

Abstract
Intravenous (IV) edaravone is approved as an amyotrophic lateral sclerosis (ALS) treatment. Because IV administration places a burden on patients, development of orally administered ALS treatments is needed. Therefore, 2 phase 1 studies of oral formulations of edaravone in healthy subjects examined the pharmacokinetics (PK), safety, racial differences, and drug‐drug interactions (DDIs) and investigated the dose of the oral formulation considered to be bioequivalent to the approved dose of the IV formulation. Study 1 was a placebo‐controlled, randomized, single‐blind study of single‐ascending‐dose oral edaravone with the dose range of 30 to 300 mg (n = 56). Study 2 was conducted in 2 cohorts (n = 84); the first assessed DDIs with multiple‐dose edaravone 120 mg/day given over 5 or 8 days (coadministered with single‐dose rosuvastatin, sildenafil, or furosemide), and the second evaluated PK and racial (Japanese/White) differences in PK parameters with doses of 100‐mg edaravone. The oral formulation of edaravone was well absorbed, and plasma concentrations of unchanged edaravone increased more than dose proportionally within the dose range of 30 to 300 mg. No effect of race on oral edaravone PK and no notable DDI effects possibly caused by orally administered edaravone were observed. The oral edaravone formulations were safe and tolerable under the assessed conditions. Mathematical modeling determined that equivalent exposures in plasma with the approved dose of the IV edaravone formulation, as reported previously, could be achieved when the oral edaravone formulation was administered at a dose of ≈100 mg, with an absolute bioavailability of ≈60%.
Keywords: clinical pharmacology, drug‐drug interactions, edaravone, oral formulation, phase 1 study, racial difference
Amyotrophic lateral sclerosis (ALS) is a progressive disorder characterized by degeneration of cerebral and spinal cord motor neurons; although the rate of progression of the disease varies greatly among individuals, death occurs in ≈3 to 5 years in patients who do not receive ventilator support. 1 The global incidence of ALS is estimated to be 1.7 per 100 000 person‐years of follow‐up. 2 The demographics and characteristics of patients with ALS are similar between Japanese and Western data sets, with the mean age of patients ranging from 55 to 66 years, the median time to diagnosis from 9 to 14 months, and with men more commonly affected. 3
Currently, few pharmacologic treatments are available for ALS, and many therapeutic approaches are only for symptomatic management; therefore, an ongoing unmet need exists for treatments that can improve outcomes for patients with ALS. 3 , 4 Edaravone is an antioxidant that works by preventing oxidative stress‐induced motor neuron death; it also inhibits nitration of tyrosine residues in the cerebrospinal fluid and improves motor functions in vivo. 4 The efficacy and safety of edaravone for ALS has been assessed in several clinical trials. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 In a pivotal phase 3 clinical study in patients with ALS, the primary efficacy end point of the 24‐week ALS Functional Rating Scale–Revised score was significantly improved in the edaravone vs placebo group (mean between‐group difference ± standard error, 2.5 ± 0.8; P = .001), indicating that patients treated with edaravone had significantly less functional loss compared with those who received placebo. 6
Edaravone has been approved for ALS treatment in several countries, including the United States and Japan. For ALS treatment, 60 mg of edaravone is administered intravenously over 60 minutes once per day. The first cycle consists of 14 consecutive dosing days followed by a 14‐day drug‐free period. Subsequent cycles consist of daily dosing for 10 days out of 14‐day periods, followed by 14‐day drug‐free periods.
The pharmacokinetic (PK) profile of intravenous (IV) edaravone has been described previously. 15 , 16 Edaravone undergoes hepatic and renal metabolism to inactive sulfate and glucuronide conjugates by sulfotransferase and multiple uridine diphosphate glucuronosyltransferase isoforms, and it is excreted mainly in urine as the glucuronide conjugate. Additionally, the PK of edaravone is not affected by age. Moreover, based on population PK analyses, no racial differences in the PK of IV edaravone are expected. 17 Population PK analyses also suggest nonlinearity in the clearance of IV edaravone and, within the IV dose range of 30 to 300 mg administered by infusion for 1 hour, an increase in exposures (area under the plasma concentration–time curve [AUC] and maximum plasma concentration [Cmax]) greater than the dose ratio was observed in recent clinical studies. 18 , 19 , 20
Importantly, a need exists for an oral formulation of edaravone for the treatment of ALS, as IV administration places a large burden on patients and caregivers, including frequent hospital visits and injections. However, although an oral formulation is being developed, to date, there is a paucity of data regarding its administration. The appropriate oral edaravone dose for ALS treatment is unknown because oral bioavailability has not been elucidated, although the plasma exposure of edaravone resulting in efficacy and with a sufficient safety margin has been established. No racial differences were observed with IV edaravone; however, since the processes involved in absorption of oral edaravone before reaching circulating blood are not completely revealed and can be altered by race or other factors, it is important to evaluate the PK profile of the oral formulation. In addition, although potential drug‐drug interactions (DDIs) were not an issue for IV administration of edaravone, clinical evaluation of DDIs was considered necessary for oral administration of edaravone.
This analysis reports the results of 2 phase 1 studies examining the PK, safety, tolerability, and DDIs associated with oral administration of edaravone in healthy subjects. Specifically, the objectives of these studies were to determine the PK profile of oral edaravone solution and suspension, confirm any racial differences in the PK profile and DDIs of oral edaravone, and to use these data in conjunction with previous IV edaravone dosage and PK information to investigate the oral equivalent dose to IV edaravone.
Subjects and Methods
Ethics
Before commencing the studies, the protocol and all other appropriate documents were reviewed and approved by the Institutional Review Board of Medical Corporation Heishinkai OPHAC Hospital (study 1) and P‐one Clinic, Keikokai Medical Corporation (study 2), as well as the Japanese Pharmaceuticals and Medical Devices Agency. All subjects in both studies provided written consent for participation. The studies were conducted at Medical Corporation Heishinkai OPHAC Hospital (study 1) and P‐one Clinic, Keikokai Medical Corporation (study 2). Additionally, the studies were conducted in accordance with the ethical principles of the Declaration of Helsinki; the Law on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices; Good Clinical Practice; and the protocol.
Subjects
Two phase 1 studies were conducted in healthy Japanese and White men between 20 and 45 years of age. Subjects in both studies were excluded if they had a current or previous history of cardiac, hepatic, renal, gastrointestinal, respiratory, psychiatric, nervous, hematopoietic, or endocrine diseases; a history of drug or food allergy; a history of alcohol or drug dependence; body mass index of <18.0 or >30.0 kg/m2 or body weight of <50 kg; a positive test for hepatitis B surface antigen, serologic test for syphilis, hepatitis C virus antibody, or HIV antigen/antibody; any clinically significant abnormalities in a laboratory test; had blood donation or sampling with a total volume of ≥400 mL within 12 weeks, ≥200 mL within 4 weeks, or ≥800 mL within a year before providing informed consent; blood component donation or blood sampling within 2 weeks before providing informed consent; had undergone any surgery; did not agree to use an effective method of contraception from the start to 14 days after study completion; had previously received edaravone; or had participated in another clinical study and received an investigational product within 12 weeks before the study start.
