

Abstract
Trastuzumab deruxtecan (T‐DXd) is a HER2‐targeting antibody‐drug conjugate composed of a novel enzyme‐cleavable linker and membrane‐permeable topoisomerase I inhibitor payload. T‐DXd has been approved for HER2‐positive metastatic breast cancer and for HER2‐positive metastatic gastric cancer. The approval in breast cancer was based on results from the DESTINY‐Breast01 (U201; NCT03248492) and J101 (NCT02564900) trials. Here, we present dose justification for the approved 5.4 mg/kg every‐3‐weeks (Q3W) dose based on exposure‐efficacy evaluated in patients with HER2‐positive breast cancer (N = 337) from these 2 trials. Exposure‐safety was assessed in patients with all tumor types (N = 639, n = 512 with breast cancer) across 5 trials, including J101 and DESTINY‐Breast01. T‐DXd doses ranged from 0.8–8.0 mg/kg Q3W; most patients received 5.4 (n = 312) or 6.4 mg/kg (n = 291). For each end point, multivariate logistic or Cox regression analysis was performed using various exposure metrics of T‐DXd and released drug. A statistically significant association was observed between intact T‐DXd area under the concentration‐time curve (AUC) and confirmed objective response rate (ORR; P = 0.028). No significant exposure‐response relationships were observed between intact T‐DXd or released drug and duration of response or progression‐free survival; however, follow‐up was limited. All evaluated safety end points demonstrated a significant (P < 0.05) relationship with either intact T‐DXd or released drug, with higher adverse event (AE) rates projected at higher exposures. Dose‐response projections suggested an increase in ORR (67.5% vs. 62.9%) and toxicity (e.g., grade ≥ 3 all‐cause treatment‐emergent AEs: 61% vs. 54%) with T‐DXd 6.4 vs. 5.4 mg/kg. Results demonstrate the benefit‐risk profile at different doses and guide clinicians in the use of the 5.4‐mg/kg Q3W dose in patients with HER2‐positive metastatic breast cancer.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Trastuzumab deruxtecan (T‐DXd), a novel antibody‐drug conjugate (ADC) composed of a HER2 antibody, cleavable tetrapeptide‐based linker, and topoisomerase I inhibitor payload, was approved in the United States and Japan for use in patients with previously treated, HER2‐positive, unresectable, or metastatic breast cancer at a dose of 5.4 mg/kg every 3 weeks.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We aimed to characterize exposure‐efficacy and exposure‐safety relationships and evaluate covariate relationships.
WHAT DOES THIS STUDY ADD TO OUR KNOW‐LEDGE?
☑ Response to T‐DXd was associated with intact drug exposure, and all safety end points were significantly correlated with either intact T‐DXd or released drug. Integrated dose‐response projections suggested modest increases in both objective response and adverse event rates when the T‐DXd dose was increased from 5.4 to 6.4 mg/kg every 3 weeks.
HOW MIGHT THIS CHANGE CLINICAL PHARMA‐COLOGY OR TRANSLATIONAL SCIENCE?
☑ This study helps to inform future modeling of ADCs, and the specific data—together with clinical analyses—support the use of the label‐approved 5.4‐mg/kg every‐3‐week dosing of T‐DXd in breast cancer.
Trastuzumab deruxtecan (T‐DXd; DS‐8201) is an antibody‐drug conjugate (ADC) composed of an anti‐HER2 antibody, cleavable tetrapeptide‐based linker, and topoisomerase I inhibitor payload (DXd). 1 T‐DXd has a drug‐to‐antibody ratio of ~ 8, with a linker that is stable in plasma and is selectively cleaved by cathepsins upregulated in tumor cells. 2 , 3 , 4 Once cleaved, the released payload is highly membrane permeable—potentially allowing for cytotoxic effects on neighboring tumor cells (known as the bystander effect)—and has a short half‐life to minimize broad systemic exposure. 3 , 5 The anti‐HER2 antibody portion of T‐DXd is anticipated to be degraded to small peptides and amino acids through catabolism much like endogenous IgG. The major elimination route of the released payload is likely to be hepatobiliary excretion of unchanged DXd (based on preclinical data) with some minor metabolism by cytochrome P450 3A4 (CYP3A4) demonstrated in vitro. 6
T‐DXd was initially evaluated in the 2‐part, phase I J101 trial (NCT02564900) in patients with HER2‐expressing solid tumors. 1 , 7 In the dose‐escalation phase, no dose‐limiting toxicities were observed and the maximum tolerated dose was not reached (with a highest tested dose of 8.0 mg/kg); 5.4 and 6.4 mg/kg every 3 weeks (Q3W) were selected as the recommended doses for expansion on the basis of the observed clinical activity and safety profile, as well as pharmacokinetic (PK)‐pharmacodynamic (PD) modeling suggesting that intact T‐DXd trough concentration at steady‐state would exceed the target exposure derived from mouse xenograft studies (4,260 ng/mL) in > 90% of patients. 8 In addition, clinical observation showed a numerical increase in hematologic adverse events (i.e., decreased platelet count and decreased neutrophil count) in the high doses compared with the doses of 3.2 mg/kg or lower. The 8.0 mg/kg was not selected because it was predicted to provide similar response and higher incidence of grade 3 hematologic adverse events based on PK‐PD modeling. The elimination half‐life of intact T‐DXd 5.4 mg/kg in the J101 study was ~ 6 days; accumulation was 35% at steady‐state (cycle 3), which was consistent with the elimination half‐life. 1 , 6 , 9 The released drug showed formation‐limited kinetics, and its apparent elimination half‐life was similar to that of intact T‐DXd. 1 , 9 In the dose‐expansion phase, an objective response rate (ORR) per investigator assessment of 59.5% (66 of 111 patients) was observed in a heavily pretreated (median of 7 prior regimens) cohort of patients with HER2‐positive unresectable or metastatic breast cancer previously treated with trastuzumab emtansine (T‐DM1) who received T‐DXd at the recommended dose of 5.4 or 6.4 mg/kg (ORR by dose: 5.4 mg/kg, 56.5%; 6.4 mg/kg, 61.5%). 7
On the basis of findings from J101, T‐DXd was further evaluated in the registrational phase II DESTINY‐Breast01 trial (U201; NCT03248492) in patients with HER2‐positive unresectable or metastatic breast cancer who had previously been treated with T‐DM1. 10 Patients enrolled in this trial were heavily pretreated, having received a median of six prior treatments. In the part I PK cohort of DESTINY‐Breast01, patients received T‐DXd 5.4, 6.4, or 7.4 mg/kg, with a new formulation, frozen liquid drug product 2. The 7.4 mg/kg dose was included to compensate for a potential reduction (estimated as ≈ 20% from a preclinical study) in T‐DXd exposure with frozen liquid drug product 2, compared with the frozen liquid drug product 1 (used in the J101 trial). 9 When these data were analyzed along with data from the J101 trial, similar PK exposures were suggested between the 2 formulations; therefore 7.4 mg/kg was not considered further, and the 5.4‐ and 6.4‐mg/kg doses were chosen for the dose‐finding stage. 9 On the basis of an exposure‐response analysis showing a balance of efficacy and safety (a significant relationship between minimum concentration (Cmin) and ORR (P = 0.035) but also significant relationships between exposure and hematologic laboratory abnormalities, dose reductions, and discontinuations due to adverse events (AEs), and the incidence of interstitial lung disease (ILD)), 11 5.4 mg/kg was recommended for the part 2 expansion. T‐DXd demonstrated a confirmed ORR by independent central review of 60.9% (112 of 184 patients treated at the recommended dose of 5.4 mg/kg Q3W); consistent responses were observed across prespecified demographic and prognostic subgroups, including those based on geographic region, Eastern Cooperative Oncology Group (ECOG) performance status at baseline, number of prior regimens, and hormone receptor status. Durable benefit was also observed with T‐DXd, with a median duration of response (DOR) of 14.8 months (95% confidence interval (CI), 13.8–16.9 months) and median progression‐free survival (PFS) of 16.4 months (95% CI, 12.7 months to not reached). The overall safety profile was acceptable and consistent with results from the phase I study. Low‐grade hematologic and gastrointestinal AEs were the most common. ILD and pneumonitis, including fatal cases, were reported (adjudicated ILD: grade 1 or 2, 10.9%; grade 3, 0.5%; grade 4, none reported; and grade 5, 2.2%). 10
As of February 2021, T‐DXd has been approved by the US Food and Drug Administration (FDA), the European Commission, and Japan’s Ministry of Health, Labour, and Welfare for HER2‐positive breast and gastric cancers, with exact indications varying by region. Clinical investigation of T‐DXd is ongoing in patients with HER2‐positive or ‐mutated metastatic non‐small cell lung cancer, HER2‐expressing advanced colorectal cancer, and other HER2‐expressing cancers.
