiponectin receptor agonist, called AdipoRon, has been identified. This small synthetic molecule has also proved to attenuate the dystrophic phenotype in mdx mice offering a promising therapeutic prospect for DMD patients [60].
In the same line, obestatin, an autocrine factor that controls the myogenic differentiation program, induces a skeletal muscle shift towards a more oxidative metabolic profile through mechanisms involving PGC1α and class II histone deacetylases (HDAC)/myocyte enhancer factor‐2 (Mef2). It has been reported that obestatin has shown activity in stabilising the sarcolemma of mdx skeletal muscle through the expression of utrophin, α‐syntrophin, β‐dystroglycan and α7β1‐integrin proteins, ameliorating the DMD phenotype [62].
Another molecule studied in preclinical assays that seems to upregulate utrophin through activation of the PGC‐1α pathway is quercetin [110]. Diet enriched with this flavonol seems to rescue dystrophic muscle in mdx mice and provide physiological cardioprotection [111, 112].
Finally, administration of the natural phenol resveratrol to mdx mice has also demonstrated stimulation of the SIRT1‐PGC‐1α pathway, a significant upregulation of utrophin expression, and activation of the slow, oxidative myogenic program in mdx mouse muscle [63].
Post‐transcriptional and translational events regulating utrophin isoform A
While utrophin upregulation at the transcriptional level has been widely investigated over the years, an increasing number of new studies support the importance of post‐transcriptional and translational regulator factors of utrophin in order to find new therapeutic targets (Figure 4).
FIGURE 4.
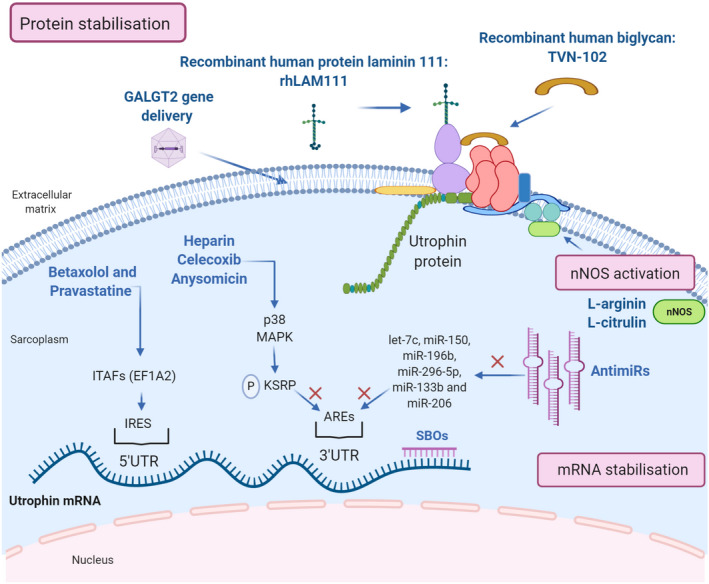
Therapeutic strategies for post‐transcriptional utrophin upregulation. Representation of the post‐transcriptional pathways to enhance utrophin expression: mRNA stabilisation, nNOS activation and protein stabilisation and the compounds acting through these mechanisms (in blue). Created with BioRender.com
Utrophin full‐length isoforms, A and B, have different 5′‐untranslated regions (5′‐UTRs). The skeletal muscle isoform, utrophin A, presents an internal ribosome entry site (IRES) at its 5′UTR that promotes expression through IRES‐dependent translational mechanisms [113]. IRES elements are thought to associate with the translational machinery, including some IRES trans‐acting factors (ITAFs). EF1A2 has been reported as a suitable ITAF able to modulate the activity of the utrophin A IRES. For clarity, we refer to utrophin isoform A as ‘utrophin’ in the manuscript.
A recent ELISA‐ based HTS assay has identified at least four FDA‐approved drugs that target eEF1A2 and cause at least a two‐fold increase in utrophin in C2C12 muscle cells. Among them, betaxolol and pravastatine, seem to improve the dystrophic phenotype of mdx mice via utrophin upregulation through IRES activation [64]. Moreover, in another study, utrophin protein levels are increased after 6α‐methylprednisolone‐21 sodium succinate (PDN) treatment of C2C12 myotubes, suggesting that glucocorticoid's mechanism in muscle cells could be at least partially explained by enhancement of utrophin translation due to IRES activation [66]. These studies highlight the increasing interest in using repurposed drugs to activate this specific pathway where endogenous utrophin levels in muscle are upregulated by promoting protein synthesis from already synthesised transcripts.