Subjects in study 2 were also excluded if they had used acetylsalicylic acid within 7 days before the study start; had alcohol or any products containing xanthine or caffeine within 24 hours before screening and visit on day −1; had nutritional supplement(s) within 7 days before the study start; had grapefruit, grapefruit juice, or any processed food(s) containing these substances within 7 days before the initiation of coadministered drug (cohort 1) or within 24 hours before each visit on day −1 (cohort 2); had tobacco or any products containing nicotine within 12 weeks before the initiation of coadministered drug (cohort 1) or within 24 hours before each visit on day −1 (cohort 2); or had food(s) containing St John's wort within 2 weeks before the initiation of coadministered drug (only in cohort 1).
Objectives
In study 1, the objectives were to evaluate the PK, safety, and tolerability of single doses of oral edaravone solution and suspension in healthy adult males; additionally, the PK profiles of edaravone suspension in different races and with different drug dissolution profiles with regard to formulations (ie, with or without xanthan gum) were assessed.
In study 2, the objectives were to evaluate drug interactions, safety, and tolerability of sildenafil, rosuvastatin, or furosemide when coadministered with oral edaravone in healthy adult males; additionally, the PK, safety, and tolerability of single and multiple doses of oral edaravone suspension, including any racial effects, were assessed.
Study Design
Study 1 was a placebo‐controlled, randomized, single‐blind study of oral edaravone. A single dose of oral edaravone was used. The initial dose of edaravone was 60 mg, and the doses for the subsequent cohorts were determined based on the results from all previous cohorts. Accordingly, edaravone doses of 30, 60, 120, 200, and 300 mg (without xanthan gum) and 300 mg (with xanthan gum) were assessed. Subjects fasted for approximately 17 hours (ie, from about 13 hours [at least 10 hours] before to 4 hours after dosing).
Study 2 was conducted in 2 cohorts; the first cohort assessed DDIs with multiple doses of oral edaravone 120 mg/day, and the second cohort evaluated PK and racial differences in PK parameters with oral administration of single‐dose 100‐mg edaravone. The dose of edaravone in cohort 2 was set as the postulated clinical oral dose that was hypothesized to yield exposure close to that of the IV formulation (ie, a clinical dose of 60 mg) based on PK results obtained in earlier studies (study 1 and a study with the IV formulation in healthy volunteers 18 ). The daily dose of edaravone in cohort 1 was set at 120 mg, which exceeds the estimated oral dose (100 mg) because the actual clinical oral dose of edaravone was not confirmed at the time of this study.
Cohort 1 of study 2 was an open‐label, add‐on study. In group 1, subjects were administered a single dose of rosuvastatin (10 mg on day 1) and sildenafil (50 mg on day 4) with multiple oral doses of edaravone (120 mg/day on days 6‐13, with coadministration of 10‐mg rosuvastatin on day 9 and 50‐mg sildenafil on day 12) after fasting for at least 10 hours. In group 2, subjects were administered a single dose of furosemide (40 mg on day 1) with multiple oral doses of edaravone (120 mg/day on days 3‐7, with coadministration of 40‐mg furosemide on day 6) after fasting for at least 10 hours (Figure S1). The choice of drugs for inclusion within the DDI studies considered the ability of edaravone to induce cytochrome P450 3A4 at high concentrations, the inhibitory effect of edaravone on breast cancer resistance protein transport activity, and the potential for inhibition of organic anion transporter 3 activity by edaravone metabolites. Therefore, sildenafil, rosuvastatin, and furosemide were chosen as typical substrates of cytochrome P450 3A, breast cancer resistance protein, and organic anion transporter 3, respectively.
Cohort 2 of study 2 was a single‐dose, open‐label study. Edaravone 100 mg was orally administered to subjects who had fasted for approximately 17 hours, from about 13 hours (at least 10 hours) before to 4 hours after dosing.
No subject received IV edaravone in either study.
Intervention
A commercially available product of edaravone for IV administration was not used in these studies. The formulation of the interventions, including the newly developed oral solution and suspension of edaravone, used in study 1 and study 2 are summarized in Table 1. In study 1, the edaravone oral solution and suspension were used, including a formulation containing xanthan gum to modify the viscosity of the suspension. In cohort 1 of study 2, the same oral suspension as in study 1, but without xanthan gum, was used, while in cohort 2 an oral suspension with various ingredients was used. In addition to edaravone, commercially available typical substrates, rosuvastatin (Crestor tablets, AstraZeneca), sildenafil (Viagra tablets, Pfizer), and furosemide (Lasix tablets, Aventis Pharma) were used in cohort 1 of study 2.
Table 1.
Composition of the Assessed Edaravone Solutions and Suspensions
| Cohort | Dose | Formulation | Race | Vehicle | |
|---|---|---|---|---|---|
| Study 1 | |||||
| 6 | 30 mg | Solution | Japanese | 50 mL of 0.1% polyvinyl alcohol solution | |
| 1 | 60 mg | Solution | Japanese | ||
| 2 | 120 mg | Suspension | Japanese | 10 mL of 0.1% polyvinyl alcohol solution | |
| 3 | 200 mg | Suspension | Japanese | ||
| 7 | 200 mg | Suspension | White | ||
| 5 | 300 mg | Suspension | Japanese | ||
| 4 | 300 mg | Suspension (with xanthan) | Japanese | 10 mL of 0.1% polyvinyl alcohol/0.5% xanthan gum solution | |
| Study 2 | |||||
| 1 | 120 mg | Suspension | Japanese | 10 mL of 0.1% polyvinyl alcohol solution | |
| 2 | 100 mg | Suspension | Japanese | Ingredient | Content |
| (with ingredients) | White | ||||
| Polyvinyl alcohol | 5 mg | ||||
| Xanthan gum | 15 mg | ||||
| Sodium bisulfite | 5 mg | ||||
| l‐cysteine hydrochloride hydrate | 2.5 mg | ||||
| Sodium hydroxide | q.s. a | ||||
| Phosphoric acid | q.s. a | ||||
| Simethicone emulsion | 2.5 mg | ||||
| d‐sorbitol | 2000 mg | ||||
| Purified water | q.s. | ||||
| Total | 5 mL |
q.s. indicates quantum sufficient.
apH adjusting for pH 4.0‐4.5 in the final formulation.
Outcomes
Assessed PK parameters included time to reach Cmax, Cmax, AUC from time 0 to 24 hours after dosing (AUC0‐24h), AUC from time 0 to infinity (AUC0‐∞), AUC from time 0 up to the last measurable concentration (AUC0‐last), and the half‐life of edaravone and its metabolites.
