

Abstract
Malaria is an infectious disease which disproportionately effects children and pregnant women. These vulnerable populations are often excluded from clinical trials resulting in one‐size‐fits‐all treatment regimens based on those established for a nonpregnant adult population. Pharmacokinetic/pharmacodynamic (PK/PD) models can be used to optimize dose selection as they define the drug exposure‐response relationship. Additionally, these models are able to identify patient characteristics that cause alterations in the expected PK/PD profiles and through simulations can recommend changes to dosing which compensate for the differences. In this review, we examine how PK/PD models have been applied to optimize antimalarial dosing recommendations for young children, including those who are malnourished, pregnant women, and individuals receiving concomitant therapies such as those for HIV treatment. The malaria field has had great success in utilizing PK/PD models as a foundation to update treatment guidelines and propose the next generation of dosing regimens to investigate in clinical trials. We propose how the malaria field can continue to use modeling to improve therapies by further integrating PK data into clinical studies and including data on drug resistance and host immunity in PK/PD models. Finally, we suggest that other disease areas can achieve similar success in applying pharmacometrics to improve outcomes by implementing three key principals.
Infectious diseases with disproportionate burdens in low resource settings create unique challenges in drug development, drug repurposing, and therapeutic evaluation. As an example, malaria, an infectious disease caused by the Plasmodium parasite, resulted in an estimated 229 million cases and 409,000 deaths in 2019. 1 Over half of all malaria cases and deaths are in children under 5 years of age in sub‐Saharan Africa. 1 Prompt and effective treatment of malaria is a cornerstone of malaria control and, like many infectious diseases, relies on historic drugs developed using empirical methods, initially studied in nonpregnant adults. The spread of drug‐resistant parasites and therefore waning efficacy of current therapeutics has created an urgent need to both quickly develop new therapies as well as repurpose approved antimalarials for treatment, prevention, and elimination.
Over the last decade, pharmacometric techniques, which use population models to define drug exposure‐response relationships, have been an essential tool for the development of malaria therapeutics. Pharmacometric models incorporate drug dose, drug concentrations, and patient outcome measures to optimize a drug’s use by defining what dose(s) are safest and result in the highest efficacy for each patient population. In this review, we describe how pharmacometric techniques, applied as pharmacokinetic/pharmacodynamic (PK/PD) models, have impacted malaria treatment, review future opportunities for the use of PK/PD models to enhance malaria treatment, and share lessons learned that could have applications to other emerging infectious diseases.
PHARMACOMETRIC PRINCIPLES
Despite being a relatively new discipline, pharmacometrics changed the drug development paradigm. PK/PD models are mathematical models, which describe a drug’s concentration‐effect relationship by estimating a drug’s PK parameters (e.g., absorption rate, bioavailability, clearance, and volume of distribution) based on drug concentrations and linking this drug exposure to clinical outcomes.
Two main types of models are used for antimalarials: (i) mechanistic and (ii) empirical, both based on preclinical and/or clinical data. Preclinical PK/PD models use in vitro drug efficacy data, parasite growth dynamics data, and animal studies to extrapolate drug exposure and efficacy for first‐in‐human studies (Figure 1 ). These preclinical models can incorporate a drug’s site of action, assess for synergy in drug combinations, and explore the role of drug resistance prior to entering human studies. Clinical PK/PD models, which have dominated antimalarial research, typically use sparse drug concentrations (1–3 samples per individual) and treatment outcomes (relapse, reinfection, and cure) collected from clinical trials. The PK/PD model framework increases the flexibility of PK sample collection times, maximizes the number of patients who can be sampled, and allows researchers to pool data from multiple studies. Once PK exposure is quantified, it can be directly linked to clinical outcomes. By understanding the relationship between drug exposure and resultant treatment outcome, one can learn what drug exposure is needed to achieve a positive treatment outcome. After the drug exposure‐response relationship is established, a pharmacometrician can explore through simulations (virtual clinical trials) what dosing regimens are needed to achieve the required drug exposure. Similarly, they can also explore the effects of patient characteristics to learn whether all individuals require the same dose or whether certain populations require different doses to be cured. The ability of pharmacometric models to account for variability from multiple sources (patient, concentration, and response) and to perform simulations are two strengths which make these tools so powerful. PK/PD models are particularly advantageous for antimalarials, as malaria impacts children, pregnant women, and patients with comorbidities, all populations which are understudied during drug development and have physiologic characteristics that frequently impact both drug PKs and drug exposure‐response relationships necessitating precision dosing.
Figure 1.
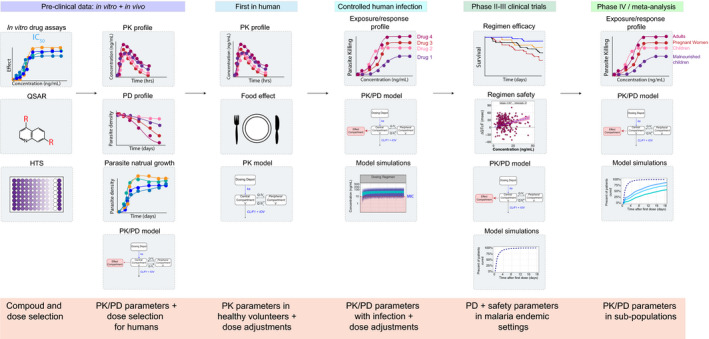
Experimental data collected over drug development that can be used for pharmacometric models. Preclinical data can be used to develop a translational platform for compound and regimen selection. High through screening can be used to select the most potent compounds and computational methods such as quantitative structure actively relationship can be used to design compounds with better PK and potency. Furthermore, preclinical experiments can be used to study development of drug resistance as well as characterize the natural parasite growth dynamics for more mechanistic models. PK/PD mouse studies can be used to develop animal PK/PD models to select clinical candidate compounds and design first in human studies. Clinical PK/PD data used to build human PK/PD models can further be used to select compounds and doses for the next trial whether phase 2,3 or post marketing studies. HTS, high‐through screening; IC50, half‐maximal inhibitory concentration; PD, pharmacodynamic; PK, pharmacokinetic; QSAR, quantitative structure actively relationship.
MALARIA BACKGROUND
Among the five species of Plasmodium that cause malaria, P. falciparum and P. vivax are the most prevalent, and P. falciparum is the most deadly. 2 , 3 , 4 Human infection begins with injection of Plasmodium sporozoites from the bite of an infected female Anopheles mosquito. Typically, there is a 1–2 week incubation period during which individuals are asymptomatic as the parasites in the merozoite stage replicate in hepatocytes. Certain species, including P. vivax, can remain dormant in the liver (hypnozoite stage) for weeks to years resulting in relapse infections. Infected hepatocytes rupture after 1–2 weeks, releasing merozoites into the bloodstream where they infect erythrocytes. In the erythrocytes, asexual reproduction continues, with parasite densities in the blood increasing. It is typically in the blood stage of infection when symptoms develop, initially presenting as a febrile or flu‐like illness, which, in some cases, can progress to organ dysfunction and death without treatment. Some merozoites will develop into gametocytes, which are ingested by mosquitoes and can undergo sexual reproduction to complete the lifecycle. 2 , 3 , 4
After repeated infections, a natural immunity develops, which can control circulating parasite densities and reduces the risk of symptomatic infection, such that older individuals in malaria endemic regions may carry circulating parasites but are unlikely to have clinical disease. 2 , 3 The presence of asymptomatic carriers of parasites poses additional challenges for eliminating disease as asymptomatic individuals are still infectious and contribute to ongoing malaria transmission. 5 Immunity is lower in travelers from nonmalaria endemic regions, and in young children and pregnant women in malaria endemic settings, making these populations most at risk of malaria complications. 6
Drugs have several important roles in malaria, including reducing the risk of morbidity and death with prompt and effective treatment, prevention of infections in high‐risk populations, such as pregnant women, children, and travelers, and assisting in malaria elimination by clearing a community’s parasites with mass drug administration. In addition to diverse purposes, antimalarial drug exposure‐response relationships depend on parasite species and stage (e.g., liver or blood stages), parasite drug resistance characteristics, and host characteristics, including background immunity. 7 , 8 Plasmodium’s complex life cycle and the natural history of malaria have introduced challenges and opportunities for drug development, as many therapeutics are only effective against certain life cycle stages of the parasite. PK/PD models are a valuable tool because they can integrate all these dynamic complexities. To demonstrate the role of PK/PD modeling in antimalarial drug development and postmarketing optimization, we will discuss models developed for malaria treatment.
MALARIA TREATMENT
The goal of malaria treatment is to initially prevent disease progression and then provide a cure. Malaria treatment is largely guided by the parasite species and drug susceptibility as well as whether the infection is uncomplicated or severe. 6 Infections are classified as severe if a patient presents with a positive blood smear for Plasmodium, along with one or more of the following: impaired consciousness, acidosis, hypoglycemia, severe anemia, renal impairment, severe hepatic impairment, hypoxia, bleeding, shock, or a parasite density > 10%. 6 , 9 Any infection not defined as severe is considered uncomplicated. Malaria treatment has largely used a one‐size‐fits‐all dosing regimen where adults, pregnant women, and children receive the same or allometrically equivalent doses, respectively. Pharmacometric models have been used to evaluate the efficacy of the original dose chosen, including testing the assumptions around using the same dose in different populations. In 2015, the World Health Organization (WHO) updated their treatment guidelines for children using a regimen derived from a PK/PD model. 10 , 11
In Africa, the current first‐line therapies for malaria are artemisinin‐based combination therapies (ACTs). There are five approved ACTs with artemether‐lumefantrine (AL) being the most widely adopted and dihydroartemisinin‐piperaquine (DP) being the newest. 1 The remaining options are artesunate‐amodiaquine, artesunate‐mefloquine, and artesunate‐sulfadoxine‐pyrimethamine. The pharmacology behind ACTs is complex and is founded on pairing a potent short‐acting artemisinin derivative (artemether, dihydroartemisinin, or artesunate) with a longer acting partner drug (lumefantrine, piperaquine (PQ), amodiaquine, mefloquine, or sulfadoxine‐pyrimethamine; Figure 2 ). 6 , 12 A standard treatment course of ACTs is 3 days with either once or twice daily dosing. Briefly, the artemisinin component, the more potent drug, rapidly clears blood stage parasites resolving clinical symptoms. 13 However, artemisinins have a very short half‐life of between 2 and 5 hours and do not remain above the minimum inhibitory concentration (MIC) beyond the standard 3 days of dosing, at which time some parasites may remain untreated. 6 , 13 The longer acting partner drug is responsible for eliminating these residual parasites to cure a patient and provides a period of post‐treatment prophylaxis against new infections (Figure 2 ). Combination therapies for malaria treatment are used to minimize treatment duration, improve adherence, and reduce the risk for selection of drug resistance. 6 , 13
Figure 2.
