

Summary
Background
Bruton tyrosine kinase (BTK) inhibition targets B‐cell and other non‐T‐cell immune cells implicated in the pathophysiology of pemphigus, an autoimmune disease driven by anti‐desmoglein autoantibodies. Rilzabrutinib is a new reversible, covalent BTK inhibitor demonstrating preclinical efficacy as monotherapy in canine pemphigus foliaceus.
Objectives
To evaluate the efficacy and safety of oral rilzabrutinib in patients with pemphigus vulgaris in a multicentre, proof‐of‐concept, phase II trial.
Methods
Patients with Pemphigus Disease Area Index severity scores 8–45 received 12 weeks of oral rilzabrutinib 400–600 mg twice daily and 12 weeks of follow‐up. Patients initially received between 0 and ≤ 0·5 mg kg−1 prednisone‐equivalent corticosteroid (CS; i.e. ‘low dose’), tapered after control of disease activity (CDA; no new lesions, existing lesions healing). The primary endpoints were CDA within 4 weeks on zero‐to‐low‐dose CS and safety.
Results
In total, 27 patients with pemphigus vulgaris were included: nine newly diagnosed (33%) and 18 relapsing (67%); 11 had moderate disease (41%) and 16 moderate to severe (59%). The primary endpoint, CDA, was achieved in 14 patients (52%, 95% confidence interval 32–71): 11 using low‐dose CS and three using no CS. Over 12 weeks of treatment, mean CS doses reduced from 20·0 to 11·8 mg per day for newly diagnosed patients and from 10·3 to 7·8 mg per day for relapsing patients. Six patients (22%) achieved complete response by week 24, including four (15%) by week 12. Treatment‐related adverse events were mostly mild (grade 1 or 2); one patient experienced grade 3 cellulitis.
Conclusions
Rilzabrutinib alone, or with much lower CS doses than usual, was safe, with rapid clinical activity in pemphigus vulgaris. These data suggest that BTK inhibition may be a promising treatment strategy and support further investigation of rilzabrutinib for the treatment of pemphigus.
Short abstract
What is already known about this topic?
Standard pemphigus treatment relies on systemic high‐dose corticosteroids (CS), rituximab and/or immunosuppressives, which are limited by delayed onset of action and potential toxicities.
Immune‐mediated mechanisms that are fast acting on both the innate and adaptive immune systems, are steroid sparing, and have safety profiles well suited for chronic administration are greatly needed for patients with pemphigus.
What does this study add?
Rilzabrutinib is an oral Bruton tyrosine kinase (BTK) inhibitor targeting B‐cell and other non‐T‐cell immune cells implicated in pemphigus pathophysiology.
Treatment with rilzabrutinib (with or without low‐dose CS) demonstrated rapid disease control and a well‐tolerated safety profile in patients with newly diagnosed and relapsing pemphigus vulgaris.
BELIEVE provides evidence for a promising treatment strategy via BTK inhibition, supporting further investigation of rilzabrutinib in other immune‐mediated diseases.
Linked Comment: A.M. Drucker and N.H. Shear. Br J Dermatol 2021; 185:691–692.
Plain language summary available online
Pemphigus is a rare, severe and potentially life‐threatening B‐cell‐mediated autoimmune disease with an estimated US prevalence of 5·2 per 100 000 people. 1 , 2 Pemphigus vulgaris (PV) is characterized by debilitating intraepithelial blisters and erosions on skin and mucous membranes. It results from IgG autoantibodies binding to the keratinocyte proteins desmoglein (Dsg)1 and Dsg3, inducing deficient keratinocyte adhesion (acantholysis). 1 , 3 While it is primarily mediated by production of B‐cell and plasma cell autoantibodies, dermal infiltrates often contain superficial interstitial and perivascular neutrophils and eosinophils resulting from innate immune response activation. 1 , 4 Thus, both innate and adaptive immunological pathways provide rational therapeutic targets. 1
First‐line PV treatment includes either high‐dose corticosteroids (CS) starting at prednisone‐equivalent doses of ≥ 1·0 mg kg−1 per day (typically ≥ 60 mg per day), or intravenous rituximab plus short‐course, intermediate‐dose oral CS for moderate‐to‐severe PV [based on results showing improved CS‐free complete response (CR) vs. CS alone]. 3 , 5 To achieve CR, newly diagnosed patients with PV still require the use of moderate‐to‐high doses of CS (0·5–1·0 mg kg−1 per day, tapered over 3 months for moderate PV or 6 months for severe PV) 6 when rituximab is included in the treatment regimen, and ≥ 1 mg kg−1 per day CS without it. In addition to the CS doses described above, premedication with intravenous methylprednisolone 100 mg (or equivalent glucocorticoid) is recommended 30 min prior to each rituximab infusion. 7 Continued or maintenance therapy to manage relapses poses a significant risk of immunosuppression and possible adverse effects (e.g. serious infections). Immune‐mediated therapies are greatly needed that are more specifically targeted, fast acting and steroid sparing, and have safety profiles well suited for chronic administration.
Rilzabrutinib (PRN1008) is a potent, oral and reversible covalent Bruton tyrosine kinase (BTK) inhibitor developed to treat autoimmune diseases. 8 BTK provides a logical ‘targeted immunosuppression’ approach, with no direct effects in T cells and plasma cells. 9 Rilzabrutinib forms both noncovalent and covalent bonds with BTK for enhanced selectivity and extended inhibition. 8 Compared with first‐ and second‐generation BTK inhibitors, rilzabrutinib exhibits minimal cross‐reactivity with other kinases, and thus lower risk for off‐target, drug‐mediated effects. 10 Rilzabrutinib intervenes in multiple immunological mechanisms including inhibiting B‐cell‐receptor signalling, IgG‐mediated Fc gamma receptor activation and phagocytosis, IgE‐mediated Fc epsilon receptor activation and degranulation, and finally activation, adhesion, recruitment and oxidative burst in neutrophils, all without directly impacting T cells or depleting B cells. 11 , 12
In vivo, rilzabrutinib improved disease symptoms and outcomes in collagen‐induced arthritis 12 , 13 and clearance of naturally occurring, new‐onset canine pemphigus foliaceus as monotherapy. 14 In healthy volunteers, oral rilzabrutinib demonstrated a large volume of distribution, a half‐life of approximately 3–4 h, and > 90% BTK occupancy within 4 h of dosing. 10 The favourable safety profiles observed in vivo 14 and in healthy volunteers, and the pharmacodynamic profile of BTK inhibition suggested durable pharmacodynamic action. 10
This open‐label, proof‐of‐concept study evaluated the efficacy and safety of rilzabrutinib, with or without concomitant low‐dose CS, in patients with PV.
