

Abstract
Nowadays the issues related to the end of life of traditional plastics are very urgent due to the important pollution problems that plastics have caused. Biodegradable plastics can help to try to mitigate these problems, but even bioplastics need much attention to carefully evaluate the different options for plastic waste disposal. In this Minireview, three different end‐of‐life scenarios (composting, recycling, and upcycling) were evaluated in terms of literature review. As a result, the ability of bioplastics to be biodegraded by composting has been related to physical variables and materials characteristics. Hence, it is possible to deduce that the process is mature enough to be a good way to minimize bioplastic waste and valorize it for the production of a fertilizer. Recycling and upcycling options, which could open up many interesting new scenarios for the production of high‐value materials, are less studied. Research in this area can be strongly encouraged.
Keywords: biodegradable plastics, composting, end-of-life options, recycling, upcycling
Biodegradable Plastics: End of life of biodegradable plastics is a strategical issue, and an understanding of the different strategies of bioplastic waste disposal is crucial. Compostability and recyclability are two viable valorizations ways where value‐added products may be obtained. Specifically, recycling and upcycling of compostable polymers, not studied in depth yet, can open new valorization routes for bioplastic waste.

1. Introduction
Today plastic pollution has become one of the most pressing environmental issue and, in light of this emergency, a more sustainable development is necessary for the near future. In this context bioplastics can play a significant role. In the last two decades bioplastics have been characterized by a notable increase in market demand and have found a variety of new applications in different fields, ranging from packaging to textiles and agriculture. Since bioplastics are relatively new materials, we cannot afford to repeat the mistakes of the past and fail to accurately design their end of life. Therefore, for bioplastics it is necessary to understand more in depth all the options and strategies for the most convenient management of waste disposal.
In order to discuss this topic, it is important to initially recapitulate some basic definitions: a polymer (or a biopolymer) is a virgin material characterized by long molecular chains, while a plastic (or a bioplastic) is a polymer that has been modified or blended with molecules, additives, fillers, and others in order to mainly meet the performances requested for a specific application. Therefore, a polymer has a very simple composition with respect to a plastic: this difference is important because research often focuses on polymers and not on the complex structures of plastics. Moreover, “bio” in biopolymers and bioplastics is used to indicate one or both of the following options: (i) the polymer is derived from biomass; (ii) it is biodegradable (i. e., degradable by the action of microorganisms).
In agreement with these definitions, bioplastics can be classified in two classes depending on their biodegradability, [1] as reported in Figure 1:
Figure 1.
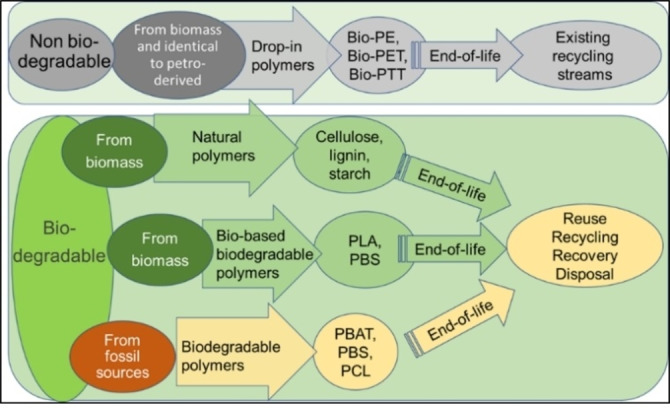
Classification of biopolymers according to their biodegradability, and some options for their end of life.
non‐biodegradable drop‐in polymers: traditional polymers, that are synthesized from fossil sources and only recently can be fully or partially prepared starting from biomass [e. g., bio‐polyethylene (PE), bio‐poly(ethylene terephthalate) (PET), bio‐poly(trimethylene terephthalate) (PTT)]
biodegradable polymers, obtained from biomass or not. They can be divided into three different groups: (i) natural polymers, such as starch, cellulose, and lignin; (ii) polymers synthesized from biomass, such as poly(lactic acid) (PLA); (iii) polymers synthesized from fossil resources, such as poly(butylene adipate‐co‐terephthalate) (PBAT) and poly(ϵ‐caprolactone) (PCL). It is notable that the biodegradable poly(butylene succinate) (PBS) can be obtained starting both from petrochemical and biomass feedstocks. The most requested and produced biodegradable polymers are PLA (18.7 % of the 2.1 million tonnes of bioplastics produced in 2020), starch‐based materials, including blends (18.7 %), PBAT and blends (13.5 %); poly(hydroxyalkanoate) (PHA) (1.7 %) is expected to strongly develop. [1]
Considering waste disposal options, drop‐in polymers do not differ from their petro‐derived counterparts. Therefore, a sustainable strategy for their end of life is recycling through the existing recycling streams, or, less sustainable, incineration or landfill.
In contrast, biodegradable polymers, regardless of their sources, have a larger spectrum of different end‐of‐life options. They are collected in Figure 2 and are characterized by a level of sustainability that tends to decrease from left to right (i. e., from reuse, recycling, recovery, to disposal) according to the waste hierarchy principles.
Figure 2.

Overview of the end‐of‐life options for biodegradable plastics.
In order to better understand the different scenarios described in Figure 2, it is necessary to make some clarifications and indicate some definitions, with specific attention to the concepts of biodegradability, compostability, and recycling/upcycling.
The concept of biodegradability, used to classify bioplastics in Figure 1, refers to a process of plastic decomposition thanks to the action of naturally occurring microorganisms, such as fungi, algae, and bacteria. It is a two‐step process that consists of a first fragmentation of the long polymer chains into oligomers and monomers, and, then, in their mineralization by microorganisms forming carbon dioxide, methane, water, and biomass. [2] This process can take place everywhere, for example, in soil, water, or physiological conditions, and is affected by a series of factors, from the chemical nature of the polymers to the availability of oxygen and light, pH temperature, humidity, microorganisms, and enzymes. The same polymer can have different rates of biodegradation under different environments, and, for these reasons, the standards for biodegradable plastics and the testing methodologies are complex and not universally identical. The most broadly recognized biodegradable standards are reported in Table 1.[ 3 , 4 ]
Table 1.
