

Abstract
Hypophosphatasia is a rare metabolic disease resulting from variant(s) in the gene‐encoding tissue‐nonspecific isozyme of alkaline phosphatase. In this 13‐week, phase 2a, multicenter, randomized, open‐label, dose‐response study (ClinicalTrials.gov: NCT02797821), the pharmacokinetics of asfotase alfa, an enzyme replacement therapy approved for the treatment of hypophosphatasia, was assessed in adult patients with pediatric‐onset hypophosphatasia. In total, 27 adults were randomly assigned 1:1:1 to a single subcutaneous dose of asfotase alfa (0.5, 2.0, or 3.0 mg/kg) during week 1. From week 3 to week 9, patients received 0.5, 2.0, or 3.0 mg/kg subcutaneously 3 times per week (equivalent to 1.5, 6.0, or 9.0 mg/kg/wk, respectively). Noncompartmental analysis revealed exposure (maximum concentration in the dosing interval and area under the concentration‐time curve from time 0 to infinity) to asfotase alfa increased between single‐ and multiple‐dose administration and with increasing doses; however, extensive interindividual variability was observed in the concentration‐time profiles within each dose cohort. Median terminal elimination half‐life was ≈5 days following multiple‐dose administration, with steady state achieved by approximately day 29. Dose‐normalized exposure data indicated that asfotase alfa activity was approximately dose‐proportional within the studied dose range. Additionally, dose‐normalized exposure was comparable across body mass index categories of <25, ≥25 to <30, and ≥30 kg/m2, indicating that asfotase alfa dosing bioavailability was consistent in these patients, including those who were obese. These data, together with previously published pharmacodynamic results in this study population, support the use of asfotase alfa at the recommended dose of 6 mg/kg/wk in adults with pediatric‐onset hypophosphatasia.
Keywords: clinical trials, diseases and disorders of/related to bone—other, disorders of calcium/phosphate metabolism—other, therapeutics—other
Hypophosphatasia is a rare, inherited, metabolic disease caused by loss‐of‐function variants in the ALPL gene, which encodes the tissue‐nonspecific isoenzyme of alkaline phosphatase (TNSALP). 1 , 2 The resulting deficiency of TNSALP activity in patients with hypophosphatasia leads to extracellular accumulation of the TNSALP substrates inorganic pyrophosphate (PPi) and pyridoxal 5′‐phosphate (PLP). 3 , 4 Elevated levels of PPi, a potent inhibitor of bone mineralization, blocks hydroxyapatite crystal formation within the skeletal matrix and can lead to skeletal hypomineralization and rickets in infants and children and osteomalacia in adults. 1 , 5 Accumulation of PLP, the major circulating form of vitamin B6, may lead to seizures in infants. 6 Neurologic symptoms, including vertigo, depression, anxiety, and neuropathy, have also been documented in adult patients with hypophosphatasia. 7
The signs, symptoms, and complications of hypophosphatasia can present from in utero to adulthood and vary widely from patient to patient. 5 Adults with hypophosphatasia may have onset in childhood or in adulthood, 8 with characteristic manifestations, including recurring/poorly healing metatarsal and long bone fractures; pseudofractures; altered mobility; muscle weakness; loss of secondary teeth; and bone, joint, and muscle pain. 9 , 10 , 11 , 12 Emerging data indicate that the clinical burden of disease in adults may also include orthopedic issues and need for assistive device/home modifications. 13
Asfotase alfa (Strensiq, Alexion Pharmaceuticals, Inc., Boston, Massachusetts) is a human recombinant TNSALP enzyme‐replacement therapy approved for the treatment of hypophosphatasia. 14 , 15 In clinical studies in infants and children, treatment with asfotase alfa reduced levels of accumulated TNSALP substrates and improved bone mineralization, growth, mobility, and survival and reduced skeletal abnormalities and respiratory symptoms, with benefits sustained for up to 7 years. 16 , 17 , 18 , 19 , 20 Data in adolescents and adults 13 to 66 years of age with hypophosphatasia have shown that asfotase alfa decreases circulating concentrations of PPi and PLP and improves functional outcomes, with improvements maintained through 5 years of treatment. 21 Additionally, pharmacodynamic results from this 13‐week, open‐label study support that the recommended asfotase alfa dose of 6 mg/kg/wk is effective at reducing TNSALP substrate concentrations to within the normal range in adults with pediatric‐onset hypophosphatasia. 22 Adults receiving asfotase alfa 6 mg/kg/wk or 9 mg/kg/wk had significantly greater reductions in circulating PPi and PLP concentrations (P < .05) compared with the lower dose of 1.5 mg/kg/wk. 22
Although previously evaluated in pediatric patients with hypophosphatasia, 14 , 18 the pharmacokinetics (PK) of asfotase alfa have not been characterized in adults with hypophosphatasia. The objective of this study was to evaluate the PK of asfotase alfa in adult patients with pediatric‐onset hypophosphatasia following administration of a range of dose regimens that encompasses the dose proven to be effective in pediatric patients (ie, 6.0 mg/kg/wk).