Patients and methods
Trial design and participants
This multicentre, single‐arm, phase II ‘BELIEVE’ study was registered at ClinicalTrials.gov (NCT02704429). All patients provided written informed consent, the study was approved by local regulatory bodies and institutional review boards or ethics committees, and it was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation guidelines for good clinical practice. The study protocol is provided in Appendix S1 (see Supporting Information).
Patients were 18–80 years of age with a clinical diagnosis of new‐onset or relapsing, moderate‐to‐severe PV based on histopathological (intraepithelial blistering with acantholysis) and immunohistological (positive direct immunofluorescence) evaluations. Newly diagnosed patients were ≤ 6 months, and relapsing patients were > 6 months to 10 years from disease onset or diagnosis at screening. Disease severity measured by the validated Pemphigus Disease Area Index (PDAI) included scores of 8 to < 45 (moderate 8 to < 15, moderate to severe ≥ 15). 15 Prednisone‐equivalent CS ≤ 0·5 mg kg−1 per day was permitted for 2 weeks prior to initiation of rilzabrutinib. Patients using recent or concomitant BTK inhibitors or immunological response modifiers, and needing ongoing CYP3A‐sensitive drugs and/or proton pump inhibitors were excluded (Appendix S2; see Supporting Information).
Treatment
Patients received an initial dose of oral rilzabrutinib 400 mg twice daily, with optional dose adjustment up to 600 mg twice daily, for 12 weeks, with an additional 12 weeks’ off‐treatment follow‐up (Figure S1; see Supporting Information). Concomitant CS doses were 0 to ≤ 0·5 mg kg−1 per day, unless higher ‘rescue’ doses were later needed. CS doses were tapered according to the tapering schedule of Werth et al. 16 and per investigator’s discretion after confirming control of disease activity (CDA) and continuing improvement at least 2 weeks later. Assessments were performed on days 1 and 2, and weeks 2, 4, 8, 12, 16, 20 and 24.
Outcomes
The primary efficacy endpoint was the proportion of patients achieving CDA within 4 weeks of starting rilzabrutinib with zero CS or a CS dose ≤ 0·5 mg kg−1 per day (defined as ‘low‐dose CS’). 17 CDA was defined as the absence of new lesions and beginning of healing of existing lesions. 17 The primary safety endpoint of treatment‐emergent adverse events (TEAEs) was reported through 24 weeks (12 weeks on treatment, 12 weeks of off‐treatment follow‐up) and were graded per the National Cancer Institute’s Common Terminology Criteria for Adverse Events, v4.0. 18
Secondary endpoints included time to CDA, CR and relapse post‐rilzabrutinib; and CS usage. CR was defined as the absence of new lesions and all lesions healed. 17 , 19 Additional and exploratory endpoints were changes from baseline in PDAI activity, 20 quality‐of‐life score (Autoimmune Bullous Disease Quality of Life, ABQOL), 21 appetite score (Simplified Nutritional Appetite Questionnaire, SNAQ), 22 pharmacokinetics, BTK occupancy in peripheral blood mononuclear cells (PBMCs), 10 and anti‐Dsg1/3 autoantibody levels (DSG1&DSG3 ELISA Test System; MBL International, Woburn, MA, USA; 23 details in Appendix S2).
Statistical analysis
All patients receiving at least one dose of rilzabrutinib were included in the safety and efficacy populations (Figure S2; see Supporting Information). Patients completing 12 weeks on rilzabrutinib and 12 weeks post‐treatment (i.e. 24 weeks) were an exploratory ‘completer population’.
Quantitative results were summarized using descriptive statistics. Efficacy and time‐to‐event results, including two‐sided 95% confidence intervals (CIs), were evaluated by the exact Clopper–Pearson method and Kaplan–Meier estimates. All statistics were calculated with SAS version 9.2 or later (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics
Between 17 February 2016 and 3 September 2018, 52 patients were screened from 13 sites in Australia, Croatia, France, Greece and Israel; 27 patients were enrolled (Figure S2). Their median age was 51 years [interquartile range (IQR) 45–60]; 56% were female and 81% were white (Tables 1 and 2). Nine patients (33%) had newly diagnosed and 18 (67%) had relapsing PV. Eleven of 18 relapsing patients had had pemphigus for > 5 years. The mean time from first diagnosis to study inclusion was 6 years (SD, 7). The mean baseline PDAI score overall was 19·1 (SD, 10·7); it was 21·2 (SD, 11·5) for newly diagnosed and 18·0 (SD, 10·4) for relapsing disease. Overall, 11 patients (41%) had moderate disease with mean baseline PDAI score 10·0 (SD, 1·2), and 16 patients (59%) had moderate‐to‐severe disease with mean baseline PDAI score 25·3 (SD, 9·7). PV was confirmed in all patients based on biopsy‐proven histopathological (intraepithelial blistering with acantholysis) and immunohistological (positive direct immunofluorescence) evaluations. Baseline anti‐Dsg antibody profiles were n = 13 anti‐Dsg3+ only, n = 10 anti‐Dsg1+/Dsg3+, n = 3 anti‐Dsg1+ only, and n = 1 double negative.
Table 1.