Relevant standards for biodegradable plastics.[3,4][a]
|
Standard |
Description |
Biodegradation environment |
|---|---|---|
|
EN 17033 : 2018 |
Biodegradable mulch films for use in agriculture and horticulture – requirements and test methods. |
Biodegradation in soil |
|
CEN/TR 17219 : 2018 |
Biodegradable thermoplastic mulch films for use in agriculture and horticulture. Guide for the quantification of alteration of films. |
|
|
ISO 23517 : 2021 |
Soil biodegradable materials for mulch films for use in agriculture and horticulture – requirements and test methods regarding biodegradation, ecotoxicity and control of constituents. |
|
|
ISO 14852 : 2018 |
Determination of the ultimate aerobic biodegradability of plastic materials in an aqueous medium |
Biodegradation in aqueous medium |
|
ASTM D6691‐17 |
Test method for determining aerobic biodegradation of plastic materials in the marine environment. |
|
|
ISO 23977‐1 : 2020 ISO 23977‐2 : 2020 |
Plastics: determination of the aerobic biodegradation of plastic materials exposed to seawater – part 1 and 2. |
Biodegradation in marine environment |
|
ISO 22404 : 2019 |
Plastics: determination of the aerobic biodegradation of non‐floating materials exposed to marine sediment. |
|
|
EN 13432 : 2000 |
Requirements for packaging to be considered recoverable through composting and biodegradation. |
Industrial composting |
|
EN 14995 : 2006 |
Evaluation of compostability of plastics. |
|
|
ISO17088 : 2021 |
Specifications for compostable plastics. |
|
|
AS4736 : 2006 |
Biodegradable plastics suitable for composting and other microbial treatment. |
|
|
ASTM D6400 |
Labeling of plastics designed to be aerobically composted in municipal or industrial facilities. |
|
|
AS 5810‐2010 |
Biodegradable plastics suitable for home composting. |
Home composting |
|
NF T51‐800 |
Specifications for plastics suitable for home composting. |
[a] EN standards refers to Europe, ASTM to United States, AS to Australia, NFT to France, ISO standards have a global character.
Biodegradability is properly exploitable in applications in which the material biodegrades after it has fulfilled its role in the same site where it was used. In particular, biodegradable plastics can be conveniently used as agricultural mulch films and vegetation mat and could include materials for fishing nets and gears. Indeed, mulch films should be displaced after use, but their removal is problematic as involves a loss of precious top‐soil, whereas the film embrittlement leads to material fragmentation and therefore soil contamination. Moreover, the film displacement is time‐consuming (about 16 h per hectare) and disposal expensive (up to 300 € per hectare). [5] In such context, a biodegradable film has the advantage to be released in the natural environment, preserving the soil: this is an example of a designed disposal. Currently, this option represents a small percentage with respect to the others, because biodegradable materials are not yet competitive with traditional polymers, but it is expected to increase in the future. It is noteworthy to underline that the choice of leaving a biodegradable plastic in the environment (soil or water), where it carries out its function, is sustainable and acceptable only for the applications mentioned above. Indeed, apart from the previous examples of applications, the leakage and dispersion of bioplastics in the natural environment must be avoided, despite the often widespread and incorrect idea that a biodegradable material can easily disappear into the environment without creating pollution.
In this context, to avoid misunderstandings, we briefly reiterate that photo‐ and oxo‐degradable plastics also exist and sometimes are marketed as “green” or “eco‐friendly” materials. They are typically petro‐derived materials containing additives that improve the polymer fragmentation under ultraviolet radiation (sunlight) and oxygen exposition. The degradation tends to be incomplete, leading to the accumulation of partially degraded plastics in the environment. In this case the dispersion of these materials is strongly discouraged.
A different option is the compostability, which occurs in an industrial plant in controlled conditions (Table 1). Industrial composting (at 58 °C) allows up to 10 % residual fragments of biodegradable plastics with a size of >2 mm within 3 months, in combination with at least 90 % mineralization, as measured against a positive control. Industrial aerobic composting is a process characterized by some consecutive phases:
one or more mechanical pretreatments of the waste;
a phase of few days during which the biological process starts;
a thermophilic phase of almost three weeks with temperature ranging between 55 and 60 °C;
a maturation phase with duration between 1 and 2 months with cooling down to room temperature;
a final refining with sieves of millimetric mesh to obtain acceptable compost quality.
In detail, during the thermophilic phase moistening and good aeration of the organic waste must be checked and favored by stirring and also by an aeration system that can also involve air blowing from the bottom. The thermophilic phase is characterized by intensive processes of degradation of faster biodegradable organic compounds, while the maturation phase leads to the stabilization of the organic matter through enrichment by means of humic acids.
Therefore, biodegradation and composting are based on a similar process of decomposition of the polymer by microorganisms, but the conditions at which the decomposition takes place are different, and the final products too. Indeed, biodegradation converts organic material in CO2, new microbial biomass, and mineral salts in aerobic conditions, whereas composting produces also an added‐value fertilizer (compost) for soil.
Bioplastic‐based items can be treated in composting plants, mixed with the organic waste, if their compostability has been proven and certified, according to the standards reported in Table 1. It is significant that in some applications, such as bags for food waste collections, where the separation between organic matter and plastics is not possible, compostability of the object is a significant advantage and the bioplastic has a sustainable end of life in the composting plant.
On the other hand, home composting is a garden procedure, very beneficial for environment but not controlled in terms of temperature and other parameters, and not regulated by international but only national standards (Table 1).
Finally, the recycling of biodegradable plastics is a relatively new challenge to valorize discarded materials by moving to an innovative circular lifecycle concept. More specifically, bioplastic upcycling, which consists of selectively deconstructing polymers into chemicals or molecular intermediates and converting them into high‐value products, seems a very interesting opportunity. However, the option of post‐consumer recycling needs a revision of the collection of the compostable plastics and/or the development of sorting strategies.
Regarding automatic sorting techniques, some progresses have been achieved on the separation of plastic flakes that are obtained from waste recycling streams after a preliminary elimination of metallic parts, washing, and mechanical grinding.
One of the most efficient technique is near‐infrared (NIR) sorting, which allows one to recognize the composition of the plastic flakes and to mechanically separate the recognized flake. NIR sorting has been successfully applied by NatureWorks to separate PLA from plastic waste containing PET bottles with an accuracy of 97.5 %. [6] This is necessary because the presence of PLA inside PET scraps to be mechanically recycled can give rise to chemical and physical degradation that can affect the properties of the recycled PET. [7]
The NIR sorting can be also used to separate biodegradable plastic mixtures. For example, Hollstein et al. [8] have compared the NIR spectra of biodegradable polymers, such as PLA, PCL, and starch‐based polymers, finding that they can be efficiently separated by NIR hyperspectral imaging.