A population PK model was previously developed based on pooled data from five clinical studies of T‐DXd. A two‐compartment model with linear elimination best described PK profiles of intact T‐DXd, whereas a one‐compartment model with time‐varying release‐rate constant and linear elimination described released‐drug PK profiles. Statistically significant covariates (country, tumor size, sex, formulation, age, body weight, albumin, total bilirubin, and aspartate aminotransferase (AST)) resulted in a < 20% change in steady‐state area under the concentration‐time curve (AUC) of intact T‐DXd and released drug, except for increased body weight (95th percentile, 86 kg) and decreased albumin (5th percentile, 31 g/L). Analysis of patients stratified by country, race, renal function, and hepatic function found no clinically meaningful differences in steady‐state exposure of intact T‐DXd or released drug. 12
Here, we characterize T‐DXd exposure‐response relationships with respect to key efficacy and safety end points to further support dose justification of T‐DXd in patients with HER2‐positive metastatic breast cancer.
METHODS
Study populations and end points
Data for these analyses were derived from 4 phase I studies (J101 (NCT02564900; data cutoff, February 1, 2019), J102 (NCT03366428; data cutoff, December 5, 2018), A103 (NCT03368196; data cutoff, September 14, 2018), 13 and A104 (NCT03383692; data cutoff, September 26, 2018)) and a phase II study (DESTINY‐Breast01 (U201; NCT03248492; data cutoff, March 21, 2019)), which are summarized in Table 1 . T‐DXd was administered Q3W, with varying doses used across studies (J101: 0.8–8.0 mg/kg; J102 and A103: 6.4 mg/kg; A104: 5.4 mg/kg; DESTINY‐Breast01: 5.4, 6.4, or 7.4 mg/kg). The median treatment duration was 6.64, 3.98, 3.95, 4.80, and 6.31 months in studies J101, J102, A103, A104, and DESTINY‐Breast01, respectively.
Table 1.
Summary of studies included in the analyses
| Study identifier | Study design | Study drug administration | No. of patients | Indication(s) |
|---|---|---|---|---|
| J101 | Phase I, 2‐part, multicenter, nonrandomized, open‐label, multiple‐dose, first‐in‐human study |
T‐DXd i.v. infusion Q3W Part 1 (dose escalation a ) 0.8, 1.6, 3.2, 5.4, 6.4, or 8.0 mg/kg Part 2 (dose expansion) 5.4 or 6.4 mg/kg |
Part 1: n = 27 Part 2: n = 262 0.8 mg/kg: n = 3 1.6 mg/kg: n = 3 3.2 mg/kg: n = 3 5.4 mg/kg: n = 91 6.4 mg/kg: n = 183 8.0 mg/kg: n = 6 |
Part 1: advanced breast cancer or gastric/GEJ adenocarcinoma Part 2a: T‐DM1–treated, HER2‐positive breast cancer Part 2b: trastuzumab‐treated, HER2‐positive gastric/GEJ adenocarcinoma Part 2c: HER2‐low breast cancer Part 2d: other HER2‐expressing solid tumors Part 2e: HER2‐expressing breast cancer |
| DESTINY‐Breast01 | Phase II, 2‐part, global, multicenter, randomized, open‐label, multiple‐dose study |
T‐DXd i.v. infusion Q3W Part 1 5.4, 6.4, or 7.4 mg/kg Part 2 5.4 mg/kg |
Part 1: n = 119 Part 2: n = 134 5.4 mg/kg: n = 184 6.4 mg/kg: n = 48 7.4 mg/kg: n = 21 |
T‐DM1–treated, HER2‐positive, unresectable and/or metastatic breast cancer |
| J102 | Phase I, multicenter, nonrandomized, open‐label, multiple‐dose study | T‐DXd 6.4‐mg/kg i.v. infusion Q3W | N = 51 | HER2‐positive, metastatic and/or unresectable breast cancer |
| A103 | Phase I, multicenter, nonrandomized, open‐label study | T‐DXd 6.4‐mg/kg i.v. infusion Q3W | N = 12 | HER2‐positive, advanced unresectable and/or refractory gastric/GEJ adenocarcinoma or breast cancer |
| A104 | Phase I, multicenter, nonrandomized, open‐label, single‐sequence, crossover DDI study |
T‐DXd 5.4‐mg/kg i.v. infusion Q3W Cohort 1 With ritonavir 200 mg b.i.d. on day 17 of cycle 2 until day 21 of cycle 3 Cohort 2 With itraconazole 200 mg b.i.d. on day 17 of cycle 2 followed by 200 mg once daily until day 21 of cycle 3 |
Cohort 1: n = 17 Cohort 2: n = 23 |
HER2‐positive advanced solid tumors |
DDI, drug‐drug interaction; GEJ, gastroesophageal junction; HER2, human epidermal growth factor receptor 2; T‐DM1, trastuzumab emtansine; T‐DXd, trastuzumab deruxtecan.
Guided by the modified continuous reassessment method using a Bayesian logistic regression model following the escalation with overdose control principle.
The efficacy data set included efficacy‐evaluable patients (those who were treated with T‐DXd and had both baseline and ≥ 1 postbaseline tumor assessments) with HER2‐positive breast cancer from the J101 and DESTINY‐Breast01 studies. Efficacy end points included confirmed ORR, DOR, and PFS per independent central review.
