

Abstract
The safety of a novel modified‐release oral capsule with orlistat and acarbose (MR‐OA) was investigated in 67 obese middle‐aged White men with a body mass index of 32 to 40 kg/m2 or 30 to 32 kg/m2 plus waist circumference >102 cm. The purpose of this investigation was to compare MR‐OA with the existing conventional orlistat regarding systemic safety defined as plasma orlistat concentration at the end of the treatment period of 14 days. Participants took the MR‐OA fixed‐dose combination formulation 3 times a day together with a major meal. Three different doses of MR‐OA were evaluated—60/20, 90/30, and 120/40 (mg orlistat/mg acarbose)—as well as 1 reference group who received the conventional orlistat, Xenical, with 120 mg of orlistat. Blood plasma was sampled on days 1 and 14. The orlistat plasma concentrations of the MR‐OA dose showed a delayed absorption and were lower compared with conventional orlistat at the end of the study. All doses were safe and well tolerated without any unexpected adverse events and no serious adverse events. The delay in the rise of orlistat plasma concentration indicates that the modified‐release properties of the MR‐OA formulation are effective. The systemic exposure of orlistat resulting from MR‐OA was similar, albeit a bit lower than the conventional orlistat with 120 mg of orlistat. We can therefore assume that the safety profile regarding the orlistat moiety of MR‐OA is comparable to the conventional orlistat and a promising approach for weight control in obese patients. Further clinical evaluation is underway.
Keywords: fixed‐dose combination, gastrointestinal tolerability, modified release, obesity treatment, weight loss product
Obesity is a major and multidimensional health problem worldwide and a major risk factor for additional health concerns such as cardiovascular diseases, diabetes mellitus, musculoskeletal disorders, and some cancers. 1 , 2 , 3 In Europe, the currently approved drugs for obesity are perorally administered orlistat (Alli and Xenical), perorally administered bupropion/naltrexone (Mysimba), and subcutaneously injected liraglutide (Saxenda). With the exception of the oral dosage forms of orlistat, all currently approved anti‐obesity pharmaceutical products have some systemic side effects. 4
We recently showed that a novel multiple‐unit modified‐release (MR) capsule with a fixed‐dose combination of orlistat and the carbohydrate‐digestion inhibitor acarbose (MR‐OA) reduced appetite and postprandial glucose. 5 We also found that the oral MR formulation enabled combining these 2 active pharmaceutical ingredients (APIs) without worsening gastrointestinal tolerability compared with the approved conventional orlistat drug product. 5
Here, we present pharmacokinetic data from the clinical study EP‐001 for obese middle‐aged men treated with the novel capsule, administered 3 times a day together with meal intake for 14 consecutive days. The aim of this study was to investigate if the systemic exposure and pharmacokinetics of MR‐OA were similar to, and therefore as safe (regarding systemic effects) as, the conventional oral dosage form containing 120 mg of orlistat.
Methods
Study Design
The clinical trial was conducted as a single‐center, controlled, randomized, parallel‐group phase IIa study with 2 weeks of daily treatment 3 times a day in the fed state with 3 different dose strengths of the novel orlistat/acarbose test product MR‐OA, namely, 60/20, 90/30, and 120/40 mg/mg (denoted MR‐OA‐60/20, MR‐OA‐90/30, and MR‐OA‐120/40, respectively). These 3 dose strengths were given to the 3 treatment groups; group I, II, and III, respectively, a conventional orlistat product, that is, Xenical, 120 mg was used as comparator (Table 1), denoted in group IV. A detailed description of the study was published previously. 5 Ethics approval was granted by the regional Ethical Review Board in Uppsala, Sweden (dnr 2016/257). All study participants signed the consent form. The trial was registered at EudraCT (2016‐001055‐50). The clinical trial was performed by the contract research organization Clinical Trial Consultants in Uppsala, Sweden.
Table 1.