Methods
Study Design
The conduct of the study was in compliance with the requirements of the World Medical Association Declaration of Helsinki and the International Conference on Harmonisation E6 Guideline for Good Clinical Practice. The protocol, informed consent form, and processes were approved by the institutional review board, independent ethics committee, or research ethics board at each of the 4 investigative sites: Vanderbilt University Medical Center, Nashville, Tennessee; Shriners Hospitals for Children, St. Louis, Missouri; Duke University Medical Center, Durham, North Carolina; Universitätsklinikum Würzburg, Würzburg, Germany. All patients provided informed consent before study initiation.
Details on the study design, inclusion and exclusion criteria, pharmacodynamic outcomes, and safety data have been previously published. 22 In brief, this 13‐week, phase 2a, multicenter, randomized, open‐label, dose‐response study (ClinicalTrials.gov NCT02797821) consisted of a screening period, a single‐dose treatment period (1 week), a washout period (1 week), a multiple‐dose treatment period (7 weeks), and a no‐dose follow‐up period (4 weeks). Adults ≥18 years of age with pediatric‐onset hypophosphatasia (defined as first sign[s] or symptom[s] of hypophosphatasia before 18 years of age) were randomly assigned 1:1:1 to a single dose of asfotase alfa (0.5, 2.0, or 3.0 mg/kg) administered subcutaneously during week 1. Patients did not receive asfotase alfa during week 2. Asfotase alfa was then administered from week 3 to week 9 at a dose of 0.5, 2.0, or 3.0 mg/kg subcutaneously 3 times per week (equivalent to 1.5, 6.0, or 9.0 mg/kg/wk, respectively). The maximum volume of medicinal product per injection was not to exceed 1.0 mL. If >1.0 mL was required, multiple injections were administered at the same time. Throughout the study, injection sites were rotated among 8 different body areas (ie, both thighs and buttocks, both sides of the abdomen, and both upper arms) and were carefully monitored for signs of any potential reactions.
Serum asfotase alfa concentrations were collected for 2 weeks following the initial dose on day 1. During weeks 3 through 8, PK samples were obtained before dosing and 6 hours after dosing on the Monday (ie, first dosing day) of each week (no samples were obtained during week 6). During week 9, the last week of dosing, a predose sample was obtained on day 57, followed by a predose trough sample on day 61, and then multiple samples after the last dose on day 61.
Bioanalytical Methods
Sample Handling
Blood samples were collected in 2.5‐mL red‐top serum separation tubes, allowed to clot for 30 minutes, and centrifuged at 3000 × g for 15 minutes, and the collected serum supernatant was immediately frozen and stored at –20°C (or colder). Serum samples were stored and analyzed within validated stability parameters (see Supplemental Appendix).
Serum Asfotase Alfa Measurement
Serum asfotase alfa concentrations were determined using a validated enzyme activity assay based on the steady‐state kinetics of asfotase alfa. A custom assay was validated in compliance with the US Food and Drug Administration's Good Laboratory Practice regulations at Charles River Laboratories (Skokie, Illinois), according to the recommendations of the Food and Drug Administration (Draft Guidance, Bioanalytical Method Validation, 2013) and European Medicines Agency (Guideline on Bioanalytical Method Validation, 2011) that were current during that time. Validation details are provided in the Supplemental Appendix. Asfotase alfa is an enzyme replacement therapy containing an alkaline phosphatase domain that is catalytically active and hydrolyzes phosphate esters. Para‐nitrophenyl phosphate (pNPP), an alkaline phosphatase substrate, was used to monitor the catalytic activity of asfotase alfa in the assay. Standard curves of asfotase alfa were prepared from 15 to 100 ng/mL in assay buffer containing 20 mM Bis‐Tris propane, 50 mM NaCl, 0.5 mM MgCl2, and 50 μM ZnCl2 with 1 g/mL bovine serum albumin at pH 9. Quality control samples (750, 450, and 300 ng/mL in serum) and study samples were handled on wet ice and diluted 10‐fold in assay buffer (or greater dilution for samples above the calibration range). The quantitation range of the assay, factoring in the minimum required dilution, was 150 to 1000 ng/mL. Standards, quality control, and study samples (20 μL) were added to a 96‐well nonbinding analytical plate, and pNPP (180 μL, 1 mM final concentration) was added to initiate the reaction. Hydrolysis of pNPP was monitored via absorbance at 405 nM (37°C for 20 minutes) using a SpectraMax Plus 384 or M5 Plate Reader (Molecular Devices, San Jose, California). The initial rates of calibrators were determined from the progress curves and fit to a linear model to generate a calibration curve using SOFTmax PRO GxP (Molecular Devices). Quality control and study samples were then interpolated from the calibration curve. Results in nanograms per milliliter were converted to units per liter based on the specific activity of the reference standard as follows:
| (1) |
Pharmacokinetic Assessments
Key assessments after both single and multiple doses of asfotase alfa included maximum concentration in the dosing interval (Cmax), time to maximum concentration in the dosing interval (tmax), terminal elimination rate constant, terminal elimination half‐life (t½), and area under the concentration‐time curve (AUC) from time 0 to 3, 7, and 14 days after a single dose and after the last dosing of the week on day 61. AUC from time 0 to infinity (AUCinf), AUC from time 0 to time of last observed concentration in dosing interval (AUClast), clearance, and volume of distribution were also evaluated following single‐dose administration. To approximate asfotase alfa accumulation with 3‐times‐per‐week dosing, the ratio of AUCinf and ratio of concentration 48 hours after first dose to the predose concentration on study day 61 after multiple dosing (CtroughR) were determined. Attainment of steady state was assessed by visual inspection of trough asfotase alfa concentrations over time.