Expression of utrophin is also regulated at its UTR 3’ end, where a series of cis‐elements, including conserved AU‐rich elements (AREs), modulate the stability of utrophin mRNA transcripts. Different proteins can bind the AU‐rich elements at the 3'‐UTR and regulate mRNA stability either negatively or positively. For example, 3'‐UTR repression has been attributed to miRNAs and K‐homology splicing regulator protein (KSRP) binding to these sites.
Several miRNAs, including let‐7c, miR‐150, miR‐196b, miR‐296‐5p, miR‐133b and miR‐206 have been shown to repress utrophin expression [114, 115], and this has led to two therapeutic approaches: targeting the microRNAs directly by using antimiRs or blocking their binding site with site‐blocking oligonucleotides (SBOs). Both mechanisms have shown to upregulate utrophin expression and improve the dystrophic phenotype in vivo. Intraperitoneal injections of specific SBOs targeted to prevent let‐7c miRNA binding to the utrophin 3’UTR resulted in higher utrophin protein expression in skeletal muscles and improvement in the dystrophic phenotype in mdx mice [116, 117]. On the other hand, a 3‐month treatment with antiMiR‐206 increases utrophin in mdx mouse muscles compared to the untreated group [118].
Activating p38 mitogen‐activated protein kinase (MAPK) reduces KSRP availability to bind utrophin's 3’UTR AREs, resulting in increased stability of existing mRNAs, increased utrophin protein production and reduction of muscle damage [67]. At least three approved drugs and activators of p38 MAPK, heparin, celecoxib and anysomicin, have demonstrated a significant utrophin upregulation efficacy in different preclinical studies.
Heparin, which is an anticoagulant commonly used in the clinic, significantly increases utrophin levels both in C2C12 [67] myoblasts and mdx mouse dystrophic fibres, leading to substantial morphological and functional improvements [68]. In addition, combinatory treatment with heparin plus AICAR (an oxidative phenotype promoter compound mentioned previously) has an additive effect, increasing utrophin protein levels nearly 3‐fold in C2C12 myoblasts and mdx mouse muscle [68].
Celecoxib is a nonsteroidal anti‐inflammatory drug (NSAID) and a specific cyclo‐oxygenase (COX)‐2 inhibitor used for osteoarthritis and rheumatoid arthritis. This drug can activate p38 MAPK signalling in skeletal muscle cells. Treated mdx mice revealed a 1.5‐ to 2‐fold increase in utrophin expression in tibialis anterior, diaphragm and heart muscles, and ameliorated the dystrophic phenotype, improving muscle strength [70].
Anisomycin is an antibiotic identified by HTS assays. In C2C12 muscle cells, it induces a 2.5‐fold increase in utrophin levels in vitro. It is also reported to significantly increase utrophin protein in the diaphragm of mdx mice treated daily with a low dose [71].
Another recent HTS screening study, targeting the 5′ and 3′ untranslated regions (UTRs), identified 27 hit compounds capable of upregulating utrophin expression [119]. In this study, trichostatin A was identified as one of these hit compounds. Previous studies had demonstrated that trichostatin A could activate the utrophin promoter [55]. It also increases utrophin levels post‐transcriptionally by interacting with the 5′ and/or 3′UTR of the utrophin mRNA, resulting in a functional improvement of the mdx mouse. The remaining hits are yet to be further studied, but this is a good starting point for additional in vitro or in vivo assays.
Utrophin‐glycoprotein complex stabilisation
Utrophin complex stabilisation is an alternative mechanism that has gained strength in the last years with promising results. One example of this approach is the extracellular matrix biglycan, a proteoglycan that plays an essential role in muscle development. Biglycan is a component of the DGC/UGC, where it regulates the expression of sarcoglycans, dystrobrevins, syntrophins and nNOS, by recruiting utrophin to the plasma membrane. In humans and mice, biglycan is most highly expressed in immature and regenerating muscle [22]. Several studies in mdx mice have shown that systemically administered recombinant human biglycan upregulates utrophin and other DGC components at the sarcolemma, while ameliorating muscle pathology and improving muscle structure and function with no obvious toxicity.[72, 73] Tivorsan Pharmaceuticals is currently developing a potential treatment for DMD and BMD called TVN‐102, a recombinant human biglycan that can be systemically administrated [120]. The FDA granted TVN‐102 orphan drug status in 2016. Meanwhile, Tivorsan Pharmaceuticals has completed pharmacological studies in rats and non‐human primates in order to determine the safe starting dose in clinical trials, planned to be initiated soon.