Characteristics of the patients with pemphigus vulgaris (PV) at baseline (n = 27)
| Age (years), median (IQR) | 51 (45–60) |
| Sex, n (%) | |
| Female | 15 (56) |
| Male | 12 (44) |
| Race, n (%) | |
| White | 22 (81) |
| Asian | 1 (4) |
| Not collected | 4 (15) |
| Baseline weight (kg), mean (SD) | 76 (18) |
| Baseline body mass index (kg m−2), mean (SD) | 27 (5) |
| Disease stage, n (%) | |
| Newly diagnoseda | 9 (33) |
| Relapsing | 18 (67) |
| Pemphigus severity at baseline, n (%)b | |
| Moderate (PDAI activity 8 to < 15) | 11 (41) |
| Moderate‐to‐severe (PDAI activity ≥ 15) | 16 (59) |
| PDAI activity, n (%)c | |
| Mucosal | 24 (89) |
| Oral | 23 (85) |
| Skin | 18 (67) |
| Scalp | 15 (56) |
| Anti‐desmoglein (Dsg) profile, n (%) | |
| Any positive | 26 (96) |
| Anti‐Dsg3 positive only | 13 (48) |
| Anti‐Dsg1 positive only | 3 (11) |
| Anti‐Dsg3 and anti‐Dsg1 positive | 10 (37) |
| Anti‐Dsg3 and anti‐Dsg1 negatived | 1 (4) |
| Total anti‐Dsg levels by ELISA, n (%) | |
| ≥ 100 units mL−1 | 20 (74) |
| < 100 units mL−1 | 7 (26) |
Data cutoff 5 March 2020. ELISA, enzyme‐linked immunosorbent assay; IQR, interquartile range; PDAI, Pemphigus Disease Area Index. aNewly diagnosed patients were diagnosed within 6 months prior to screening. bSeverity score based on Shimizu et al. 2014. 15 cPemphigus Disease Area Index (PDAI) activity score is a total combined activity score, including skin, scalp and mucous membranes, ranging from 0 to 250 (higher score indicates higher severity). 20 dThis patient with a positive diagnosis for PV was referred into the trial with a 9‐year history of relapsing PV mainly on the scalp. No anti‐Dsg titres had been done before and it was not a screening requirement for the trial. The anti‐Dsg1 and anti‐Dsg3 results returned later from abroad were negative. The patient responded to treatment.
Table 2.
Duration of pemphigus and corticosteroid dose at baseline
| All patients (n = 27) | Newly diagnosed (n = 9) | Relapsing (n = 18) | |
|---|---|---|---|
| Duration of pemphigus since first diagnosis (years) | |||
| Mean (SD) | 6·0 (7·0) | 0·1 (0·1) | 8·9 (6·8) |
| Median (IQR) | 3·4 (0·2–10·4) | 0 (0–0·2) | 7·2 (3·4–11·6) |
| Corticosteroid dose (mg per day), mean (SD) | |||
| At screening | 13·9 (15·4) | 20·0 (13·0) | 10·9 (15·9) |
| At baseline (week 1, day 1) | 13·5 (11·1) | 20·0 (9·7) | 10·3 (10·6) |
IQR, interquartile range.
Rilzabrutinib treatment
Patients initiated treatment with rilzabrutinib 400 mg twice daily. Six patients entered the study without any CS treatment; four patients (one newly diagnosed; three chronic relapsing, with durations of PV prior to study initiation of 3·4, 3·5 and 8·2 years) received rilzabrutinib only (no CS) for the entire first 12 weeks. The starting low CS dose (≤ 0·5 mg kg−1 per day) was determined by clinical need and screening period dose. Three relapsing patients dose escalated rilzabrutinib due to increasing disease activity; one patient achieved CDA on day 17 and increased rilzabrutinib to 500 mg twice daily with CS (0·23 mg kg−1 per day) on day 35. The other two patients increased the rilzabrutinib dose to 600 mg twice daily: the first increased on day 24 and achieved CDA on day 30 with CS (0·27 mg kg−1 per day since baseline) and the second dose escalated on day 58 (no CS) and did not achieve CDA. BTK occupancy levels just prior to dose escalations were below the 70% target for these three patients (66%, 43% and 23%, respectively).
The mean duration of rilzabrutinib exposure was 80 days (SD, 21). Three patients achieved CDA on rilzabrutinib alone (no CS) by 2 weeks; two patients relapsed off rilzabrutinib at week 20 and received low‐dose CS. The other 24 patients (89%) received concomitant low‐dose CS at some time during the first 12 weeks, including 11 (46%) who tapered to minimal CS of < 10 mg per day. CS doses were stable during screening, indicated by similar mean screening and baseline CS doses: screening 20·0 mg per day (SD, 13·0) for newly diagnosed and 10·9 mg per day (SD, 15·9) for relapsing patients; and at baseline: 20·0 mg per day (SD, 9·7) for newly diagnosed and 10·3 mg per day (SD, 10·6) for relapsing patients.
Pharmacokinetics and Bruton tyrosine kinase occupancy
Pharmacokinetic and BTK occupancy in PBMCs indicated rapid rilzabrutinib absorption and clearance from plasma, with high levels of durable BTK inhibition (target threshold of ≥ 70%) observed 2 h after the first rilzabrutinib dose and maintained throughout the dosing interval, reflecting the slow dissociation rate of rilzabrutinib from BTK (Figure S3; see Supporting Information).
Efficacy
The primary endpoint of CDA with zero‐to‐low‐dose CS at or prior to week 4 was achieved in 14 of 27 patients (52%, 95% CI 32–71; Figure 1a), including three patients not initially taking CS. In 14 patients achieving CDA, the primary endpoint response was accompanied by low CS doses compared with guideline recommendations for medium‐to‐high‐dose CS: 3 mean day 1 dose of 18 mg per day in five newly diagnosed patients and 13 mg per day in nine relapsing patients. CDA was achieved rapidly, with a median time to first CDA of 33 days (95% CI 29–62) of rilzabrutinib based on Kaplan–Meier estimates (Figure 2a). The CDA rate improved over time: 19 (70%) by week 12, and 23 (85%) after an additional 12 weeks off rilzabrutinib. The median time to relapse per Kaplan–Meier estimates in patients achieving CDA and after treatment completion or discontinuation was 96 days (IQR 6 to not estimable, range 27–99, 95% CI 57 to not estimable; Figure 2b).
Figure 1.