Statistical Analysis
Analyses were performed using the PK analysis set, which included all treated patients for whom a PK profile was adequately characterized with complete concentration‐time profiles. PK parameter values were estimated using the noncompartmental analysis module in Phoenix WinNonlin v7.0 (Certara, Princeton, New Jersey). The data set for noncompartmental analysis was derived from the NONMEM population PK‐pharmacodynamic data set. Concentrations that were below the assay lower limit of quantification (LLOQ) and were associated with time points before the first quantifiable concentration in a profile were replaced with 0 for the noncompartmental analysis. Data are summarized using descriptive statistics (mean, standard deviation [SD], median, minimum, maximum, and count [n]). AUC parameters were calculated using the linear‐log trapezoidal method.
Results
Patients
All 27 patients who were randomized into the study were included in the PK analysis set. The number of patients in the 0.5, 2.0, or 3.0 mg/kg cohorts were 8, 10, and 9, respectively. Key patient demographics are summarized in Table 1. The median (min‐max) patient age was 45 (18‐77) years, and the majority were women (59%) and White (96%). In total, there were 942 (83%) quantifiable serum asfotase alfa concentrations included in the analysis; 198 (17%) were below the assay LLOQ.
Table 1.
Demographic and Baseline Disease Characteristics
| Asfotase Alfa 0.5‐mg/kg Cohort (n = 8) | Asfotase Alfa 2.0‐mg/kg Cohort (n = 10) | Asfotase Alfa 3.0‐mg/kg Cohort (n = 9) | Overall (N = 27) | |
|---|---|---|---|---|
| Age at enrollment, y, median (range) | 44.5 (18‐64) | 42.5 (24‐77) | 55.0 (24‐69) | 45.0 (18‐77) |
| Sex, n (%) | ||||
| Female | 5 (63) | 6 (60) | 5 (56) | 16 (59) |
| Male | 3 (38) | 4 (40) | 4 (44) | 11 (41) |
| Race, n (%) | ||||
| White | 7 (88) | 10 (100) | 9 (100) | 26 (96) |
| Multiple | 1 (13) | 0 | 0 | 1 (4) |
| Height, cm, median (min‐max) | 160.2 (135‐180) | 163.1 (148‐174) | 161.0 (150‐175) | 162.0 (135‐180) |
| Weight, kg, median (min‐max) | 91.0 (48‐121) | 72.7 (51‐89) | 78.0 (62‐116) | 80.5 (48‐121) |
| BMI, kg/m2, median (min‐max) | 32.6 (22‐52) | 27.4 (20‐33) | 31.3 (24‐40) | 30.3 (20‐52) |
BMI, body mass index.
All 27 patients enrolled in the study used concomitant medications during the course of study drug treatment. The most commonly reported medications were nonsteroidal anti‐inflammatory and antirheumatic drugs (7/8 [87.5%] patients in the 0.5‐mg/kg cohort, 8/10 [80.0%] patients in the 2.0‐mg/kg cohort, and 7/9 [77.8%] patients in the 3.0‐mg/kg cohort); vitamins A and D, individually and in combination (4/8 [50.0%] patients in the 0.5‐mg/kg cohort, 4/10 [40.0%] patients in the 2.0‐mg/kg cohort, and 5/9 [55.6%] patients in the 3.0‐mg/kg cohort); and other analgesics and antipyretics (4/8 [50.0%] patients in the 0.5‐mg/kg cohort, 4/10 [40.0%] patients in the 2.0‐mg/kg cohort, and 3/9 [33.3%] patients in the 3.0‐mg/kg cohort).
No patients in the 0.5‐mg/kg cohort had >1 injection per dose. All patients in the 2.0‐mg/kg cohort had 2 injections per dose, except for 1 patient who received some doses as 1 injection and 1 patient who received some doses as 3 injections. All patients in the 3.0‐mg/kg cohort had 2 to 4 injections per dose, except 1 patient who received all doses as 5 injections. Overall, compliance, defined as (total number of injections received through week 9 / number of injections expected) × 100, was high in all 3 dose cohorts, ranging from a mean of 97.7% to 99.5%.