Similarly, the recombinant human protein laminin‐111 (rhLAM111), another extracellular matrix protein, has shown to upregulate other proteins such as utrophin and α7β1 integrin, both capable of restoring muscle cell adhesion and stimulating muscle regeneration in DMD patients. Research in mdx mouse has demonstrated that rhLAM111 can strengthen muscles and improve muscle function. The underlying mechanisms of action reported involved elevated levels of different compensatory proteins and utrophin increases of 1.3‐fold. However, it is not completely clear if this increase in utrophin is sufficient to induce a phenotypical improvement [76, 77]. Indeed, some studies claim that higher utrophin concentrations (1.5/2‐fold increase) are necessary to achieve a therapeutic effect [38]. In any case, recent results show that laminin prevents muscle disease progression in the golden retriever muscular dystrophy (GRMD) dog model of DMD and, thus, it could be a novel protein therapy for DMD patients [120].
Overexpression of CT‐GalNAc 2 (cytotoxic T‐cell N‐acetylgalactosamine transferase), or Galgt2 protein, has been shown to increase synapse‐associated proteins, including utrophin, and enhances its transportation to the sarcolemma [75]. AAV‐mediated GALGT2 gene delivery has shown protection in both wild‐type and dystrophin‐mdx skeletal myofibres from eccentric contraction‐induced injury. It also prevents muscular dystrophy and ameliorates the phenotype in different animal models [121, 122]. Following these studies, the first clinical trial of AAVrh74‐mediated GALGT2 gene delivery in DMD boys began recruiting in 2018.
CONCLUDING REMARKS
Utrophin upregulation is a promising therapeutic approach, applicable for all DMD and BMD patients, that has demonstrated functionally compensation for the lack of dystrophin, improving the pathological phenotype in different dystrophic models.
Many pathways involved in utrophin expression are currently being explored, and some of them have only started to be elucidated. There are high expectations in many compounds that have demonstrated efficacy in activating utrophin expression in preclinical assays. However, the amount of utrophin required by dystrophic patients to achieve a relevant clinical benefit remains to be determined. Hopefully, soon some of these molecules will reach clinical studies and become therapeutic options for the Duchenne community.
CONFLICT OF INTERESTS
A. V.‐I. and V. A.‐G. are shareholders of Miramoon Pharma SL, a company developing DMD treatments, but not related to the focus of this manuscript. All authors report no known competing financial interests or personal relationships that could have appeared to influence the work reported in this manuscript.
AUTHOR CONTRIBUTIONS
P. S.‐M., investigation, writing of original draft, review and editing, visualisation. L. d.‐l.‐P.‐O., investigation, writing ‐ review and editing. A. L.‐M., investigation, writing ‐ review and editing. A. V.‐I. writing ‐ review and editing. V. A.‐G.: conceptualisation, writing or original draft, review and editing, visualisation, supervision, project administration and funding acquisition.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12735.
Soblechero‐Martín P, López‐Martinez A, de la Puente‐Ovejero L, Vallejo‐Illarramendi A, Arechavala‐Gomeza V. Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol Appl Neurobiol. 2021;47:711–723. 10.1111/nan.12735
Funding information
This work was supported by funding from Health Institute Carlos III (ISCIII, Spain) and the European Regional Development Fund, (ERDF/FEDER), ‘A way of making Europe’: Grant PI15/00333; Basque Government (grants 2016111029, 2018222035 and 2020333012) and Duchenne Parent Project Spain (grant 05/2016). P. S‐M holds a Rio Hortega Fellowship from ISCIII (CM19/00104). V.A‐G holds a Miguel Servet Fellowship from the ISCIII (CPII17/00004), part‐funded by ERDF/FEDER. A. L‐M acknowledges funding by Biocruces Bizkaia Health Research Institute (BC/I/DIV/19/001). V.A‐G also acknowledges funding from Ikerbasque (Basque Foundation for Science). None of this funding represents a conflict of interest with the content of this review.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.