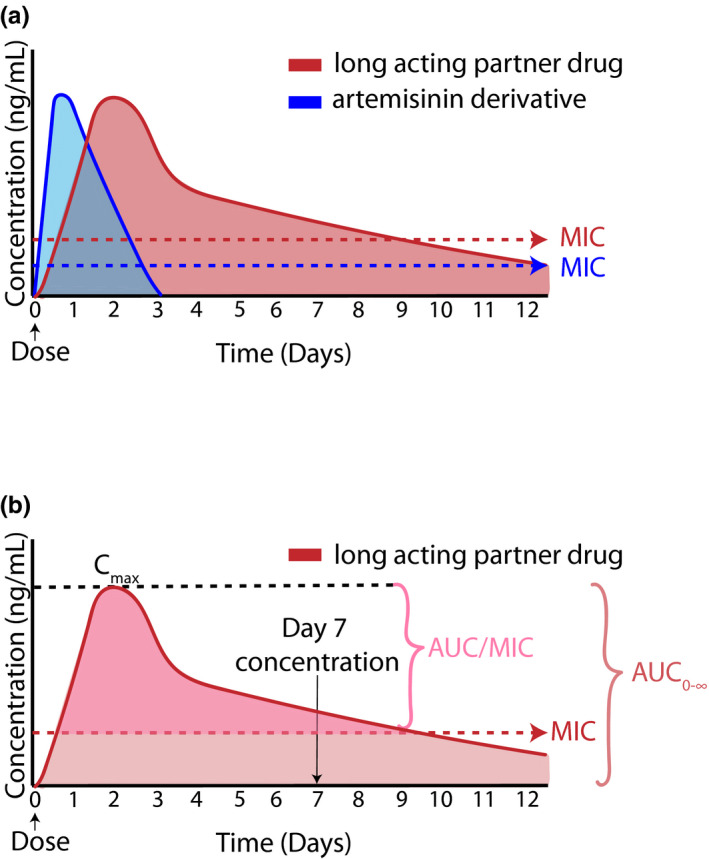
Pharmacokinetic profile of ACTs. (a) Schematic representation of the plasma profile of both artemisinin and long‐acting partner drug. Although the artemisinin is quickly eliminated, the partner drug has elevated concentrations able to kill parasites for a much longer period of time. (b) Pharmacokinetic markers predictive of treatment outcome. Both day 7 concentration and the area under the concentration time curve (AUC) are associated with malaria treatment outcomes. The Cmax represents the maximum concentration and the minimum inhibitor concentration able to kill parasites is the MIC. Concentration and MIC are set to arbitrary values.
Since their introduction into sub‐Saharan Africa, ACTs have maintained a > 95% treatment efficacy in standard treatment efficacy studies, however, the duration of the post‐treatment prophylactic effect varies widely. 14 Despite high treatment efficacy in aggregate, incorporating PK/PD modeling approaches to standard treatment efficacy studies has identified populations at particular risk of malaria treatment failure with current ACT regimens. In this review, we will focus on two of the most commonly prescribed ACTs, AL and DP. Lumefantrine and PQ, in particular, are the most widely studied using PK/PD modeling approaches and these analyses have resulted in changes to dosing guidelines for vulnerable populations or new model informed dosing regimens currently under study. 15 , 16
EXPOSURE RESPONSE BIOMARKER: DAY 7 CONCENTRATIONS
Some measure of drug exposure is required to understand a drug’s concentration‐response relationship. Typical measures of drug exposure are area under the concentration time curve (AUC), maximum concentration (Cmax), or time above the minimum inhibitory concentration (AUC/MIC; Figure 2b ). Day 7 concentrations of the long‐lasting partner drugs have been adopted as a surrogate measure of AUC (Figure 2b ). Multiple studies using lumefantrine and PQ have shown this measure is highly correlated to both AUC and treatment outcomes. 10 , 17 , 18 Although this value was not derived from PK/PD models, it has been used extensively for evaluating the efficacy of model‐simulated dosing regimens.
Whereas artemisinins are extremely potent with parasite reduction ratios of ~ 104‐fold the 3‐day treatment regimen only exposes 2 asexual life cycles to artemisinins. 12 This results in the presence of residual parasites after the artemisinins are below effective concentrations. By day 7, only the partner drug is present at concentrations above the MIC and so this concentration reflects the amount of drug present to kill residual parasites. If the concentrations are high enough, a patient will be cured. However, if these concentrations are too low, relapse infections will occur. Studies have proposed target concentrations of ≥ 57 ng/mL PQ and lumefantrine concentrations of ≥ 200 ng/mL to ensure malaria cure. 10 , 18 , 19 The target concentration for lumefantrine is somewhat less certain with values of 175 ng/mL to 500 ng/mL being cited. 20 , 21 These target concentrations have provided a valuable benchmark and facilitated comparisons between studies. Finally, researchers have argued that in addition to cost effectiveness, this measure is also pragmatic as all patients return to the clinic on day 7 for a blood draw to monitor parasite density. Given there are many possible reasons for why treatment failure can occur, day 7 concentrations can help elucidate whether altered PK may be involved.
PEDIATRIC MALARIA TREATMENT WITH DIHYDROARTEMISININ PIPERAQUINE
As previously described, PK/PD models are well‐suited to determine the correct dose of a medication. Because young children (< 5 years) are one of the most vulnerable groups for contracting malaria and also for worse treatment outcomes, this population has been the focus of many recent PK/PD studies. 1 Children are known to have a higher body weight adjusted clearance compared with adults and receive a higher mg/kg dose to counteract these developmental changes. 22 In addition, the PK of infants is particularly difficult to scale due to age‐based maturation of metabolizing enzymes such as CYP3A4. 22 DP combines an artemisinin derivative with PQ, a long acting aminoquinoline derivative with the longest half‐life (~ 3 weeks) of approved partner drugs. In sub‐Saharan Africa, DP provides excellent treatment efficacy > 95% and provides a 1 month post‐treatment prophylactic effect. DP, like many ACTs, is dosed according to weight‐bands, and day 7 PQ concentrations of 57 ng/mL (capillary) in children < 5 years of age has been associated with a decreased risk of recurrent infection after treatment. 10 In one study, day 7 PQ concentration was the only significant predictor associated with the risk of recurrent malaria. A 5.9% increase risk of new infection for every 1 ng/mL decrease in day 7 PQ concentration was reported. 10 As PQ concentrations have been a key predictor of outcomes, obtaining optimal PQ concentrations in young children has been a major focus of PK/PD modeling efforts.
Although DP is a highly effective therapy, studies which investigated PQ PK revealed that children had reduced PQ exposure compared with adults and older children. 10 , 23 , 24 , 25 Two studies reported than 40–60% of children did not achieve day 7 PQ concentrations above 57 ng/mL after standard WHO dosing. 10 , 23 When exploring the underlying cause for lower PQ exposure, the majority of PK models found that including allometric scaling as a covariate on all clearance and volume parameters was the only significant covariate. One study enrolled both adults and children and reported children had a substantially increased PQ clearance (1.85 vs. 0.9 L/h*kg) compared with adults. 25 However, studies that enrolled infants, 6 months to 2 years of age, detected a relationship between age and PQ clearance ((AGEi/12)0.35) where older children had higher clearance than predicted by weight alone. 23 Taken together, the effect of weight and age resulted in 6‐month‐old children having half the clearance value of a 2‐year‐old. 23 The authors attributed this finding to represent maturation of the drug metabolizing enzymes (CYPs; CYP3A4 for PQ) responsible for PQ metabolism. 26 By more accurately quantifying the impacts of age and weight on PQ PK, it was found that the approved weight‐based dosing for DP was insufficient for low weight children (< 10.5 kg) 1–2 years of age. These findings indicated that the dose children were receiving was not large enough to account for the ontogenetic changes. This was especially true for younger and underweight children.