For PK analyses of edaravone and its metabolites, ≈4 or 5.5 mL of blood was drawn from the vein into a vacuum tube with heparin. In study 1, blood samples were taken before dosing and at 0.25, 0.5, 1.5, 2, 4, 6, 8, 12, 24, 36, and 48 hours after dosing. In cohort 1 of study 2, the time points of blood sampling were on days 6, 9, and 12 for group 1 and days 3 and 6 for group 2 (before and 0.083, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, and 12 hours after dosing); days 7, 8, 10, 11, and 13 for group 1 and days 4, 5, and 7 for group 2 (before dosing); and day 14 for group 1 and day 8 for group 2 (24 hours after dosing). In cohort 2 of study 2, blood samples were taken before dosing and at 0.083, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 24, 36, and 48 hours after dosing. For PK analyses of rosuvastatin, sildenafil, and furosemide in cohort 1 of study 2, approximately 3, 3, and 4 mL of blood, respectively, were drawn to a vacuum tube with heparin on days 1 and 9 (before and 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, and 48 hours after dosing) for rosuvastatin and days 4 and 12 (before and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 48 hours after dosing) for sildenafil in group 1, and on days 1 and 6 (before and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 48 hours after dosing) for furosemide in group 2.
For edaravone and its metabolites, the blood was transferred into a tube with a stabilizer and the tube was centrifuged at 4°C, 1500 × g for 10 minutes. The specified amount of plasma was placed into a tube with the fixed amount of internal standard, stabilizer, and buffer and stored at −70°C or lower before conducting the assay. For other drugs, plasma was isolated and stored soon after blood sampling. Plasma drug concentrations were assessed using validated liquid chromatography–mass spectrometry methodologies. The details of bioanalytical assays are shown in Table S1. Concentrations of edaravone sulfate conjugate were determined as the edaravone concentration after hydrolysis; the determined concentrations were then converted to concentrations of edaravone sulfate conjugate. The values below the lower limit of quantification were designated as 0. Assays to determine plasma concentrations of all analytes were conducted by Sumika Chemical Analysis Service, Ltd. (Osaka, Japan).
Safety end points included adverse events (AEs) and treatment‐related AEs, 12‐lead electrocardiogram, vital signs, laboratory tests, and (only in study 2) sensory tests.
Statistical Methods
The sample size in cohort 1 of study 2 was calculated so that the 90% confidence interval (CI) of the mean ratio of Cmax and AUC0‐∞ of rosuvastatin, sildenafil, or furosemide between administrations alone and with edaravone would fall within the limits of 0.80 to 1.25. This determination used the geometric coefficient of variation percentage (CV%; intra‐individual) of each parameter hypothesized on the basis of available data from the literature 21 , 22 and with a power of ≥80% in 2 one‐sided tests with a significance level of 5%, assuming estimated mean ratios of 1, as no information about PK parameters for coadministration of rosuvastatin, sildenafil, or furosemide with edaravone was available at the time. The sample size in study 1 and cohort 2 of study 2 was not based on statistical calculations but was chosen to allow a sufficient number of subjects to meet the study objectives. Study 1 was to include 56 subjects (6 in the edaravone group and 2 in the placebo group in each cohort). Study 2 was to include 32 subjects for group 1 and 34 subjects for group 2 in cohort 1 and 18 subjects (9 Japanese and 9 White) in cohort 2.
In both studies, PK analyses were conducted on all subjects who received ≥1 dose of the study drug and had evaluable data, and safety analyses were conducted on all subjects who received ≥1 dose of the study drug. The PK parameters in plasma were determined from the individual concentration vs time data by noncompartmental analysis using WinNonlin professional version 6.3 (Certara, Princeton, New Jersey). Statistical analyses for rosuvastatin, sildenafil, furosemide, and edaravone and its metabolites were performed using data from subjects whose PK data were available at ≥1 treatment period.
In study 1, dose proportionality testing on Cmax, AUC0‐last, and AUC0‐∞ of unchanged edaravone, and the sulfate and glucuronide conjugates were performed with log‐transformed doses by power model analysis. In this model, ln(Y) = α + β × ln(X) (where Y is the pharmacokinetic parameter and X is the dose) was described as a simple regression. The effect of differences in formulation was investigated by comparing Cmax, AUC0‐24h and AUC0‐∞ of unchanged edaravone and its metabolites between the presence and absence of xanthan gum. In both study 1 and study 2, the AUC0‐24h, AUC0‐∞, and Cmax of unchanged edaravone, and the sulfate and glucuronide conjugates of Japanese and White subjects were compared to assess the effect on PK by race.
In study 2, AUC0‐∞, AUC0‐last, and Cmax of rosuvastatin, sildenafil, and furosemide alone and in the presence of edaravone were analyzed for the assessments of effect on PK of rosuvastatin, sildenafil, and furosemide by edaravone. A linear mixed‐effects model was fitted to log‐transformed PK parameters. Estimates of least squares mean difference between treatments were obtained with their 2‐sided 90%CIs for the difference. These estimates and limits were then back‐transformed to obtain ratios of least squares geometric means. If the 90%CIs lay entirely within the limits of 0.80 to 1.25 for the ratio of AUC0‐∞ and AUC0‐last, as well as Cmax, this would provide evidence of no effect of edaravone on the PK of rosuvastatin, sildenafil, and furosemide.
Nonlinearity in plasma exposure, as determined from the single‐dose PK analyses, made it difficult to determine the dose of the edaravone oral suspension that is bioequivalent to 60 mg of the IV formulation. Therefore, it was necessary to determine the dose using a mathematical model. Bioequivalent doses of the edaravone oral suspension to the edaravone IV formulation were estimated with a 4‐parameter logistic model, using log‐transformed Cmax or AUC0‐∞ obtained in the oral dose range of 30 to 300 mg in study 1 and study 2 using the following nonlinear regression equation: ln(Y) = δ + (α−δ) / [1 + (X / γ)β], where Y is the PK parameter and X is the dose. These were calculated to yield Cmax or AUC0‐∞ equivalent to those after infusion of the approved 60 mg/60 min edaravone IV formulation (ie, 1195 ng/mL or 1738 ng·h/mL, respectively) obtained in a previous study of the edaravone IV formulation in healthy Japanese subjects at the therapeutic dose. 18
Results
Subject Disposition and Characteristics
All study groups were generally comparable regarding baseline demographic characteristics (Table S2). Study 1 included 56 subjects with a mean ± standard deviation (SD) age of 25.8 ± 5.0 years and weight of 65.7 ± 9.7 kg. Study 2 included 84 subjects: 32 and 34 subjects in groups 1 and 2 of cohort 1, respectively, with respective mean ± SD ages of 32.6 ± 6.2 years and 31.0 ± 7.3 years, and weights of 67.3 ± 7.5 kg and 68.1 ± 8.9 kg; 9 subjects each in the Japanese and White groups of cohort 2 with mean ± SD ages of 32.3 ± 9.1 years and 33.9 ± 8.5 years, and weights of 67.8 ± 7.2 and 73.4 ± 9.0 kg, respectively. In both studies, subject medical history, complications, allergic history, drinking status, and smoking status were consistent with the inclusion and exclusion criteria.
Single‐Dose Edaravone Pharmacokinetics
The single‐dose PK findings of unchanged edaravone in study 1 are summarized in Table 2 and Figure 1A. After oral administration in a fasted condition, edaravone was well absorbed with a time to reach Cmax of 0.3 to 0.8 hours. The Cmax and AUC of unchanged edaravone increased as the dose increased within the range of 30 to 300 mg. Using a power model, the Cmax (slope, 1.55; 95%CI, 1.36‐1.73), AUC0‐last (slope, 1.60; 95%CI, 1.48–1.73), and AUC0‐∞ (slope, 1.59; 95%CI, 1.47–1.72) of unchanged edaravone increased more than the dose ratio over the dose range of 30 to 300 mg.