Another technique that can be used to separate biodegradable plastics is triboelectric sorting, which consists of electrically charging the surface of the particles of different polymers by friction of the mixture particles moving in a tribocharger and/or by friction of the particles against tribocharger walls. The charged particles are then separated by electrostatic action. [9] The ability of the plastic materials to be electrically charged is listed in triboelectric series. Żenkiewicz et al. [10] report that PLA, PCL, and PHBV are located in the triboelectric series close to its positive end, and that the electrostatic separation of a PLA/PCL mixture provides fractions of these polymers of very high purity. On the other hand, the separation of PLA/PHBV and PCL/PHBV mixtures was less efficient, since purities of the PHBV fractions in both cases were approximately 80 %.
In light of these first considerations, this Minireview aims at focusing on the state of the art of the basic research concerning composting, recycling, and upcycling of bioplastics, in order to evidence the opportunities and limits of these different end‐of‐life strategies. Implications of environmental and energy impacts of these options will not be discussed. Biodegradable polymers, specifically PLA, PHA, PBAT, and starch‐based materials, will be the focus of this analysis.
2. Composting versus Re/Upcycling for Specific Biodegradable Polymers: State of the Art
2.1. PLA
2.1.1. Composting
The degradation of PLA in compost only occurs in favorable environmental conditions of high temperature and humidity and in the presence of appropriate microorganisms. The degradation process proceeds via a sequential mechanism, wherein in the first step the molecular weight of PLA is reduced by chemical hydrolysis in thermophilic conditions, and, then, the lactic acid oligomers are assimilated by microorganisms as an energy source.
Temperature is the first important limiting parameter, since only above PLA glass transition temperature (55 °C) the increased flexibility of the chains facilitates the accessibility to chemical and biological degradation. [11] So, PLLA does not degrade at mesophilic temperatures (25 and 37 °C), while at 60 °C its degradation degree reaches 90 % within 120 days. [12] For this reason, PLA is not suitable for home composting degradation. Then, moisture‐rich environments favor chemical hydrolysis, [13] the growth and the reproduction of microorganisms; alkaline conditions enhance hydrolytic degradation,[ 14 , 15 ] even if, for most microorganisms and enzymes, a pH‐neutral medium is optimal. The concentration of PLA in the composting mixture is another relevant parameter: chemical hydrolysis of PLA in a garden waste/PLA 70 : 30 wt% mixture lowers the pH because of the huge amount of lactic acid generated, thus reducing the degrading action of composting microorganisms. [16] Also, the addition of hydrophilic or catalytic compounds, such as thioridazine, [17] hydroxyapatite, [18] β‐tricalcium phosphate, [19] or lauric acid, [20] is known to enhance the hydrolytic degradation of PLA.
Additionally, Tsuji [21] provides a good overview of the material factors that affect degradation behavior of PLA, such as molecular weight and crystallinity. The effect of molecular weight on the degradation rate is significant at M n lower than 4×104 g mol−1 since the enhanced molecular mobility and the increased density of hydrophilic terminal groups increase the degradation rate of PLA. On the other hand, the degradation rate decreases with an increase in crystallinity (Χ c). The chains in the crystalline regions are more resistant to hydrolysis than those in the amorphous regions. The relevance of the material thickness has been introduced by Ruggero et al.: [22] a 500 μm PLA film only degraded by 3 % under thermophilic conditions [58 °C and 50–55 relative humidity (RH%)] for 20 days followed by a maturation phase (37 °C and 50–55 RH%) for 40 days. Therefore, it appears that the combination of a 20 day thermophilic phase and the 500 μm thickness of the material prevents a complete degradation within 2 months, and recirculation of macro‐residues of bioplastics is necessary.
2.1.2. Recycling
PLA cannot be mechanically recycled several times due to the worsening of physical properties and functional quality because of PLA thermal degradation. [23] In particular, top quality is crucial in the case of thin films, for which optical and barrier properties among others play an important role. [24] Żenkiewicz et al. [25] report that after 10 extrusion processes on PLA, the impact strength decreases from 2.6 to 2.08 kJ m−2 (20.2 %), tensile strength from 72.3 to 68.5 MPa (5.2 %), and tensile strength at break from 62.6 to 57.5 MPa (8.3 %), with the largest reduction occurring in the first step, although the Young's modulus remains unvaried. Solutions might include the addition of oxidative stabilizers to prevent free‐radical‐induced chain scission [26] or of chain extenders, which help partially recover the impaired molecular weight. [27]
Chemical recycling of PLA consists of obtaining chemicals from thermal or solvolytic depolymerization processes. Thermal decomposition requires high temperatures (200–400 °C) and produces a complex mixture of products.[ 28 , 29 , 30 , 31 ] Solvolytic depolymerization is preferable because it results in lactic acid production while alcoholysis allows obtaining lactate esters.[ 32 , 33 ]
PLA can be hydrolyzed with 95 % conversion to lactic acid within 2 h at 160 °C [34] or 20 min at 250 °C.[ 35 , 36 ] The reaction times decrease with temperature increase; however, excessively high temperatures (above 250 °C) are not recommended because they induce racemization and decomposition of the lactic acid formed. [35] On the contrary, temperatures lower than the melting point result in much longer times to get high yields because the crystalline regions are more difficult to hydrolytically depolymerize since they are impermeable to water diffusion. It is also important to note that the hydrolysis of PLA is autocatalytic since the carboxyl groups generated with each hydrolytic cleavage further catalyze the reaction. [37]
The sustainability of chemical recycling of PLA to obtain lactic acid has been shown to be preferable to other options, such as glucose fermentation, from an energetic point of view. It has been estimated that the energy required for producing lactic acid from corn is 55 MJ kg−1, which is by far higher than the 14 MJ kg−1 necessary for producing lactic acid from PLA hydrolysis. [38]
2.1.3. Upcycling
PLA can also be depolymerized by alcoholysis to obtain value‐added products. Various alcohols in the presence of transesterification catalyst were demonstrated to completely depolymerize PLA into lactate esters such as methyl, ethyl, and propyl lactate.[ 39 , 40 , 41 ] Lactate esters have been assessed as green solvents because of their bio‐derivation, biodegradability, and low toxicity.[ 42 , 43 , 44 ] Moreover, there is the potential to transform lactate esters into lactide, which could then be converted into PLA, resulting in a circular economy. [45]
A different approach aims at partially degrading the macromolecular structure, obtaining oligomers for specific applications. A remarkable example is the fabrication of bio‐based adhesives derived from partially depolymerized PLA. [46]
2.2. Starch‐based materials
2.2.1. Composting
Starch‐based materials were first developed in the 1970s. [47] In particular, thermoplastic starch (TPS) polymers were produced from starch through the structural modification occurring inside the starch granule when processed at temperature in the presence of specific plasticizers (e. g., water, glycerol).