The safety data set included evaluable patients (those who had received ≥ 1 dose of T‐DXd) from the J101, J102, A103, A104, and DESTINY‐Breast01 studies, and was not limited to patients with breast cancer. Safety end points included anemia, neutropenia, and thrombocytopenia (all laboratory test based), ILD (adjudicated by an independent adjudication committee as being drug related), decrease in left ventricular ejection fraction (LVEF; AE and echocardiogram [ECHO]/multigated acquisition [MUGA] based), discontinuations associated with AEs, dose reductions associated with AEs, drug interruptions associated with AEs, grade ≥ 3 AEs, and serious AEs. Unless otherwise specified, AEs were treatment‐emergent events regardless of causality.
PK exposure measures
The PK exposure metrics of T‐DXd and its payload were estimated for individual patients based on empirical Bayes estimates of PK parameters and patients’ specific dosing history from a pooled population PK analysis. 12 Exposure metrics included maximum concentration (Cmax), Cmin, and AUC at cycle 1 and steady‐state as well as average concentration (Cavg) during treatment or Cavg up to time of event. Cavg was calculated as cumulative AUC divided by duration of treatment, or cumulative AUC divided by the time from the start of treatment to time of event, taking into account dose interruption and dose reduction. The exposure metrics were tested for each end point, as described below, and the most statistically significant exposure metric was generally selected in the exposure‐response model; however, consistency across end points and correlation between exposure metrics were also considered.
Exposure‐efficacy analysis
Initial exploratory plots were used to assess exposure‐response relationships. For binary variables (confirmed ORR), data were split by exposure quantiles and the mean response probability was plotted vs. mean exposure together with a fitted logistic regression line. For time‐to‐event variables (PFS and DOR), Kaplan‐Meier curves were plotted by quantile, together with a log rank test.
Following graphical exploration, a logistic regression model with linear or nonlinear function was evaluated for confirmed ORR, and Cox proportional hazards regression model was considered for PFS and DOR (see Supplement for details).
Once the exposure metric was selected into the model, a covariate analysis was conducted according to the standard stepwise forward‐addition and backward‐elimination processes. Covariates were tested on both the intercept and the slope of the exposure‐response relationship (see Supplement for details).
Exposure‐safety analysis
The exposure‐safety analysis was performed in evaluable patients from the aforementioned five studies. Because ILD has been identified as an important risk, an additional sensitivity analysis was performed in patients from J101 and DESTINY‐Breast01 only, due to a shorter treatment duration in the other 3 trials. ILD was modeled using time‐to‐event–based Cox proportional hazards regression to gain a thorough understanding of the time course and incidence of ILD events. Other safety end points were modeled using linear logistic regression to characterize the relationship between drug exposure and event rates. As was done for the exposure‐efficacy analysis, covariates were tested on both the intercept and the slope of the exposure‐response relationship (see Supplement for details).
Dose‐response projection
Based on the developed exposure‐response models, dose‐response projections were made for the 5.4‐ and 6.4‐mg/kg doses. For each end point, 1,000 exposure‐covariate groups were sampled with replacement from the 5.4‐ and 6.4‐mg/kg groups and 1,000 models were simulated from the variance‐covariance matrix of the exposure‐response model. Model‐estimated response rate and 90% CI were then calculated for each dose.
RESULTS
Exposure‐efficacy analysis
Of 493 efficacy‐evaluable patients from the J101 (n = 258) and DESTINY‐Breast01 (n = 235) trials, 337 (J101, n = 106; DESTINY‐Breast01, n = 231) with HER2‐positive breast cancer were included in this analysis. A total of 152 patients from J101 were excluded from this analysis because they did not have HER2‐positive breast cancer (n = 151) or had taken prohibited medication (CYP3A4 inhibitor) prior to starting treatment (n = 1). Four patients from the DESTINY‐Breast01 trial were excluded because they did not have HER2‐positive breast cancer (n = 1) or had missing PK samples (n = 3). Among 337 patients, 2 and 1 received the lower doses of 0.8 and 1.6 mg/kg, respectively, whereas 211, 100, 21, and 2 received the higher doses of 5.4, 6.4, 7.4, and 8.0 mg/kg, respectively.
A statistically significant relationship was observed between intact T‐DXd Cavg to time of response and confirmed central ORR (P = 0.028; Figure 1a ), with no significant covariates identified. In the current analysis data set, 25% and 37% of patients had a dose interruption and dose reduction, respectively. The relative dose intensity was 92% and was slightly numerically lower in the 6.4‐mg/kg group than in the 5.4‐mg/kg group (Table S3 ). The predicted ORR in patients with median Cavg was 64.9%; the predicted ORR in patients with Cavg at the 5th and 95th percentiles was 55.7% and 74.5%, respectively (Figure 1b ). A numerical predictive check of the final model demonstrated that across all quartiles of intact T‐DXd Cavg, there was agreement between model‐predicted and observed ORR, and the observed ORR was contained within the 90% prediction interval. Although not statistically significant, a similar trend was observed in the relationship between steady‐state Cmin of intact T‐DXd and confirmed central ORR (Figure 1c ). No statistically significant relationship was observed between released drug exposure and confirmed central ORR.
Figure 1.
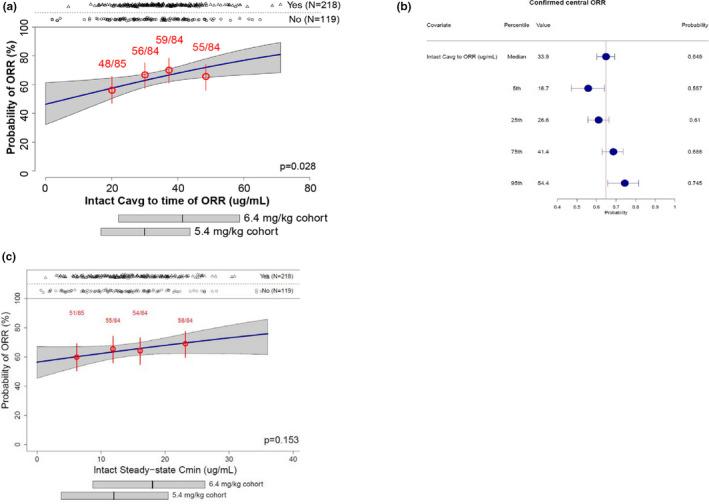
Exposure‐response plot (a) and forest plot of confirmed central ORR by Cavg (b) and exposure‐response plot of confirmed central ORR by Cmin (c). In panels a and c, numbers in red represent n/N; horizontal bars below the plot illustrate the 5th, 50th, and 95th percentiles of exposures. Cavg, average concentration; Cmin, minimum concentration; ORR, objective response rate.