Summary of Noncompartmental PK Parameters
The estimated PK parameters are summarized in Table 2. Overall, exposure (Cmax, AUClast, and AUCinf) increased with increasing dose and between single‐ and multiple‐dose administrations; however, a large portion of concentrations in the 0.5‐mg/kg cohort were below the LLOQ following day 1 dosing, and as such, several parameters were not evaluable in this cohort following the single‐dose administration. On day 61, for all 3 dose cohorts, λz (and thus t½) was calculated (Table 2). Individual asfotase alfa concentration‐time profiles provided sufficient data points for defining the terminal elimination phase and thus the estimation of the terminal elimination rate constant and half‐life (Figure 1). However, AUClast, AUCinf, apparent clearance (CL/F), and apparent volume of distribution (Vz/F) on day 61 were not evaluated because of the dosing regimen (interval) being given in the study. Because asfotase alfa was given in once‐every‐other‐day dosing (for 3 times every week), in the last week of the study (week 9), doses were typically given on Monday (day 57 dosing), Wednesday (day 59 dosing), and Friday (day 61 dosing). Due to nonsymmetric dosing interval during chronic dosing (ie, every other day from Monday to Friday and a 3‐day gap between Friday and the following Monday), we have chosen not to compare day 61 PK with day 1 PK. One comparison that we could have pursued is day 57 dose‐normalized AUC from time 0 to 168 hours vs day 1 dose‐normalized AUCinf. However, due to lack of dense PK/pharmacodynamic sample collection on day 57 and to avoid any confusion, we have left such a comparison out. Thus, it would be misleading to calculate and compare the day 61 and day 1 PK parameters, such as AUClast, AUCinf, CL/F, and Vz/F, that were impacted by the dosing interval schedule in this study on day 61. Geometric mean values for t½ ranged from 3.7 to 5.5 days. Median tmax occurred earlier following multiple‐dose (0.7‐1.2 days) vs single‐dose (1.9‐3.0 days) administration. Geometric mean values for CtroughR ranged from 3.1 to 4.7 across the 3 dose cohorts, indicating marked accumulation of asfotase alfa at all dose levels following multiple‐dose administration.
Table 2.
Summary of Noncompartmental Pharmacokinetic Parameters
| Parameter | Asfotase Alfa 0.5 mg/kg (n = 8) | Asfotase Alfa 2.0 mg/kg (n = 10) | Asfotase Alfa 3.0 mg/kg (n = 9) | |||
|---|---|---|---|---|---|---|
| Day 1 | Day 61 | Day 1 | Day 61 | Day 1 | Day 61 | |
| Cmax, U/L | 218 (24.1) | 743 (63.6) | 617 (51.8) | 1900 (53.6) | 915 (35.3) | 3600 (43.7) |
| tmax, d | 1.9 (0.5‐3.0) | 0.7 (0.0‐2.5) | 2.3 (1.5‐5.0) | 1.2 (0.0‐2.5) | 3.0 (1.5‐6.0) | 1.0 (0.3‐1.3) |
| AUClast, U × d/L | 588 (77.0) | NE | 3310 (60.6) | NE | 5620 (63.3) | NE |
| AUCinf, U × d/L | NE | NE | 5350 (45.6) | NE | 9030 (40.3) | NE |
| t½, d | NE | 5.1 (10.2) | 3.7 (16.4) | 4.6 (28.0) | 4.7 (17.9) | 5.5 (18.3) |
| λz, d−1 | NE | 0.135 (10.2) | 0.189 (16.4) | 0.150 (28.0) | 0.148 (17.9) | 0.125 (18.3) |
| CL/F, U/L | NE | NE | 18.8 (37.2) | NE | 22.1 (41.2) | NE |
| Vz/F, L | NE | NE | 99.1 (25.9) | NE | 150 (36.2) | NE |
| CtroughR | NE | 4.15 (35.3) | NE | 3.14 (65.5) | NE | 4.68 (33.6) |
λz, terminal elimination rate constant; AUCinf, area under the concentration‐time curve from time 0 to infinity; AUClast, area under the concentration‐time curve from time 0 to time of the last observed concentration in a dosing interval; CL/F, apparent clearance after subcutaneous dosing; Cmax, maximum concentration in the dosing interval; CtroughR, ratio of concentration 48 hours after first dose to predose concentration on day 61 after multiple dosing; CV%, coefficient of variation; NE, not evaluable; t½, terminal elimination half‐life; tmax, time of maximum concentration in the dosing interval; Vz/F, apparent volume of distribution after subcutaneous dosing.
Geometric mean and geometric CV% were provided for all parameters except tmax, where median and range were provided.
NE: Due to the limited available data, several parameters were not evaluable or not evaluated. For all 3 dose cohorts, CtroughR was not evaluated because it is not a relevant PK parameter after a single dose else; see the Results section for explanations.
Figure 1.