Literature reports examples of good compostability of starch. For example, 100 % of corn starch at 58 °C was mineralized after 44 days of incubation under aerobic conditions. [48] Effects of concentration were reported by German et al.: [49] based on both lab and in‐situ experiments, the authors concluded that starch degradation rate decreased by up to 50 % when the starch was <10 % of the soil organic matter. This was attributed to a decrease in enzymatic activity, since the cost for microorganisms to produce extracellular enzymes would be too high compared to the metabolic energy yield when a small amount of starch is available.
Torres et al. [50] reported the degradation of 12 starch films from different crops in organic compost, with 50 % of moisture to enable aerobic conditions, and pH range 7.0–8.0. The first stage of degradation, mainly associated with the leaching of plasticizer, corresponded to a weight loss of around 30 % in 24 h. The second stage, mainly due to the biological activity and the glycosidic bonds scission, was slower, resulting in a gradual decrease of weight, until 90 % of the original weight in around 20 days. Then, the rate of degradation decreased dramatically, but in 10 days 95 % of weight loss was achieved.
Literature reports also examples of effective biodegradation of different starch‐based TPS films and foams.[ 51 , 52 , 53 ]
2.2.2. Recycling
Few examples of recycling are reported, just related to starch‐based blends. Lactic acid can be produced by fungus R. Orizae NRRL395 from Mater Bi powder. [54] Moreover, Mater Bi can be recycled into the same product. [55] In particular, the authors subjected an injection moulding grade of Mater Bi series Z (ZIO1U, 50 % of corn starch and 50 % of PCL) to multiple steps in an extruder, simulating recycling and processing operations. They concluded that only after five extrusions some decrease of rheological and mechanical properties was measured. [55]
2.3. PBAT and blends
2.3.1. Compostability
The terephthalate group in PBAT can be recalcitrant in terms of material compostability. Indeed, aromatic polyesters PET or poly(buthylene terephthalate) (PBT) were found to be resistant to hydrolysis at mild conditions and to significant attacks by microorganisms. However, the presence of aliphatic components along the aromatic chain of PBAT increases hydrolytic susceptibility and biological degradability. A content of about 30–50 mol% of terephthalate units results a good compromise to still meet the requirements for compostability,[ 56 , 57 ] correlated also to the molecular weight and especially to the crystalline content. Indeed, biodegradability is enhanced as crystallinity decreases.
The biodegradation of PBAT is structured in an initial hydrolytic degradation, followed by microbial assimilation and mineralization. During the initial hydrolysis step the action of microbial enzymes promotes the degradation of the non‐crystalline portion of aliphatic units. The degradation is significantly enhanced as the temperature increases. In the case of non‐enzymatic degradation, PBAT undergoes hydrolytic degradation due to the breakage of ester links caused by the reaction between water and the carbonyl groups, while β‐C−H hydrogen transfer reactions occur randomly along the chain. [58] Then, oligomers and monomers generated from the hydrolysis pass through the microbial cellular membranes and are assimilated to produce energy, carbon dioxide, water, and new biomass. [22]
De Hoe et al. [59] report also that light‐induced crosslinking reactions can take place along the PBAT macromolecular structures. In this case, the biodegradation rate tends to decrease with the increasing of crosslinking density beyond a certain limit threshold.
Many studies focused on the bacteria able to degrade PBAT under soil, home, and industrial composting conditions. If the thermophilic bacteria (typically belonging to actinomycetes strain) are able to rapidly cleavage the ester bonds at relatively high temperatures (50–60 °C), at mild temperatures (25–30 °C) the PBAT degradation, operated by fungal strains or mesophilic bacteria such as firmicutes and proteobacteria, is very slow.[ 60 , 61 , 62 ]
The presence of fillers can be an interesting way to accelerate the biodegradation rate of PBAT. Indeed, aerobic bacteria, which act in the degradation of fillers in soil (controlled compost according to Standard ASTM D5988‐12), are hydrophilic. Thus, when the composites are exposed to the soil, microorganisms consume the fillers, leaving the polymer matrix more porous and therefore accelerating the rate of biodegradation of the material. [57]
Concerning PBAT/PLA blends, recent studies confirm the excellent compostability of these materials, able to lose 75 wt% in 90 days under composting conditions. This rate of degradation can be further enhanced employing fillers exerting catalytic activity, such as ZnO particles. [63]
Ruggero et al. [64] reported the degradation study of a Mater Bi film [PBAT (70 wt%), starch (20 wt%), unknown additive (10 wt%)] under simulated industrial composting (20 days of thermophilic phase followed by 40 days of maturation). They found that the starch grain in Mater Bi, a natural polysaccharide, degrades first generating cavities, which enhances the degradation of the whole polymer by increasing the surface area. The PBAT component, instead, takes a longer period to be completely assimilated by microorganisms and transformed into stable products, and it is much more sensitive to process conditions like an insufficient moistening content and short thermophilic phase.[ 22 , 64 ]
2.3.2. Recycling
Thermal degradation during recycling generally occurs through hydrolysis due to moisture and high temperature. Addition of nanocarbon‐based fillers with scavenging activity or natural antioxidants, such as polyphenols, essential oils, lignin nanocrystals, or vitamins, may limit their degradation, but a more recent strategy takes into account the preparation of multilayers by film blowing of different biopolymers through coextrusion. [65] Scaffaro et al. studied the possibility of recycling PBAT‐based materials by exploiting the tendency of crosslinking under UV exposure, which usually limits their biodegradability. More precisely, they found that the addition of organoclays under UV irradiation enhances this phenomenon, leading to the formation of porous structure, which may be further reused to fabricate high‐value materials. [66]
No examples of upcycling of PBAT are reported in literature.