REFERENCES
- 1. Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919‐928. [DOI] [PubMed] [Google Scholar]
- 2. Ryder S, Leadley RM, Armstrong N, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet journal of rare diseases. 2017;12:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifiro G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta‐analysis. Orphanet J Rare Dis. 2020;15:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45:498‐506. [PMC free article] [PubMed] [Google Scholar]
- 5. Marden JR, Freimark J, Yao Z, Signorovitch J, Tian C, Wong BL. Real‐world outcomes of long‐term prednisone and deflazacort use in patients with Duchenne muscular dystrophy: experience at a single, large care center. J Comp Eff Res. 2020;9:177‐189. [DOI] [PubMed] [Google Scholar]
- 6. Mendell JR, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro‐dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurology. 2020;77(9):1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McDonald CM, Campbell C, Torricelli RE, et al. Clinical Evaluator Training G, Group ADS. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390:1489‐1498. [DOI] [PubMed] [Google Scholar]
- 8. Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;90:e2146‐e2154. [DOI] [PubMed] [Google Scholar]
- 9. Frank DE, Schnell FJ, Akana C, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94:e2270‐e2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Viltolarsen DS. First approval. Drugs. 2020;80:1027‐1031. [DOI] [PubMed] [Google Scholar]
- 11. Godfrey C, Desviat LR, Smedsrod B, et al. Delivery is key: lessons learnt from developing splice‐switching antisense therapies. EMBO Mol Med. 2017;9:545‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aartsma‐Rus A, Arechavala‐Gomeza V. Why dystrophin quantification is key in the eteplirsen saga. Nat Rev Neurol. 2018;14:454‐456. [DOI] [PubMed] [Google Scholar]
- 13. Love DR, Hill DF, Dickson G, et al. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989;339:55‐58. [DOI] [PubMed] [Google Scholar]
- 14. Weir AP, Burton EA, Harrod G, Davies KE. A‐ and B‐utrophin have different expression patterns and are differentially up‐regulated in mdx muscle. J Biol Chem. 2002;277:45285‐45290. [DOI] [PubMed] [Google Scholar]
- 15. Burton EA, Tinsley JM, Holzfeind PJ, Rodrigues NR, Davies KE. A second promoter provides an alternative target for therapeutic up‐regulation of utrophin in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1999;96:14025‐14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malik D, Basu U. Repression‐free utrophin‐A 5'UTR variants. Mol Biol Res Commun. 2019;8:129‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lidov HG, Selig S, Kunkel LM. Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet. 1995;4:329‐335. [DOI] [PubMed] [Google Scholar]
- 18. Pillers D‐A, Bulman DE, Weleber RG, et al. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat Genet. 1993;4:82‐86. [DOI] [PubMed] [Google Scholar]
- 19. Wilson J, Putt W, Jimenez C, Edwards YH. Up71 and up140, two novel transcripts of utrophin that are homologues of short forms of dystrophin. Hum Mol Genet. 1999;8:1271‐1278. [DOI] [PubMed] [Google Scholar]
- 20. Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:108‐117. [DOI] [PubMed] [Google Scholar]
- 21. Matsumura K, Campbell KP. Dystrophin‐glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve. 1994;17:2‐15. [DOI] [PubMed] [Google Scholar]
- 22. Bowe MA, Mendis DB, Fallon JR. The small leucine‐rich repeat proteoglycan biglycan binds to alpha‐dystroglycan and is upregulated in dystrophic muscle. J Cell Biol. 2000;148:801‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin‐related proteins in muscle. Physiol Rev. 2002;82:291‐329. [DOI] [PubMed] [Google Scholar]
- 24. Belhasan DC, Akaaboune M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci Lett. 2020;722:134833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rajaganapathy S, McCourt JL, Ghosal S, et al. Distinct mechanical properties in homologous spectrin‐like repeats of utrophin. Sci Rep. 2019;9:5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li D, Bareja A, Judge L, et al. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci. 2010;123:2008‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love DR, Morris GE, Ellis JM, et al. Tissue distribution of the dystrophin‐related gene product and expression in the mdx and dy mouse. Proc Natl Acad Sci USA. 1991;88:3243‐3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Clerk A, Morris GE, Dubowitz V, Davies KE, Sewry CA. Dystrophin‐related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J. 1993;25:554‐561. [PubMed] [Google Scholar]
- 29. Lin S, Burgunder JM. Utrophin may be a precursor of dystrophin during skeletal muscle development. Brain Res Dev Brain Res. 2000;119:289‐295. [DOI] [PubMed] [Google Scholar]