Table 2.
Plasma PK Parameters of Unchanged Edaravone in Studies 1 and 2
| Study 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cohort | Dose (mg) | Formulation | Race | Statistics | tmax (h) | Cmax (ng/mL) | AUC0‐24h (ng·h/mL) | AUC0‐∞ (ng·h/mL) | t1/2 (h) |
| 6 (N = 6) | 30 | Solution | Japanese | Mean (SD) | 0.4 (0.1) | 208 (112) | 285 (113) | 284 (111) | 2.4 (0.7) |
| CV% | 36.5 | 53.8 | 39.6 | 39.0 | 30.6 | ||||
| 1 (N = 6) | 60 | Solution | Japanese | Mean (SD) | 0.3 (0.1) | 755 (356) | 816 (231) | 814 (235) | 3.2 (0.9) |
| CV% | 35.0 | 47.1 | 28.3 | 28.9 | 28.7 | ||||
| 2 (N = 6) | 120 | Suspension | Japanese | Mean (SD) | 0.4 (0.1) | 1735 (738) | 2242 (911) | 2259 (929) | 5.1 (1.5) |
| CV% | 36.5 | 42.5 | 40.6 | 41.1 | 28.4 | ||||
| 3 (N = 6) | 200 | Suspension | Japanese | Mean (SD) | 0.4 (0.1) | 4933 (1268) | 6254 (1236) | 6313 (1246) | 9.1 (2.4) |
| CV% | 31.0 | 25.7 | 19.8 | 19.7 | 26.2 | ||||
| 7 (N = 6) | 200 | Suspension | White | Mean (SD) | 0.6 (0.3) | 3692 (1529) | 4935 (1980) | 4979 (2012) | 6.5 (1.7) |
| CV% | 49.0 | 41.4 | 40.1 | 40.4 | 26.7 | ||||
| 5 (N = 6) | 300 | Suspension | Japanese | Mean (SD) | 0.8 (0.4) | 5426 (2496) | 9034 (2738) | 9121 (2755) | 9.1 (3.3) |
| CV% | 55.8 | 46.0 | 30.3 | 30.2 | 36.5 | ||||
| 4 (N = 6) | 300 | Suspension | Japanese | Mean (SD) | 0.6 (0.3) | 8805 (933) | 11319 (1053) | 11431 (1057) | 11.8 (4.1) |
| (with xanthan) | CV% | 45.0 | 10.6 | 9.3 | 9.2 | 34.8 | |||
| Study 2, Cohort 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Dose (mg) | Formulation | Race | Statistics | tmax (h) | Cmax (ng/mL) | AUC0‐24h (ng·h/mL) | AUC0‐∞ (ng·h/mL) | t1/2 (h) |
| 9 | 100 | Suspension | Japanese | Mean (SD) | 0.4 (0.3) | 1810 (850) | 1603 (414) | 1647 (433) | 9.3 (4.9) |
| (with ingredients) | CV% | 70.2 | 46.9 | 25.8 | 26.3 | 52.2 | |||
| 9 | 100 | Suspension | White | Mean (SD) | 0.4 (0.2) | 1484 (920) | 1362 (362) | 1423 (391) | 16.2 (15.8) |
| (with ingredients) | CV% | 54.7 | 62.0 | 26.6 | 27.5 | 97.3 | |||
AUC, area under the plasma concentration–time curve; AUC0‐24h, AUC from time 0 to 24 hours after dosing; AUC0‐∞, AUC from time 0 to infinity; Cmax, maximum plasma concentration; CV%, coefficient of variation percentage; PK, pharmacokinetic; SD, standard deviation; t1/2, half‐life; tmax, time to reach maximum plasma concentration.
Figure 1.
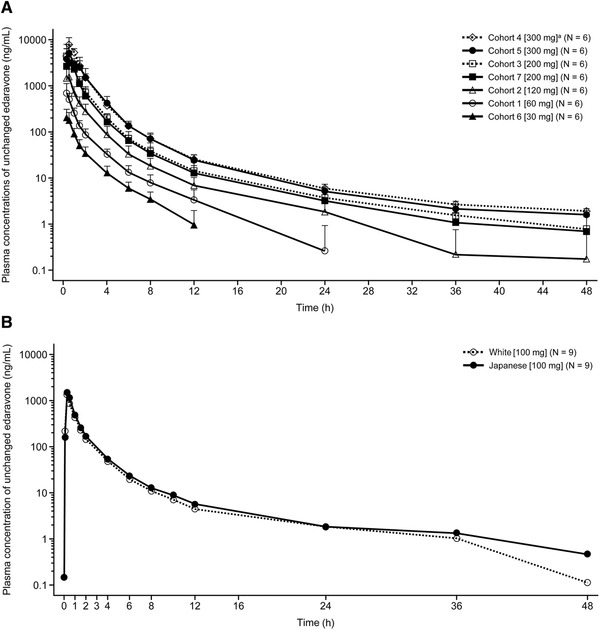
Mean plasma concentrations of unchanged edaravone over time after single‐dose administration of edaravone in study 1 (A) and cohort 2 in study 2 (B). Data are shown as mean ± standard deviation. The number of subjects in each cohort is denoted by N. In (A), Japanese subjects were included in all cohorts apart from cohort 7, which included White subjects. aWith xanthan gum.
In study 2, single‐dose administration of 100‐mg edaravone under fasted conditions resulted in generally similar PK parameters as those obtained at a comparable dose in study 1 (Table 2; Figure 1B).
Effect of Differences in Formulation on Edaravone Pharmacokinetics
The findings regarding the assessment of plasma PK parameters of unchanged edaravone after oral administration of 300‐mg edaravone suspension with xanthan gum vs without xanthan gum in a fasted condition in Japanese healthy subjects in study 1 are summarized in Table 2. The AUC0‐24h and AUC0‐∞ of unchanged edaravone for the suspension with xanthan gum were slightly greater than without xanthan gum, and the Cmax for the suspension with xanthan gum was greater than without xanthan gum.
Effect of Race on Edaravone Pharmacokinetics
The findings regarding the effect of race on unchanged edaravone PK after oral administration in studies 1 and 2 are summarized in Table 2. No remarkable differences between White and Japanese subjects under fasted conditions were observed at doses of 200 mg or 100 mg of edaravone. However, the CV% tended to be greater in White than in Japanese subjects in study 1.
Drug‐Drug Interactions with Edaravone
The DDI findings from cohort 1 of study 2 are summarized in Table 3 and Figure 2. No clinically relevant differences in plasma concentrations of rosuvastatin, sildenafil, and furosemide administered alone or coadministered with oral edaravone were observed. Notably, for each of these combinations, the 90%CI for the ratio was within the range of 0.8 to 1.25, which is the criterion for declaration of no effect on rosuvastatin, sildenafil, and furosemide PK from edaravone coadministration.