Cave, Cmax, Clast, AUClast (Arithmetic Mean ± Standard Deviation) and tmax Values for Orlistat (Median and Interquartile Range) Determined at Day 1 (Visit 2) and Day 14 (Visit 4), Respectively, During the 14‐Day Study
| Treatment Group Orlistat/Acarbose, mg | Group I MR‐OA 60/20 (n = 17) | Group II MR‐OA 90/30 (n = 17) | Group III MR‐OA 120/40 (n = 16) | Group IV Conventional Orlistat 120 (n = 17) |
|---|---|---|---|---|
| Cave, ng/mL‐V 2 | 0.38 ± 0.32 a | 0.64 ± 0.63 a | 0.53 ± 0.35 a | 1.21 ± 0.67 |
| Cave, ng/mL‐V 4 | 0.35 ± 0.21 a | 0.60 ± 0.40 a | 0.54 ± 0.35 a | 1.45 ± 1.10 |
| Cmax, ngl/mL‐V 2 | 0.65 ± 0.57 a | 1.26 ± 1.42 a | 1.05 ± 0.87 a | 3.32 ± 1.93 |
| Cmax, ng/mL‐V 4 | 0.88 ± 0.69 a | 1.55 ± 1.13 a | 1.24 ± 1.10 a | 3.19 ± 2.28 |
| Clast , ng/mL‐V 2 | 0.62 ± 0.59 a | 1.24 ± 1.43 a | 1.01 ± 0.87 a | 2.86 ± 1.52 |
| Clast, ng/mL‐V 4 | 0.80 ± 0.76 a | 1.54 ± 1.14 a | 1.24 ± 1.10 a | 2.89 ± 1.99 |
| AUClast, ng/mL × h‐V 2 | 0.57 ± 0.46 a | 1.27 ± 1.19 a | 1.13 ± 1.05 a | 3.42 ± 3.48 |
| AUClast, ng/mL × h‐V 4 | 0.95 ± 0.52 a | 1.53 ± 1.10 a | 1.51 ± 1.36 a | 4.08 ± 3.78 |
| tmax, h‐V 2 | 6.00 (5.88‐6.00) | 6.00 (6.00‐6.00) | 6.00 (6.00‐6.00) | 6.00 (6.00‐6.00) |
| tmax, h‐V 4 | 6.00 (4.90‐6.00) | 6.00 (6.00‐6.00) | 6.00 (6.00‐6.00) | 6.00 (6.00‐6.00) |
AUClast,: area under the concentration–time curve from time 0 to the last measurable concentration; Cave, individual mean concentration; Clast, last measurable concentration; Cmax, maximum plasma concentration; tmax, time to maximum plasma concentration.
Significantly different from conventional orlistat group, adjusted for multiple comparisons according to Holm.11 “V 2” = visit 2 (day 1), “V 4” = visit 4 (day 14).
Study Subjects
Included in the study were obese male White subjects 24‐60 years old with either a body mass index (BMI) of 32–40 kg/m2 or BMI of 30–32 kg/m2 together with waist circumference >102 cm. Subjects were screened for eligibility as per study‐specific inclusion/exclusion criteria within 5 weeks before (days –35 to –4) randomization and the first oral administration of the test and reference products. 5 Additional inclusion criteria were based on medical history, physical findings, vital signs, electrocardiogram (ECG), and blood chemistry values at the time of screening. All subjects had an adequate plasma glucose control (ie, without previous diagnosis of diabetes mellitus type 2); serum creatinine levels <1.5 times the upper limit of normal; serum aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, and gamma‐glutamyl transferase levels <2.5 times the upper limit of normal, and bilirubin levels <1.5 times the upper limit of normal.
Treatments
Eligible and consenting subjects arrived at the research clinic in a fasted condition (at least 8 hours since a meal) on the morning of the first study day (visit 2, day 1) and were randomly assigned to 1 of 4 parallel treatment groups. Drug products were taken orally concomitant with the 3 main daily meals (fed state); these were either 1 of the dose strengths of the test product or the reference product. Each subject was instructed to take the drug product halfway through each meal.