Kaplan‐Meier analysis stratified by quartile for intact T‐DXd Cavg to time of response demonstrated no statistically significant relationship between DOR and intact T‐DXd Cavg response (Figure 2a ). Likewise, no statistically significant relationship was observed between DOR and other intact T‐DXd exposure metrics or released drug exposure. Notably, 88 of 218 patients (40%) remained at risk beyond 6 months. The effect of T‐DXd exposure on PFS was also evaluated and, although there was a trend toward an increased probability of PFS with increasing intact T‐DXd Cavg to time of response (P = 0.127), no intact T‐DXd exposure–PFS relationships achieved statistical significance (P > 0.05; Figure 2b ). In this analysis, 188 of 337 patients (56%) remained at risk beyond 6 months. Because no significant exposure‐response relationships were observed for DOR and PFS, no further covariate assessment was performed for these end points.
Figure 2.
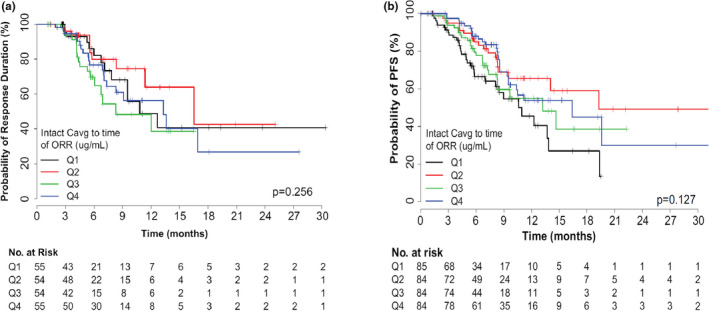
Kaplan‐Meier curves of duration of response (a) and progression‐free survival (b) stratified by intact T‐DXd exposure. Cavg, average concentration; ORR, objective response rate; PFS, progression‐free survival; Q, quartile; T‐DXd, trastuzumab deruxtecan.
Exposure‐safety analysis
A total of 645 patients (n = 512 with breast cancer) were evaluable for safety across the 5 studies; 639 were included in this exposure‐safety analysis and 6 were excluded (DESTINY‐Breast01, n = 3 (missing PK samples); J101, n = 3 (protocol deviation for taking prohibited medication (CYP3A4 inhibitors) prior to starting treatment)). Among 639 patients, 3 each received the lower doses of 0.8, 1.6, and 3.2 mg/kg, respectively, whereas 312, 291, 21, and 6 received the higher doses of 5.4, 6.4, 7.4, and 8.0 mg/kg, respectively. A summary of the observed incidences of events for the safety end points is provided in Table S4 . All safety end points were correlated with intact T‐DXd and/or released drug exposure. Selected exposure metrics together with significant covariates included in the final model for each end point are summarized in Table 2 .
Table 2.
Summary of significant (P < 0.05) exposure‐response relationships identified for safety end points
| Safety end point | Exposure variable | Slope effect |
Covariates (increase [+]/decrease [−] in event rate) |
|---|---|---|---|
| Discontinuations due to AEs | Intact steady‐state AUC | + |
Asian‐Japan on slope (+) a Asian–non‐Japan on slope (−) a ≥ 6 prior cancer therapies on intercept (+) |
| Dose reductions due to AEs | Released drug Cavg through event cycle | + |
HER2‐negative status on intercept (−) b HER2 unknown/other status on intercept (−) b Body weight ≥ 60 kg on slope (−) ≥ 6 prior cancer therapies on slope (−) |
| Dose interruptions due to AEs | + |
Asian‐Japan on intercept (+) a Asian–non‐Japan on intercept (+) a ≥ 6 prior cancer therapies on intercept (−) |
|
| Grade ≥ 3 AEs | + | Body weight ≥ 60 kg on slope (−) | |
| Serious AEs | + | ECOG PS ≥ 1 on slope (+) | |
| Anemia (any grade) | + |
Body weight ≥ 60 kg on slope (−) Male sex on intercept (+) Baseline hemoglobin level < 120 g/L on intercept (+) |
|
| Anemia (grade ≥ 3) | + |
Asian‐Japan on slope (+) a Asian–non‐Japan on slope (+) a ECOG PS ≥ 1 on intercept (−) Baseline hemoglobin level < 120 g/L on intercept (+) Age ≥ 60 years on intercept (+) |
|
| Neutropenia (any grade) | + |
Asian‐Japan on slope (+) a Asian–non‐Japan on slope (+) a Male sex on slope (−) Baseline neutrophil count < 3.6 × 109/L on intercept (+) |
|
| Neutropenia (grade ≥ 3) | + |
Asian‐Japan on slope (+) a Asian–non‐Japan on slope (+) a Baseline neutrophil count < 3.6 × 109/L on intercept (+) |
|
| Thrombocytopenia (any grade) | + |
Asian‐Japan on slope (+) a Asian–non‐Japan on slope (+) a Baseline platelet count < 220 × 109/L on intercept (+) |
|
| Thrombocytopenia (grade ≥ 3) | + |
Asian‐Japan on slope (+) a Asian–non‐Japan on slope (+) a ≥ 6 prior cancer therapies on intercept (+) ≥ 6 prior cancer therapies on slope (−) |
|
| LVEF decrease by ECHO (grade ≥ 2) | Intact steady‐state Cmax | + | None |
| ILD (any grade) | Intact steady‐state AUC | + |
Asian‐Japan (+) a Asian–non‐Japan (−) a |
| ILD (grade ≥ 3) | Intact steady‐state Cmax | + | None |
AE, adverse event; AUC, area under the concentration‐time curve; Cavg, average concentration; Cmax, maximum concentration; ECHO, echocardiogram; ECOG PS, Eastern Cooperative Oncology Group performance status; HER2, human epidermal growth factor receptor 2; ILD, interstitial lung disease; LVEF, left ventricular ejection fraction.
Reference category is non‐Asian.
Reference category is HER2‐positive status.
Intact T‐DXd exposures were correlated with discontinuations associated with AEs (steady‐state AUC), ILD (any grade (steady‐state AUC) and grade ≥ 3 (steady‐state Cmax)), and LVEF decrease by ECHO/MUGA (grade ≥ 2 (steady‐state Cmax)). LVEF decrease by ECHO was used as an end point due to low patient numbers when LVEF was included as an investigator‐reported AE (any‐grade LVEF decrease, n = 14; grade ≥ 3, n = 2). The Cavg of released drug up to the end of the event cycle was correlated with the remaining AEs. Figure 3 shows example logistic regression plots for discontinuations associated with AEs (Figure 3a ) and anemia (Figure 3b ) and Cox regression fits for ILD (Figure 3c ) demonstrating increased event probabilities with increasing exposure of intact T‐DXd or released drug. Numerical predictive checks supported the final model for each of the safety end points.
Figure 3.