Table 3.
Pharmacokinetic Parameters and Statistical Analysis of Rosuvastatin, Sildenafil, and Furosemide Alone or Coadministered With Edaravone in Study 2
| Parameter (Unit) | Mean (SD) [CV%] | LS Mean | LS Mean Ratio (90%CI) | ||
|---|---|---|---|---|---|
| Rosuvastatin + Edaravone (N = 31 a ) | Rosuvastatin Alone (N = 32) | Rosuvastatin + Edaravone (N = 31 a ) | Rosuvastatin Alone (N = 32) | (Rosuvastatin + Edaravone / Rosuvastatin Alone) | |
| Cmax (ng/mL) | 11.3 (5.0) [44.3] | 11.3 (5.0) [43.8] | 10.4 | 10.6 | 0.98 (0.91‐1.06) |
| AUC0‐last (ng·h/mL) | 156 (58.9) [37.7] | 149 (63.7) [42.6] | 147 | 140 | 1.05 (0.99‐1.11) |
| AUC0‐∞ (ng·h/mL) | 165 (60.2) [36.5] | 161 (66.8) [41.6] | 155 | 152 | 1.02 (0.97‐1.08) |
| t1/2 (h) | 12.1 (2.2) [18.2] | 14.4 (4.0) [27.5] | – | – | – |
| Sildenafil + Edaravone (N = 31 a ) | Sildenafil Alone (N = 31 a ) | Sildenafil + Edaravone (N = 31 a ) | Sildenafil Alone (N = 31 a ) | (Sildenafil + Edaravone / Sildenafil Alone) | |
|---|---|---|---|---|---|
| Cmax (ng/mL) | 201 (97.9) [48.7] | 232 (135) [58.3] | 183 | 194 | 0.94 (0.80‐1.10) |
| AUC0‐last (ng·h/mL) | 471 (193) [41.1] | 519 (233) [44.8] | 439 | 470 | 0.93 (0.88‐1.00) |
| AUC0‐∞ (ng·h/mL) | 482 (195) [40.5] | 532 (236) [44.2] | 450 | 483 | 0.93 (0.88‐0.99) |
| t1/2 (h) | 3.5 (1.4) [40.5] | 2.8 (1.1) [37.3] | – | – | – |
| Furosemide + Edaravone (N = 34) | Furosemide Alone (N = 34) | Furosemide + Edaravone (N = 34) | Furosemide Alone (N = 34) | (Furosemide + Edaravone / Furosemide Alone) | |
|---|---|---|---|---|---|
| Cmax (ng/mL) | 1701 (506.5) [29.8] | 1676 (777.7) [46.4] | 1627 | 1503 | 1.08 (0.96‐1.23) |
| AUC0‐last (ng·h/mL) | 3883 (743.6) [19.1] | 3826 (1041) [27.2] | 3814 | 3684 | 1.04 (0.98‐1.10) |
| AUC0‐∞ (ng·h/mL) | 3906 (736.2) [18.8] | 3861 (1031) [26.7] | 3839 | 3724 | 1.03 (0.98‐1.09) |
| t1/2 (h) | 5.8 (3.8) [65.1] | 6.9 (3.5) [51.3] | – | – | – |
AUC, area under the plasma concentration–time curve; AUC0‐last, AUC from time 0 to the last measurable concentration; AUC0‐∞, AUC from time 0 to infinity; CI, confidence interval; Cmax, maximum plasma concentration; CV%, coefficient of variation percentage; LS, least squares; SD, standard deviation; t1/2, half‐life.
One subject withdrew from the study before the administration of sildenafil.
Figure 2.
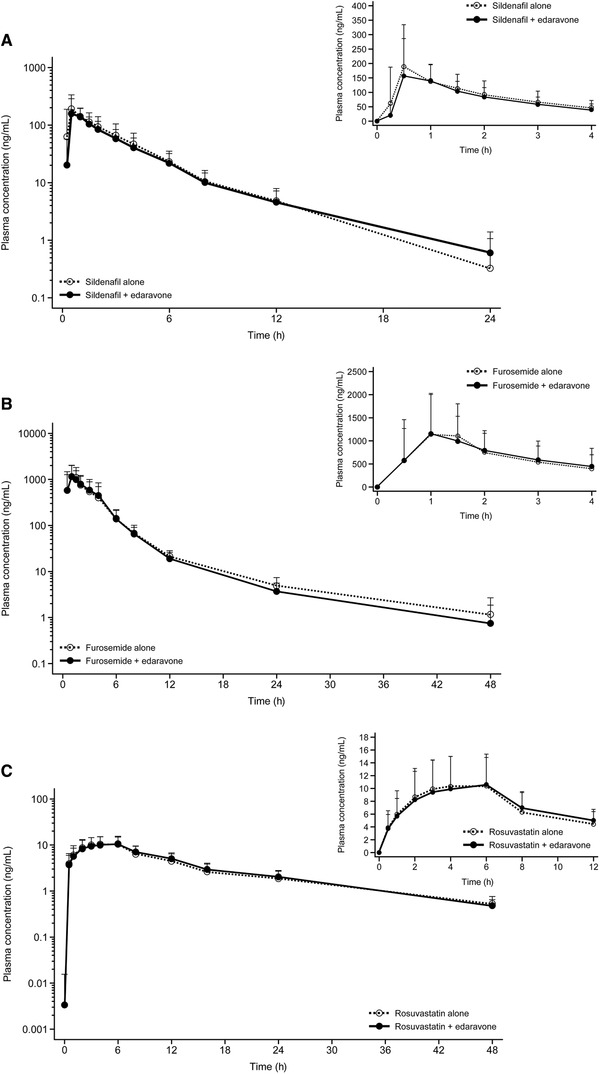
Mean plasma concentrations of sildenafil alone and in combination with edaravone (A), furosemide alone and in combination with edaravone (B), and rosuvastatin alone and in combination with edaravone (C) in study 2. Data are shown as mean ± standard deviation. Each panel includes data for 0 to 24 or 48 hours as well as 0 to 4 or 12 hours.
Multiple‐Dose Edaravone PK
The multiple‐dose PK results for unchanged edaravone in cohort 1 of study 2 are summarized in Figure 3 and Table 4. No accumulation in Cmax and AUC were observed between the first dose and the repeated administration of edaravone.
Figure 3.
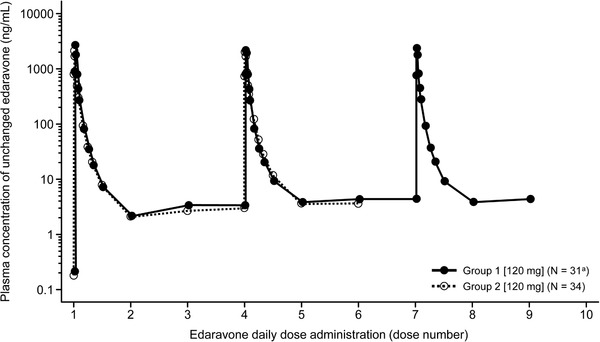
Plasma concentration profiles of unchanged edaravone over time after multiple administrations of edaravone in groups 1 and 2 of cohort 1 in study 2. In group 1, edaravone was administered once daily for 8 days from days 6 to 13 with coadministration of 10 mg of rosuvastatin on day 9 (fourth edaravone dosing) and 50 mg of sildenafil on day 12 (seventh edaravone dosing). In group 2, edaravone was administered once daily for 5 days from days 3 to 7 with coadministration of 40 mg of furosemide on day 6 (fourth edaravone dosing). aOne subject withdrew from the study before the administration of sildenafil and edaravone.