Study Outline
The clinical trial consisted of 5 visits to the clinic, including screening and follow‐up. 5 The total duration of the experiments was 2 weeks for each subject with no overnight stays. Visit 1 included screening and a health investigation and occurred 1 to 5 weeks before the study start. On days –1 through –3, baseline questionnaires were completed by each subject outside the clinic. On study days 1 and 14 (visits 2 and 4), the drug product was administered at the research clinic together with standardized meals (breakfast and lunch) at specific times. For the remaining days, the drug product was self‐administered three times a day by each subject, together with all 3 daily main meals. There were no dietary restrictions during the study days at home except that subjects had to abstain from alcohol for 24 hours and from food and drink for at least 8 hours before visits 1, 2, and 4. On day 21 (visit 5), a final follow‐up visit was performed at the clinic. Visit 3, day 7 of the experimental period included a safety check‐up in which plasma/serum concentrations of liver enzymes were assessed. At visit 5, about 1 week (4‐10 days) after end of the experimental period, another safety follow‐up was performed.
Diet During Visits 2 and 4
On study days 1 and 14, each subject received 2 meals during the visit. Drug products were given concomitant with food throughout the study. Breakfast was rye bread with butter, cheese, and salami (846 kcal, 16 energy percent [E%] protein, 50 E% fat, and 34 E% carbohydrates). Lunch was ham and cheese pie, and white bread with butter (1053 kcal, 14 E% protein, 55 E% fat, and 31 E% carbohydrates). Halfway through the meal, about 50 mL of tap water was consumed together with the drug product. Water consumption ad libitum was allowed between 2 and 3 hours after breakfast as well as 2 hours after lunch.
Fixed‐Dose MR Formulation of 2 Active Drugs (MR‐OA) and Pharmacological Rationale
The test drug product was a solid, oral, multiple‐unit, MR capsule with a fixed‐dose combination of 2 APIs, orlistat and acarbose. The capsule contained 3 different types of pellets or granules but of a consistent size fraction (≤700 μm) that allowed a continuous postprandial gastric emptying rate. 6 , 7 Each type of pellet had a specified and different API release rate. Orlistat and acarbose are respectively classified as class IV and III drugs according to the Biopharmaceutical Classification System, 8 , 9 and their biopharmaceutical properties were central for the test product, which was designed to optimize the physiological enteroendocrine‐mediated gastrointestinal brake feedback satiety response. 10
Plasma Pharmacokinetics
Blood samples were collected from a cubital vein in heparinized Vacutainer tubes (Becton Dickinson, East Rutherford, New Jersey) at –15, –14, 30, 60, 90, 120, 225, 270, 300, 330, and 360 minutes on study day 1 (visit 2) and day 14 (visit 4). Capsules were taken in the fed state at time 0 and 240 minutes. The limited sampling of blood for pharmacokinetic assessment was due to extensive blood samples for a number of biochemical and neuroendocrine biomarkers and other safety markers. Meals were initiated at time points –5 (breakfast) and 235 minutes (lunch) on each study day. Each subject had 10 minutes to finish their meals. The blood samples were kept on ice and then centrifuged for 10 minutes at 1500 g. Plasma was stored at –70°C until analysis.
Orlistat Assay
Plasma concentrations of orlistat were quantified by the Swedish National Veterinary Institute using a validated ultra‐performance liquid chromatography–tandem mass spectrometry method (Acquity ultra‐performance liquid chromatography systems coupled to Xevo‐TQS micro tandem quadrupole mass spectrometers [Waters Corporation, Milford, Massachusetts]) in accordance with the OECD Principles of Good Laboratory Practice 1997. All samples were thawed, vortex‐mixed, and centrifuged. The samples were then treated with acetonitrile to precipitate the proteins followed by liquid‐liquid extraction using hexane, isolation, and evaporation of the organic phase after the samples were reconstituted. They were then vortex‐mixed and centrifuged before being injected into the ultra‐performance liquid chromatography–tandem mass spectrometry method system. The ionization technique was positive electrospray. The chromatographic column was a YMC‐Triart Phenyl (50 × 2.1 mm length × inner diameter, particle diameter 1.9 μm). The chromatographic elution was carried out with a mobile phase consisting of components A, 2.0 mM ammonium acetate in water, and B, acetonitrile. The total run time was 5.0 minutes after washing and equilibrating the column. The autosampler was programmed to inject 10 μL of sample. The data collection was performed in the multiple reaction monitoring mode. The multiple reaction monitoring transitions were m/z 496 > 319 for orlistat and m/z 499 > 319 for orlistat‐d3. The orlistat‐d3 (Toronto Research Chemicals, Toronto, Canada) was used as an internal standard. The calibration curve range for orlistat was 0.05 to 25 ng/mL. The lower limit of quantification was 0.05 ng/mL. In each analytical batch, at least two‐thirds of the quality control samples must have determined concentrations within ± 10% of their respective nominal concentrations.