- 30. Galvagni F, Cantini M, Oliviero S. The utrophin gene is transcriptionally up‐regulated in regenerating muscle. J Biol Chem. 2002;277:19106‐19113. [DOI] [PubMed] [Google Scholar]
- 31. Arechavala‐Gomeza V, Kinali M, Feng L, et al. Immunohistological intensity measurements as a tool to assess sarcolemma‐associated protein expression. Neuropathol Appl Neurobiol. 2010;36:265‐274. [DOI] [PubMed] [Google Scholar]
- 32. Anthony K, Arechavala‐Gomeza V, Ricotti V, et al. Biochemical characterization of patients with in‐frame or out‐of‐frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurology. 2014;71:32‐40. [DOI] [PubMed] [Google Scholar]
- 33. Ruiz‐Del‐Yerro E, Garcia‐Jimenez I, Mamchaoui K, Arechavala‐Gomeza V. Myoblots: dystrophin quantification by in‐cell western assay for a streamlined development of Duchenne muscular dystrophy (DMD) treatments. Neuropathol Appl Neurobiol. 2018;44:463‐473. [DOI] [PubMed] [Google Scholar]
- 34. Kleopa KA, Drousiotou A, Mavrikiou E, Ormiston A, Kyriakides T. Naturally occurring utrophin correlates with disease severity in Duchenne muscular dystrophy. Hum Mol Genet. 2006;15:1623‐1628. [DOI] [PubMed] [Google Scholar]
- 35. Vainzof M, Passos‐Bueno MR, Man N, Zatz M. Absence of correlation between utrophin localization and quantity and the clinical severity in Duchenne/Becker dystrophies. Am J Med Genet. 1995;58:305‐309. [DOI] [PubMed] [Google Scholar]
- 36. Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome‐linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349‐353. [DOI] [PubMed] [Google Scholar]
- 38. Tinsley J, Deconinck N, Fisher R, et al. Expression of full‐length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441‐1444. [DOI] [PubMed] [Google Scholar]
- 39. Weir AP, Morgan JE, Davies KE. A‐utrophin up‐regulation in mdx skeletal muscle is independent of regeneration. Neuromuscul Disord. 2004;14:19‐23. [DOI] [PubMed] [Google Scholar]
- 40. Deconinck AE, Rafael JA, Skinner JA, et al. Utrophin‐dystrophin‐deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717‐727. [DOI] [PubMed] [Google Scholar]
- 41. Guiraud S, Edwards B, Babbs A, et al. The potential of utrophin and dystrophin combination therapies for Duchenne muscular dystrophy. Hum Mol Genet. 2019;28:2189‐2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guiraud S, Squire SE, Edwards B, et al. Second‐generation compound for the modulation of utrophin in the therapy of DMD. Hum Mol Genet. 2015;24:4212‐4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gilbert R, Nalbantoglu J, Petrof BJ, et al. Adenovirus‐mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum Gene Ther. 1999;10:1299‐1310. [DOI] [PubMed] [Google Scholar]
- 44. Kennedy TL, Moir L, Hemming S, et al. Utrophin influences mitochondrial pathology and oxidative stress in dystrophic muscle. Skelet Muscle. 2017;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen HC, Chin YF, Lundy DJ, et al. Utrophin compensates dystrophin loss during mouse spermatogenesis. Sci Rep. 2017;7:7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sonnemann KJ, Heun‐Johnson H, Turner AJ, Baltgalvis KA, Lowe DA, Ervasti JM. Functional substitution by TAT‐utrophin in dystrophin‐deficient mice. PLoS Med. 2009;6:e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Call JA, Ervasti JM, Lowe DA. TAT‐muUtrophin mitigates the pathophysiology of dystrophin and utrophin double‐knockout mice. J Applied Physiol. 1985;2011(111):200‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Duan D. Micro‐utrophin therapy for duchenne muscular dystrophy. Mol Ther. 2019;27:1872‐1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Corbi N, Libri V, Fanciulli M, Tinsley JM, Davies KE, Passananti C. The artificial zinc finger coding gene ‘Jazz’ binds the utrophin promoter and activates transcription. Gene Ther. 2000;7:1076‐1083. [DOI] [PubMed] [Google Scholar]
- 50. Onori A, Desantis A, Buontempo S, et al. The artificial 4‐zinc‐finger protein Bagly binds human utrophin promoter A at the endogenous chromosomal site and activates transcription. Biochem Cell Biol. 2007;85:358‐365. [DOI] [PubMed] [Google Scholar]
- 51. Onori A, Pisani C, Strimpakos G, et al. UtroUp is a novel six zinc finger artificial transcription factor that recognises 18 base pairs of the utrophin promoter and efficiently drives utrophin upregulation. BMC Mol Biol. 2013;14:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pisani C, Strimpakos G, Gabanella F, et al. Utrophin up‐regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1172‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilkinson IVL, Perkins KJ, Dugdale H, et al. Chemical proteomics and phenotypic profiling identifies the aryl hydrocarbon receptor as a molecular target of the utrophin modulator ezutromid. Angew Chem Int Ed Engl. 2020;59:2420‐2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tinsley JM, Fairclough RJ, Storer R, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6:e19189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moorwood C, Lozynska O, Suri N, Napper AD, Diamond SL, Khurana TS. Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PLoS One. 2011;6:e26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gramolini AO, Angus LM, Schaeffer L, et al. Induction of utrophin gene expression by heregulin in skeletal muscle cells: role of the N‐box motif and GA binding protein. Proc Natl Acad Sci USA. 1999;96:3223‐3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Basu U, Gyrd‐Hansen M, Baby SM, et al. Heregulin‐induced epigenetic regulation of the utrophin‐A promoter. FEBS Lett. 2007;581:4153‐4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rodova M, Brownback K, Werle MJ. Okadaic acid augments utrophin in myogenic cells. Neurosci Lett. 2004;363:163‐167. [DOI] [PubMed] [Google Scholar]
- 59. Lecompte S, Abou‐Samra M, Boursereau R, Noel L, Brichard SM. Skeletal muscle secretome in Duchenne muscular dystrophy: a pivotal anti‐inflammatory role of adiponectin. Cell Mol Life Sci. 2017;74:2487‐2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Abou‐Samra M, Selvais CM, Boursereau R, Lecompte S, Noel L, Brichard SM. AdipoRon, a new therapeutic prospect for Duchenne muscular dystrophy. J Cachexia, Sarcopenia Muscle. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ljubicic V, Jasmin BJ. Metformin increases peroxisome proliferator‐activated receptor gamma Co‐activator‐1alpha and utrophin A expression in dystrophic skeletal muscle. Muscle Nerve. 2015;52:139‐142. [DOI] [PubMed] [Google Scholar]
- 62. Gonzalez‐Sanchez J, Sanchez‐Temprano A, Cid‐Diaz T, et al. Improvement of Duchenne muscular dystrophy phenotype following obestatin treatment. J Cachexia Sarcopenia Muscle. 2018;9:1063‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ljubicic V, Burt M, Lunde JA, Jasmin BJ. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1‐PGC‐1α axis. Am J Physiol Cell Physiol. 2014;307:C66‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Peladeau C, Adam N, Bronicki LM, et al. Identification of therapeutics that target eEF1A2 and upregulate utrophin A translation in dystrophic muscles. Nat Commun. 2020;11:1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miura P, Coriati A, Belanger G, et al. The utrophin A 5'‐UTR drives cap‐independent translation exclusively in skeletal muscles of transgenic mice and interacts with eEF1A2. Hum Mol Genet. 2010;19:1211‐1220. [DOI] [PubMed] [Google Scholar]
- 66. Miura P, Andrews M, Holcik M, Jasmin BJ. IRES‐mediated translation of utrophin A is enhanced by glucocorticoid treatment in skeletal muscle cells. PLoS One. 2008;3:e2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Amirouche A, Tadesse H, Lunde JA, Belanger G, Cote J, Jasmin BJ. Activation of p38 signaling increases utrophin A expression in skeletal muscle via the RNA‐binding protein KSRP and inhibition of AU‐rich element‐mediated mRNA decay: implications for novel DMD therapeutics. Hum Mol Genet. 2013;22:3093‐3111. [DOI] [PubMed] [Google Scholar]
- 68. Peladeau C, Ahmed A, Amirouche A, et al. Combinatorial therapeutic activation with heparin and AICAR stimulates additive effects on utrophin A expression in dystrophic muscles. Hum Mol Genet. 2016;25:24‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cramer ML, Xu R, Martin PT. Soluble heparin binding epidermal growth factor‐like growth factor is a regulator of GALGT2 expression and GALGT2‐dependent muscle and neuromuscular phenotypes. Mol Cell Biol. 2019;39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Peladeau C, Adam NJ, Jasmin BJ. Celecoxib treatment improves muscle function in mdx mice and increases utrophin A expression. FASEB J. 2018;32:5090‐5103. [DOI] [PubMed] [Google Scholar]
- 71. Hadwen J, Farooq F, Witherspoon L, Schock S, Mongeon K, MacKenzie A. Anisomycin activates utrophin upregulation through a p38 signaling pathway. Clin Transl Sci. 2018;11:506‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Amenta AR, Yilmaz A, Bogdanovich S, et al. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci USA. 2011;108:762‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Young MF, Fallon JR. Biglycan: a promising new therapeutic for neuromuscular and musculoskeletal diseases. Curr Opin Genet Dev. 2012;22:398‐400. [DOI] [PubMed] [Google Scholar]
- 74. Ito M, Ehara Y, Li J, Inada K, Ohno K. Protein‐anchoring therapy of biglycan for Mdx mouse model of duchenne muscular dystrophy. Hum Gene Ther. 2017;28:428‐436. [DOI] [PubMed] [Google Scholar]