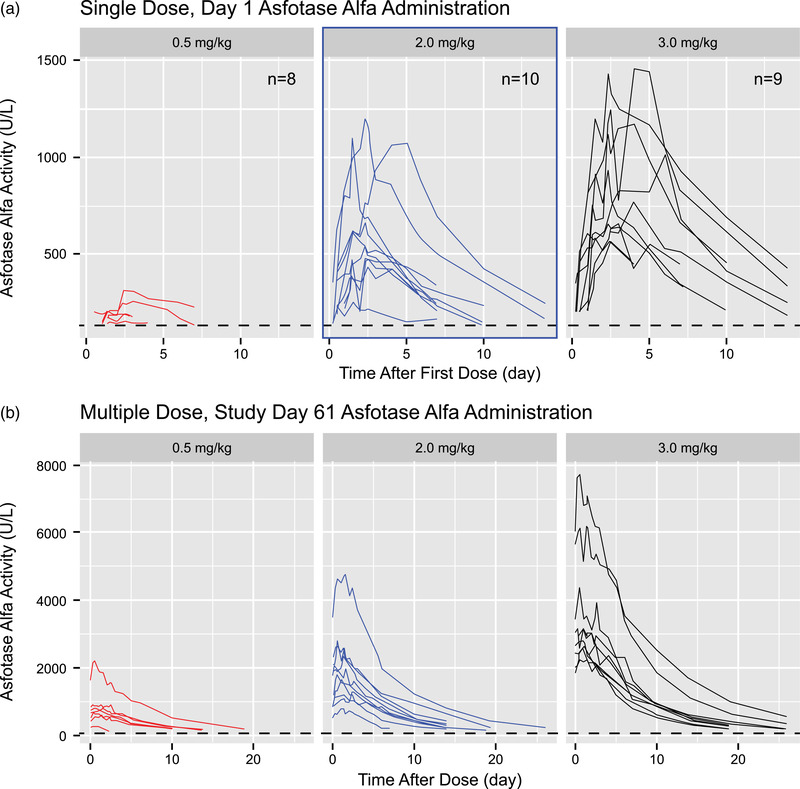
Individual pharmacokinetic profiles after (A) single‐dose, day 1, administration of asfotase alfa (semi‐log scale) and (B) multiple‐dose, day 61, administration of asfotase alfa (linear scale).
Individual PK Profiles
Individual PK profiles after single and multiple doses in the 0.5, 2.0, and 3.0 mg/kg cohorts are shown in Figure 1. These composite plots show increasing asfotase alfa exposure with increasing dose following both single‐ and multiple‐dose administration. Notably, extensive interindividual variability was observed in the concentration‐time profiles within each dose cohort, although all profiles seemed to display a mono‐exponential decline when viewed on a semilog graph.
Mean (SD) PK Profiles
Figure 2 shows mean (SD) asfotase alfa activity across the 3 dose cohorts following single‐dose (Figure 2A) and multiple‐dose (Figure 2B) administration. Similar to the individual PK profiles, plots of mean (SD) PK serum asfotase alfa concentration‐time data show increasing asfotase alfa exposure with increasing dose (Figure 2). Trough concentrations from day 15 to day 61 are shown in Figure 2C. Individual and mean trough concentrations appeared relatively stable following day 29, indicating that steady state was reached at this time point of the multiple‐dosing period. Mean trough concentrations in the 3.0 mg/kg cohort increased slightly after day 29, although marked interindividual variability was observed in this treatment group.
Figure 2.
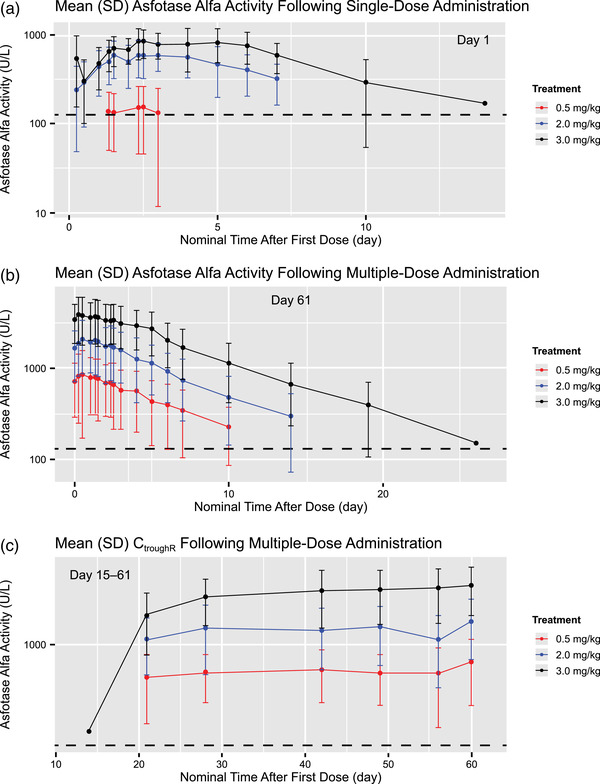
Arithmetic mean (± standard deviation [SD]) plasma concentration–time profiles after (A) single‐dose, day 1, and (B) multiple‐dose, day 61, administration of asfotase alfa and (C) trough concentrations on days 15 to 61 after administration of asfotase alfa. CtroughR, ratio of concentration 48 hours after first dose to the predose concentration on study day 61 after multiple dosing.
Dose‐Normalized PK Profiles
Dose‐normalized mean PK profiles after single‐ and multiple‐dose administration of asfotase alfa are shown in Figure 3, and show comparable asfotase alfa activity across dose cohorts on days 1 and 61. These plots indicate that asfotase alfa activity was approximately dose‐proportional within the 0.5‐ to 3.0‐mg/kg dose range.
Figure 3.