2.4. PHA
2.4.1. Composting
The compostability of commercially relevant PHAs, such as poly‐3‐hydroxybutyrate (PHB) and poly(hydroxybutyrate‐co‐valerate) (PHBV), is confirmed in controlled industrial compost conditions.[ 67 , 68 , 69 ] The mechanism of degradation of PHA‐based films was extensively studied by several research groups. Luo et al. [70] and Rutkowska et al. [71] demonstrated that PHB and PHBV samples are subjected to enzymatic degradation rather than hydrolytic processes occurring from the surface with the concomitant erosion of the external layers. In both of the cases the samples disintegrate in about 50 days with a degradation of 80 %.
A relationship between chemical composition of the materials and biodegradation in compost has been found. Solvent casted films of PHB and PHBV presenting a different amount of valerate content biodegrade with a rate that increases with the valerate monomer content. This effect is most likely associated with a lower crystalline character of the materials presenting higher amount of valeric acid, providing an easier target for the enzymatic action. [72]
While the addition of a hydrolytic compound such as triethylcitrate slightly enhances the hydrolytic degradation of PHB, the addition of a hydrophobic compound such as butyryltrihexyl citrate was demonstrated to decelerate the degradation of PHB. [73]
Common PHAs undoubtedly demonstrate enhanced degradation in industrial compost; however, under home composting conditions, the extent of degradation is variable and difficult to be reproduced. This behavior is the result of the variation of key parameters, such as temperature, humidity, and acidity, changing during the compost maturation. For example, Gunning et al. report a PHB degradation by 50 % in 84 days in organic waste home compost with 74–89 % humidity and a temperature of 34–66 °C, [74] while Mergaert et al. highlight the biodegradation of PHB, PHBV (10 % HV) and PHBV (20 % HV) to be 4, 6–17, and 67 %, respectively, during a period of 151 days with a temperature ranging between 5 and 30 °C. [75]
2.4.2. Recycling
Two main issues emerge when considering mechanical recycling: the scarcity of mono‐material end‐products based on PHAs and their thermomechanical stability. The first one is due to the brittleness of a material such as PHB, which very often needs to be formulated in blends with other biopolymers or additives; the second issue is associated with thermodegradative processes degrading the macromolecule under processing conditions. Rivas et al., for example, studied the effect of multiple reprocessing cycles of PHB via extrusion and compression molding on the thermomechanical properties of the material. [76] Tensile strength at break of PHB is suffering a massive reduction of more than 50 % after just three cycles with a concomitant increase of crystallinity and a decreasing molecular weight through a β‐chain scission mechanism, as described in Scheme 1.
Scheme 1.

Chain scission mechanism.
A similar set of experiments were developed by Zaverl et al. to study the effect of multiple extrusion cycles on PHBV. [77] In this case, the mechanical properties are maintained for four cycles while, after the fifth cycle, the material presents a decrease of 7.1 and 8.3 % for tensile and flexural strength, respectively. Gel permeation chromatography (GPC) analysis confirms the detrimental effect of processing on the molecular weight, starting from the third cycle and culminating with a 16.6 % decrease after the fifth cycle. In any case, the introduction of proper additives or chain extenders opens a vast scenario of promising opportunities for extending the mechanical recycling and applications of PHAs.[ 78 , 79 ]
As mechanical recycling aims at preserving the macromolecular structure of PHAs, the chemical recycling approach proposes its complete degradation. While in classical polycondensation materials the chemical recycling allows to recover monomeric structures suitable for a new cycle of polymerization, in the case of PHAs it refers to the production of carbon‐based compounds suitable for a second microbial fermentation from post‐consumer PHAs. A notable example of this approach was developed by Sato et al. [80] studying the closed‐loop recycling of poly(hydroxy decanoate) (PHD) performed by metabolically engineered Escherichia coli. Interestingly, the authors demonstrated the possibility to synthesize PHD from a mixture of 2‐alkenoic acids, which are the main constituents of the pyrolysis mixture obtained by PHD thermal treatment, thereby establishing a fully close‐loop process (Scheme 2).
Scheme 2.
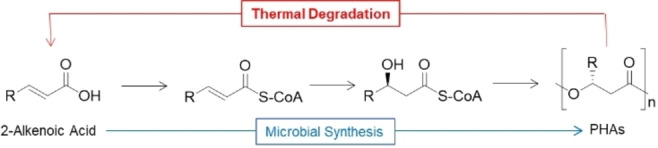
Proposed chemical recycling of PHAs via 2‐alkenoic acids synthesis.
2.4.3. Upcycling
Physical‐chemical degradative processes, such as pyrolysis, cracking, gasification, chemolysis, or enzymatic hydrolysis, can be considered valuable methods for the upcycling of PHAs into valuable products, such as chemicals or new monomers, opening new applications and markets.[ 81 , 82 , 83 ]
Ariffin et al., for example, studied the controlled pyrolysis of PHB and PHBV and their conversion into crotonic acid and 2‐pentenoic acid. [84] The thermal degradation of pure PHAs in presence of alkali earth compound catalysts has been optimized, obtaining 85 % yield of crotonic acid, 11 % yield of 2‐pentenoic acid, and only 4 % of remaining oligomers. Furthermore, crotonic acid has been successfully co‐polymerized with acrylic acid for the production of materials demonstrating potential applications as enzyme‐stabilizing agents and hydrogels for biosorbents, wastewater treatment, and agriculture.
It is worth mentioning that chemical upcycling not only derives from complete depolymerization of PHAs but may involve the controlled degradation of the polymer in order to obtain oligomers. Kaihara et al., [85] for example, performed a controlled enzymatic digestion on poly(hydroxybutyrate‐co‐hexanoate) (PHBH) and PHBV with Candida Antarctica Lipase B at 70 °C to obtain low‐M n cyclic oligomers (400 Da). Cyclic oligomers can be further re‐polymerized by the same lipase with caprolactone to fabricate PBH‐co‐PCL copolymers with a M w of 21000 Da. Finally, Yang et al. [86] managed to thermally degrade PHB by simply extruding it at 220 °C for 45 min to obtain crotonic acid‐terminated PHB oligomers (1600 Da). Such oligomers can be successfully grafted onto PLA chains by reactive extrusion, introducing an impressive ductility into the system. As a result, the introduction of 20 wt% of the oligomers produced a boosting of about 66 times in the elongation at break in comparison with pure PLA.