Table 4.
Plasma PK Parameters of Unchanged Edaravone After Multiple Doses in Cohort 1 of Study 2
| Group | Dose (mg) | Day (Dose Number) | Statistics | tmax (h) | Cmax (ng/mL) | AUC0‐24h (ng·h/mL) | t1/2 (h) |
|---|---|---|---|---|---|---|---|
| 1 (N = 31 a ) | 120 | Day 6 (first) b | Mean (SD) | 0.3 (0.1) | 2724 (1533) | 2599 (725) | 5.2 (1.1) |
| CV (%) | 33.6 | 56.3 | 27.9 | 21.3 | |||
| Day 9 (fourth) b | Mean (SD) | 0.3 (0.1) | 2302 (943) | 2554 (659) | 6.6 (1.2) | ||
| CV (%) | 38.5 | 41.0 | 25.8 | 17.9 | |||
| Day 12 (seventh) b | Mean (SD) | 0.3 (0.2) | 2455 (1111) | 2618 (702) | 6.7 (1.3) | ||
| CV (%) | 54.3 | 45.3 | 26.8 | 19.5 | |||
| 2 (N = 34) | 120 | Day 3 (first) c | Mean (SD) | 0.3 (0.2) | 2180 (1035) | 2522 (912) | 4.7 (0.9) |
| CV (%) | 57.2 | 47.5 | 36.2 | 19.7 | |||
| Day 6 (fourth) c | Mean (SD) | 0.3 (0.2) | 2074 (1051) | 2718 (1004) | 5.5 (0.6) | ||
| CV (%) | 51.7 | 50.7 | 36.9 | 10.9 |
AUC, area under the plasma concentration–time curve; AUC0‐24h, AUC from time 0 to 24 hours after dosing; Cmax, maximum plasma concentration; CV%, coefficient of variation percentage; PK, pharmacokinetic; SD, standard deviation; t1/2, half‐life; tmax, time to reach maximum plasma concentration.
One subject withdrew from the study before the administration of sildenafil and edaravone.
Edaravone was administered once daily for 8 days from days 6 to 13 with coadministration of 10 mg of rosuvastatin on day 9 and 50 mg of sildenafil on day 12.
Edaravone was administered once daily for 5 days from days 3 to 7 with coadministration of 40 mg of furosemide on day 6.
Edaravone Metabolite Concentrations and Pharmacokinetic Parameters Over Time
The PK parameters of edaravone metabolites after oral edaravone administration in study 1 and study 2 are summarized in Tables S3 and S4, respectively. In study 1, the Cmax and AUC of sulfate conjugate were higher than those of unchanged edaravone in all cohorts. Although the AUC of glucuronide conjugate was higher than that of unchanged edaravone in all cohorts, the Cmax was lower in cohorts receiving 200 mg or 300 mg of edaravone. Analysis in study 1 of dose proportionality of the metabolites found that both AUC0‐last and AUC0‐∞ increased dose proportionally (slope, 0.99; 95%CI, 0.88‐1.10 for the sulfate conjugate; slope, 1.08; 95%CI, 0.97‐1.19 for the glucuronide conjugate; same slope values for AUC0‐last and AUC0‐∞ for each metabolite), while the Cmax increased less than the dose ratio (slope, 0.69; 95%CI, 0.57‐0.81 for the sulfate conjugate; slope, 0.81; 95%CI, 0.71‐0.90 for the glucuronide conjugate) within the dose range of 30 to 300 mg. The Cmax of both metabolites increased dose proportionally within the dose range of 30 to 120 mg.
Additionally, the PK parameters of the metabolites were similar between Japanese and White subjects and between the formulation with and without xanthan gum. In study 2, no remarkable changes were observed in the PK parameters of sulfate and glucuronide conjugates in the oral administration of edaravone after repeated administrations.
Modeling to Determine the Bioequivalent Oral Dose to Intravenous Edaravone
Based on the regression curve obtained with the 4‐parameter logistic model, oral edaravone doses producing an AUC0‐∞ equivalent to that of the approved 60 mg/60 min edaravone IV formulation were calculated to fall in the range of 100 to 105 mg (Figure 4). The Cmax after administration of 100 to 105 mg oral edaravone was predicted to be higher than the Cmax after infusion of the 60 mg/60 min edaravone IV formulation since the oral edaravone dose producing a Cmax equivalent to that of the edaravone IV was calculated to be approximately 85 mg.
Figure 4.
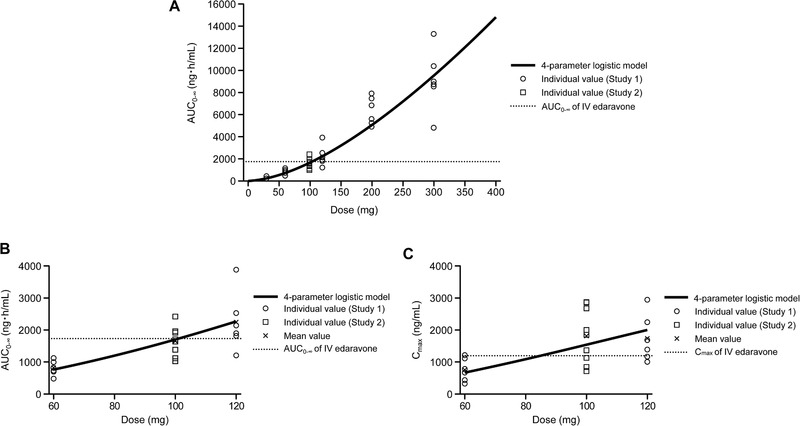
Mathematical 4‐parameter logistic modeling of area under the curve (AUC) vs edaravone dose (A, B) and maximum plasma concentration (Cmax) vs edaravone dose (C) obtained in pharmacokinetic studies in healthy subjects. (B) shows data magnified from (A). The x‐axis in (C) is also magnified. AUC0‐∞ and Cmax of IV edaravone are the values for the IV formulation of edaravone at a dose of 60 mg/60 min. (A and B) AUC0‐∞ of IV edaravone = 1738 ng·h/mL. (C) Cmax of IV edaravone = 1195 ng/mL. AUC, area under plasma concentration–time curve; AUC0‐∞, AUC from time 0 to infinity; Cmax, maximum plasma concentration; IV, intravenous.
Safety
In study 1, no serious AE was reported, and one treatment‐emergent AE led to study discontinuation (moderate conjunctivitis in a subject who received 200 mg of edaravone; not considered related to edaravone by the investigator). One treatment‐emergent AE was considered reasonably related to edaravone (mild headache in a subject who received 200 mg of edaravone and recovered without sequelae).