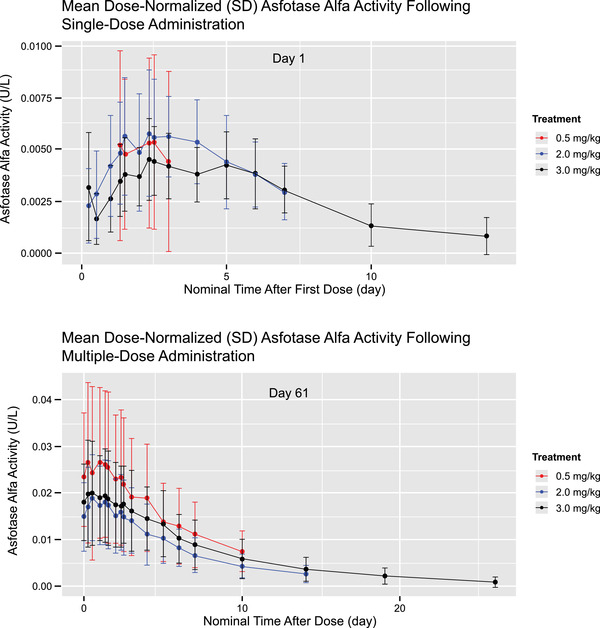
Dose‐normalized arithmetic mean (± standard deviation [SD]) plasma concentration–time profiles after (A) single‐dose, day 1, and (B) multiple‐dose, day 61, administration of asfotase alfa.
Dose Normalization by Body Mass Index
Baseline body mass index (BMI) data indicated that of the 27 patients in the study population, 8 (30%) were overweight (BMI, 25 to <30 kg/m2) and 14 (52%) were obese (BMI, ≥30 kg/m2). 23 To determine whether patient BMI impacted the PK profile of asfotase alfa under the total body weight–based dosing regimen, we conducted a post hoc analysis to evaluate dose‐normalized exposure data by patient BMI categories (<25, ≥25 to <30, and ≥30 kg/m2). As shown in Figure 4, the median dose‐normalized AUC from time 0 to 14 days was generally comparable across the 3 BMI groups following both single‐ and multiple‐dose administration of asfotase alfa.
Figure 4.
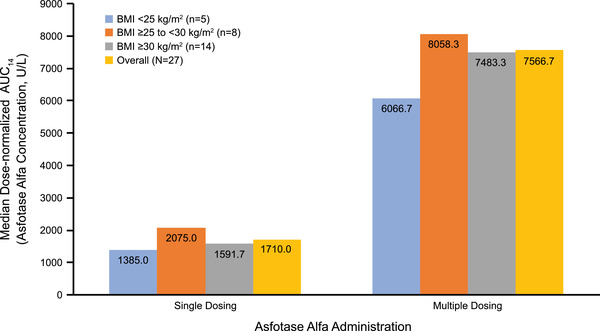
Dose‐normalized median AUC14, categorized by patient BMI categories (<25, ≥25 to <30, and ≥30 kg/m2) after single dose, day 1, and multiple dose, day 61, administration of asfotase alfa. AUC14, area under the concentration‐time curve from time 0 to 14 days; BMI, body mass index.
Discussion
Data from this phase 2 study are the first to describe the PK profile of asfotase alfa in adults with pediatric‐onset hypophosphatasia. The noncompartmental analysis results suggest asfotase alfa exposure increases over the 0.5‐ to 3.0‐mg/kg dose range after single‐ and multiple‐dose administration. Concentration‐time profiles for all cohorts suggest a relatively slow absorption and a mono‐exponential decline in the terminal phase. While there were no signs of time‐dependent elimination in this study, the rate of absorption, as assessed by tmax, appeared to be faster with multiple‐dose administration. Because of a relatively long half‐life (≈5 days) and dosing frequency (3 times per week), CtroughR values indicated marked accumulation of asfotase alfa at all dose levels.
Our results are generally consistent with previously conducted PK analyses using observed data from pediatric patients with hypophosphatasia. 14 Reported Cmax values following multiple doses of asfotase alfa (2.0 mg/kg 3 times a week) in pediatric patients (mean, 1566 U/L and 1840 U/L) 14 were comparable to those observed following multiple‐dose administration in adult patients in our study receiving the same dose (median, 1900 U/L). However, tmax following multiple‐dose administration of 2.0 mg/kg asfotase alfa in our adult population (median, 2.1 days) was longer than previously reported for pediatric patients receiving the same dose (mean, 0.6‐0.9 days). 14 Possible explanations for this difference include variabilities due to study and patient population.
In this study, multiple injections of asfotase alfa were recommended for patients requiring an injection volume of >1.0 mL for a single dose. Injection site rotation with subcutaneous administration has been recommended as a common practice for various biologics such as insulin. 24 Although the potential impact of injection site rotation on asfotase alfa PK parameters has not been evaluated, a PK study on the rotation of subcutaneous injection sites with the biologic golimumab has supported the flexibility in the choice of various injection sites, such as upper arm, abdomen, and thigh. 25 In our study, the relatively comparable variabilities among the 3 dosing cohorts (Table 2) support the lack of apparent impact of subcutaneous injection site rotation on asfotase alfa multiple‐dose PK (eg, dose‐response findings).