3. Summary and Outlook
The choice of the most convenient way for the waste management of biodegradable plastics depends on a series of factors, such as material characteristics and specific applications. For example, biodegradability in soil and/or in water is a good option for some specific applications where recovery of the material is not convenient or possible, such as for agricultural (mulch films) and sea activities. In these cases, soil and water can be preserved, in contrast to the situations in which the use of the traditional durable plastics can cause serious pollution problems, including microplastics release. However, this practice must be carefully controlled not only in terms of the impact of the amount of bioplastics accumulated and microplastic eventually dispersed in soil or water, [2] but also by adopting all actions to avoid that bioplastics are commonly dispersed in natural environments.
For other uses, the opportunity to transform wastes into value added products has to be pursued.
Composting has the advantage to convert organic waste and compostable plastics into a fertilizer for soil. With regard to the compostability of biopolymers, literature reports studies that mainly focus on the industrial process (i. e., a controlled process), where some variables, such as temperature, pH, humidity, concentration of bioplastics in the waste mixture, characteristics of the polymers (i. e., molecular weight and crystallinity), or dimensions and thickness of the plastics items, are correlated to the rate of polymer degradation. Relationships between chemical structure of the biopolymer and its compostability in industrial plants are also discussed. Therefore, the advances in the comprehension of the biodegradation processes are notable and the progresses from a technological point of view make this approach an actual opportunity with a certain degree of maturity, in spite of some problems such as pollution of non‐compostable plastics, accumulation of plastics with long degradation times, or unclear effect of additives on biodegradation rate of plastics. It is important to underline that compostable materials are the unique solutions for some applications, such as bags for food waste collections, where the separation between organic matter and plastics is not possible.
On the other hand, recycling/upcycling options show the favorable possibilities of producing materials, polymers, oligomers, and monomers available for further valorization routes and applications. Examples of the success of these approaches, such as mechanical and chemical recycling, are available for traditional plastics. Recycling and upcycling of plastics present outstanding benefits in terms of saving materials, designing multiple recycling cycles, expanding plastic lifetime, and mitigating the consumption of energy and raw materials for their synthesis as well as the impact of their extraction process.
However, for bioplastics the processes tend to have a lower degree of maturity, mainly because studies on biopolymer end of life tend to not consider the feasibility of recycling when the polymer are compostable. Moreover, bioplastics often suffer from low thermal stability that limits mechanical reprocessing. Finally, even if primary pre‐consumer recycling can be a favorable option also for bioplastics, the option of post‐consumer recycling needs a revision of the collection of the compostable plastics and/or the development of sorting strategies.
Indeed, sorting and cleaning processes are necessary, and a new collection policy must be defined. Additionally, research and strategies must be implemented because at the moment they are very limited for some materials, such as PBAT, starch‐based materials, and blends. Some problems, such as thermal degradation and loss of properties for mechanical recycling, have to be solved, and new strategical eco‐routes for the recovery of oligomers, monomers, and molecules that can be exploited to prepare new materials and products have to be studied.
As a conclusion, based only on the state of the art of the scientific research, without considering the implications of environmental and energy impact, the scenario appears in evolution and the basic research can surely have a strong influence in defining future developments.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Prof. Annamaria Celli has a Ph.D. in Materials Engineering, and she is expert in biopolymers, synthesis and characterization, mainly in polymers derived from biomass, composites, and nanocomposites. Her activities focus on the valorization of by‐products of agro‐industries and agro‐wastes and on the end of life of plastics. She is responsible for UNIBO of the H2020 projects NoAW, Agrimax, Usable Packaging and Preserve. She is WP leader in PROLiFiC and involved in the Terminus project.

Biographical Information
Dr. Grazia Totaro has a degree in Chemistry (University of Ferrara) and a Ph.D. in Materials Engineering at the School of Engineering (University of Bologna). She carried out postdoctoral research (2010–2019) and is currently junior researcher at the same school. Her research interests include the preparation and characterization of polycondensates, multifunctional nanocomposites, and composites from renewable resources. She is involved in PROLiFiC and Terminus H2020 projects.

Acknowledgements
Open Access Funding provided by Universita di Bologna within the CRUI‐CARE Agreement.
C. Gioia, G. Giacobazzi, M. Vannini, G. Totaro, L. Sisti, M. Colonna, P. Marchese, A. Celli, ChemSusChem 2021, 14, 4167.
Contributor Information
Dr. Grazia Totaro, Email: grazia.totaro@unibo.it.
Prof. Annamaria Celli, Email: annamaria.celli@unibo.it.
References
- 1. https://www.european-bioplastics.org.
- 2. Agarval S., Macromol. Chem. Phys. 2020, 221, 2000017. [Google Scholar]
- 3. https://www.en-standard.eu.
- 4. Rujnic Havstad M. in Plastic Waste and Recycling, Biodegradable plastics (Ed.: Letcher T. M.), Academic Press Elsevier, 2020, chap. 5, pp. 97–129. [Google Scholar]
- 5. https://www.ows.be/wp-content/uploads/2017/08/Expert-statement-mulching-films.pdf.