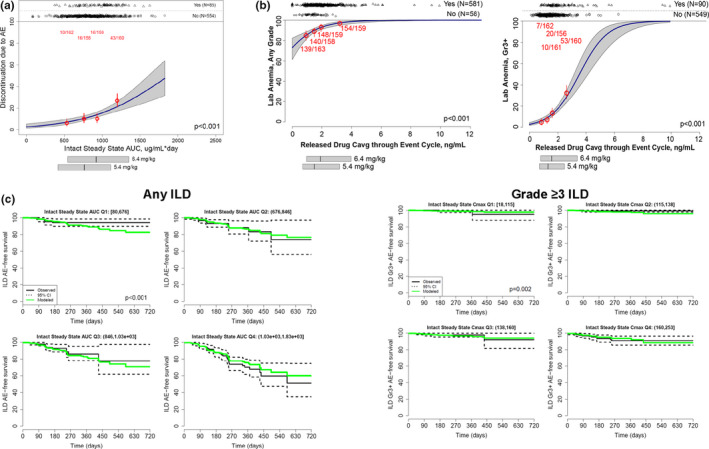
Logistic regression plots of discontinuations associated with AEs (a) and anemia (b) and Cox regression plots of ILD (c). In panels a and b, numbers in red represent n/N; horizontal bars below the plot illustrate the 5th, 50th, and 95th percentiles of exposures. In panel c, the solid black line represents the estimated Kaplan‐Meier curves, the dashed lines are 95% CIs, and the solid green line represents the estimated Kaplan‐Meier curve determined using multivariate Cox regression. AE, adverse event; AUC, area under the concentration‐time curve; Cavg, average concentration; CI, confidence interval; Cmax, maximum concentration; ILD, interstitial lung disease; Q, quartile.
Analysis of covariates identified race‐country (Asian‐Japan, Asian–non‐Japan, and non‐Asian) as the most common covariate—it was observed to be significantly associated with eight safety end points, including discontinuations and dose interruptions due to AEs, most hematologic end points (grade ≥ 3 anemia, neutropenia (any grade and grade ≥ 3), and thrombocytopenia (any grade and grade ≥ 3)), and any‐grade (but not ≥ grade 3) ILD (Table 2 ). For these eight end points, the modeled event rates in patients in the Asian‐Japan group were greater than those in the non‐Asian group. Example forest plots of event probabilities for discontinuations associated with AEs, anemia, and ILD are shown in Figure 4 and final model parameters are shown in Table S5 . There was a higher probability of discontinuations associated with AEs in the Asian‐Japan group and a lower probability in the Asian–non‐Japan group than in the non‐Asian group. The probability of discontinuations associated with AEs was also higher in patients who had received greater than or equal to six prior nonhormonal cancer therapies than in patients who had received less than six (Figure 4a ). The probability of anemia of any grade was decreased in patients weighing ≥ 60 kg and was increased in men or patients with baseline hemoglobin level < 120 g/L (Figure 4b ). The probability of grade ≥ 3 anemia was increased in patients in the Asian‐Japan and Asian–non‐Japan groups relative to the non‐Asian group and was also increased in patients aged ≥ 60 years and those with baseline hemoglobin level < 120 g/L but was decreased in patients with an ECOG performance status ≥ 1 (Figure 4b ). With respect to ILD, characteristics appeared to differ based on severity. Any‐grade ILD was projected to occur more frequently in the Asian‐Japan group than in the non‐Asian group. Conversely, high‐grade ILD (grade ≥ 3) was projected to occur at a similar frequency regardless of race‐country (Figure 4c ). A sensitivity analysis of ILD using data from the J101 and DESTINY‐Breast01 trials identified the same covariates and similar parameter estimates.
Figure 4.
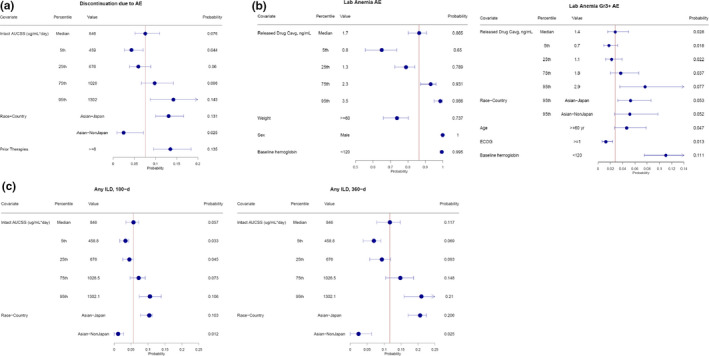
Forest plots of probability of discontinuations associated with AEs (a), anemia (b), and ILD (c). AE, adverse event; AUCSS, area under the concentration‐time curve at steady state; Cavg, average concentration; ECOG PS, Eastern Cooperative Oncology Group performance status; ILD, interstitial lung disease.
Integrated dose‐response projections
Model‐projected event rates were plotted and compared for T‐DXd doses of 5.4 and 6.4 mg/kg (Figure 5 ). Dose‐response projections suggested a numerical increase in efficacy with 6.4 vs. 5.4 mg/kg, with an absolute increase of 4.6% in ORR (67.5% (90% CI, 58.4–76.0%) vs. 62.9% (90% CI, 55.4–69.5%)). However, the 6.4‐mg/kg dose was also associated with a corresponding increase in projected grade ≥ 3 AEs (6.9% projected absolute increase; 60.6% (90% CI, 57.3%–63.6%) vs. 53.7% (90% CI, 50.2–57.1%)) and discontinuations due to AEs (3.1% projected absolute increase; 14.4% (90% CI, 12.4–17.2%) vs. 11.3% (90% CI, 9.5–13.9%)). Additionally, the incidence of some specific AEs, including grade ≥ 2 LVEF decrease (by ECHO; 4.0% projected absolute increase; 17.2% (90% CI, 14.6–20.1%) vs. 13.2% (90% CI, 11.1–15.9%)), any‐grade ILD (3.0% projected absolute increase; 17.0% (90% CI, 14.8–19.2%) vs. 14.0% (90% CI, 11.5–15.6%)), and grade ≥ 3 ILD at 360 days of treatment (1.6% projected absolute increase; 4.0% (90% CI, 2.6–5.1%) vs. 2.4% (90% CI, 1.7–3.2%)), were projected to be higher with 6.4 vs. 5.4 mg/kg. Dose‐response projections stratified by race‐country showed a similar pattern of benefit‐risk in the Asian‐Japan, Asian–non‐Japan, and non‐Asian groups. Higher rates of some hematologic laboratory abnormalities were projected in Asian‐Japan and Asian–non‐Japan patients compared with non‐Asian patients at both 5.4‐ and 6.4‐mg/kg doses (Figure 5 ). Notably, projections for efficacy are based on data from patients with metastatic breast cancer only, whereas those for safety are based on patients with various solid tumors.
Figure 5.