- 75. Yoon JH, Chandrasekharan K, Xu R, Glass M, Singhal N, Martin PT. The synaptic CT carbohydrate modulates binding and expression of extracellular matrix proteins in skeletal muscle: partial dependence on utrophin. Mol Cell Neurosci. 2009;41:448‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Goudenege S, Lamarre Y, Dumont N, et al. Laminin‐111: a potential therapeutic agent for Duchenne muscular dystrophy. Mol Ther. 2010;18:2155‐2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rooney JE, Gurpur PB, Burkin DJ. Laminin‐111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2009;106:7991‐7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Peter AK, Marshall JL, Crosbie RH. Sarcospan reduces dystrophic pathology: stabilization of the utrophin‐glycoprotein complex. J Cell Biol. 2008;183:419‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Marshall JL, Oh J, Chou E, et al. Sarcospan integration into laminin‐binding adhesion complexes that ameliorate muscular dystrophy requires utrophin and alpha7 integrin. Hum Mol Genet. 2015;24:2011‐2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gregorevic P, Blankinship MJ, Allen JM, et al. Systemic delivery of genes to striated muscles using adeno‐associated viral vectors. Nat Med. 2004;10:828‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mendell JR, Campbell K, Rodino‐Klapac L, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363:1429‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Le Guiner C, Servais L, Montus M, et al. Long‐term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun. 2017;8:16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yoshimura M, Sakamoto M, Ikemoto M, et al. AAV vector‐mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol Ther. 2004;10:821‐828. [DOI] [PubMed] [Google Scholar]
- 84. Gregorevic P, Allen JM, Minami E, et al. rAAV6‐microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Duan D. Micro‐dystrophin gene therapy goes systemic in duchenne muscular dystrophy patients. Hum Gene Ther. 2018;29:733‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ebihara S, Guibinga GH, Gilbert R, et al. Differential effects of dystrophin and utrophin gene transfer in immunocompetent muscular dystrophy (mdx) mice. Physiol Genomics. 2000;3:133‐144. [DOI] [PubMed] [Google Scholar]
- 87. Cerletti M, Negri T, Cozzi F, et al. Dystrophic phenotype of canine X‐linked muscular dystrophy is mitigated by adenovirus‐mediated utrophin gene transfer. Gene Ther. 2003;10:750‐757. [DOI] [PubMed] [Google Scholar]
- 88. Kennedy TL, Guiraud S, Edwards B, et al. Micro‐utrophin improves cardiac and skeletal muscle function of severely affected D2/mdx mice. Mol Ther Methods Clin Dev. 2018;11:92‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Filareto A, Parker S, Darabi R, et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun. 2013;4:1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Song Y, Morales L, Malik AS, et al. Non‐immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med. 2019;25:1505‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dennis CL, Tinsley JM, Deconinck AE, Davies KE. Molecular and functional analysis of the utrophin promoter. Nucleic Acids Res. 1996;24:1646‐1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gyrd‐Hansen M, Krag TO, Rosmarin AG, Khurana TS. Sp1 and the ets‐related transcription factor complex GABP alpha/beta functionally cooperate to activate the utrophin promoter. J Neurol Sci. 2002;197:27‐35. [DOI] [PubMed] [Google Scholar]
- 93. Chakkalakal JV, Stocksley MA, Harrison MA, et al. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc Natl Acad Sci USA. 2003;100:7791‐7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Strimpakos G, Corbi N, Pisani C, et al. Novel adeno‐associated viral vector delivering the utrophin gene regulator jazz counteracts dystrophic pathology in mdx mice. J Cell Physiol. 2014;229:1283‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Moorwood C, Khurana TS. Duchenne muscular dystrophy drug discovery – the application of utrophin promoter activation screening. Expert Opin Drug Discov. 2013;8:569‐581. [DOI] [PubMed] [Google Scholar]
- 96. Krag TO, Bogdanovich S, Jensen CJ, et al. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc Natl Acad Sci USA. 2004;101:13856‐13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tinsley J, Robinson N, Davies KE. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2‐arylbenzoxazole utrophin modulator, following single‐ and multiple‐dose administration to healthy male adult volunteers. J Clin Pharmacol. 2015;55:698‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Muntoni F, Tejura B, Spinty S, et al. A phase 1b trial to assess the pharmacokinetics of ezutromid in pediatric duchenne muscular dystrophy patients on a balanced diet. Clin Pharmacol Drug Dev. 2019;8:922‐933. [DOI] [PubMed] [Google Scholar]
- 99. Chatzopoulou M, Claridge TDW, Davies KE, et al. Isolation, structural identification, synthesis, and pharmacological profiling of 1,2‐trans‐dihydro‐1,2‐diol metabolites of the utrophin modulator ezutromid. J Med Chem. 2020;63:2547‐2556. [DOI] [PubMed] [Google Scholar]