Pharmacokinetic Data Analysis
For purposes of data analysis, a plasma orlistat concentration of 0.025 ng/mL was used in the calculations for plasma samples below the lower limit of quantification at 0.05 ng/mL. The following pharmacokinetic parameters were calculated: the individual mean concentration, maximum plasma concentration, time to maximum plasma concentration (tmax), and area under the concentration–time curve from time 0 to the last measurable concentration. Calculations were performed with Python 3.0 (Python Software Foundation, Beaverton, Oregon).
Statistical Analysis
Differences among the 3 MR‐OA groups (60/20, 90/30, and 120/40) and the reference product group (conventional orlistat, Xenical) were tested with Welch's t test for continuous data. Data are presented as mean ± standard deviation. Significance was set at α = .05. Adjustments for multiple comparisons were made according to Holm. 11 Statistical analyses were performed with R Commander version 2.4‐2 (R Foundation for Statistical Computing, Vienna, Austria). 12
Results
As has been shown previously, 5 the randomized men were aged 42.9 ± 9.1 (mean ± standard deviation) and had a BMI of 34.8 ± 2.7 kg/m2. Mean concentration and highest maximum plasma concentration of orlistat were <5 ng/mL for all doses of MR‐OA as well as in conventional orlistat at both visit 2 and visit 4 (Table 1). Moreover, a delay in the rise in plasma concentrations in the afternoon was observed for the test products (groups I‐III) compared with the conventional orlistat product (group IV; Figure 1). The last measurable concentration and area under the concentration–time curve from time 0 to the last measurable concentration were lower in the MR‐OA groups at both visit 2 and visit 4 compared with the conventional orlistat group (P < .05; Table 1). There were no differences in tmax (Table 1). The orlistat plasma concentrations were higher after the second dose of the day compared with the initial morning dose for all 4 treatments (Figure 1). The individual variation in orlistat concentration for MR‐OA 120/40 and conventional orlistat 120 mg are presented in Figure 2. The pharmacokinetics were linear, and there were no signs of accumulation of the drug in plasma. No serious adverse events (SAEs) occurred, and the frequency of non‐SAEs was low (Table S1).
Figure 1.
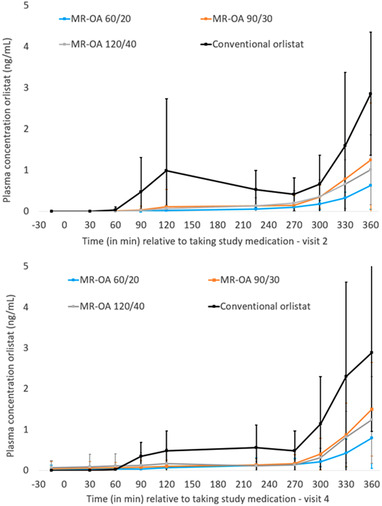
Plasma concentrations of orlistat (mean ± standard deviation) during day 1 (visit 2) and day 14 (visit 4). Drug products taken at 0 and 240 minutes. Breakfast initiated 5 minutes before the first dose, and lunch initiated 5 minutes before the second dose. MR‐OA, modified‐release oral capsule with orlistat and acarbose.
Figure 2.
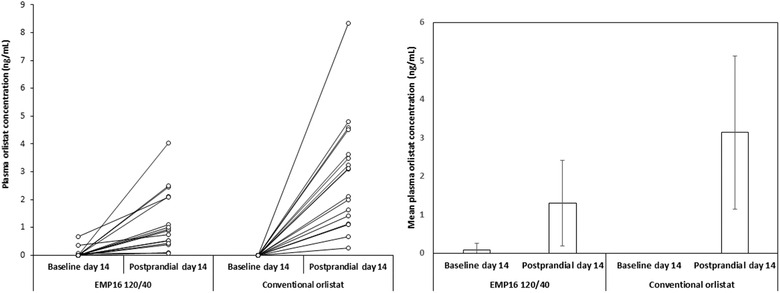
Baseline (before breakfast and first dose of the day) and postprandial (highest concentration after lunch and second dose) individual (left) and mean ± standard deviation (right) orlistat plasma concentrations at day 14 for test product modified‐release oral capsule with orlistat and acarbose (MR‐OA; “EMP16 120/40”) and reference product with orlistat 120 mg (“Conventional orlistat”).