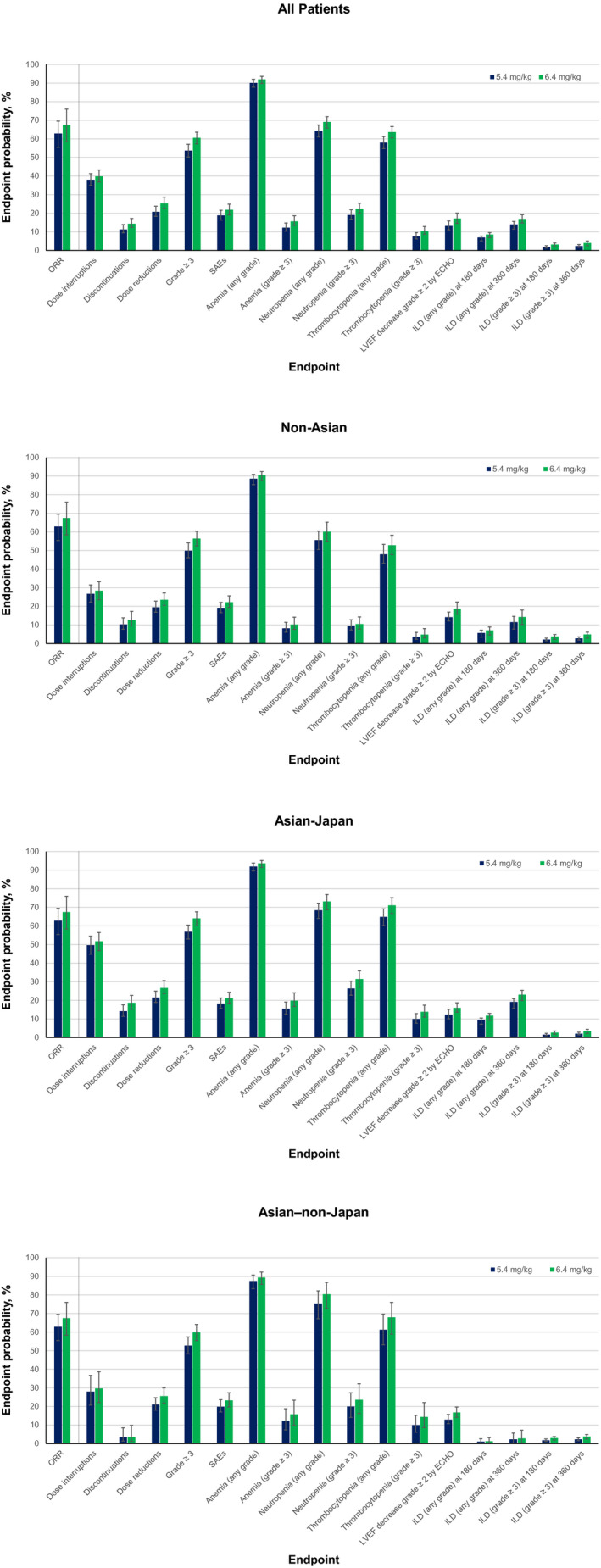
Model‐projected event rates for T‐DXd 5.4‐ and 6.4‐mg/kg doses in the overall population (all patients), non‐Asian group, Asian‐Japan group, and Asian–non‐Japan group. Error bars represent 90% CI. No effect of race‐country was observed on ORR; therefore, the ORR on all race‐country plots represents the ORR for the overall population. Discontinuations, dose interruptions, and dose reductions plotted are those due to adverse events. Anemia, neutropenia, and thrombocytopenia are laboratory test based. ECHO, echocardiogram; ILD; interstitial lung disease; LVEF, left ventricular ejection fraction; ORR, objective response rate; SAE, serious adverse event.
DISCUSSION
This is the first report of exposure‐response relationships in patients treated with the novel HER2‐directed ADC T‐DXd. This analysis demonstrated that objective response to T‐DXd in patients with HER2‐positive metastatic breast cancer was significantly correlated with intact drug exposure and that all safety end points were significantly correlated with either intact T‐DXd or released drug exposure. Integrated dose‐response projections suggested modest increases in both ORRs and AE rates when the T‐DXd dose was increased from 5.4 to 6.4 mg/kg Q3W.
Exposure‐efficacy analysis showed that ORR was significantly correlated with intact T‐DXd Cavg in patients with metastatic breast cancer. This result may not be surprising, because Cavg reflects the actual average exposure in individual patients. Because 25% and 37% of patients had dose interruptions or dose reductions, respectively, during treatment and these events occurred more often in the 6.4‐ vs. 5.4‐mg/kg group, evaluation of Cavg as the exposure metric is important because it considers the impact of dose interruptions or reductions and therefore represents the actual average exposure in individual patients. The model predicted an absolute increase in ORR of 4.6% with 6.4 vs. 5.4 mg/kg (67.5% vs. 62.9%), which was consistent with observed clinical data in patients with HER2‐positive metastatic breast cancer treated with T‐DXd in the DESTINY‐Breast01 (ORR (6.4 vs. 5.4 mg/kg), 68.8% vs. 60.9%) and J101 (ORR, 61.5% vs. 56.5%) trials. 7 , 10 Exposure‐response analysis of T‐DM1 in patients with HER2‐positive metastatic breast cancer showed a similar correlation between T‐DM1 exposure (Cmin at cycle 1, day 21) and ORR. 14 Previous studies have demonstrated that higher disease burden at baseline can impact the clearance of monoclonal antibodies, leading to higher target‐mediated clearance, which can confound exposure‐efficacy relationships. 15 , 16 Importantly, as described in a population PK analysis, target‐mediated clearance was not observed at a clinically relevant dose of T‐DXd (nonlinear clearance was observed at dose levels ≤ 1.6 mg/kg). 10 Tumor size (sum of diameters of target tumors) was included in the assessed covariates in this analysis and was not associated with the T‐DXd exposure–ORR relationship. Overall, no significant covariates were identified in the exposure‐ORR relationship.
Despite the observed relationship between intact T‐DXd and ORR, no significant correlation was observed between exposure and DOR or PFS. Exposure‐response analysis of T‐DM1 demonstrated significantly greater PFS and OS with higher T‐DM1 exposure. 14 One potential reason for lack of a significant exposure‐DOR or exposure‐PFS association in the current analysis was the limited duration of patient follow‐up at the time of this analysis, with only 88 of 218 (40%) and 188 of 337 (56%) patients remaining at risk beyond 6 months for DOR and PFS, respectively. In addition, the relatively narrow range of intact T‐DXd (3‐fold) and released drug (4‐fold) exposure due to the limited dose levels available for this analysis may limit a more complete picture of the exposure‐response relationships. As a limitation, immortal time bias may exist for time‐to‐event measures. This potential bias has not been accounted for in the Cox regression analysis of DOR and PFS because a consistent early landmark exposure or treatment predictor could not be readily identified.