In general, a concern with body weight–based dosing of medications is the potential for underdosing or overdosing patients with obesity, depending on the properties of the drug and the individual patient's characteristics. 26 , 27 As asfotase alfa dosing is based on total body weight, there may be a concern in the clinical setting as to whether overweight or obese patients are receiving treatment doses that are higher than necessary. If patients with high BMI were being overdosed when receiving asfotase alfa doses based on total body weight, we would anticipate observing the highest dose‐adjusted AUC values in the highest BMI category (≥30 kg/m2) or a categorical increase in the dose‐adjusted AUC values from <25 kg/m2 BMI to 25 to <30 kg/m2 and then again to ≥30 kg/m2. However, we did not observe these systemic trends, and exposure was comparable across the 3 BMI categories. Therefore, dosing of asfotase alfa by total body weight is considered to be appropriate for patients with hypophosphatasia, and we expect that dosing by BMI would not obtain better or more harmonious PK exposures.
As previously reported, 15 patients from this study tested positive for anti–asfotase alfa antibodies after baseline (median titer, 0 [min‐max, 0‐4]), with no patients testing positive for neutralizing antibodies at baseline or at any time during the study. 22 Given the small number of patients within each dose cohort that tested positive for anti–asfotase alfa antibodies and the short treatment duration of the study, it was not possible to further evaluate the impact of antidrug antibody status on PK outcomes. However, in clinical trials of asfotase alfa with treatment durations of up to 7 years, the development of antidrug antibodies has not been reported to have a clinically relevant impact on either safety or efficacy outcomes. 16 , 17 , 19 , 21
There were several limitations to this study. First, the small sample size limited the analysis. Specifically, in the 0.5‐mg/kg cohort, a large proportion of concentrations were below the LLOQ following day 1 dosing, and as such, several parameters were not evaluable, including AUCinf, CL/F, Vz/F, and λz (and thus t½). In addition, as noted in the Results section, given the irregular dosing intervals (ie, 3 times a week dosing results in 2 intervals of 2 days and 1 interval of 3 days in a 7‐day period), AUC for a dosing interval at steady state was not determined at week 9 and could not be used to calculate a steady‐state clearance estimate or an accumulation ratio based on AUC values. Steady state was instead estimated by visual inspection of trough asfotase alfa concentrations over time. Also, extensive interindividual variability was observed in each cohort in the concentration‐time profiles, which could have been a result of individual patient disease status, allometrically scaled weight effects on CL/F and Vz/F after subcutaneous dosing, or immunogenicity. Finally, while the risk of assay interference from other medications was not assessed, the risk is low based on the mechanism of action of asfotase alfa and specificity of the catalytic domain, and thus, no concomitant medications with the potential for interference were identified in use or tested as part of this study.
Conclusion
In this open‐label study of data from adult patients with pediatric‐onset hypophosphatasia, noncompartmental analysis revealed accumulation of asfotase alfa at all 3 dose levels tested. Individual and mean plots of the serum asfotase alfa concentration‐time data showed increasing exposure with increasing doses following both single‐ and multiple‐dose administration. Asfotase alfa activity was approximately dose proportional within the studied range, and there was no evidence of an influence of BMI on asfotase alfa exposure. Taken together with the previously published pharmacodynamic results in this patient population, these PK data further support the use of asfotase alfa at the recommended approved dose of 6 mg/kg/wk in adults with pediatric‐onset hypophosphatasia.
Conflicts of Interest
W.‐J. Pan, R. Pradhan, and R. Pelto are employees of Alexion Pharmaceuticals, Inc., the study sponsor, and may own stock and stock options in the company. L. Seefried is a clinical study investigator and has received consultancy fees, institutional research funding, and grant support from Alexion Pharmaceuticals, Inc.
Funding
This study was funded by Alexion Pharmaceuticals, Inc. Pharmacokinetic data analysis for the study was performed by Jeannine Fisher of Metrum Research Group, LLC, and was funded by Alexion Pharmaceuticals, Inc. Editorial and writing support was provided by Bina J. Patel, PharmD, CMPP, of Peloton Advantage, LLC, an OPEN Health company, and was funded by Alexion Pharmaceuticals, Inc.
Data Sharing Statement
Alexion will consider requests for disclosure of clinical study participant‐level data provided that participant privacy is assured through methods like data deidentification, pseudonymization, or anonymization (as required by applicable law), and if such disclosure was included in the relevant study informed consent form or similar documentation. Qualified academic investigators may request participant‐level clinical data and supporting documents (statistical analysis plan and protocol) pertaining to Alexion‐sponsored studies. Further details regarding data availability and instructions for requesting information are available in the Alexion Clinical Trials Disclosure and Transparency Policy at http://alexion.com/research‐development; link to Data Request Form: https://alexion.com/contact‐alexion/medical‐information).
Supporting information
Supporting Information
Acknowledgments
The authors thank the patients who participated in the study and the investigators and clinical personnel for their valued contribution to obtaining the samples for this analysis. The authors also thank Jeannine Fisher of Metrum Research Group, LLC, for conducting pharmacokinetic data analysis for this study and Shanggen Zhou of Covance Inc. for conducting the biostatical analysis.