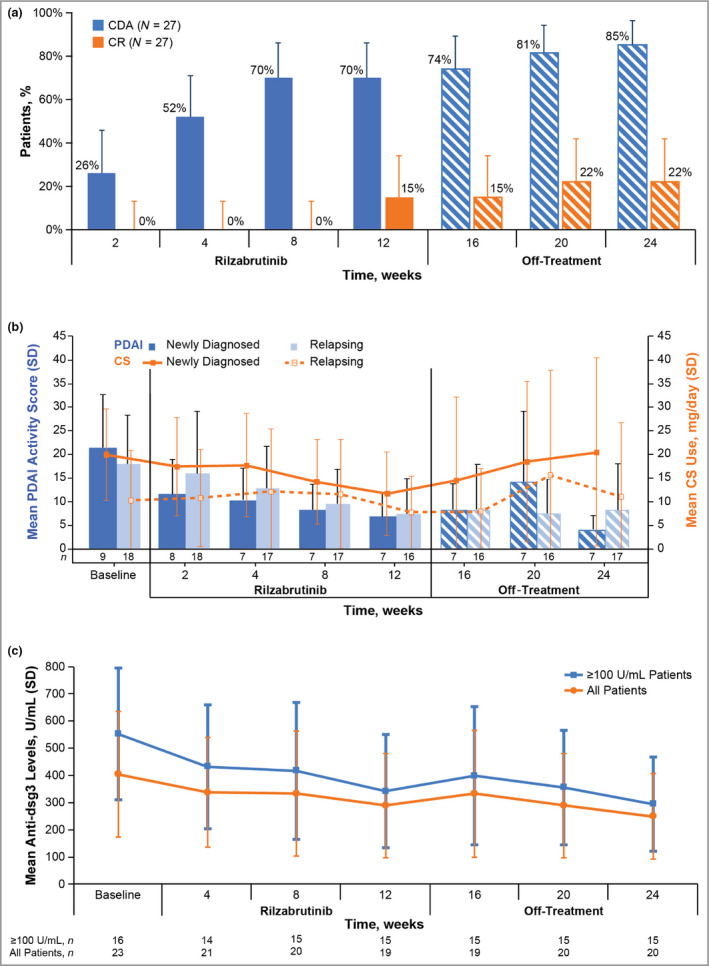
Efficacy of rilzabrutinib over time based on control of disease activity (CDA) and complete response (CR) (a); Pemphigus Disease Area Index (PDAI) scores and corticosteroid (CS) use (b); and anti‐desmoglein (Dsg)3 autoantibody levels (c) (intent to treat). (a) Percentage of patients with CDA and CR over time through the rilzabrutinib treatment and off‐treatment periods (n = 27). The bars represent the 95% confidence intervals. (b) Mean PDAI activity scores and CS use over time. The bars indicate SDs. (c) Mean anti‐Dsg3 levels in all patients and in patients with ≥ 100 units mL−1 anti‐Dsg3 at baseline; levels are shown from baseline through rilzabrutinib treatment (weeks 4–12) and off‐treatment (weeks 16–24). The bars indicate SDs. Data cutoff 5 March 2020.
Figure 2.
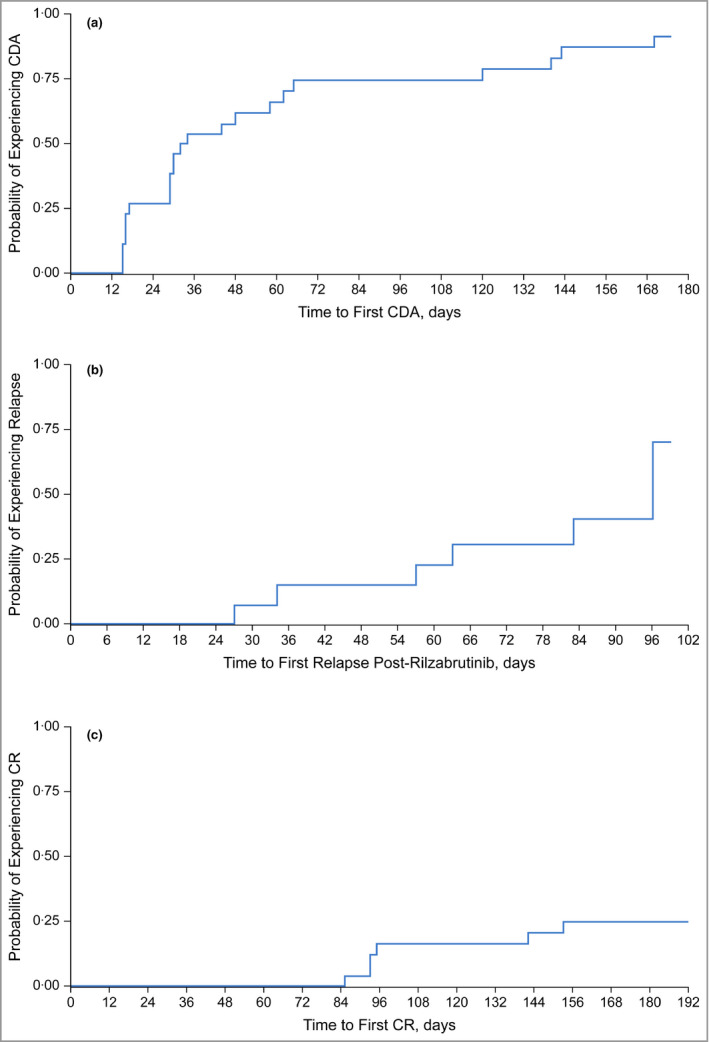
Kaplan–Meier estimates for time to first control of disease activity (CDA) (a); time to relapse following completion or discontinuation of rilzabrutinib (b); and time to first complete response (CR) (c). Data cutoff 5 March 2020.
The CR endpoint was met in six of 27 patients (22%) by week 24. Four patients (15%) achieved CR by week 12 and an additional two by week 20 without increased doses of CS (Figure 2c). The mean CS dose at the time of CR was 7·2 mg per day (range 1–20). The same six patients were included in the completers set population (n = 24; Table S2; see Supporting Information). Among the six patients who achieved CR, the median duration of CR (from achievement to last follow‐up visit) was 65 days (IQR 22–85). Three patients maintained CR for ≥ 80 days, and three for between 23 and 60 days.
Disease severity, measured by PDAI scores, improved more rapidly, and from a higher starting point, in newly diagnosed patients than in relapsing patients (Figure 1b). PDAI score reduction was evident as early as the week 2 visit in both groups.
Overall CS use decreased more over time in newly diagnosed than relapsing patients. The mean CS dose in newly diagnosed patients fell from 20·0 mg per day (SD, 9·7) at baseline to 11·8 mg per day (SD, 8·9) at 12 weeks (Figure 1b). In relapsing patients, the mean CS dose was reduced from 10·3 mg per day (SD, 10·6) at baseline to 7·8 mg per day (SD, 7·7) at 12 weeks. For all patients over 12 weeks of rilzabrutinib, the mean dose of CS was 13 mg per day (or 0·18 mg kg−1 per day). During follow‐up off rilzabrutinib, four patients required increased rescue doses of CS (> 0·5 mg kg−1 per day) to control flare‐ups.