- 6. Alaerts L., Augustinus M., Van Acker K., Sustainability 2018, 10, 1487. [Google Scholar]
- 7. La Mantia F. P., Botta L., Morreale M., Scaffaro R., Polym. Degrad. Stab. 2021, 97, 21–24. [Google Scholar]
- 8. Hollstein F., Wohllbe M., Arnaiz S., Manjon D. in Optical Characterization of Materials (Eds.: Beyerer J., Leon F. P., Langle T.), KIT Scientific Publishing, Karlsruhe, 2015, pp. 71–87. [Google Scholar]
- 9. Wu G., Li J., Xu Z., Waste Manage. 2013, 33, 585–597. [DOI] [PubMed] [Google Scholar]
- 10. Żenkiewicz M., Żuk T., Markiewicz E., Polym. Test. 2015, 42, 192–198. [Google Scholar]
- 11. Brdlik P., Boruvka M., Behalek L., Lenfeld P., Polymer 2021, 13, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Itävaara M., Karjomaa S., Selin J. F., Chemosphere 2002, 46, 879–885. [DOI] [PubMed] [Google Scholar]
- 13. Ho K.-L. G., Pometto A. L. III, Hinz P. N., J. Environ. Polym. Degrad. 1999, 7, 83–92. [Google Scholar]
- 14. Makino K., Ohshima H., Kondo T., J. Microencapsulation 1986, 3, 203–212. [DOI] [PubMed] [Google Scholar]
- 15. Cam D., Hyon S.-h, Ikada Y., Biomaterials 1995, 16, 833–843. [DOI] [PubMed] [Google Scholar]
- 16. Ghorpade V. M., Gennadios A., Hanna M. A., Bioresour. Technol. 2001, 76, 57–61. [DOI] [PubMed] [Google Scholar]
- 17. Kishida A., Yoshioka S., Takeda Y., Uchiyama M., Chem. Pharm. Bull. 1989, 37, 1954–156. [Google Scholar]
- 18. Shikinami Y., Okuno M., Biomaterials 1999, 20, 859–877. [DOI] [PubMed] [Google Scholar]
- 19. Ignatius A. A., Augat P., Claes L. E., J. Biomater. Sci. Polym. Ed. 2001, 12, 185–194. [DOI] [PubMed] [Google Scholar]
- 20. Renouf-Glauser A. C., Rose J., Farrar D., Cameron R. E., Biomaterials 2005, 26, 2415–2422. [DOI] [PubMed] [Google Scholar]
- 21. Tsuji H. in Bio-Based Plastics: Materials and Applications (Ed.: Kabasci S.), Wiley, 2014, pp. 171–239. [Google Scholar]
- 22. Ruggero F., Onderwater R. C. A., Carretti E., Roosa S., Benali S., Raquez J. M., Gori R., Lubello C., Wattiez R., J. Polym. Environ. 2021, 29, 3015–3028. [Google Scholar]
- 23. Carrasco F., Pagès P., Gámez-Pérez J., Santana O. O., Maspoch M. L., Polym. Degrad. Stab. 2010, 95, 116–125. [Google Scholar]
- 24. Badia J. D., Ribes-Greus A., Eur. Polym. J. 2016, 84, 22–39. [Google Scholar]
- 25. Żenkiewicz M., Richert J., Rytlewski P., Moraczewski K., Stepczynska M., Karasiewicz T., Polym. Test. 2009, 28, 412–418. [Google Scholar]
- 26. Pillin I., Montrelay N., Bourmaud A., Grohens Y., Polym. Degrad. Stab. 2008, 93, 321–328. [Google Scholar]
- 27. Cosate de Andrade M. F., Fonseca G., Morales A. R., Innocentini Mei L. H., Adv. Polym. Technol. 2018, 37, 2053–2060. [Google Scholar]
- 28. Westphal C., Perrot C., Karlsson S., Polym. Degrad. Stab. 2001, 73, 281–287. [Google Scholar]
- 29. Fan Y., Nishida H., Shirai Y., Endo T., Green Chem. 2003, 5, 575–579. [Google Scholar]
- 30. Fan Y., Nishida H., Mori T., Shirai Y., Endo T., Polymer 2004, 45, 1197–1205. [Google Scholar]
- 31. Herrera-Kao W. A., Loria-Bastarrachea M. I., Pedez-Padilla Y., Cauich-Rodriguez J. V., Vasquez-Torres H., Cervantes-Uc J. M., Polym. Bull. 2018, 75, 4191–4205. [Google Scholar]
- 32.P. Coszach, J.-C. Bogaert, J. Willocq, US 2012/0142958 A1, 2012.
- 33.P. Coszach, J.-C. Bogaert, J. Willocq, US 2012/0204308 A1, 2012.
- 34. Piemonte V., Gironi F., J. Polym. Environ. 2013, 21, 313–318. [Google Scholar]
- 35. Tsuji H., Daimon H., Fujie K., Biomacromolecules 2003, 4, 835–840. [DOI] [PubMed] [Google Scholar]
- 36. Faisal M., Saeki T., Tsuji H., Daimon H., Fujie K., WIT Trans. Ecol. Environ. 2006, 92, 225–233. [Google Scholar]
- 37. Mohd-Adnan A.-F., Nishida H., Shirai Y., Polym. Degrad. Stab. 2008, 93, 1053–1058. [Google Scholar]
- 38. Piemonte V., Sabatini S., Gironi F., J. Polym. Environ. 2013, 21, 640–647. [Google Scholar]
- 39. Román-Ramírez L. A., McKeown P., Jones M. D., Wood J., ACS Catal. 2019, 9, 409–416. [Google Scholar]
- 40. Petrus R., Bykowski D., Sobota P., ACS Catal. 2016, 6, 5222–5235. [Google Scholar]
- 41. Leibfarth F. A., Moreno N., Hawker A. P., Shand J. D., J. Polym. Sci. Part A 2012, 50, 4814–4822. [Google Scholar]
- 42. Calvo-Flores F. G., Monteagudo-Arrebola M. J., Dobado J. A., Isac-García J., Top. Curr. Chem. 2018, 376, 1–40. [DOI] [PubMed] [Google Scholar]
- 43. Bowmer C. T., Hooftman R. N., Hanstveit A. O., Venderbosch P. W. M., van der Hoeven V., Chemosphere 1998, 37, 1317–1333. [DOI] [PubMed] [Google Scholar]
- 44. Pereira C. S. M., Silva V. M. T. M., Rodrigues A. E., Green Chem. 2011, 13, 2658–2671. [Google Scholar]
- 45. Upare P. P., Hwang Y. K., Chang J. S., Hwang D. W., Ind. Eng. Chem. Res. 2012, 51, 4837–4842. [Google Scholar]
- 46. Li Y., Wang D., Sun X. S., RSC Adv. 2015, 5, 27256–27265. [Google Scholar]
- 47. Mercier C., Feillet P., Cereal Chem. 1975, 52, 283–297. [Google Scholar]
- 48. Degli-Innocenti F., Tosin M., Bastioli C., J. Environ. Polym. Degrad. 1998, 6, 197–202. [Google Scholar]