Discussion
The plasma pharmacokinetics of orlistat in the MR‐OA treatment groups were comparable to treatment with conventional orlistat in this phase IIa study. The mean plasma concentration of orlistat was <5 ng/mL in subjects receiving MR‐OA and the conventional orlistat, which is similar to other studies. 13 , 14 , 15 The slightly higher plasma concentrations observed in the conventional orlistat is explained by the difference in dissolution. The conventional orlistat is dissolved already in the stomach and therefore has a longer retention time for the slow dissolution process. The plasma concentration profiles of orlistat clearly showed that the absorption and systemic exposure of orlistat was higher after the lunch meal than after breakfast. Food often increases the absorption of a drug compound with low solubility due to increased luminal drug solubilization, improved wetting of solid‐state particles, and longer gastric retention time for dissolution (due to a reduced gastric emptying rate). 16 In this case, the fat content of lunch was ≈30% higher than breakfast, which may partly explain the somewhat higher plasma concentration of orlistat following lunch. Another possible explanation for the plasma concentration profile pattern observed during the day might be diurnal factors, 7 as the postprandial metabolism of fat varies during the day. 17
The pharmacokinetic time‐concentration profile indicates that the MR properties of MR‐OA behave according to the theoretical calculations and projections. The tmax was similar for all the tested formulations (Table 1), but this could be due to the limited sampling. We observed a delay in the rise of orlistat plasma concentration compared with the conventional orlistat product (Figure 1), most probably due to the delayed and slower release of APIs from the MR test product. The low morning predose concentration at Day 14 (Figures 1 and 2) indicates that in both the MR‐OA groups as well as the conventional group, orlistat reaches its peak concentrations after the dinner dose and is cleared from the body during the night, to be back at <5 ng/mL (not measurable) at breakfast the following day. This is in accordance with the 120‐mg orlistat clearance of ≈8 hours described in the literature. 15 This shows that there was no blood plasma accumulation of orlistat from the MR‐OA formulation.
For acarbose, a low fraction of absorption can be expected due to a low intestinal permeability. 16 Acarbose plasma concentrations are difficult to measure due to a lack of ionized functional chemical groups within the molecule; therefore, bioequivalence in glucose‐lowering capability is the usual way of determining an acarbose effect. 18 , 19
In the previous study, 5 we showed an expected effect on plasma glucose concentration, which was similar to that reported with conventional oral product of acarbose, 20 a sign that the MR‐OA formulation does not affect acarbose absorption.
Finally, no signs were found that the well‐known safety of orlistat and acarbose were affected. No SAEs were reported, and the AEs were mild. The frequency of pharyngitis was similar in the groups and, in our opinion, not related to the study medications, as has been shown previously (unpublished data available on the product label).
Conclusions
The pharmacokinetic profile of orlistat indicates that the MR‐OA formulations produce a delayed and slow release of the APIs in vivo. Since the plasma concentration of orlistat was low after 2 weeks of treatment (in the same range as the approved conventional orlistat product), this novel MR dosage form with orlistat and acarbose can be regarded as pharmacokinetically safe. We are now investigating higher dose strengths of MR‐OA for an extended period of time in a larger and more clinically diverse population to determine optimal dose strength of the drug product.
Conflicts of Interest
The authors declare the following competing financial interests: S.G., A.F., A.S., G.A., and U.H. have equity interests in Empros Pharma AB and have acted as employees or consultants for the company.
Funding
This study was funded by Empros Pharma AB.