In study 2, no serious AE was reported and no AE led to study discontinuation. All AEs were mild in severity and resolved or were resolving without medical treatment. The one subject withdrawal from this study before edaravone administration was not related to safety issues.
Discussion
These 2 phase 1 studies determined the PK profile of oral edaravone formulations, confirmed that there were no remarkable racial differences in the PK profile and a minimal risk of DDIs with oral edaravone, and determined the oral equivalent dose to IV edaravone. Importantly, single‐dose oral edaravone up to a dose of 300 mg and multiple‐dose oral edaravone at 120 mg once daily over 8 days were well tolerated, and no safety concerns were identified.
The oral suspension of edaravone was well absorbed, and plasma concentrations of unchanged edaravone increased more than dose proportionally within the dose range of 30 to 300 mg based on the dose proportionality test. The dose proportionality analysis on Cmax between unchanged edaravone and its metabolites showed that the slope of unchanged edaravone was greater than the proportional slope (ie, 1) and those of the metabolites were lower than the proportional slope. This suggests that greater increases in plasma exposures of unchanged edaravone relative to the dose ratio were attributable to metabolic saturation of sulfate and glucuronide conjugates occurring in the first‐pass metabolism. As for the dose‐proportional increases in AUCs of the metabolites, when higher doses of edaravone were administered, which led to saturated metabolism, continuous generation of the metabolites due to maintained higher concentrations of unchanged edaravone, as suggested by the delayed time to Cmax, was considered to compensate for the less‐than‐dose‐proportional increase in Cmax. Additionally, when edaravone was orally administered once daily for up to 8 days, no significant accumulation in plasma concentration was observed for unchanged edaravone or the sulfate and glucuronide conjugates. Steady state was considered achieved within 4 repeated administrations based on the comparisons of plasma concentrations at the end of the dosing interval over the dosing durations and of PK parameters between the first dose and with repeated administration of edaravone. Observed half‐lives supported that no significant accumulation occurred and that a small number of doses are required to reach steady state.
Racial differences in the PK of unchanged edaravone and its metabolites were examined at oral edaravone doses of 200 mg and 100 mg, the latter of which is closer to the dose that provides equivalent plasma exposures to approved IV edaravone of 60 mg. 18 Notably, there were no remarkable differences in the PK profile of unchanged edaravone, or the sulfate and glucuronide conjugates between healthy Japanese and White subjects. The slight differences in PK parameters by race could be associated with body weight differences because the differences became smaller when adjusted for body weight. The relatively high CV% observed in the White population might be caused by the individual differences in body weight. However, since no marked differences were observed, it is unlikely that there are clinically meaningful alterations in PK parameters based on race.
The Cmax and AUC of unchanged edaravone in the suspension with xanthan gum, which was added to increase the viscosity of the suspension, were greater and slightly greater, respectively, than without xanthan gum. This showed that increasing viscosity by the addition of xanthan gum does not interfere with the absorption of edaravone. Notably, the addition of xanthan gum is pharmaceutically favorable because it can maintain high dispersibility via increasing viscosity in the formulation. In addition, increased viscosity of the oral formulation can also prevent accidental aspiration during swallowing and is therefore favorable for patients with ALS.
No clinically relevant differences in the plasma concentration of rosuvastatin, sildenafil, and furosemide administered alone or coadministered with edaravone were observed. Edaravone, rosuvastatin, sildenafil, and furosemide alone and rosuvastatin, sildenafil, or furosemide coadministered with edaravone were well tolerated, and there were no safety concerns identified. These findings suggest that, as with IV edaravone, there is minimal risk of DDIs caused by oral edaravone coadministered with various therapeutic agents. Although the PK of oral edaravone was found not to be affected by dosing in combination with coadministered drugs used in this study, the direct DDI effects on edaravone PK have not been evaluated when used in combination with specific inhibitors and inducers that may be encountered in clinical practice. Nonetheless, we consider that the effect on edaravone PK would not be significant with such coadministered compounds, considering the elimination route of edaravone via sulfate and glucuronide conjugation followed by renal elimination because it is reported that sulfate conjugation and glucuronide conjugation are less susceptible to drug interactions. 23 , 24 , 25
Overall, by comparing the data from our study of oral edaravone and a prior study of the IV formulation, 18 we can see that following Cmax, oral edaravone elimination was triphasic, as was observed for IV edaravone. Sulfate and glucuronide conjugates of oral edaravone were the main metabolites observed, which is consistent with the observation after IV infusion. 15 Moreover, the abundance ratio of the metabolites in plasma following oral dosing was similar to that observed after IV dosing 16 ; as a result, we can deduce that there is no new metabolic pathway specific to the absorption process, including first‐pass metabolism, with oral edaravone.
Nonlinearity observed in the relationship between dose and PK necessitated using a mathematical model to determine the dose of the edaravone oral suspension that provides equivalent plasma exposures to that of the IV formulation observed in the previous study. Accordingly, oral edaravone doses producing an AUC0‐∞ equivalent to that of the approved 60 mg/60 min edaravone IV formulation were calculated to fall in the range of 100 to 105 mg. By comparing these doses to provide postulated equivalent AUC0‐∞, the absolute bioavailability is estimated to be 57% to 60%, which is attributed to absorption and first‐pass metabolism; however, the absolute bioavailability may vary depending on the oral dosage because of nonlinear plasma exposure. Oral edaravone at these doses can be expected to provide sufficient efficacy to patients when compared with the IV formulation because the postulated plasma concentrations for oral edaravone were not less than those of the IV formulation with an equivalent AUC0‐∞ and higher Cmax. However, actual comparisons of plasma exposures between the approved 60 mg/60 min edaravone IV formulation and oral edaravone at estimated equivalent doses in additional clinical studies are necessary because of the uncertainty of a mathematical model for oral dose estimation. In addition, this estimation is based on the PK results in healthy subjects, not in patients with ALS. Although no significant alteration in the PK of edaravone caused by ALS is expected, the edaravone PK profile in patients with ALS has not yet been examined. Clinical studies in patients with ALS are also necessary to further determine the safety profile of oral edaravone, particularly because the effect of higher Cmax at the postulated oral clinical dose compared with the IV formulation on the safety profile should be investigated, and because only a small number of subjects were included in our study to establish the safety outcomes with oral edaravone at the postulated clinical dose.
In conclusion, the findings from these 2 phase 1 studies in healthy subjects showed no effect of race on oral edaravone PK or notable DDI effects of oral edaravone, and this formulation appears safe and tolerable under the assessed conditions with a safety profile that is comparable to that of IV edaravone assessed in previous clinical trials. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 These studies also show that plasma exposure of oral edaravone that is not less than the IV edaravone formulation with an equivalent AUC0‐∞ can be achieved when the oral suspension of edaravone is administered at a dose of ≈100 mg.
Conflicts of Interest
All authors are employees of Mitsubishi Tanabe Pharma Corporation, which manufactures and markets edaravone.
Funding
This study was funded by Mitsubishi Tanabe Pharma Corporation. Tricia Newell, PhD, and Sally‐Anne Mitchell, PhD, of Edanz Evidence Generation provided medical writing support, which was funded by Mitsubishi Tanabe Pharma Corporation.