In an exploratory analysis, mean anti‐Dsg3 antibody levels decreased from 404 units mL−1 (SD, 462) at baseline to 289 units mL−1 (SD, 384) after 12 weeks of rilzabrutinib (with or without low‐dose CS). In 16 patients with high baseline anti‐Dsg3 autoantibodies (≥ 100 units mL−1), mean anti‐Dsg3 levels decreased from 552 (SD, 486) to 342 (SD, 416) after 12 weeks of rilzabrutinib, with a trending further reduction during 12 weeks off‐treatment [week 24: mean, 295 units mL−1 (SD, 346); Figure 1c].
CDA rates were similar across all prespecified subgroups (Figure 3). By week 4, CDA rates were 55% in moderate and 50% in moderate‐to‐severe pemphigus, 56% for newly diagnosed and 50% for relapsing and chronic patients, and 43% and 55% for patients with baseline total anti‐Dsg antibody titres < 100 and ≥ 100 units mL−1, respectively.
Figure 3.
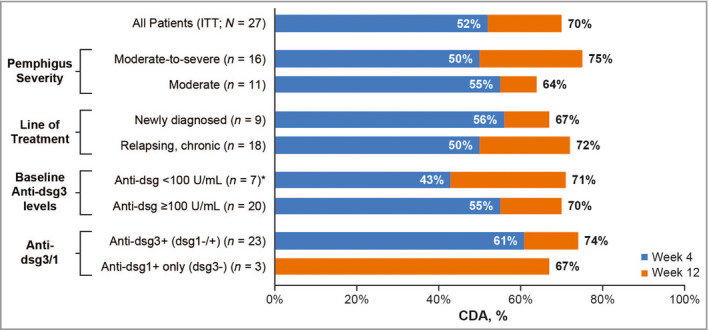
Subgroup analysis of control of disease activity (CDA) rates at 4 weeks (primary endpoint) and 12 weeks (intent to treat: ITT; n = 27). Data cutoff 5 March 2020. One patient with only anti‐dsg‐1+ (dsg‐3‐) had 0% CDA rate at week 4. *Included one patient who was negative for anti‐desmoglein (Dsg) antibodies and who did not achieve CDA.
Safety
TEAEs independent of causality occurred in 20 (74%) patients; the most common were nausea (22%) and headache (15%; Table 3). Twelve of 27 patients (44%) experienced a treatment‐related TEAE, with the most common being nausea (15%) and upper abdominal pain (11%); all others occurred in no more than two patients. The majority of treatment‐related TEAEs were grade 1/2 and transient.
Table 3.
Summary of treatment‐emergent adverse events by system organ class and preferred term (in at least two patients)
| Rilzabrutinib (n = 27) | Independent of causality | Treatment relateda |
|---|---|---|
| Any treatment‐emergent adverse event | 20 (74) | 12 (44) |
| Gastrointestinal disorders | 11 (41) | 7 (26) |
| Nausea | 6 (22) | 4 (15) |
| Upper abdominal pain | 3 (11) | 3 (11) |
| Diarrhoea | 3 (11) | 2 (7) |
| Dry mouth | 2 (7) | 2 (7) |
| Vomiting | 2 (7) | 1 (4) |
| Abdominal pain | 2 (7) | 0 |
| Nervous system disorders | 10 (37) | 6 (22) |
| Headache | 4 (15) | 2 (7) |
| Dizziness | 2 (7) | 1 (4) |
| Paraesthesia | 2 (7) | 0 |
| Infections and infestationsb | 10 (37) | 3 (11)b |
| Upper respiratory infection | 2 (7) | 2 (7) |
| Staphylococcal skin infection | 2 (7) | 0 |
| General disorders or administration‐site conditions | 7 (26) | 2 (7) |
| Fatigue | 2 (7) | 1 (4) |
| Peripheral swelling | 2 (7) | 0 |
| Musculoskeletal and connective tissue disorders | 7 (26) | 2 (7) |
| Arthralgia | 2 (7) | 1 (4) |
| Pain in extremity | 2 (7) | 1 (4) |
| Muscle spasms | 2 (7) | 0 |
| Respiratory, thoracic and mediastinal disorders | 4 (15) | 2 (7) |
| Epistaxis | 2 (7) | 2 (7) |
| Ear and labyrinth disorders | 3 (11) | 2 (7) |
| Vertigo | 2 (7) | 1 (4) |
| Psychiatric disorders | 2 (7) | 0 |
| Anxiety | 2 (7) | 0 |
| Skin and subcutaneous tissue disorders | 2 (7) | 2 (7) |
| Erythema | 2 (7) | 2 (7) |
Values are n (%). Data cutoff 5 March 2020. aAll were grade 1 or 2 unless noted otherwise. bOne patient had a treatment‐related serious adverse event (grade 3 cellulitis of the leg). Treatment with rilzabrutinib at 400 mg twice daily was briefly interrupted and was resumed for a further 2 months without event recurrence and the patient completed the study.
Three patients experienced serious adverse events. One 69‐year‐old man with relapsing PV for 9 years, long‐standing type 2 diabetes mellitus, and a prior history of recurrent cellulitis, developed grade 3 cellulitis on day 26 (deemed treatment related by the investigator). After hospitalization for a 3‐day course of intravenous antibiotics, during which rilzabrutinib was suspended, the patient completed 12 weeks of rilzabrutinib and low‐dose CS. The second patient, a 41‐year‐old woman, developed pneumonitis (day 9 of the trial) due to inflammation of a previously undiagnosed congenital pulmonary sequestration; she died from complications of lung surgery on the 34th day after her last rilzabrutinib exposure. The investigator believed this was not related to rilzabrutinib. The third patient, a 46‐year‐old man, had a pancreatic pseudocyst discovered on day 29 during routine clinical examination, after which the patient withdrew from the study for elective surgery. The investigator regarded this as not related to rilzabrutinib.