Supporting information
Supporting Information
References
- 1. Maula A, Kai J, Woolley AK, et al. Educational weight loss interventions in obese and overweight adults with type 2 diabetes: a systematic review and meta‐analysis of randomized controlled trials. Diabet Med. 2020;37:623‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Opio J, Croker E, Odongo GS, Attia J, Wynne K, McEvoy M. Metabolically healthy overweight/obesity are associated with increased risk of cardiovascular disease in adults, even in the absence of metabolic risk factors: a systematic review and meta‐analysis of prospective cohort studies. Obes Rev Off J Int Assoc Study Obes. 2020;21:e13127. [DOI] [PubMed] [Google Scholar]
- 3. Lin C‐J, Chang Y‐C, Cheng T‐Y, Lo K, Liu S‐J, Yeh TL. The association between metabolically healthy obesity and risk of cancer: a systematic review and meta‐analysis of prospective cohort studies. Obes Rev Off J Int Assoc Study Obes. 2020;21:e13049. [DOI] [PubMed] [Google Scholar]
- 4. Krentz AJ, Fujioka K, Hompesch M. Evolution of pharmacological obesity treatments: focus on adverse side‐effect profiles. Diabetes Obes Metab. 2016;18:558‐570. [DOI] [PubMed] [Google Scholar]
- 5. Holmbäck U, Forslund A, Grudén S, et al. Effects of a novel combination of orlistat and acarbose on tolerability, appetite, and glucose metabolism in persons with obesity. Obes Sci Pract. 2020;6:313‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rhie JK, Hayashi Y, Welage LS, et al. Drug marker absorption in relation to pellet size, gastric motility and viscous meals in humans. Pharm Res. 1998;15:233‐238. [DOI] [PubMed] [Google Scholar]
- 7. Nyholm D, Lennernäs H, Johansson A, Estrada M, Aquilonius S‐M. Circadian rhythmicity in levodopa pharmacokinetics in patients with Parkinson disease. Clin Neuropharmacol. 2010;33:181‐185. [DOI] [PubMed] [Google Scholar]
- 8. Ahr HJ, Boberg M, Krause HP, et al. Pharmacokinetics of acarbose. Part I: absorption, concentration in plasma, metabolism and excretion after single administration of [14C]acarbose to rats, dogs and man. Arzneimittelforschung. 1989;39:1254‐1260. [PubMed] [Google Scholar]
- 9. Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413‐420. [DOI] [PubMed] [Google Scholar]
- 10. Steinert RE, Feinle‐Bisset C, Asarian L, Horowitz M, Beglinger C, Geary N. Ghrelin, CCK, GLP‐1, and PYY(3‐36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol Rev. 2017;97:411‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6(2):65‐70. [Google Scholar]
- 12. Fox J, Bouchet‐Valat M. Rcmdr: R Commander. R package version 2.4‐4. 2018.
- 13. Henness S, Perry CM. A review of its use in the management of obesity. Drugs. 2006;66:1625‐1656. [DOI] [PubMed] [Google Scholar]
- 14. McClendon KS, Riche DM, Uwaifo GI. Orlistat: current status in clinical therapeutics. Expert Opin Drug Saf. 2009;8:727‐744. [DOI] [PubMed] [Google Scholar]
- 15. Zhi J, Melia AT, Eggers H, Joly R, Patel IH. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol. 1995;35:1103‐1108. [DOI] [PubMed] [Google Scholar]
- 16. Welling PG. Influence of food and diet on gastrointestinal drug absorption: a review. J Pharmacokinet Biopharm. 1977;5:291‐334. [DOI] [PubMed] [Google Scholar]
- 17. Holmbäck U, Forslund A, Forslund J, et al. Metabolic responses to nocturnal eating in men are affected by sources of dietary energy. J Nutr. 2002;132:1892‐1899. [DOI] [PubMed] [Google Scholar]
- 18. Laube H. Acarbose. Clin Drug Investig. 2002;22:141‐156. [Google Scholar]
- 19. Zhang M, Yang J, Tao L, Li L, Ma P, Fawcett JP. Acarbose bioequivalence: exploration of new pharmacodynamic parameters. AAPS J. 2012;14:345‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang W, Kim D, Philip E, et al. A multinational, observational study to investigate the efficacy, safety and tolerability of acarbose as add‐on or monotherapy in a range of patients: the Gluco VIP study. Clin Drug Investig. 2013;33:263‐274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information