The two largest PK/PD studies of PQ conducted in African children performed simulations and concluded that children should receive 1.5 to 2 times the current DP dose to achieve day 7 PQ levels of 57 ng/mL. 10 , 23 The exact percentage of children attaining the target concentration differed (82–86% 10 and 50% 23 ) between studies but both indicated that double the number of children would be protected by increasing the dose. In addition to potentially improving treatment outcomes, the authors argued that increasing the dose could have other benefits, such as reducing the selective pressure for drug resistance in parasites and extending the post‐treatment prophylaxis period. 10
The end result of all these studies was that many were combined into individual patient meta‐analyses conducted by the WorldWide Antimalarial Resistance Network (WWARN). 11 , 16 One analysis was focused on defining PQ’s PK parameters with population modeling and using their model to optimize dosing regimens. 16 This analysis was built from 11 clinical trials and included 8,776 PQ samples from 728 patients. The PK model confirmed the previous findings that young small children were being underdosed. Children < 25 kg were predicted to have a median day 7 PQ concentration of 29.4 ng/mL compared with children and adults > 25 kg who had concentrations of 38.1 ng/mL. Additionally, this model was also able to show in young children enzyme maturation reaches 50% at 6 months of age. By pooling data from multiple studies, this analysis highlighted that underdosing was universal across continents. Due in part to the large dataset size, this analysis was able to refine the previously proposed dosing guidelines and recommend a minimum dose of 64 mg/kg PQ for children 5–15 kg paying close attention to young children and low weight populations. The necessity of dosing changes for DP in young children was confirmed by a second WWARN analysis, which included data from 7,072 patients, with 136 recrudescent infections. This analysis found that after controlling for dose and baseline parasitemia, children 1–5 years of age had a 3.71 increased risk of recrudescence compared with children > 12 years of age and that every 5 mg/kg increase in dose was associated with a 13% decrease in risk of recrudescence. Ultimately, WWARN’s work, guided by PK/PD modeling, was the foundation for an update to DP dosing recommendations in young children by the WHO. This is a powerful example of how pharmacometrics and statistical approaches to data integration and analysis led to a high impact policy change.
PEDIATRIC MALARIA TREATMENT WITH ARTEMETHER LUMEFANTRINE
Lumefantrine concentrations and other measures of drug exposure are quite variable. 15 , 27 , 28 Similar to PQ, lumefantrine exposure is predictive of treatment outcomes. 21 , 29 , 30 Multiple studies have documented lower lumefantrine exposure in children and pregnant women compared with adults and, in some cases, these lower lumefantrine exposures have been linked to higher risks of recurrent infection after treatment with AL (Table 1 ). 15 , 29 , 30 , 31 The most comprehensive study on this topic, an individual patient data meta‐analysis conducted by WWARN, which included PK data from 4,122 participants including pregnant women, children, and nonpregnant adults from across multiple continents, reported that pregnant women and children < 25 kg had lower day 7 lumefantrine levels compared with nonpregnant adults. 15 After allometrically scaling clearance and volume for weight, children < 15 kg had 24% lower day 7 lumefantrine levels compared with adults, and children 15–24 kg had 13% lower day 7 levels. As the investigators considered optimized AL regimens to improve lumefantrine exposure in children, it was noted that lumefantrine was associated with dose limited absorption, such that longer treatment courses rather than increased daily doses were predicted to improve lumefantrine exposure. This robust PK meta‐analysis led to recommendations to extend twice daily dosing for 5 days, which was predicted to better match the distribution of adult and pediatric day 7 concentrations. It also predicted that 75% of children would maintain lumefantrine concentrations over the 200 ng/mL previously recommended threshold.
Table 1.
Population PK and PK/PD models of partners drugs in vulnerable population after malaria treatment with ACTs
| Country, year | Population | No. of participants | PK covariate(s) found | Drug exposure target used for dose optimization | PK/PD relationship from study data | Dosing recommendations |
|---|---|---|---|---|---|---|
| Lumefantrine | ||||||
| Multi‐country (meta‐analysis), 2018 15 | Adults and children including pregnant women | 4,122 | Lumefantrine bioavailability decreased when parasitemia was detected and there was dose‐limiting absorption | Day 7 LUM concentrations from non‐pregnant adults for dose optimization | Insufficient power for recrudescent infection to identify population specific associations | Twice daily dosing for 5 days would improve day 7 LUM levels for children |
| Uganda, 2016 30 | Children 6 months to 2 years | 105 | CL allometrically increased with weight, lower age had lower relative bioavailability | A day 7 LUM capillary concentration < 200 ng/mL | A day 7 LUM concentration < 200 increased hazard of 28‐day recurrent parasitemia by 3‐fold | Children < 2 had suboptimal concentrations (median 7‐day of 216) but new regimens were not recommended |
| Tanzania, 2014 64 | Pregnant and nonpregnant women | 55 | 34% Lower bioavailability and 78% higher clearance during pregnancy | Literature derived LUM targets used (50, 175, 280, 600 ng/mL) for day 7 simulations. | Four‐fold increased odds of recurrent malaria in pregnant women after AL | 6 doses over 5 days predicted to decrease the number of individuals below the LUM targets |
| Tanzania, 2013 27 | Adults, pregnant women, and children (1–78 years) | 143 | CL allometrically increased with weight | Literature derived LUM targets used (50, 175, 280, 600 ng/mL) for day 7 | Not evaluated | 6 doses over 5 days predicted to decrease the number of individuals below the LUM targets |
| Papua New Guinean, 2011 29 | Children 5 to 10 years | 13 | Weight effect on CL, lower LUM exposure was observed in small children (15–35 kg) | Not evaluated | Lower average LUM AUC among children with recurrent infection children | Not evaluated |
| Mali & Niger, 2019 73 | Children age 5‐59 months with and without severe acute malnutrition | 397 |
Malnutrition measured by MUAC lowered LUM bioavailability by 25.4% per decrease in MUAC Age (maturation effect) and weight (allometric scaling) increased CL |
LUM AUC, day 7 concentration and Cmax values reported in non‐SAM children | SAM children had reduced LUM exposure and increased risk of new infections | 6 doses over 5 days or 9 doses over 3 days predicted to result in equivalent exposure in non‐SAM and SAM children |
| Multi‐country (meta‐analysis) 2007 54 |
Nonpregnant adults HIV‐malaria co‐infected, malaria‐infected and HIV‐infected |
793 |
CL allometrically increased with weight Lopinavir/ritonavir: 50.1% slower clearance 67.2% increased bioavailability 47.6% reduction in absorption rate Efavirenz: 89.9% increased clearance |
Literature derived LUM target of 200 ng/mL for day 7 simulations. | Not evaluated |
Extending treatment over 5 or 6 days was predicted to increase lumefantrine exposure for patients on efavirenz No changed need for lopinavir/ritonavir |
| Tanzania, 2015 53 | HIV‐malaria co‐infected adults | 269 |
Efavirenz 58% lower bioavailability Nevirapine: 32% increased bioavailability |
Literature derived LUM target of 280 ng/mL for day 7 simulations. | Not evaluated | 6 doses over 5 days predicted to decrease the number of individuals below the LUM targets for efavirenz patients |
| Uganda, 2015 51 | HIV‐infected adults | 89 |
Efavirenz 72.6% increased clearance Lopinavir/ritonavir: 62.1% decreased clearance Nevirapine: 24.8% decreased clearance |
Literature derived LUM targets used (175, 280, ng/mL) for day 7 | Not evaluated | Extending treatment over 7 days was recommended to increase lumefantrine exposure for patients on efavirenz |
| Piperaquine | ||||||
| Multi‐country (meta‐analysis) 2017 16 | Adults and children including pregnant women | 728 | CL increased by age (maturation effect) and allometrically by weight. Dose‐occasion impacted bioavailability. | Non‐pregnant adult median day 7 PQ concentrations | Not evaluated | Increased dose and increased bands for weight‐band dosing of children < 25 kg |
| Uganda, 2015 23 | Children 6 months to 2 years | 107 | CL increase by age (maturation effect) and allometrically by weight. There was lower exposure in low weight for age children. | Literature derived 57 ng/mL capillary PQ concentration on day 7 | Not evaluated | 1.5‐2 times dosing for each weight band from 6 months to 2 years, but recommended further QT evaluation of these regimens |
| Cambodia, 2013 27 | Adults and children (7–53 years) | 60 | CL allometrically increased with weight | Not evaluated | Not evaluated | Not evaluated |
| Burkina Faso, 2012 10 | Children 2 to 10 years | 236 | CL allometrically increased with weight, despite increased weight‐normalized PQ dose, young children had lower day 7 PQ concentrations. | Literature derived 30 ng/mL venous transformed to 57 ng/mL capillary PQ concentration on day 7 | 5.9% increased risk of recurrent malaria for each 1 ng/m decrease in day 7 capillary drug concentrations. | 30 mg/kg/day dose in children < 34 kg decreased percentage of children with day 7 PQ concentration < 57 ng/mL from up to 45% to < 20% |
| Papua New Guinean, 2012 24 | Children 5 to 10 years | 34 | CL allometrically increased with weight | Not evaluated | Lower PQ average AUC among children with recurrent infection (n = 8 children) | No model‐based changes recommended |
| Cambodia, 2004 25 | Children 2 to 10 years and adults > 16 years | 85 | Separate models for adults and children, clearance 2 times higher for children compared with adults. | Not evaluated | Underpowered (high cure rates) to detect associations. | No model‐based changes recommended. |
ACT, artemisinin‐based combination therapy; AL, artemether‐lumefantrine; AUC, area under the curve; CL, clearance; Cmax, peak concentration; LUM, lumefantrine; MUAC, mid‐upper arm circumference; PD, pharmacodynamic; PK, pharmacokinetic; PQ, piperaquine; SAM, severe acute malnutrition.
Multiple smaller studies have also investigated lumefantrine PK in pediatric populations (Table 1 ). One challenge with these studies and the WWARN meta‐analysis is identification of a consistent lumefantrine target associated with treatment response. Despite the large patient population from the WWARN study, the low rate of treatment failure prevented identification of population specific day 7 lumefantrine targets associated with efficacy, so a conservative 596 ng/mL nonpregnant adult level was used. Other proposed day 7 targets have included 50 ng/mL, 175 ng/mL, and 200 ng/mL, which were associated with lower risks of recurrent malaria (i.e., combined recrudescence and new infections after treatment). When using these targets for dose optimization, different AL regimens were identified, including spreading the standard 6 doses of AL over 5 days. Currently, twice daily AL for 5 days is being explored for malaria treatment in pediatric populations (NCT03453840). 15 A consensus on which outcomes and PK targets are most relevant for antimalarial dosing, which is still needed.