Quality‐of‐life and appetite scores
Mean ABQOL scores 21 decreased slightly from 19·3 at baseline to 14·8 at 12 weeks: mean alteration −3·7 (SD, 7·0) (Table 4). In newly diagnosed and relapsing patients, respectively, mean ABQOL scores at baseline to 12 weeks decreased from 20·1 to 12·6 (mean change −6·6) and from 18·9 to 15·8 (mean change −2·5). The SNAQ score 22 increased slightly after 12 weeks of treatment [mean change +1·1 (SD, 2·5)], indicating that appetite was not suppressed.
Table 4.
Quality of life (ABQOL) for all patients and newly diagnosed and relapsing patients
| Date of visit | All patients | Newly diagnosed | Relapsing |
|---|---|---|---|
| Baseline | n = 27 | n = 9 | n = 18 |
| 19·3 (8·4) | 20·1 (7·6) | 18·9 (8·9) | |
| Week 4 | n = 26 | n = 8 | n = 18 |
| 17·3 (10·4) | 14·1 (7·5) | 18·7 (11·3) | |
| Week 12 | n = 24 | n = 7 | n = 17 |
| 14·8 (10·0) | 12·6 (9·2) | 15·8 (10·5) | |
| Change from baseline to end of treatment (week 12) | −3·7 (7·0) | −6·6 (4·4) | −2·5 (7·6) |
| Week 24 (off‐treatment) | n = 2415·6 (9·7) | n = 7 13·7 (10·6) | n = 1716·4 (9·5) |
Values are the mean (SD). Data cutoff 5 March 2020. ABQOL (Autoimmune Bullous Disease Quality of Life) is a quality‐of‐life questionnaire designed and validated specifically for patients with autoimmune bullous disease. 21
Discussion
The BELIEVE study is the first trial to demonstrate clinical activity of BTK inhibition in human autoimmune skin disease, and extends the findings in pemphigus reported for rilzabrutinib monotherapy in dogs. 14 Outcomes were observed in a global population, including a majority with chronic, relapsing disease, and efficacy was consistent across subgroups.
Rilzabrutinib showed rapid and clinically meaningful efficacy, with most patients achieving CDA by 4 weeks on the background of zero‐to‐low‐dose CS. Attribution of therapeutic effect to rilzabrutinib comes from the three patients using no CS who achieved CDA in 4 weeks, from significant flare of disease in six responders after cessation of rilzabrutinib, and from the consistently low doses of CS used in other CDA or CR responders. Relative to current clinical guidelines 17 and the recent Ritux3 study, 5 patients here were on much lower CS doses than recommended, likely subtherapeutic without supplementation. For example, newly diagnosed patients with moderate PV assigned to rituximab in the Ritux3 trial 5 had a starting dose of concomitant CS 0·5 mg kg−1 per day for a month, in addition to any unaccounted for pretreatment with intravenous methylprednisolone 100 mg (or equivalent) given before each of the two infusions. 7 The concomitant CS dose was around twofold higher than the mean 20 mg per day starting dose used here for similar patients.
Excitingly, the rapid achievement of CDA (median 33 days) by patients without moderate‐to‐high CS doses suggests the possibility of a CS‐free rilzabrutinib acute control regimen for future patients, or at least one with a rapid CS taper to minimize CS‐related adverse effects. Notably, newly diagnosed patients with more severe disease were able to achieve clinical responses and still taper CS doses from a mean of 20·0 to 11·8 mg per day during 12 weeks of treatment. The fact that a high proportion of patients achieved CDA, despite limited, initial anti‐Dsg3 reduction, suggests a faster effect of rilzabrutinib on the innate immune system. Delayed anti‐Dsg3 reductions were likely due to rilzabrutinib’s indirect depletion of short‐lived plasma cells due to B‐lymphocyte inhibition. This is consistent with BTK‐mediated inhibition of multiple non‐T‐cell immune cells, rather than delayed adaptive immune suppression, reflected better by later changes in PDAI score and partial reductions in autoantibody formation. 24 The rapidity of onset of response, coupled with the fact that rilzabrutinib has no direct effect on plasma cells, suggests that its early efficacy in pemphigus is partly due to its effects on the innate immune system.
CR was observed beginning at 12 weeks in four patients, increasing further to six patients by 20 weeks, a promising result given the limited study duration and because CR on minimal therapy is considered a late‐occurring endpoint (around 1–2 years), 17 as observed at a median beyond 36 weeks in the rituximab‐containing arm of the randomized controlled Ritux3 trial. 5 The overall mean PDAI score reduction of 56% by week 12 was consistent with the achievement of clinical response endpoints. ABQOL scores decreased at a lower gradient than PDAI scores, as expected with quality‐of‐life scores. Recently, the ABQOL score was shown to be sensitive to change (although less sensitive than objective severity scores) in newly diagnosed patients with autoimmune bullous disease (including pemphigus). 25
The safety results indicated a favourable risk‐to‐benefit profile for rilzabrutinib. Most treatment‐related TEAEs were mild (grade 1 or 2) and transient, with no reported cases of AEs commonly associated with marketed irreversible BTK inhibitors (e.g. major haemorrhage, atrial fibrillation, or thrombocytopenia and neutropenia). 26 , 27 Additionally, rilzabrutinib enabled reduced initial CS doses and facilitated rapid tapering, a major goal in pemphigus therapies. 6 Unlike B‐cell‐depleting treatments such as rituximab, with a risk for sustained immunosuppression until B‐cell reconstitution, an advantage of rilzabrutinib is rapid clearance and reversible pharmacodynamic effects after stopping treatment. Although there was a death reported due to complications of surgery for a pulmonary sequestration, it was believed to be unrelated to rilzabrutinib, which had ceased a month earlier.
Study limitations include those associated with open‐label, single‐arm designs lacking a randomized, double‐blind control group. The cohort design limitations are somewhat mitigated by the disease’s natural history not to remit without initial CS doses ≥ 0·5 mg kg−1 per day, and prior probability established by the efficacy of BTK inhibitor monotherapy in canine pemphigus. 14 , 28 Study strengths include a broad patient population, starting dose validation based on BTK inhibition in PBMCs, and robust clinical endpoints of CDA, CR and CS use. 17 Despite a treatment duration of 12 weeks with rilzabrutinib being too short to adequately assess long‐term CR rates, an encouraging 22% CR rate was observed, with ongoing trends towards CS reduction. Rapid and sustained improvement in pemphigus activity was observed by PDAI score, a sensitive clinical instrument with high accuracy and reproducibility. 29
In conclusion, this study confirms rilzabrutinib as the first BTK inhibitor showing a favourable risk‐to‐benefit profile in PV. Rilzabrutinib was effectively and safely combined with zero or low‐dose CS for a rapid response. Following this proof‐of‐concept trial, a phase III pivotal study (PEGASUS; NCT03762265) of rilzabrutinib vs. placebo with CS taper is underway for PV.