All evaluated safety end points demonstrated an exposure‐response relationship with intact T‐DXd and/or released drug, with higher AE rates projected at 6.4 mg/kg vs. 5.4 mg/kg T‐DXd. Intact T‐DXd exposure was most significantly correlated with discontinuations associated with AEs, ILD, and grade ≥ 2 LVEF decreases by ECHO, whereas the Cavg of released drug was most significant for all other safety end points. Similar exposure‐response profiles regarding ILD and discontinuations associated with AEs were expected because discontinuation was mandated in patients with grade ≥ 2 ILD, and ILD/pneumonitis was the most common AE leading to discontinuation. 7 , 10 Previous studies have suggested that AEs associated with ADCs can be driven by non–target‐mediated pinocytosis of the intact ADC or by the released payload. 17 , 18 , 19 The mechanism of some AEs associated with T‐DXd is unknown and may warrant further evaluation to clarify whether they are driven by released drug or by intact T‐DXd. For instance, ILD was most strongly associated with intact T‐DXd exposure. However, pulmonary toxicity has been observed with HER2‐directed therapies, including trastuzumab and T‐DM1, 20 , 21 and topoisomerase I inhibitors. 22 Therefore, further evaluation is currently ongoing in order to understand the mechanism of T‐DXd–associated ILD. LVEF dysfunction has previously been associated with anti‐HER2 therapy, including trastuzumab. 23 However, data from T‐DXd clinical trials, including DESTINY‐Breast01 10 and J101, 7 , 24 have demonstrated no clinically significant cardiotoxicity in patients treated with T‐DXd. This is further evidenced by the very limited incidence of LVEF dysfunction reported as an AE across T‐DXd trials, and thus the end point assessed was amended to grade ≥ 2 LVEF decrease by ECHO/MUGA for this analysis. The relatively low incidence of cardiotoxicity across T‐DXd trials may limit the interpretability of any associations observed in these analyses.
In a pooled population PK analysis, exposures of intact T‐DXd and released drug were similar between Asian vs. non‐Asian and Japanese vs. non‐Japanese patients. 12 Despite similar PK exposures, discontinuations associated with AEs, dose interruptions associated with AEs, and hematologic AEs were generally higher in the Asian‐Japan group than in the non‐Asian group at both the 5.4‐ and 6.4‐mg/kg doses. The Asian‐Japan group had more any‐grade ILD events than the non‐Asian group. One possible explanation for the higher rate of any‐grade ILD in the Asian‐Japan group could be differences in ILD monitoring and/or physician experience with drug‐induced pulmonary toxicities. Alternatively, there is evidence that Japanese patients may have a higher susceptibility to drug‐induced ILD than non‐Japanese patients. 25 , 26 In addition, a recent study identified a potential genetic polymorphism linked to the pathophysiology of ILD in Japanese patients with antineutrophil cytoplasmic antibody–associated vasculitis, suggesting that there could be some genetic basis to increased ILD susceptibility in Japanese patients. 27 The mechanisms of T‐DXd–associated ILD require further evaluation, and it remains unknown whether genetic variants play a role in the modeled increase observed in the Asian‐Japan group. The limited population in the non‐Asian group in the assessed studies unfortunately precluded further assessment of potential differential safety signals in other race groups.
Dose‐response projections in the overall population suggested a modest 4.6% increase in ORR but also an increase in most AEs with T‐DXd 6.4 vs. 5.4 mg/kg Q3W. Notably, the majority of 90% CIs for model‐projected event rates, including those for ORR, overlapped between the 5.4‐ and 6.4‐mg/kg doses. One exception was the increased rate of grade ≥ 3 AEs, for which the 90% CIs did not overlap. Clinical data from the J101 trial support these model projections, with an ORR of 56.5% in patients treated with 5.4 mg/kg and 61.5% in patients treated with 6.4 mg/kg, but with an increase in grade ≥ 3 AEs (5.4 vs. 6.4 mg/kg, 39% vs. 58%). 7 In line with clinical observations, the current analysis also suggested a lower rate of any‐grade ILD and grade ≥ 3 ILD with T‐DXd 5.4 vs. 6.4 mg/kg. Overall, exposure‐response analysis in patients with HER2‐positive metastatic breast cancer supported clinically meaningful efficacy of T‐DXd at both the 5.4‐ and 6.4‐mg/kg doses, but the 5.4‐mg/kg dose was considered to have a more positive benefit‐risk profile due to a lower modeled incidence of safety events without a meaningful reduction in efficacy compared with the 6.4‐mg/kg dose.
No significant covariates were identified for the exposure‐ORR relationship, whereas race‐country was identified as the most common and relevant covariate for the safety end points evaluated. The slope of exposure‐response relationships for the hematologic laboratory abnormalities appeared to be greater in the Asian−non‐Japan group than in the non‐Asian group, with a similar pattern observed for Asian‐Japan patients. Although serious AEs, grade ≥ 2 LVEF reductions by ECHO, and grade ≥ 3 ILD events were similar between the Asian‐Japan and non‐Asian groups, discontinuations due to AEs, dose interruption associated with AEs, hematologic laboratory abnormalities, and ILD of any grade were higher, and the slope of exposure‐response relationships for these AEs was greater in Asian‐Japan patients. Dose‐response projections stratified by race‐country suggested similar overall benefit‐risk profiles among non‐Asian, Asian‐Japan, and Asian−non‐Japan patients except for higher hematologic laboratory abnormalities in the Asian−non‐Japan and Asian‐Japan vs. non‐Asian group at both the 5.4‐ and 6.4‐mg/kg doses. These results supported the selection of 5.4 mg/kg as the recommended dose in Asian−non‐Japan and Asian‐Japan patients.
In conclusion, the results of these analyses demonstrate the benefit‐risk profile at different doses and guide clinicians in the use of the T‐DXd 5.4 mg/kg Q3W dose in patients with HER2‐positive metastatic breast cancer; this corresponds to the dose regimen recommended in the recently approved FDA prescribing information.
FUNDING
The studies included in this analysis were funded by Daiichi Sankyo, Inc. and AstraZeneca Pharmaceuticals, LP.
Conflicts of Interest
O.Y., T.G., M.A.T., L.Z., J.S., and F.L. report employment with and equity ownership in Daiichi Sankyo. C.L. reports employment with Daiichi Sankyo. H.I. reports honoraria from AstraZeneca, Chugai/Roche, Daiichi Sankyo, Eisai, Lilly, Novartis, Pfizer, and Taiho, and consulting fees from AbbVie, AstraZeneca, Chugai/Roche, Daiichi Sankyo, Kyowa Hakko Kirin, Lilly, Novartis, Odonate, and Pfizer. C.‐C.L. reports travel support from BeiGene and Daiichi Sankyo, advisory fees from Blueprint Medicines, Boehringer Ingelheim, Daiichi Sankyo, and Novartis, and honoraria from Boehringer Ingelheim, Lilly, Novartis, and Roche. K.T. reports receiving research funding from Daiichi Sankyo, Eisai, Lilly, and Pfizer. J.W. reports personal fees from Daiichi Sankyo related to the current work and lecture fees from AstraZeneca, Chugai, Eisai, Eli Lilly, Novartis, and Pfizer outside of the submitted work. R.W. and H.K. report consulting fees from Daiichi Sankyo related to the current work and consulting fees from undisclosed sponsors outside of the submitted work.