PK/PD modeling has also played an important role in understanding the food requirements for AL. Many studies have investigated lumefantrine’s PKs to identify the sources of variability in hopes of defining strategies to improve drug exposure. As a lipophilic hydrophobic compound, poor bioavailability was quickly identified as one area limiting drug exposure. 28 , 32 , 33 Researchers noted increased bioavailability when lumefantrine was given with food and performed different modeling studies to understand the food effects. 34 , 35 Dedicated studies indicated that regardless of the food type, lumefantrine bioavailability increased and, for milk, increased by an estimated 57% and 65% for crushed tablets and dispersible tablets compared with no food, respectively. 34 It has also been shown that only 36 mL of milk or 1.2 g of fat is needed to achieve 90% bioavailability. 35 These findings were significant as they indicated that AL should be administered with food and increasing lumefantrine exposure may not be as simple as increasing the dose.
ACT DRUG‐DRUG INTERACTIONS
Drug‐drug interactions (DDIs), if clinically significant, can put a patient at risk for malaria treatment failure if concentrations are reduced or for adverse events if concentrations are elevated. Malaria endemic regions in sub‐Saharan Africa have a large population of HIV‐infected individuals receiving antiretroviral therapy (ART). 1 , 36 Many ACTs have common mechanisms of metabolism with ARTs, namely CYP450 isoenzymes, including CYP3A4/5, CYP2B6, and CYP2C9. 20 , 37 , 38 The CYP450 enzymes responsible for ACT metabolism are also those which are induced or inhibited by ARTs. 39 , 40 , 41 Artemisinin components, such as artemether and artesunate, whereas active against P. falciparum, are also pro‐drugs which undergo metabolic activation by CYP3A4 and CYP2B6 to form dihydroartemisinin. 42 Lumefantrine is also metabolized by CYP3A4 to form desbutyl‐lumefantrine. 43 Although studies report desbutyl‐lumefantrine is more potent than the parent, it has relatively low exposure making its contribution to treatment outcomes unclear. 44 Both dihydroartemisinin (DHA) and PQ are metabolized by UDP‐glucuronosyltransferases and CYP3A4, respectively, to inactive metabolites. 26 , 45 The impact of co‐administration of AL with several commonly used ARTs in malaria endemic regions, lopinavir/ritonavir, efavirenz, and nevirapine, have been studied. 46 , 47 , 48 , 49 , 50 A clear understanding of the extent of these interactions and benefits of dose adjustments have been explored with PK/PD models.
Three studies utilized a population approach to identify interactions between efavirenz and nevirapine and two of these further documented changes by lopinavir/ritonavir. 49 , 50 , 51 , 52 , 53 These studies used data from clinical trials conducted in various populations ranging from healthy volunteers, HIV‐infected but not malaria infected individuals, and HIV‐malaria co‐infected patients from Africa and the United States. Only one of these studies included artemether and dihydroartemisinin in their analysis. 51 Across all three studies, efavirenz reduced lumefantrine exposure (AUC) by 47–70% in comparison to patients not receiving ART. In two of the studies, this effect was due to 72.6–89.9% increased lumefantrine clearance with the remaining study indicating a 58% reduced bioavailability. All studies attributed these findings to induction of intestinal and/or hepatic CYP3A4 by efavirenz. 54 Interestingly, each study reports a different effect of concomitant nevirapine use, which is a weak CYP3A4 and moderate CYP2B6 inducer. The effect ranged from a 25% reduction to a 32% increase in lumefantrine bioavailability in comparison with patients who did not receive ART. The reason(s) for these conflicting results are unclear but may be due to differences in study design or factors not measured. Ritonavir is a CYP3A4 inhibitor and is responsible for any ACT PK differences noted. 55 Lopinavir/ritonavir was found to increase lumefantrine exposure; both studies reported a 50–62% reduced clearance and the larger study of the two (a pooled analysis of 62 individuals) also identified a 67% increase in bioavailability and a 47.6% slower absorption rate in comparison with patients not receiving ART. The pooled analysis, also tested for a disease effect on PK, confirmed that neither HIV nor malaria altered lumefantrine concentrations. An understanding of what parameters are most effected by covariates can help explain possible mechanisms underlying these DDIs.
A single study assessed artemether and DHA PK among individuals with HIV receiving different ARTs (efavirenz, nevirapine, and lopinavir/ritonavir). 51 Lopinavir/ritonavir increased artemether clearance by 32% and DHA clearance by 143% compared with patients not receiving ARTs. Both efavirenz and nevirapine were found to reduce artemether bioavailability by 71% and 66%, respectively, compared with patients not receiving ARTs. Nevirapine was additionally found to decrease DHA clearance by 44%. However, the lower clearance was not sufficient to compensate for lower artemether levels, and there was a net decreased DHA exposure (AUC0–894hrs). These interactions all led to reduced artemether exposure and are of concern as these may result in reduced parasite clearance potentially putting patients at risk for malaria treatment failure.
These PK/PD studies concluded that artemether‐lumefantrine treatment with lopinavir/ritonavir should be monitored for any toxicities due to increased exposure but appeared safe and effective. 51 , 52 However, researchers agree that the reduced AL exposure seen with concomitant efavirenz is concerning and warrants trials to investigate dose adjustments. 51 , 52 , 53 Clinical trial simulations based on the final PK/PD models found that extending treatment to 5 or 7 days would equalize lumefantrine exposure between those receiving an interacting ART regimen, and those who do not take these drugs. Although the studies discussed above enrolled adult patients, similar results have been seen in children and pregnant women indicating the universal benefit of dosing changes for all patient populations. 50 , 51 , 52 , 53 , 54 , 55 , 56 In addition to testing these novel regimens, the importance of designing the next set of clinical trials to collect and integrate treatment outcome data should not be overlooked. Along with understanding how DDIs impact antimalarial PK exposure, it is equally important to understand their effects on parasite clearance rates and treatment outcomes.
PREGNANCY
Malaria during pregnancy poses a serious risk for the health of both the mother and developing fetus. 57 , 58 Pregnant women have reduced immunity against malaria and immunity is acquired over subsequent pregnancies. 59 Pregnant women require both effective malaria treatment and prevention options. However, the physiological changes, including increased plasma and body water volume, reduced plasma protein concentrations, altered expression of drug metabolizing enzymes, and increased gastric transit time that occur during pregnancy can alter the PK/PD of therapeutics, including antimalarials. 60 , 61 Pharmacometric models have been used to quantify the effects of pregnancy on treatment and prevention regimens.
Artemether‐lumefantrine PKs have been evaluated extensively in pregnant women. Although the results are somewhat conflicting, 62 , 63 the majority of studies found pregnancy reduced lumefantrine exposure (Table 1 ). 17 , 64 , 65 , 66 , 67 Only three studies, however, recorded treatment outcomes. One study conducted in pregnant women reported a 12% increased odds of treatment failure for women who were enrolled later in pregnancy measured by estimated gestational age (EGA; in weeks). 67 This difference in response was attributed largely to altered PKs. Pregnancy was found to increase the volume of the central compartment by 7.2% per increase in EGA. The second study reported a 4.04 increased odds of treatment failure in pregnant women (categorical covariate) compared with nonpregnant controls. 64 The parameter covariate relationships identified were pregnancy decreased bioavailability by 34% and increased intercompartmental clearance by 78%. The final study only enrolled pregnant women. 65 The population PK model identified that pregnancy decreased the rate of absorption (EGA/median EGA)‐0.715) and linearly decreased the intercompartmental clearance (EGA‐(median EGA)*‐2.71). A time to event model was built to capture therapeutic outcomes and identified a maximum effect (Emax) relationship between lumefantrine concentrations and the hazard of relapse infection (Emax fixed to 1 half‐maximal effective concentration 169 ng/mL). The differences in parameters, which pregnancy was reported to effect, may speak to the many and complex changes that pregnancy can create. Pregnancy can change body composition, gastrointestinal motility, and CYP expression, all plausible explanations for the results detailed. 64 , 65 , 67 The differences in study design, namely the enrollment of comparator arms, may also have influenced these findings. Only Mosha et al. included nonpregnant women as the comparator arm in their study and may be the best positioned to comment on pregnancy’s effect on PK parameters. 64 However, this study reported a very low number of new and relapse infections (6 in pregnant women and 1 in nonpregnant women). The remaining two studies were better powered to investigate outcomes (38 67 and 39 65 new and relapse infections). All three analyses suggested that elongating the dosing interval over 5 or more days would increase lumefantrine concentrations and provide equivalent exposure to that seen in nonpregnant controls (Figure 3 ). A strength of these studies was that they indicated reduced exposure in both African and Asian women suggesting these findings have broad applications.
Figure 3.
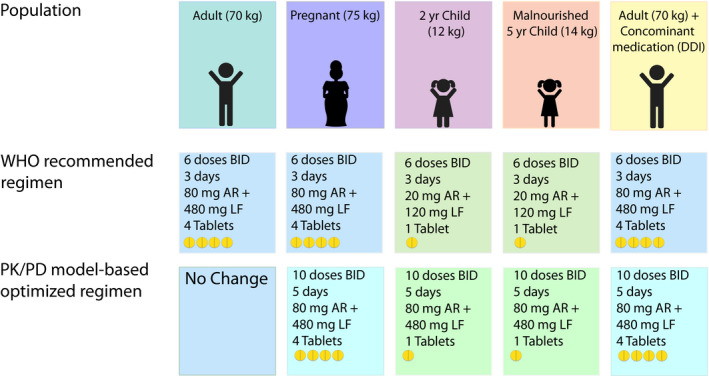
Artemether‐lumefantrine dosing in different populations. The World Health Organization (WHO) endorsed treatment guidelines are based solely on a patient’s weight with adults receiving 4 tablets (80 mg artemether (AR) and 480 mg lumefantrine (LF). Young children < 15 kg receive 1 tablet of 20 mg artemether and 120 mg lumefantrine. Pharmacokinetic/pharmacodynamic (PK/PD) models have proposed that artemether‐lumefantrine dosing be extended over 5 instead of 3 days in special populations, including pregnant women, young and underweight children, as well as HIV co‐infected patients receiving efavirenz based antiretroviral therapy or other CYP450 inducers.