- 100. Casper RF, Quesne M, Rogers IM, et al. Resveratrol has antagonist activity on the aryl hydrocarbon receptor: implications for prevention of dioxin toxicity. Mol Pharmacol. 1999;56:784‐790. [PubMed] [Google Scholar]
- 101. Babbs A, Chatzopoulou M, Edwards B, et al. From diagnosis to therapy in Duchenne muscular dystrophy. Biochem Soc Trans. 2020;48:813‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC‐1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ljubicic V, Miura P, Burt M, et al. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet. 2011;20:3478‐3493. [DOI] [PubMed] [Google Scholar]
- 104. Miura P, Chakkalakal JV, Boudreault L, et al. Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum Mol Genet. 2009;18:4640‐4649. [DOI] [PubMed] [Google Scholar]
- 105. Garbincius JF, Michele DE. Dystrophin‐glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc Natl Acad Sci USA. 2015;112:13663‐13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mantuano P, Sanarica F, Conte E, et al. Effect of a long‐term treatment with metformin in dystrophic mdx mice: A reconsideration of its potential clinical interest in Duchenne muscular dystrophy. Biochem Pharmacol. 2018;154:89‐103. [DOI] [PubMed] [Google Scholar]
- 107. Hafner P, Bonati U, Klein A, et al. Effect of combination l‐citrulline and metformin treatment on motor function in patients with duchenne muscular dystrophy: a randomized clinical trial. JAMA Network Open. 2019;2:e1914171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vianello S, Yu H, Voisin V, et al. Arginine butyrate: a therapeutic candidate for Duchenne muscular dystrophy. FASEB J. 2013;27:2256‐2269. [DOI] [PubMed] [Google Scholar]
- 109. Abou‐Samra M, Lecompte S, Schakman O, et al. Involvement of adiponectin in the pathogenesis of dystrophinopathy. Skelet Muscle. 2015;5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ballmann C, Hollinger K, Selsby JT, Amin R, Quindry JC. Histological and biochemical outcomes of cardiac pathology in mdx mice with dietary quercetin enrichment. Exp Physiol. 2015;100:12‐22. [DOI] [PubMed] [Google Scholar]
- 111. Ballmann C, Denney TS, Beyers RJ, et al. Lifelong quercetin enrichment and cardioprotection in Mdx/Utrn+/‐ mice. Am J Physiol Heart Circ Physiol. 2017;312:H128‐H140. [DOI] [PubMed] [Google Scholar]
- 112. Hollinger K, Shanely RA, Quindry JC, Selsby JT. Long‐term quercetin dietary enrichment decreases muscle injury in mdx mice. Clin Nutr. 2015;34:515‐522. [DOI] [PubMed] [Google Scholar]
- 113. Miura P, Thompson J, Chakkalakal JV, Holcik M, Jasmin BJ. The utrophin A 5'‐untranslated region confers internal ribosome entry site‐mediated translational control during regeneration of skeletal muscle fibers. J Biol Chem. 2005;280:32997‐33005. [DOI] [PubMed] [Google Scholar]
- 114. Morgoulis D, Berenstein P, Cazacu S, et al. sPIF promotes myoblast differentiation and utrophin expression while inhibiting fibrosis in Duchenne muscular dystrophy via the H19/miR‐675/let‐7 and miR‐21 pathways. Cell Death Dis. 2019;10:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Basu U, Lozynska O, Moorwood C, Patel G, Wilton SD, Khurana TS. Translational regulation of utrophin by miRNAs. PLoS One. 2011;6:e29376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mishra MK, Loro E, Sengupta K, Wilton SD, Khurana TS. Functional improvement of dystrophic muscle by repression of utrophin: let‐7c interaction. PLoS One. 2017;12:e0182676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sengupta K, Loro E, Khurana TS. PMO‐based let‐7c site blocking oligonucleotide (SBO) mediated utrophin upregulation in mdx mice, a therapeutic approach for Duchenne muscular dystrophy (DMD). Sci Rep. 2020;10:21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bulaklak K, Xiao B, Qiao C, et al. MicroRNA‐206 downregulation improves therapeutic gene expression and motor function in mdx mice. Mol Ther Nucleic Acids. 2018;12:283‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Loro E, Sengupta K, Bogdanovich S, et al. High‐throughput identification of post‐transcriptional utrophin up‐regulators for Duchenne muscle dystrophy (DMD) therapy. Sci Rep. 2020;10:2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Barraza‐Flores P, Fontelonga TM, Wuebbles RD, et al. Laminin‐111 protein therapy enhances muscle regeneration and repair in the GRMD dog model of Duchenne muscular dystrophy. Hum Mol Genet. 2019;28:2686‐2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nguyen HH, Jayasinha V, Xia B, Hoyte K, Martin PT. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci USA. 2002;99:5616‐5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Durko M, Allen C, Nalbantoglu J, Karpati G. CT‐GalNAc transferase overexpression in adult mice is associated with extrasynaptic utrophin in skeletal muscle fibres. J Muscle Res Cell Motil. 2010;31:181‐193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.