AUTHOR CONTRIBUTIONS
O.Y., H.I., C.‐C.L, J.W., R.W., H.K., C.L., L.Z., and F.L. wrote the manuscript. O.Y., R.W., H.K., M.A.T., T.G., C.L., L.Z., J.S., and F.L. designed the research. O.Y., H.I., C.C.L, K.T., and J.W. performed the research. O.Y., R.W., H.K., M.A.T., T.G., L.Z., J.S., and F.L. analyzed the data.
Supporting information
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Table S5
Acknowledgments
The authors thank Michael Demars, PhD, of ArticulateScience LLC, Hamilton, NJ, USA, for providing medical writing/editorial support, which was funded by AstraZeneca Pharmaceuticals, LP, Gaithersburg, MD, USA, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
References
- 1. Doi, T. et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS‐8201), a HER2‐targeting antibody‐drug conjugate, in patients with advanced breast and gastric or gastro‐oesophageal tumours: a phase 1 dose‐escalation study. Lancet Oncol. 18, 1512–1522 (2017). [DOI] [PubMed] [Google Scholar]
- 2. Aggarwal, N. & Sloane, B.F. Cathepsin B: multiple roles in cancer. Proteomics Clin. Appl. 8, 427–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nakada, T. , Sugihara, K. , Jikoh, T. , Abe, Y. & Agatsuma, T. The latest research and development into the antibody‐drug conjugate, [fam‐] trastuzumab deruxtecan (DS‐8201a), for HER2 cancer therapy. Chem. Pharm. Bull. (Tokyo) 67, 173–185 (2019). [DOI] [PubMed] [Google Scholar]
- 4. Ruan, J. , Zheng, H. , Fu, W. , Zhao, P. , Su, N. & Luo, R. Increased expression of cathepsin L: a novel independent prognostic marker of worse outcome in hepatocellular carcinoma patients. PLoS One 9, e112136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogitani, Y. et al. DS‐8201a, a novel HER2‐targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T‐DM1. Clin. Cancer Res. 22, 5097–5108 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Enhertu (fam‐trastuzumab deruxtecan‐nxki) [package insert]. Basking Ridge, NJ: Daiichi Sankyo, Inc; 2020.
- 7. Tamura, K. et al. Trastuzumab deruxtecan (DS‐8201a) in patients with advanced HER2‐positive breast cancer previously treated with trastuzumab emtansine: a dose‐expansion, phase 1 study. Lancet Oncol. 20, 816–826 (2019). [DOI] [PubMed] [Google Scholar]
- 8. Yin, O. et al. Population pharmacokinetic analysis of DS‐8201a ([Fam‐] trastuzumab deruxtecan), a HER2‐targeting antibody‐drug conjugate, in patients with HER2‐positive breast cancer or other solid tumors. Clin. Pharmacol. Ther. 105(suppl 1) (2019). 10.1002/cpt.1344 [DOI] [Google Scholar]
- 9. Center for Drug Evaluation and Research . Multi‐discipline review: Enhertu <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761139Orig1s000MultidisciplineR.pdf> (2019). Accessed April 20, 2021.
- 10. Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2‐positive breast cancer. N. Engl. J. Med. 382, 610–621 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tamura, K. et al. Dose justification for DS‐8201a, a HER2‐targeted antibody‐drug conjugate, for HER2‐positive breast cancer: observed clinical data and exposure‐response analyses. Cancer Res. 79(4 suppl), abstract P6–17‐10 (2019). 10.1158/1538-7445.SABCS18-P6-17-10 [DOI] [Google Scholar]
- 12. Yin, O. et al. Population pharmacokinetics of trastuzumab deruxtecan in patients with HER2‐positive breast cancer and other solid tumors. Clin. Pharmacol. Ther. 109, 1314–1325 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang, D.‐Y. et al. Safety and pharmacokinetic results from a phase 1, multicenter, open‐label study of [fam‐] trastuzumab deruxtecan (T‐DXd; DS‐8201a) in subjects with advanced HER2‐positive breast cancer. Mol. Cancer Ther. 18, abstract C041 (2019). [Google Scholar]
- 14. Center for Drug Evaluation and Research . Clinical pharmacology and biopharmaceutics review: Kadcyla <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/125427Orig1s000ClinPharmR.pdf> (2012). Accessed January 17, 2020.
- 15. Bajaj, G. , Wang, X. , Agrawal, S. , Gupta, M. , Roy, A. & Feng, Y. Model‐based population pharmacokinetic analysis of nivolumab in patients with solid tumors. CPT Pharmacometrics Syst. Pharmacol. 6, 58–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, Y. , Booth, B. , Rahman, A. , Kim, G. , Huang, S.M. & Zineh, I. Toward greater insights on pharmacokinetics and exposure‐response relationships for therapeutic biologics in oncology drug development. Clin. Pharmacol. Ther. 101, 582–584 (2017). [DOI] [PubMed] [Google Scholar]
- 17. Ait‐Oudhia, S. , Zhang, W. & Mager, D.E. A mechanism‐based PK/PD model for hematological toxicities induced by antibody‐drug conjugates. AAPS J. 19, 1436–1448 (2017). [DOI] [PubMed] [Google Scholar]
- 18. Thon, J.N. , Devine, M.T. , Jurak Begonja, A. , Tibbitts, J. & Italiano, J.E. Jr. High‐content live‐cell imaging assay used to establish mechanism of trastuzumab emtansine (T‐DM1)–mediated inhibition of platelet production. Blood 120, 1975–1984 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uppal, H. et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T‐DM1). Clin. Cancer Res. 21, 123–133 (2015). [DOI] [PubMed] [Google Scholar]
- 20. Herceptin (trastuzumab) [package insert]. South San Francisco, CA: Genentech, Inc; 2017.
- 21. Kadcyla (ado‐trastuzumab emtansine) [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
- 22. Dimopoulou, I. , Bamias, A. , Lyberopoulos, P. & Dimopoulos, M.A. Pulmonary toxicity from novel antineoplastic agents. Ann. Oncol. 17, 372–379 (2006). [DOI] [PubMed] [Google Scholar]
- 23. Ewer, S.M. & Ewer, M.S. Cardiotoxicity profile of trastuzumab. Drug Saf. 31, 459–467 (2008). [DOI] [PubMed] [Google Scholar]
- 24. Shitara, K. et al. Trastuzumab deruxtecan (DS‐8201a) in patients with advanced HER2‐positive gastric cancer: a dose‐expansion, phase 1 study. Lancet Oncol. 20, 827–836 (2019). [DOI] [PubMed] [Google Scholar]
- 25. Azuma, A. & Kudoh, S. High prevalence of drug‐induced pneumonia in Japan. Japan Med. Assoc. J. 50, 405–411 (2007). [Google Scholar]
- 26. Kudoh, S. et al. Interstitial lung disease in Japanese patients with lung cancer. Am. J. Respir. Crit. Care Med. 177, 1348–1357 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Namba, N. et al. Association of MUC5B promoter polymorphism with interstitial lung disease in myeloperoxidase‐antineutrophil cytoplasmic antibody‐associated vasculitis. Ann. Rheum. Dis. 78, 1144–1146 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Table S5