Based on the findings from PK/PD models, one group has studied the extended 5‐day regimen in pregnant women in the Democratic Republic of Congo with other trials planned. 68 The investigators found for both the standard 3 day and extended 5 day regimen minimal differences in outcome measures (100% polymerase chain reaction‐corrected clinical and parasitological response in both populations) and lumefantrine exposure (AUC: 3 day regimen 531 h*µg/mL nonpregnant, 586 pregnant; 5 day regimen 933 h*µg/mL nonpregnant, 853 pregnant) between pregnant and nonpregnant controls. It is unexpected that this study did not detect any effect of pregnancy on lumefantrine PK as this differs from the majority of studies reported in the literature. However, artemether and DHA exposure was reduced by 1.2% per increase in each gestational week, due to lower artemether bioavailability compared with nonpregnant adults. The study also identified that pregnant women had longer parasite clearance rates (3.3 hours vs. 2.43 hours) compared with nonpregnant controls. This finding was not associated with artemether or DHA exposure and the authors suggest this may be due to reduced splenic clearance instead. 68 Most importantly, this study confirmed that extending AL dosing over 5 days increased exposure to artemether, DHA, and lumefantrine in both pregnant and nonpregnant populations. The extended regimen was safe for both mother and fetus and well‐tolerated. This proof‐of‐concept clinical trial confirmed that extending AL dosing over 5 days is a promising alternative for areas where AL treatment efficacy is waning due to low immunity or high levels of drug resistance.
MALNUTRITION
While multiple studies have evaluated malnutrition as a risk factor for malaria, 69 , 70 , 71 , 72 few dedicated studies have been conducted to investigate malnutrition’s effects on ACT PK/PD. 73 Only one study to date was specifically designed to analyze the interplay between severe acute malnutrition and lumefantrine PK/PD in African children. 73 After controlling for ontogenetic changes with allometric scaling and enzyme maturation, mid‐upper arm circumference was found to alter bioavailability whereby for every 1 cm decrease in mid‐upper arm circumference, there was a corresponding 25.4% decrease in bioavailability. Decreased absorption was predicted to reduce lumefantrine exposure when measured as both AUC0‐28days and day 7 concentrations. Malnutrition was not independently associated with increased risk of malaria when tested as a covariate on the PD model parameters. It is due to reduced exposure that malnourished children had an increased risk of reinfection compared with nourished children. Similar to the lumefantrine studies discussed above, simulations for this study population supported extending AL treatment to 5 days (Figure 3 ). This study highlights the importance of conducting dedicated PK/PD analyses in subgroups, such as malnourished children who are at particular risk of modified PK exposure. PK/PD models, by quantifying drug exposure‐response relationships, have increased power to detect PK differences in understudied populations.
LESSONS LEARNED AND FUTURE DIRECTIONS
Evaluation of drug exposure‐response relationships in large, diverse populations receiving antimalarials has led to important dosing and guidance changes for ACTs, such as DP, and has been instrumental in identifying optimized regimens for clinical trials in vulnerable populations, such as pregnant women and children. 1 However, we argue that with adjustments to future antimalarial trials and studies, PK/PD models could be leveraged to have an even more rapid and broader impact on antimalarial policy. We recommend: (i) studies of new and established antimalarials enrich enrollment for populations at high risk of altered PK exposure (e.g., pregnant women, young children, and those with comorbidities such as severe malnutrition, HIV, and tuberculosis), (ii) PK/PD studies be designed to assess for a broader range of drug exposure‐response relationships (e.g., high sensitivity assays for parasitemia, rigorous assessment of nutritional status, and birth outcomes), and (iii) PK/PD modeling studies expand their analyses to include biomarkers of immunity and drug resistance.
Quantifying antimalarial PK in high‐risk subpopulations
PK/PD models increase the power to detect drug exposure‐response relationships by quantifying variability in drug exposure and treatment response in the population. This is particularly helpful for small clinical studies focused on under‐represented subgroups. Malaria endemic regions have significant burdens of malnutrition, HIV, and tuberculosis. 74 As a result, antimalarials are frequently used in individuals at especially high risk of malaria in combination with physiologic conditions (e.g., chronic inflammation or low protein binding) or DDIs known to reduce antimalarial exposure. 75 However, these populations are often excluded from standard clinical trials or are difficult to recruit. As a result, there have been delays in quantifying PK/PD relationships for antimalarials in malnourished individuals and those with DDIs. Recent data in these populations, nearly 20 years after the introduction of ACTs, suggests that longer treatment durations (5 days for AL) or higher daily doses of antimalarials (DP) may be needed for these groups to achieve target treatment outcomes. 15 , 49 Early inclusion of high‐risk subgroups into clinical trials designed to incorporate PK/PD modeling techniques can help us more rapidly identify and characterize the needs of high‐risk subpopulations and devise precision dosing regimens that are acceptable for low resource settings.
Optimizing clinical study designs for PK/PD modeling approaches
Another challenge faced by clinical PK/PD models for antimalarials has been accurate measurement of covariates, which impact PK exposure and outcomes during the study. The mismatch between self‐reported and actual adherence is an example of this limitation. Lower than reported adherence can limit the generalizability of PK/PD models and can bias dose optimization recommendations. 76 Antimalarials pose a particular challenge as drug absorption for ACTs can be variable between individuals and dosing occasions 20 even when all doses are directly observed. This confounding factor limits our ability to differentiate between physiologic and behavioral effects impacting pharmacology and identification of safe and effective antimalarial regimens. Incorporation of more rigorous adherence measures into clinical studies can improve the generalizability of PK/PD models and inform novel dosing regimens which maintain effectiveness despite imperfect adherence. 76 , 77 We encourage clinical studies to more robustly collect data on adherence and other covariates (e.g., nutritional status, concomitant drug concentrations, and markers of parasite drug resistance) of importance for PK and PD.
In addition to measuring clinical covariates that could impact drug exposure and treatment response, we can improve our understanding of drug efficacy by utilizing the more sensitive and quantitative measures of malaria outcomes which are now being developed in malaria research. These include use of ultrasensitive quantitative polymerase chain reaction to quantify parasite densities, including submicroscopic parasitemia, or measurement of placental malaria using a severity grading metric for histopathology. 78 In conjunction with measuring biomarkers of immunity and drug resistance, as described in the section below, these more sensitive and quantitative measures of malaria outcomes in PK/PD studies can enhance our mechanistic understanding of drug response and will allow us to consider goals for malaria control beyond preventing symptomatic disease, such as malaria elimination. It can also allow us to develop population‐specific drug exposure targets for children, pregnant women, and those with comorbid conditions.
Finally, PK/PD models can be utilized to explore the impact of pharmacologic interventions beyond antimalarial efficacy. To achieve this valuable goal, measurement of select nonmalarial outcomes must be included in clinical studies. As an example, despite widespread antifolate resistance in east Africa, monthly intermittent preventative treatment in pregnant women (IPTp) with the antifolate sulfadoxine‐pyrimethamine increased birth weight at delivery. 79 Antibacterial benefits may mediate this effect, 79 but bacterial outcomes have been poorly characterized in malaria IPT studies. In addition, in longitudinal malaria chemoprevention studies, although birth outcomes or progression of malnutrition are often measured in clinical studies, PK data is rarely available in a sufficient number of study participants to detect associations between drug exposure and these outcomes. Highly sensitive drug quantification methods using low volume plasma or blood spots are likely to improve our ability to concurrently quantify PK and outcomes, including rare events, in large trials.
However, birth outcomes and child morbidity are drivers of malaria control policy. In fact, the WHO has made clear that any new IPTp regimens should not only prevent malaria but must also decrease adverse birth outcomes. 80 PK/PD modeling is perfectly positioned to clarify relationships between drug exposures and these key outcomes and to leverage these relationships for dose optimization. PK data and comprehensive outcomes data must be collected concurrently in large study populations and dedicated trials must be carefully designed.
Quantifying the impacts of drug resistance and immunity
Drug efficacy and drug resistance
An important goal of antimalarial drug development and policy is to select antimalarial combinations with high barriers of drug resistance and to identify and react to a failure of a treatment regimen due to drug resistance early. Antimalarial drug resistance develops stepwise, with mutations accumulating, which decrease but do not fully eliminate activity against parasites. Decreased sensitivity to artemisinins and PQ is spreading in southeast Asia, 81 and AL treatment failure rates > 10% have been reported in sub‐Saharan Africa. 82 Ideal antimalarial treatment regimens would achieve high efficacy while minimizing dosing frequency, toxicity, and selection for drug resistance. Unfortunately, these goals can be at odds when long acting antimalarials prolong efficacy but can also result in long subtherapeutic trials that can select for more resistant parasites. 83 , 84 Capturing the dynamics of how drug exposure selects for drug resistance with PK/PD models is a valuable contribution to antimalarial regimen selection and dose optimization. Drug resistance in PK/PD models has been influential in simulation studies, guiding selection of the triple ACT regimens, 85 and predicting failure of malaria chemoprevention regimens. 86 However, relationships between antimalarial exposure and biomarkers of drug resistance have rarely been quantified and validated with clinical data. Higher drug concentrations can overcome decreased antimalarial sensitivity and with PD targets, drug regimens can be optimized to minimize malaria recrudescence after treatment or malaria infection after chemoprevention. 8 , 87 It will be important for PK/PD models to incorporate drug resistance biomarkers as they become available, and that these biomarkers be incorporated into clinical trials that include PK data.