References
- 1. Conti F, Ciullini L, Pugliese G. Hypophosphatasia: clinical manifestation and burden of disease in adult patients. Clin Cases in Miner Bone Metab. 2017;14(2):230‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weiss MJ, Cole DE, Ray K, et al. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc Natl Acad Sci U S A. 1988;85(20):7666‐7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whyte MP, Mahuren JD, Vrabel LA, Coburn SP. Markedly increased circulating pyridoxal‐5'‐phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J Clin Invest. 1985;76(2):752‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Russell RG. Excretion of inorganic pyrophosphate in hypophosphatasia. Lancet. 1965;2(7410):461‐464. [DOI] [PubMed] [Google Scholar]
- 5. Rockman‐Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev. 2013;10(suppl 2):380‐388. [PubMed] [Google Scholar]
- 6. Balasubramaniam S, Bowling F, Carpenter K, et al. Perinatal hypophosphatasia presenting as neonatal epileptic encephalopathy with abnormal neurotransmitter metabolism secondary to reduced co‐factor pyridoxal‐5'‐phosphate availability. J Inherit Metab Dis. 2010;33(suppl 3):S25‐S33. [DOI] [PubMed] [Google Scholar]
- 7. Colazo JM, Hu JR, Dahir KM, Simmons JH. Neurological symptoms in hypophosphatasia. Osteoporos Int. 2019;30(2):469‐480. [DOI] [PubMed] [Google Scholar]
- 8. Högler W, Langman C, da Silva HG, et al. Diagnostic delay is common among patients with hypophosphatasia: initial findings from a longitudinal, prospective, global registry. BMC Musculoskelet Disord. 2019;20(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berkseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013;54(1):21‐27. [DOI] [PubMed] [Google Scholar]
- 10. Weber TJ, Sawyer EK, Moseley S, Odrljin T, Kishnani PS. Burden of disease in adult patients with hypophosphatasia: results from two patient‐reported surveys. Metabolism. 2016;65:1522‐1530. [DOI] [PubMed] [Google Scholar]
- 11. Barvencik F, Beil FT, Gebauer M, et al. Skeletal mineralization defects in adult hypophosphatasia—a clinical and histological analysis. Osteoporos Int. 2011;22(10):2667‐2675. [DOI] [PubMed] [Google Scholar]
- 12. Mori M, DeArmey SL, Weber TJ, Kishnani PS. Case series: odontohypophosphatasia or missed diagnosis of childhood/adult‐onset hypophosphatasia? Call for a long‐term follow‐up of premature loss of primary teeth. Bone Rep. 2016;5:228‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seefried L, Dahir K, Petryk A, et al. Burden of illness in adults with hypophosphatasia: data from the Global Hypophosphatasia Patient Registry. J Bone Miner Res. 2020;35(11):2171‐2178. [DOI] [PubMed] [Google Scholar]
- 14. Strensiq [package insert]. Boston, MA: Alexion Pharmaceuticals, Inc.; 2020. [Google Scholar]
- 15. Strensiq (asfotase alfa) [EMEA summary of product characteristics]. Levollois‐Perret, France: Alexion Europe; 2020. [Google Scholar]
- 16. Hofmann CE, Harmatz P, Vockley J, et al. Efficacy and safety of asfotase alfa in infants and young children with hypophosphatasia: a phase 2 open‐label study. J Clin Endocrinol Metab. 2019;104(7):2735‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whyte MP, Madson KL, Phillips D, et al. Asfotase alfa therapy for children with hypophosphatasia [with online only supplement]. JCI Insight. 2016;1(9):e85971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whyte MP, Greenberg CR, Salman NJ, et al. Enzyme‐replacement therapy in life‐threatening hypophosphatasia [with supplementary appendix]. N Engl J Med. 2012;1366(10):904‐913. [DOI] [PubMed] [Google Scholar]
- 19. Whyte MP, Simmons JH, Moseley S, et al. Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single‐arm, open‐label, phase 2 extension trial [with supplementary appendix]. Lancet Diabetes Endocrinol. 2019;7(2):93‐105. [DOI] [PubMed] [Google Scholar]
- 20. Whyte MP, Rockman‐Greenberg C, Ozono K, et al. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016;101(1):334‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kishnani PS, Rockman‐Greenberg C, Rauch F, et al. Five‐year efficacy and safety of asfotase alfa therapy for adults and adolescents with hypophosphatasia. Bone. 2019;121:149‐162. [DOI] [PubMed] [Google Scholar]
- 22. Seefried L, Kishnani PS, Moseley S, et al. Pharmacodynamics of asfotase alfa in adults with pediatric‐onset hypophosphatasia. Bone. 2021;142:115664. [DOI] [PubMed] [Google Scholar]
- 23. Defining adult overweight and obesity. 2020. https://www.cdc.gov/obesity/adult/defining.html. Accessed October 6, 2020.
- 24. Frid AH, Kreugel G, Grassi G, et al. New insulin delivery recommendations. Mayo Clin Proc. 2016;91(9):1231‐1255. [DOI] [PubMed] [Google Scholar]
- 25. Xu Z, Wang Q, Zhuang Y, et al. Subcutaneous bioavailability of golimumab at 3 different injection sites in healthy subjects. J Clin Pharmacol. 2010;50(3):276‐284. [DOI] [PubMed] [Google Scholar]
- 26. Erstad BL. Improving medication dosing in the obese patient. Clin Drug Investig. 2017;37(1):1‐6. [DOI] [PubMed] [Google Scholar]
- 27. Leykin Y, Miotto L, Pellis T. Pharmacokinetic considerations in the obese. Best Pract Res Clin Anaesthesiol. 2011;25(1):27‐36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information