Author Contribution
Dedee F Murrell: Conceptualization (lead); Data curation (lead); Formal analysis (lead); Investigation (lead); Methodology (lead); Writing‐original draft (lead); Writing‐review & editing (lead). Aikaterini Patsatsi: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Panagiotis Stavropoulos: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). sharon baum: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Tal Zeeli: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Johannes S Kern: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Angeliki‐Viktoria Roussaki‐Schulze: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Rodney Sinclair: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Ioannis Bassukas: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Dolca Thomas: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Ann Neale: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (supporting); Methodology (equal); Supervision (equal); Validation (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Puneet Arora: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (supporting); Methodology (equal); Supervision (equal); Validation (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Frédéric Caux: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). Victoria Werth: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Investigation (equal); Writing‐review & editing (equal). S. G. Gourlay: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Writing‐review & editing (equal). Pascal Joly: Conceptualization (lead); Data curation (equal); Formal analysis (equal); Investigation (equal); Methodology (equal); Writing‐original draft (equal); Writing‐review & editing (equal).
Supporting information
Appendix S1 Study protocol.
Appendix S2 Supplementary methods, results and references.
Figure S1 BELIEVE study design.
Figure S2 BELIEVE CONSORT diagram.
Figure S3 (a) Rilzabrutinib pharmacokinetics and (b) Bruton tyrosine kinase occupancy.
Table S1 List of BELIEVE study investigators.
Table S2 Efficacy results of rilzabrutinib in intent‐to‐treat (n = 27) and completers set for complete response (n = 24) patient populations.
Powerpoint S1 Journal Club Slide Set.
Acknowledgments
We thank the patients who participated in the trial, their caregivers, the trial investigators (Table S1), members of the BELIEVE‐PV data and safety monitoring board and adjudication committee, and members of the BELIEVE‐PV trial team. Thank you to Professor Branka Marinovic for providing clinical expertise that supported the conception of the study and for screening patients, although none were enrolled at her institution. We also thank Fiona Liu for statistical support, and Julie Kern, PhD, CMPP and Kristin MacIntosh Teasell, MS, CMPP of Second City Science LLC for their medical writing and editorial assistance (funded by Principia Biopharma Inc., a Sanofi Company). This study was supported by Principia Biopharma Inc., a Sanofi Company. Statistical analyses of the data were provided by Principia Biopharma Inc., a Sanofi Company.
Conflicts of interest
D.F.M. reports advisory board and principal investigator or investigator roles for Principia Biopharma and Roche; is a principal investigator for clinical studies for or is an advisor supported by AbbVie, Amgen, ArgenX, AstraZeneca, Botanix, Dermira, Eli Lilly, Galderma, Janssen, LEO, Lilly, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma and UCB; and has a patent for treatment of epidermolysis bullosa with topical sirolimus. A.P. reports personal fees from Principia Biopharma. P.S. reports research funding from Principia Biopharma; and compensation for travel, accommodation and expenses from Frezyderm SA. J.S.K. reports fees to the institution for consulting from CSL and Exopharm; for an advisory board from Novartis; and for conducting clinical trials from AbbVie, Amgen, Amicus, Amryt, Arena, BMS, Boehringer, Cutanea, CSL, Dermira, Eli Lilly, Galderma, Novartis, Pfizer, Principia Biopharma, Regeneron and UCB Biopharma. R.S. reports participating as a principal investigator or investigator for clinical studies conducted by AbbVie, Arcturus, AstraZeneca, Bayer, Boston Pharmaceutical, Botanix, Dermira, Eli Lilly, Galderma, Janssen, LEO Pharma, Principia Biopharma, Regeneron, Reistone and UCB Biopharma; reports grant and investigator support from Sun Pharma; and is principal investigator, director and founder of Samson Clinical, with a patent for the treatment of autoimmune disease (#62811353 related to a different medication – tofacitinib). I.D.B. reports personal fees and nonfinancial support from Genesis Pharma, LEO Pharma and Novartis; and nonfinancial support from Roche. D.T., A.N. and P.A. are or were employees of and received stock from Principia Biopharma Inc., a Sanofi Company, at the time of the study. F.C. reports consulting or advisory roles and honoraria from Pierre Fabre Dermatologie, Principia Biopharma, LEO Pharma and Novartis; and participated as a principal investigator for clinical trials conducted by Principia Biopharma, Regeneron and Roche Laboratories. V.P.W. reports personal fees from Principia Biopharma; grant and personal fees and nonfinancial support from Genentech; grant and personal fees from AstraZeneca, Janssen and Syntimmune; and personal fees and nonfinancial support from ArgenX; and holds copyright for PDAI with no cost to license. S.G.G. is a former employee and stockholder of Principia Biopharma. P.J. reports consulting or advisory roles and honoraria from ArgenX, AstraZeneca, Eli Lilly, Novartis and Sanofi; and compensation for travel, accommodation and expenses from AbbVie, Eli Lilly and Novartis. S.B., T.Z. and A.‐V.R‐S. declare they have no conflicts of interest.
Funding sources This study was supported by Principia Biopharma Inc., a Sanofi company.
Conflicts of interest Conflicts of interest statements can be found in the Appendix.
Data availability Qualified researchers may request access to patient‐level data and related documents (including, e.g., the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications). Patient‐level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data sharing criteria, eligible studies, and process for requesting access can be found at https://www.clinicalstudydatarequest.com.
A complete list of investigators in the BELIEVE trial is provided in Table S1 (see Supporting Information).
The study results were previously presented in part as a poster presentation at the 2018 Medical Dermatology Society meeting, and in oral presentations at the late breaker sessions at the 2018 European Academy of Dermatology and Venereology meeting in Geneva, Switzerland, and the 2019 American Academy of Dermatology and European Academy of Dermatology and Venereology meetings.