Malaria immunity
Naturally acquired immunity to malaria develops with increasing age and following repeated exposure to malaria parasites. This immunity is characterized by a decreasing likelihood that blood‐stage parasite infections are associated with symptoms, and thought to be comprised of two distinct but complementary processes: (i) anti‐parasite immunity, which helps control blood‐stage infection such that in highly endemic settings older (more immune) individuals carry lower parasite densities than younger individuals; and (ii) anti‐disease immunity, which allows individuals to tolerate high parasite densities without developing a fever. 88 Importantly, pre‐existing antimalarial immunity can influence malaria treatment outcomes and PK/PD relationships by accelerating parasite clearance and reducing the risk of recrudescence following treatment. 89 Antibodies specific for blood‐stage malaria antigens have been associated with a reduced risk of treatment failure. 7 , 90 , 91 , 92 A recent paper further found that functional characteristics of the Ig subclass of antimalarial antibodies—the ability to both fix complement and mediate opsonic phagocytosis—were also associated with faster parasite clearance. 93 These data show the importance for future malaria PK/PD models to include biomarkers of antimalarial immunity, particularly as malaria vaccines are studied in conjunction with pharmacological‐based malaria control interventions.
Model‐informed drug development
With variability in drug response predicted due to drug resistance, immunity, and potentially other comorbidities, it is not surprising that predicting dose, response, and the best combinations of antimalarial drugs from preclinical data using PK/PD models has been challenging.
Translational PK/PD
Developing a translational mechanistic model for malaria to accelerate drug combination and dose selection has been challenging (Figure 1 ). Standard 48 hour in vitro parasite sensitivity experiments with synchronized parasite cultures are likely a poor surrogate for drug efficacy over a 1–2 week treatment duration with drug combinations. In vitro systems to assess P. falciparum drug sensitivity and a mouse model with P. berghei (rodent specific parasite species) allowed for a pipeline of new antimalarial drug candidates, but mechanistic PK/PD models based on preclinical data have overpredicted clinical benefits. 94 , 95 , 96 Recent developments have focused on creating a mouse model able to sustain P. falciparum infections and now a humanized mouse model engrafted with human erythrocytes exists. 97 Unfortunately, this advanced model still overpredicts the clinical benefit of drug candidates. 98 Modeling parasite dynamics with and without drugs from preclinical data requires the reliable transformation of in vitro drug efficacy to in vivo drug activity. Furthermore, our understanding of human parasite burden and dynamics without drug pressure continues to evolve to incorporate polyclonal infections, immunity, infection timing, age, and size. We expect that by incorporating longitudinal parasite density measurements from murine and human infection volunteer data, we may improve predictions of mechanistic PK/PD models for drug efficacy and/or develop a superior method to rank compounds allowing for more rapid translation of drugs from discovery to development.
Controlled human malaria infection models
PK/PD models are an indispensable tool for drug development and drug repurposing. Achieving safe single dose malaria treatment and prevention regimens would transform the malaria therapeutic landscape. To fill this gap, a controlled human P. falciparum infection model has been developed for human studies, and is being used to quantify the initial PK/PD relationships. These studies are conducted in a hospital settling which allows for the collection of intensive PK and PD data. Non‐immune healthy volunteers are infected with malaria either by mosquito bites (sporozoite‐induced) or by direct injection of blood‐stage parasites. 99 If the sporozoite‐induced infection is used, researchers can study if the candidate drugs have any effect on liver stage parasites. Typically, studies will define a set parasite density threshold at which treatment will begin. Parasite densities are closely monitored before treatment is provided and this data can be used to establish the natural growth dynamics of P. falciparum. Controlled human malaria infection studies provide the malaria community with a unique opportunity to understand the PK/PD of new therapeutics early in develop and make informed decisions on the next dose to be used.
LESSONS LEARNED
Antimalarial PK/PD relationships have been some of the best quantified among anti‐infectives in the global health arena. Pharmacometric methods, including PK/PD modeling, led directly to dosing changes for ACTs, have facilitated selection of repurposed drug regimens for malaria treatment and prevention, and are becoming an integral component of mathematical models which guide malaria control policy. Other global health disease areas can improve treatment, prevention, and elimination efforts by following the example set and lessons learned within the malaria field.
Standardized clinical trial design, biomarker and outcomes measurement, and PK data collection enhances the quality of clinical studies
Malaria clinical trials are conducted in many countries, with diverse populations, drugs, and sample sizes. However, the research community has been able to maximize the reach and scientific conclusions from these trials owing to the standardized manner in which they are conducted. As an example, malaria treatment efficacy studies use directly observed therapy, conduct follow‐up for clinical or parasitological relapse at standard intervals through 28 to 42 days after treatment, and when PK data is collected, usually obtain day 7 PK concentrations. Outcome measures have standard definitions, including adequate clinical and parasitological response, recrudescence infection (treatment failure), or reinfection. Both designing and conducting trials in a similar manner has facilitated comparison of results across studies, including the ability to pool data to identifying subpopulations at risk and in indicating to regulators how pervasive dosing issues are globally. Infectious disease clinical trials could greatly benefit from standardization to facilitate post hoc data analyses especially as it pertains to PK/PD measures.
Strong systems to share clinical, molecular, and PK data enhances our ability to identify optimal antimalarial regimens for vulnerable populations
WWARN has been an instrumental organization in collecting, standardizing, and generating PK/PD databases for malaria research. They have demonstrated the power of pooled individual patient data meta‐analyses by aggregating historic data and leveraging the large number of patients and observations to answer important questions about understudied populations. WWARN has encouraged investigators and set a precedent that clinical data be shared. As described above, these studies have helped to identify high‐risk subpopulations and used PK/PD modeling to recommend new dosing regimens. Although not all of these studies have ultimately resulted in changes to dosing guidelines, they have helped indicate the next steps in dosing regimens, which are currently being explored in clinical trials.
Clear translation of findings from PK/PD analyses into predicted improvements in treatment outcomes has led to policy changes in antimalarial dosing guidelines
PK/PD model‐informed dosing of antimalarials has become a valued tool for antimalarial research and policy. Pharmacometricians have presented the results of their PK/PD modeling work in terms of clinical impact and have identified dosing regimens that consider safety, efficacy, and implementation. Some of these changes have been enacted directly (e.g., DP dosing in pediatric populations) whereas others are already in clinical trials (AL in young children and pregnancy, triple ACT regimens in South East Asia). Although we note that PK/PD studies for antimalarials could be improved by diversifying population specific outcomes to include those that are highest priority for regulators, such as birth outcomes among pregnancy, pharmacometricians in antimalarial research have made significant progress in translating scientific findings into action. As the value of PK/PD modeling has been demonstrated, it has opened the door to more advanced applications, and a more rapid translation of scientific discovery to policy.
CONCLUSIONS
Pharmacometric modeling has played an instrumental role in improving malaria treatment by generating dosing regimens and new drug candidates. Malaria investigators have used PK/PD modeling to extensively study dosing in high‐risk groups and identified that pregnant women, young and underweight children, as well as individuals receiving concomitant therapy with CYP450 inducers could all benefit from dose adjustments. As PK/PD modeling becomes more widespread in clinical studies, we expect to see more updates to malaria treatment and prevention guidelines. By understanding how the malaria field has experienced success in applying pharmacometrics to improve outcomes, we suggest these successes can be achieved in other disease areas.
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
All authors declared no competing interests for this work.