Author Contributions
Hidetoshi Shimizu and Yoshinobu Nakamaru were involved in study design, data analysis, and interpretation. Yukiko Nishimura, Yoichi Shiide, and Makoto Akimoto were involved in study design, study conduct, and interpretation of the data. Hideaki Matsuda was involved in study conduct and data collection. Munetomo Matsuda was involved in study design and conduct. Yuichiro Kato was involved in data analysis. Kazuoki Kondo was involved in data interpretation and providing medical expertise. All authors were involved in writing or reviewing the manuscript draft and provided final approval for the submitted manuscript.
Supporting information
Supplementary information
Acknowledgments
The authors acknowledge the subjects participating in these studies and the investigators and staff of the study sites, including Dr Hiroshi Mikami of Medical Corporation Heishinkai OPHAC Hospital and Dr Kenichi Furihata of P‐one Clinic, Keikokai Medical Corporation.
Clinical trial registration: Study 1, NCT04481750; Study 2, NCT04481789
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
- 1. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162‐172. [DOI] [PubMed] [Google Scholar]
- 2. Marin B, Boumediene F, Logroscino G, et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta‐analysis. Int J Epidemiol. 2017;46(1):57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takei K, Tsuda K, Takahashi F, Hirai M, Palumbo J. An assessment of treatment guidelines, clinical practices, demographics, and progression of disease among patients with amyotrophic lateral sclerosis in Japan, the United States, and Europe. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):88‐97. [DOI] [PubMed] [Google Scholar]
- 4. Bhandari R, Kuhad A, Kuhad A. Edaravone: a new hope for deadly amyotrophic lateral sclerosis. Drugs Today (Barc). 2018;54(6):349‐360. [DOI] [PubMed] [Google Scholar]
- 5. Abe K, Itoyama Y, Sobue G, et al. Confirmatory double‐blind, parallel‐group, placebo‐controlled study of efficacy and safety of edaravone (MCI‐186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7‐8):610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Writing Group on Behalf of the Edaravone ALS 19 Study Group . Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2017;16(7):505‐512. [DOI] [PubMed] [Google Scholar]
- 7. Writing Group on Behalf of the Edaravone ALS 19 Study Group . Open‐label 24‐week extension study of edaravone (MCI‐186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):55‐63. [DOI] [PubMed] [Google Scholar]
- 8. Kalin A, Medina‐Paraiso E, Ishizaki K, et al. A safety analysis of edaravone (MCI‐186) during the first six cycles (24 weeks) of amyotrophic lateral sclerosis (ALS) therapy from the double‐blind period in three randomized, placebo‐controlled studies. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):71‐79. [DOI] [PubMed] [Google Scholar]
- 9. Writing Group on Behalf of the Edaravone ALS 17 Study Group . Exploratory double‐blind, parallel‐group, placebo‐controlled extension study of edaravone (MCI‐186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):20‐31. [DOI] [PubMed] [Google Scholar]
- 10. Edaravone ALS 16 Study Group . A post‐hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI‐186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):11‐19. [DOI] [PubMed] [Google Scholar]
- 11. Takei K, Tsuda K, Takahashi F, Palumbo J. Post‐hoc analysis of open‐label extension period of study MCI186‐19 in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):64‐70. [DOI] [PubMed] [Google Scholar]
- 12. Writing Group on Behalf of the Edaravone ALS 18 Study Group . Exploratory double‐blind, parallel‐group, placebo‐controlled study of edaravone (MCI‐186) in amyotrophic lateral sclerosis (Japan ALS severity classification: Grade 3, requiring assistance for eating, excretion or ambulation). Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):40‐48. [DOI] [PubMed] [Google Scholar]
- 13. Takahashi F, Takei K, Tsuda K, Palumbo J. Post‐hoc analysis of MCI186‐17, the extension study to MCI186‐16, the confirmatory double‐blind, parallel‐group, placebo‐controlled study of edaravone in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):32‐39. [DOI] [PubMed] [Google Scholar]
- 14. Takei K, Takahashi F, Liu S, Tsuda K, Palumbo J. Post‐hoc analysis of randomised, placebo‐controlled, double‐blind study (MCI186‐19) of edaravone (MCI‐186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):49‐54. [DOI] [PubMed] [Google Scholar]
- 15. Cruz MP. Edaravone (Radicava): a novel neuroprotective agent for the treatment of amyotrophic lateral sclerosis. P T. 2018;43(1):25‐28. [PMC free article] [PubMed] [Google Scholar]
- 16. Center for Drug Evaluation and Research . Radicava (edaravone) injection. Mitsubishi Tanabe Pharma Development America, Inc; Summary Review. Application no.: 209176. Approval date: May 5, 2017. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209176Orig1s000ClinPharmR.pdf. Accessed January 7, 2021.
- 17. Nakamaru Y, Kinoshita S, Kawaguchi A, Takei K, Palumbo J, Suzuki M. Pharmacokinetic profile of edaravone: a comparison between Japanese and Caucasian populations. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(supp1):80‐87. [DOI] [PubMed] [Google Scholar]
- 18. Shimizu H, Inoue S, Endo M, et al. A randomized, single‐blind, placebo‐controlled, 3‐way crossover study to evaluate the effect of therapeutic and supratherapeutic doses of edaravone on QT/QTc interval in healthy subjects. Clin Pharmacol Drug Dev. 2021;. 10(1):46‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakamaru Y, Kakubari M, Yoshida K, et al. Open‐label, single‐dose studies of the pharmacokinetics of edaravone in subjects with mild, moderate, or severe hepatic impairment compared to subjects with normal hepatic functioning. Clin Ther. 2020;42(8):1467‐1482.e4. [DOI] [PubMed] [Google Scholar]
- 20. Nakamaru Y, Kakubari M, Yoshida K, Akimoto M, Kondo K. An open‐label, single‐dose study to evaluate the pharmacokinetic variables of edaravone in subjects with mild, moderate, or no renal impairment. Clin Ther. 2020;42(9):1699‐1714. [DOI] [PubMed] [Google Scholar]
- 21. Stopfer P, Giessmann T, Hohl K, et al. Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin Pharmacol Ther. 2016;100(3):259‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menon RM, Badri PS, Wang T, et al. Drug‐drug interaction profile of the all‐oral anti‐hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. Journal of Hepatology. 2015;63(1):20‐29. [DOI] [PubMed] [Google Scholar]
- 23. Bohnert T, Patel A, Templeton I, et al. Evaluation of a new molecular entity as a victim of metabolic drug‐drug interactions ‐ an industry perspective. Drug Metab Dispos. 2016;44(8):1399‐1423. [DOI] [PubMed] [Google Scholar]
- 24. Williams JA, Hyland R, Jones BC, et al. Drug‐drug interactions for UDP‐glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201‐1208. [DOI] [PubMed] [Google Scholar]
- 25. Kiang TK, Ensom MH, Chang TK. UDP‐glucuronosyltransferases and clinical drug‐drug interactions. Pharmacol Ther. 2005;106(1):97‐132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.