Plain language summary available online
References
- 1. Didona D, Maglie R, Eming R et al. Pemphigus: current and future therapeutic strategies. Front Immunol 2019; 10:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wertenteil S, Garg A, Strunk A et al. Prevalence estimates for pemphigus in the United States: a sex‐ and age‐adjusted population analysis. JAMA Dermatol 2019; 155:627–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murrell DF, Pena S, Joly P et al. Diagnosis and management of pemphigus: recommendations of an international panel of experts. J Am Acad Dermatol 2020; 82:575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kempf W, Hantschke M, Kutzner H et al. Dermatopathology. Heidelberg, Germany: Steinkopff‐Verlag Heidelberg, 2008. [Google Scholar]
- 5. Joly P, Maho‐Vaillant M, Prost‐Squarcioni C et al. First‐line rituximab combined with short‐term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel‐group, open‐label randomised trial. Lancet 2017; 389:2031–40. [DOI] [PubMed] [Google Scholar]
- 6. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet 2019; 394:882–94. [DOI] [PubMed] [Google Scholar]
- 7. Rituxan (rituximab) [prescribing information]. South San Francisco, CA: Genentech, Inc., 2019. [Google Scholar]
- 8. Bradshaw JM, McFarland JM, Paavilainen VO et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 2015; 11:525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer 2018; 17:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith PF, Krishnarajah J, Nunn PA et al. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br J Clin Pharmacol 2017; 83:2367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langrish CL, Francesco MR, Xing Y et al. Rilzabrutinib (PRN1008) shows BTK‐mediated mechanisms of action supporting clinical development for immune‐mediated diseases. J Invest Dermatol 2020; 140:S78 (abstract 569). [Google Scholar]
- 12. Langrish CL, Bradshaw JM, Francesco MR et al. Preclinical efficacy and anti‐inflammatory mechanism of action of Bruton tyrosine kinase inhibitor rilzabrutinib for immune‐mediated disease. J Immunol 2021; 206:1454–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hill RJ, Bradshaw MJ, Bisconte A et al. Preclinical characterization of PRN1008, a novel reversible covalent inhibitor of BTK that shows efficacy in a rat model of collagen‐induced arthritis. Ann Rheum Dis 2015; 74:216 (abstract THU0068). [Google Scholar]
- 14. Goodale EC, White SD, Bizikova P et al. Open trial of Bruton’s tyrosine kinase inhibitor (PRN1008) in the treatment of canine pemphigus foliaceus. Vet Dermatol 2020; 31:410–e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimizu T, Takebayashi T, Sato Y et al. Grading criteria for disease severity by pemphigus disease area index. J Dermatol 2014; 41:969–73. [DOI] [PubMed] [Google Scholar]
- 16. Werth VP, Fivenson D, Pandya AG et al. Multicenter randomized, double‐blind, placebo‐controlled, clinical trial of dapsone as a glucocorticoid‐sparing agent in maintenance‐phase pemphigus vulgaris. Arch Dermatol 2008; 144:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murrell DF, Dick S, Ahmed AR et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol 2008; 58:1043–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. CTCAE . Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. US Department of Health and Human Services; National Institutes of Health, National Cancer Institute, v4.03: 14 June 2010.
- 19. Hertl M, Jedlickova H, Karpati S et al. Pemphigus. S2 Guideline for diagnosis and treatment – guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol 2015; 29:405–14. [DOI] [PubMed] [Google Scholar]
- 20. Rosenbach M, Murrell DF, Bystryn JC et al. Reliability and convergent validity of two outcome instruments for pemphigus. J Invest Dermatol 2009; 129:2404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sebaratnam DF, Hanna AM, Chee SN et al. Development of a quality‐of‐life instrument for autoimmune bullous disease: the Autoimmune Bullous Disease Quality of Life questionnaire. JAMA Dermatol 2013; 149:1186–91. [DOI] [PubMed] [Google Scholar]
- 22. Wilson MM, Thomas DR, Rubenstein LZ et al. Appetite assessment: simple appetite questionnaire predicts weight loss in community‐dwelling adults and nursing home residents. Am J Clin Nutr 2005; 82:1074–81. [DOI] [PubMed] [Google Scholar]
- 23. Ishii K, Amagai M, Hall RP et al. Characterization of autoantibodies in pemphigus using antigen‐specific enzyme‐linked immunosorbent assays with baculovirus‐expressed recombinant desmogleins. J Immunol 1997; 159:2010–17. [PubMed] [Google Scholar]
- 24. Rip J, Van Der Ploeg EK, Hendriks RW et al. The role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit Rev Immunol 2018; 38:17–62. [DOI] [PubMed] [Google Scholar]
- 25. Ferries L, Gilibert A, Duvert‐Lehembre S et al. Sensitivity to change and correlation between the autoimmune bullous disease quality‐of‐life questionnaires ABQOL and TABQOL, and objective severity scores. Br J Dermatol 2020; 183:944–5. [DOI] [PubMed] [Google Scholar]
- 26. Calquence (acalabrutinib) [prescribing information]. Wilmington, DE: AstraZeneca Pharmaceuticals LP, 2019. [Google Scholar]
- 27. Imbruvica (ibrutinib) [prescribing information]. Sunnyvale, CA: Pharmacyclics LLC, 2020. [Google Scholar]
- 28. Goodale EC, Varjonen KE, Outerbridge CA et al. Efficacy of a Bruton’s tyrosine kinase inhibitor (PRN473) in the treatment of canine pemphigus foliaceus. Vet Dermatol 2020; 31:291–e71. [DOI] [PubMed] [Google Scholar]
- 29. Hebert V, Boulard C, Houivet E et al. Large international validation of ABSIS and PDAI pemphigus severity scores. J Invest Dermatol 2019; 139:31–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Study protocol.
Appendix S2 Supplementary methods, results and references.
Figure S1 BELIEVE study design.
Figure S2 BELIEVE CONSORT diagram.
Figure S3 (a) Rilzabrutinib pharmacokinetics and (b) Bruton tyrosine kinase occupancy.
Table S1 List of BELIEVE study investigators.
Table S2 Efficacy results of rilzabrutinib in intent‐to‐treat (n = 27) and completers set for complete response (n = 24) patient populations.
Powerpoint S1 Journal Club Slide Set.