References
- 1. World Health Organization . World Malaria Report 2020 (Geneva, Switzerland, 2020). https://www.who.int/publications/i/item/9789240015791 [Google Scholar]
- 2. Phillips, M.A. , Burrows, J.N. , Manyando, C. , van Huijsduijnen, R.H. , Van Voorhis, W.C. & Wells, T.N.C. Malaria. Nat. Rev. Dis. Primers 3, 17050 (2017). [DOI] [PubMed] [Google Scholar]
- 3. White, N.J. , Pukrittayakamee, S. , Hien, T.T. , Faiz, M.A. , Mokuolu, O.A. & Dondorp, A.M. Malaria.Lancet 383, 723–735 (2014). [DOI] [PubMed] [Google Scholar]
- 4. Ashley, E.A. , Pyae Phyo, A. & Woodrow, C.J. Malaria. Lancet 391, 1608–1621 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Slater, H.C. et al. The temporal dynamics and infectiousness of subpatent Plasmodium falciparum infections in relation to parasite density. Nat. Commun. 10, 1433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. World Health Organization . Guidelines for the treatment of malaria, 3rd ed. 1‐317 (Geneva, Switzerland, 2015). [Google Scholar]
- 7. Greenhouse, B. et al. Decreasing efficacy of antimalarial combination therapy in Uganda is explained by decreasing host immunity rather than increasing drug resistance. J. Infect. Dis. 199, 758–765 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wallender, E. et al. Modeling prevention of malaria and selection of drug resistance with different dosing schedules of dihydroartemisinin‐piperaquine preventive therapy during pregnancy in Uganda. Antimicrob. Agents Chemother. 63, e01393‐18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Severe malaria. Trop. Med. Int. Health 19 (Suppl 1), 7–131 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Tarning, J. et al. Population pharmacokinetics and pharmacodynamics of piperaquine in children with uncomplicated falciparum malaria. Clin. Pharmacol. Ther. 91, 497–505 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. WorldWide Antimalarial Resistance Network (WWARN) DP Study Group. The effect of dosing regimens on the antimalarial efficacy of dihydroartemisinin‐piperaquine: a pooled analysis of individual patient data. PLoS Medicine 10, e1001564; discussion e1001564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. White, N.J. , Stepniewska, K. , Barnes, K. , Price, R.N. & Simpson, J. Simplified antimalarial therapeutic monitoring: using the day‐7 drug level? Trends Parasitol. 24, 159–163 (2008). [DOI] [PubMed] [Google Scholar]
- 13. German, P.I. & Aweeka, F.T. Clinical pharmacology of artemisinin‐based combination therapies. Clin. Pharmacokinet. 47, 91–102 (2008). [DOI] [PubMed] [Google Scholar]
- 14. Conrad, M.D. & Rosenthal, P.J. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect. Dis. 19, e338–e351 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Kloprogge, F. et al. Artemether‐lumefantrine dosing for malaria treatment in young children and pregnant women: a pharmacokinetic‐pharmacodynamic meta‐analysis. PLoS Medicine 15, e1002579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoglund, R.M. et al. Population pharmacokinetic properties of piperaquine in falciparum malaria: an individual participant data meta‐analysis. PLoS Medicine 14, e1002212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mutagonda, R.F. , Kamuhabwa, A.A. , Minzi, O.M. , Massawe, S.N. , Maganda, B.A. & Aklillu, E. Malaria prevalence, severity and treatment outcome in relation to day 7 lumefantrine plasma concentration in pregnant women. Malar. J. 15, 278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. WorldWide Antimalarial Resistance Network (WWARN) Lumefantrine PK/PD Study Group . Artemether‐lumefantrine treatment of uncomplicated Plasmodium falciparum malaria: a systematic review and meta‐analysis of day 7 lumefantrine concentrations and therapeutic response using individual patient data. BMC Med. 13, 227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Price, R.N. et al. Clinical and pharmacological determinants of the therapeutic response to dihydroartemisinin‐piperaquine for drug‐resistant malaria. Antimicrob. Agents Chemother. 51, 4090–4097 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White, N.J. , van Vugt, M. & Ezzet, F. Clinical pharmacokinetics and pharmacodynamics and pharmacodynamics of artemether‐lumefantrine. Clin. Pharmacokinet. 37, 105–125 (1999). [DOI] [PubMed] [Google Scholar]
- 21. Price, R.N. et al. Molecular and pharmacological determinants of the therapeutic response to artemether‐lumefantrine in multidrug‐resistant Plasmodium falciparum malaria. Clin. Infect. Dis. 42, 1570–1577 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kearns, G.L. , Abdel‐Rahman, S.M. , Alander, S.W. , Blowey, D.L. , Leeder, J.S. & Kauffman, R.E. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 349, 1157–1167 (2003). [DOI] [PubMed] [Google Scholar]
- 23. Sambol, N.C. et al. Population pharmacokinetics of piperaquine in young Ugandan children treated with dihydroartemisinin‐piperaquine for uncomplicated malaria. Clin. Pharmacol. Ther. 98, 87–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salman, S. et al. Pharmacokinetic comparison of two piperaquine‐containing artemisinin combination therapies in Papua New Guinean children with uncomplicated malaria. Antimicrob. Agents Chemother. 56, 3288–3297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hung, T.Y. et al. Population pharmacokinetics of piperaquine in adults and children with uncomplicated falciparum or vivax malaria. Br. J. Clin. Pharmacol. 57, 253–262 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee, T.M. et al. In vitro metabolism of piperaquine is primarily mediated by CYP3A4. Xenobiotica 42, 1088–1095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Staehli Hodel, E.M. et al. Population pharmacokinetics of mefloquine, piperaquine and artemether‐lumefantrine in Cambodian and Tanzanian malaria patients. Malar. J. 12, 235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ezzet, F. , Mull, R. & Karbwang, J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br. J. Clin. Pharmacol. 46, 553–561 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salman, S. et al. Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob. Agents Chemother. 55, 5306–5313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tchaparian, E. et al. Population pharmacokinetics and pharmacodynamics of lumefantrine in young Ugandan children treated with artemether‐lumefantrine for uncomplicated malaria. J. Infect. Dis. 214, 1243–1251 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mwesigwa, J. et al. Pharmacokinetics of artemether‐lumefantrine and artesunate‐amodiaquine in children in Kampala, Uganda. Antimicrob. Agents Chemother. 54, 52–59 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ashley, E.A. et al. Pharmacokinetic study of artemether‐lumefantrine given once daily for the treatment of uncomplicated multidrug‐resistant falciparum malaria. Trop. Med. Int. Health 12, 201–208 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Ezzet, F. , van Vugt, M. , Nosten, F. , Looareesuwan, S. & White, N.J. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob. Agents Chemother. 44, 697–704 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Borrmann, S. et al. The effect of food consumption on lumefantrine bioavailability in African children receiving artemether‐lumefantrine crushed or dispersible tablets (Coartem) for acute uncomplicated Plasmodium falciparum malaria. Trop. Med. Int. Health 15, 434–441 (2010). [DOI] [PubMed] [Google Scholar]
- 35. Ashley, E.A. et al. How much fat is necessary to optimize lumefantrine oral bioavailability? Trop. Med. Int. Health 12, 195–200 (2007). [DOI] [PubMed] [Google Scholar]
- 36. Kwenti, T.E. Malaria and HIV coinfection in sub‐Saharan Africa: prevalence, impact, and treatment strategies. Res. Rep. Trop. Med. 9, 123–136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grace, J.M. , Aguilar, A.J. , Trotman, K.M. , Peggins, J.O. & Brewer, T.G. Metabolism of beta‐arteether to dihydroqinghaosu by human liver microsomes and recombinant cytochrome P450. Drug Metab. Dispos. 26, 313–317 (1998). [PubMed] [Google Scholar]
- 38. Simonsson, U.S. , Jansson, B. , Hai, T.N. , Huong, D.X. , Tybring, G. & Ashton, M. Artemisinin autoinduction is caused by involvement of cytochrome P450 2B6 but not 2C9. Clin. Pharmacol. Ther. 74, 32–43 (2003). [DOI] [PubMed] [Google Scholar]
- 39. Fellay, J. et al. Variations of CYP3A activity induced by antiretroviral treatment in HIV‐1 infected patients. Eur. J. Clin. Pharmacol. 60, 865–873 (2005). [DOI] [PubMed] [Google Scholar]
- 40. Barry, M. , Mulcahy, F. , Merry, C. , Gibbons, S. & Back, D. Pharmacokinetics and potential interactions amongst antiretroviral agents used to treat patients with HIV infection. Clin. Pharmacokinet. 36, 289–304 (1999). [DOI] [PubMed] [Google Scholar]
- 41. Ma, Q. et al. Pharmacokinetic drug interactions with non‐nucleoside reverse transcriptase inhibitors. Expert. Opin. Drug Metab. Toxicol. 1, 473–485 (2005). [DOI] [PubMed] [Google Scholar]
- 42. Navaratnam, V. , Mansor, S.M. , Sit, N.W. , Grace, J. , Li, Q. & Olliaro, P. Pharmacokinetics of artemisinin‐type compounds. Clin. Pharmacokinet. 39, 255–270 (2000). [DOI] [PubMed] [Google Scholar]
- 43. Coartem . Package Insert. (Novartis Pharmaceuticals Corporation, East Hanover, NJ, 2009). [Google Scholar]
- 44. Wong, R.P. , Salman, S. , Ilett, K.F. , Siba, P.M. , Mueller, I. & Davis, T.M. Desbutyl‐lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial activity that may influence artemether‐lumefantrine treatment outcome. Antimicrob. Agents Chemother. 55, 1194–1198 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ilett, K.F. et al. Glucuronidation of dihydroartemisinin in vivo and by human liver microsomes and expressed UDP‐glucuronosyltransferases. Drug Metab. Dispos. 30, 1005–1012 (2002). [DOI] [PubMed] [Google Scholar]
- 46. World Health Organization . Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection 1‐480 (WHO, Geneva, Switzerland, 2016). [PubMed] [Google Scholar]
- 47. Banda, C.G. et al. Impact of efavirenz‐, ritonavir‐boosted lopinavir‐, and nevirapine‐based antiretroviral regimens on the pharmacokinetics of lumefantrine and safety of artemether‐lumefantrine in plasmodium falciparum‐negative HIV‐infected malawian adults stabilized on antiretroviral therapy. Antimicrob. Agents Chemother. 62, e01162‐18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Byakika‐Kibwika, P. et al. Significant pharmacokinetic interactions between artemether/lumefantrine and efavirenz or nevirapine in HIV‐infected Ugandan adults. J. Antimicrob. Chemother. 67, 2213–2221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wallender, E. et al. Predicting optimal dihydroartemisinin‐piperaquine regimens to prevent malaria during pregnancy for human immunodeficiency virus‐infected women receiving efavirenz. J. Infect. Dis. 217, 964–972 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parikh, S. et al. Antiretroviral choice for HIV impacts antimalarial exposure and treatment outcomes in Ugandan children. Clin. Infect. Dis. 63, 414–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoglund, R.M. et al. Artemether‐lumefantrine co‐administration with antiretrovirals: population pharmacokinetics and dosing implications. Br. J. Clin. Pharmacol. 79, 636–649 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Francis, J. et al. An individual participant data population pharmacokinetic meta‐analysis of drug‐drug interactions between lumefantrine and commonly used antiretroviral treatment. Antimicrob. Agents Chemother. 64, e01162‐18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maganda, B.A. , Ngaimisi, E. , Kamuhabwa, A.A. , Aklillu, E. & Minzi, O.M. The influence of nevirapine and efavirenz‐based anti‐retroviral therapy on the pharmacokinetics of lumefantrine and anti‐malarial dose recommendation in HIV‐malaria co‐treatment. Malar. J. 14, 179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Faucette, S.R. et al. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 320, 72–80 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yeh, R.F. et al. Lopinavir/ritonavir induces the hepatic activity of cytochrome P450 enzymes CYP2C9, CYP2C19, and CYP1A2 but inhibits the hepatic and intestinal activity of CYP3A as measured by a phenotyping drug cocktail in healthy volunteers. J. Acquir. Immune Defic. Syndr. 42, 52–60 (2006). [DOI] [PubMed] [Google Scholar]