- 49. German D. P., Chacon S. S., Allison S. D., Ecology 2011, 92, 1471–1480. [DOI] [PubMed] [Google Scholar]
- 50. Torres F. G., Troncoso O. P., Torres C., Diaz D.A, Amaya E., Int. J. Biol. Macromol. 2011, 48, 603–606. [DOI] [PubMed] [Google Scholar]
- 51. Maran J. P., Sivakumar V., Thirugnanasambandham K., Sridhar R., Carbohydr. Polym. 2014, 101, 20–28. [DOI] [PubMed] [Google Scholar]
- 52. de M. Neto B. A., J. Fornari C. C. M., Paranhos da Silva E. G., Franco M., dos Santos Reis N., Ferreira Bonomo R. C., de Almeida P. F., Pontes K. V., Int. J. Food Prop. 2018, 20, S2429–S2440. [Google Scholar]
- 53. Engel J. B., Ambrosi A., Tessaro I. C., Carbohydr. Polym. 2019, 225, 115234. [DOI] [PubMed] [Google Scholar]
- 54. Accinelli C., Saccà M. L., Mencarelli M., Vicari A., Chemosphere 2012, 89, 136–143. [DOI] [PubMed] [Google Scholar]
- 55. La Mantia F. P., Scaffaro R., Bastioli C., Macromol. Symp. 2002, 180, 133–140. [Google Scholar]
- 56. Jian J., Xiangbin Z., Xianbo H., Adv. Ind. Eng. Polym. Res. 2020, 3, 19–26. [Google Scholar]
- 57. Ferreira F. V., Cividanes L. S., Gouveia R. F., Lona L. M. F., Polym. Eng. Sci. 2019, E7–E15. [Google Scholar]
- 58. Fu Y., Wu G., Bian X. C., Zeng J. B., Weng Y. X., Molecules 2020, 25, 15. [Google Scholar]
- 59. De Hoe G. X., Zumstein M. T., Getzinger G. J., Ruegsegger I. R., Kohler H−P. E., Maurer-Jones M. A., Sander M., Hillmyer M. A., McNeill K., Environ. Sci. Technol. 2019, 53, 2472–2481. [DOI] [PubMed] [Google Scholar]
- 60. Soulenthone P., Tachibana Y., Muroi F., Suzuki M., Ishii N., Ohta Y., Kasuya K. I., Polym. Degrad. Stab. 2020, 181, 109481. [Google Scholar]
- 61. Witt U., Einig T., Yamamoto M., Kleeberg I., Deckwer W. D., Muller R. J., Chemosphere 2001, 44, 289–299. [DOI] [PubMed] [Google Scholar]
- 62. Muller R. J., Witt U., Rantze E., Deckwer W. D., Polym. Degrad. Stab. 1998, 59, 203–208. [Google Scholar]
- 63. del Campo A., de Lucas-Gil E., Rubio-Marcos F., Arrieta M. P., Fernández-García M., Fernández J. F., Munoz-Bonilla A., Polym. Degrad. Stab. 2021, 185, 109501. [Google Scholar]
- 64. Ruggero F., Carretti E., Gori R., Lotti T., Lubello C., Chemosphere 2020, 246, 125770. [DOI] [PubMed] [Google Scholar]
- 65. Scaffaro R., Maio A., Sutera F., Gulino E. F., Morreale M., Polymer 2019, 11, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Scaffaro R., Maio A., Gammino M., La Mantia F. P., Polym. Degrad. Stab. 2020, 187, 109549. [Google Scholar]
- 67. Greene J., SciEnvironm 2018, 1, 104.. [Google Scholar]
- 68. Muniyasamy S., Ofosu O., John M. J., Anandjiwala R. D., J. Renewable Mater. 2016, 4, 133–145. [Google Scholar]
- 69. Weng Y. X., Wang Y., Wang X. L., Wang Y. Z., Polym. Test. 2010, 29, 579–587. [Google Scholar]
- 70. Luo S., Netravali A. N. A., Polym. Degrad. Stab. 2003, 80, 59–66. [Google Scholar]
- 71. Rutkowska M., Krasowska K., Heimowska A., Adamus G., Sobota M., Musioł M., Janeczek H., Sikorska W., Krzan A., Žagar E., Kowalczuk M., J. Polym. Environ. 2008, 16, 183–191. [Google Scholar]
- 72. Weng Y. X., Wang X. L., Wang Y. Z., Polym. Test. 2011, 30, 372–380. [Google Scholar]
- 73. Freirer T., Kunze C., Nischan C., Kramer S., Sternberg K., Saß M., Hopt U. T., Shmitz K.-P., Biomaterials 2002, 23, 2649–2657. [DOI] [PubMed] [Google Scholar]
- 74. Gunning M. A., Geever L. M., Killion J. A., Lyons J. G., Higginbotham C. L., Polym. Test. 2013, 32, 1603–1611. [Google Scholar]
- 75. Mergaert J., Anderson C., Wouters A., Swings J., Kersters K., FEMS Microbiol. Lett. 1992, 103, 317–321. [DOI] [PubMed] [Google Scholar]
- 76. Rivas L., Casarin S., Nepomuceno N., Alencar M., Agnelli J., De Medeiros E., De Oliveira Wanderley Neto A., De Oliveira M., De Medeiros A., Ferreira Santos A., Polimeros 2017, 27, 122–128. [Google Scholar]
- 77. Zaverl M., Seydibeyoglu O., Misra M., Mohanty A., J. Appl. Polym. Sci. 2012, 125, E324–E331. [Google Scholar]
- 78. Duangphet S., Szegda D., Song J., Tarverdi K., J. Polym. Environ. 2014, 22, 1–8. [Google Scholar]
- 79. Arza C. R., Jannasch P., Johansson P., Magnusson P., Werker A., Maurer F. H. J., J. Appl. Polym. Sci. 2015, 132, 6–11. [Google Scholar]
- 80. Sato S., Ishii N., Hamada Y., Abe H., Tsuge T., Polym. Degrad. Stab. 2012, 97, 329–336. [Google Scholar]
- 81. Sohn Y. J., Kim H. T., Baritugo K. A., Jo S. Y., Song H. M., Park S. Y., Park S. K., Pyo J., Cha H. G., Kim H., et al., Biotechnol. J. 2020, 15, 1–16. [Google Scholar]
- 82. Lamberti F. M., Román-Ramírez L. A., Wood J., J. Polym. Environ. 2020, 28, 2551–2571. [Google Scholar]
- 83. Feghali E., Tauk L., Ortiz P., Vanbroekhoven K., Eevers W., Polym. Degrad. Stab. 2020, 179, 109241. [Google Scholar]
- 84. Ariffin H., Nishida H., Hassan M. A., Shirai Y., Biotechnol. J. 2010, 5, 484–492. [DOI] [PubMed] [Google Scholar]
- 85. Kaihara S., Osanai Y., Nishikawa K., Toshima K., Doi Y., Matsumura S., Macromol. Biosci. 2005, 5, 644–652. [DOI] [PubMed] [Google Scholar]
- 86. Yang X., Clénet J., Xu H., Odelius K., Hakkarainen M., Macromolecules 2015, 48, 2509–2518. [Google Scholar]