- 56. Hughes, E. et al. Efavirenz‐based antiretroviral therapy reduces artemether‐lumefantrine exposure for malaria treatment in HIV‐infected pregnant women. J. Acquir. Immune Defic. Syndr. 83, 140–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Briggs, J. et al. Impact of microscopic and submicroscopic parasitemia during pregnancy on placental malaria in a high‐transmission setting in Uganda. J. Infect. Dis. 220, 457–466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Steketee, R.W. , Nahlen, B.L. , Parise, M.E. & Menendez, C. The burden of malaria in pregnancy in malaria‐endemic areas. Am. J. Trop. Med. Hyg. 64, 28–35 (2001). [DOI] [PubMed] [Google Scholar]
- 59. Cutts, J.C. et al. Pregnancy‐specific malarial immunity and risk of malaria in pregnancy and adverse birth outcomes: a systematic review. BMC Med. 18, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Anderson, G.D. Pregnancy‐induced changes in pharmacokinetics: a mechanistic‐based approach. Clin. Pharmacokinet. 44, 989–1008 (2005). [DOI] [PubMed] [Google Scholar]
- 61. Isoherranen, N. & Thummel, K.E. Drug metabolism and transport during pregnancy: how does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab. Dispos. 41, 256–262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lohy Das, J. et al. Population pharmacokinetics of artemether, dihydroartemisinin, and lumefantrine in Rwandese pregnant women treated for uncomplicated Plasmodium falciparum Malaria. Antimicrob. Agents Chemother. 62(10), e00518‐18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nyunt, M.M. et al. Artemether‐lumefantrine pharmacokinetics and clinical response are minimally altered in pregnant Ugandan women treated for uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 60, 1274–1282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mosha, D. et al. Population pharmacokinetics and clinical response for artemether‐lumefantrine in pregnant and nonpregnant women with uncomplicated Plasmodium falciparum malaria in Tanzania. Antimicrob. Agents Chemother. 58, 4583–4592 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kloprogge, F. et al. Lumefantrine and desbutyl‐lumefantrine population pharmacokinetic‐pharmacodynamic relationships in pregnant women with uncomplicated Plasmodium falciparum malaria on the Thailand‐Myanmar border. Antimicrob. Agents Chemother. 59, 6375–6384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kloprogge, F. et al. Population pharmacokinetics of lumefantrine in pregnant and nonpregnant women with uncomplicated Plasmodium falciparum malaria in Uganda. CPT Pharmacometrics. Syst. Pharmacol. 2, e83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tarning, J. et al. Population pharmacokinetics of lumefantrine in pregnant women treated with artemether‐lumefantrine for uncomplicated Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 53, 3837–3846 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Onyamboko, M.A. et al. A randomized controlled trial of three‐ versus five‐day artemether‐lumefantrine regimens for treatment of uncomplicated Plasmodium falciparum malaria in pregnancy in Africa. Antimicrob. Agents Chemother. 64, e01140‐19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Arinaitwe, E. et al. The association between malnutrition and the incidence of malaria among young HIV‐infected and ‐uninfected Ugandan children: a prospective study. Malar. J. 11, 90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oldenburg, C.E. , Guerin, P.J. , Berthe, F. , Grais, R.F. & Isanaka, S. Malaria and nutritional status among children with severe acute malnutrition in Niger: a prospective cohort study. Clin. Infect. Dis. 67, 1027–1034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sumbele, I.U. , Bopda, O.S. , Kimbi, H.K. , Ning, T.R. & Nkuo‐Akenji, T. Nutritional status of children in a malaria meso endemic area: cross sectional study on prevalence, intensity, predictors, influence on malaria parasitaemia and anaemia severity. BMC Public Health 15, 1099 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Friedman, J.F. et al. Malaria and nutritional status among pre‐school children: results from cross‐sectional surveys in western Kenya. Am. J. Trop. Med. Hyg. 73, 698–704 (2005). [PubMed] [Google Scholar]
- 73. Chotsiri, P. et al. Severe acute malnutrition results in lower lumefantrine exposure in children treated with artemether‐lumefantrine for uncomplicated Malaria. Clin. Pharmacol. Ther. 106, 1299–1309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. GBD 2017 Causes of Death Collaborators . Global, regional, and national age‐sex‐specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392, 1736–1788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kajubi, R. et al. Antiretroviral therapy with efavirenz accentuates pregnancy‐associated reduction of dihydroartemisinin‐piperaquine exposure during malaria chemoprevention. Clin. Pharmacol. Ther. 102, 520–528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Challenger, J.D. , Bruxvoort, K. , Ghani, A.C. & Okell, L.C. Assessing the impact of imperfect adherence to artemether‐lumefantrine on malaria treatment outcomes using within‐host modelling. Nat. Commun. 8, 1373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Permala, J. , Tarning, J. , Nosten, F. , White, N.J. , Karlsson, M.O. & Bergstrand, M. Prediction of improved antimalarial chemoprevention with weekly dosing of dihydroartemisinin‐piperaquine. Antimicrob. Agents Chemother. 61, e02491‐16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ategeka, J. et al. Relationships between measures of malaria at delivery and adverse birth outcomes in a high‐transmission area of Uganda. J. Infect. Dis. 222, 863–870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Roh, M.E. et al. Overall, anti‐malarial, and non‐malarial effect of intermittent preventive treatment during pregnancy with sulfadoxine‐pyrimethamine on birthweight: a mediation analysis. Lancet Glob. Health 8, e942–e953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. World Health Organization . Meeting report of the WHO Evidence Review Group on Malaria in Pregnancy. (ed. Meeting, M.P.A.C.) (Geneva, Switzerland, 2017). [Google Scholar]
- 81. Hamilton, W.L. et al. Evolution and expansion of multidrug‐resistant malaria in southeast Asia: a genomic epidemiology study. Lancet Infect. Dis. 19, 943–951 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dimbu, P.R. et al. Continued low efficacy of artemether‐lumefantrine in Angola, 2019. Antimicrob. Agents Chemother. 65, e01949‐20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hastings, I.M. & Hodel, E.M. Pharmacological considerations in the design of anti‐malarial drug combination therapies ‐ is matching half‐lives enough? Malar. J. 13, 62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hastings, I.M. , Watkins, W.M. & White, N.J. The evolution of drug‐resistant malaria: the role of drug elimination half‐life. Philos. Trans. R Soc. Lond. B Biol. Sci. 357, 505–519 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zaloumis, S.G. et al. In silico investigation of the decline in clinical efficacy of artemisinin combination therapies due to increasing artemisinin and partner drug resistance. Antimicrob. Agents Chemother. 62, e01292‐18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bergstrand, M. , Nosten, F. , Lwin, K.M. , Karlsson, M.O. , White, N.J. & Tarning, J. Characterization of an in vivo concentration‐effect relationship for piperaquine in malaria chemoprevention. Sci. Transl. Med. 6, 260ra147 (2014). [DOI] [PubMed] [Google Scholar]
- 87. Dzinjalamala, F.K. et al. Association between the pharmacokinetics and in vivo therapeutic efficacy of sulfadoxine‐pyrimethamine in Malawian children. Antimicrob. Agents Chemother. 49, 3601–3606 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rodriguez‐Barraquer, I. et al. Quantification of anti‐parasite and anti‐disease immunity to malaria as a function of age and exposure. Elife 7, e35832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. White, N.J. The parasite clearance curve. Malar. J. 10, 278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Staedke, S.G. , Sendagire, H. , Lamola, S. , Kamya, M.R. , Dorsey, G. & Rosenthal, P.J. Relationship between age, molecular markers, and response to sulphadoxine‐pyrimethamine treatment in Kampala, Uganda. Trop. Med. Int. Health 9, 624–629 (2004). [DOI] [PubMed] [Google Scholar]
- 91. Keh, C.E. et al. Associations between antibodies to a panel of Plasmodium falciparum specific antigens and response to sub‐optimal antimalarial therapy in Kampala, Uganda. PLoS One 7, e52571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pinder, M. et al. Immunoglobulin G antibodies to merozoite surface antigens are associated with recovery from chloroquine‐resistant Plasmodium falciparum in Gambian children. Infect. Immun. 74, 2887–2893 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. O'Flaherty, K. et al. Contribution of functional antimalarial immunity to measures of parasite clearance in therapeutic efficacy studies of artemisinin derivatives. J. Infect. Dis. 220, 1178–1187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Patel, K. , Batty, K.T. , Moore, B.R. , Gibbons, P.L. & Kirkpatrick, C.M. Predicting the parasite killing effect of artemisinin combination therapy in a murine malaria model. J. Antimicrob. Chemother. 69, 2155–2163 (2014). [DOI] [PubMed] [Google Scholar]
- 95. Rottmann, M. et al. Preclinical antimalarial combination study of M5717, a Plasmodium falciparum elongation factor 2 inhibitor, and pyronaridine, a hemozoin formation inhibitor. Antimicrob. Agents Chemother. 64, e02181‐19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zaloumis, S. et al. Assessing the utility of an anti‐malarial pharmacokinetic‐pharmacodynamic model for aiding drug clinical development. Malar. J. 11, 303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ito, R. , Takahashi, T. , Katano, I. & Ito, M. Current advances in humanized mouse models. Cell. Mol. Immunol. 9, 208–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. McCarthy, J.S. et al. Linking murine and human Plasmodium falciparum challenge models in a translational path for antimalarial drug development. Antimicrob. Agents Chemother. 60, 3669–3675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stanisic, D.I. , McCarthy, J.S. & Good, M.F. Controlled human malaria infection: applications, advances, and challenges. Infect. Immun. 86, e00479‐17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]