

Abstract
XRCC1 protein is essential for viability in mammals and is required for efficient DNA single-strand break repair and genetic stability following DNA base damage. We report here that XRCC1-dependent strand break repair in G1 phase of the cell cycle is abolished by mutations created within the XRCC1 BRCT domain that interact with DNA ligase III. In contrast, XRCC1-dependent DNA strand break repair in S phase is largely unaffected by these mutations. These data describe a cell cycle-specific role for a BRCT domain, and we conclude that the XRCC1-DNA ligase III complex is required for DNA strand break repair in G1 phase of the cell cycle but is dispensable for this process in S phase. The S-phase DNA repair process can remove both strand breaks induced in S phase and those that persist from G1 and can in part compensate for lack of repair in G1. This process correlates with the appearance of XRCC1 nuclear foci that colocalize with Rad51 and may thus function in concert with homologous recombination.
DNA strand breakage can result in chromosomal rearrangement and is a major threat to genetic stability. Of particular threat are breaks that arise from damaged DNA bases, several thousand of which occur spontaneously per cell each day (20). The most common such breaks are single-strand breaks, which are formed as intermediates of base excision repair. The threat to genetic stability from DNA strand breaks that arise from base damage is illustrated by the phenotype of rodent cells that harbor mutations within the DNA repair gene XRCC1. XRCC1 is essential for embryonic development in mice (35), and XRCC1 mutant mouse or CHO cells that possess little or no XRCC1 protein exhibit increased frequencies of spontaneous sister chromatid exchange and chromosomal aberration (6, 7, 12, 29, 35, 39). XRCC1 mutant cells appear unable to efficiently rejoin DNA single-strand breaks resulting from either endogenous base damage (35) or that induced by ionizing radiation or alkylating agents (36, 37, 39). Sequence analysis has not provided any indication of the role of XRCC1 in single-strand break repair (SSBR). However, biochemical studies have revealed that this protein interacts with DNA ligase III and DNA polymerase β (6–8, 18). Thus, it is possible that XRCC1 functions as a molecular chaperone or scaffold protein, stabilizing and/or modifying the activity of other polypeptides. For example, the interaction of XRCC1 with DNA ligase III appears to be required for normal cellular levels of the latter, and reduced levels of DNA ligase III can result in inefficient SSBR during the excision repair of abasic sites in vitro (6, 7, 11). This interaction is mediated by a C-terminal BRCT domain in XRCC1, designated BRCT II (23, 34). BRCT domains have been identified in more than 40 polypeptides, defining a novel protein superfamily, and typically span 80 to 100 amino acids (5, 10). The function of these structures appears to involve, but may not be restricted to, the mediation of protein-protein interactions (5, 10, 23, 34). In the present study, we have examined the importance of the XRCC1 BRCT II domain and its interaction with DNA ligase III to SSBR in Chinese hamster ovary (CHO) cells.
MATERIALS AND METHODS
Expression constructs, cell lines, and cell synchrony.
A mutant XRCC1pmBRCT open reading frame (ORF) was generated by subcloning constructs described previously (34) and was inserted into the mammalian expression vector pcD2E (17). All XRCC1 ORFs encode a decahistidine tag at the C terminus. XRCC1 mutant EM9 cells were transfected with pcD2E or pcD2E harboring XRCC1 by electroporation, and >50 independent G418r clones were pooled to generate the cell lines EM9-X, EM9-XpmBRCT, and EM9-V. Synchrony in G1 was achieved via serum starvation and incubation in mimosine (25). Cells were incubated sequentially in α-MEM plus 10% fetal bovine serum (FBS) for 12 to 15 h, α-MEM plus 0.1% FBS for 48 h, and α-MEM plus 10% FBS and 300 μM mimosine (Sigma) for 24 h. To maintain G1 synchrony, mimosine was retained in the medium for up to 7 h. To release into S phase or G2, incubation was continued in mimosine-free medium for 5 to 8 h (untreated cells) or 7 to 24 h (ethyl methanesulfonate [EMS]-treated cells). Mammalian cell survival assays were conducted as described previously (7).
Drosophila melanogaster XRCC1.
A number of D. melanogaster EST cDNA clones (GenBank accession no. AA392201, AA246970, AA264299, AA735260, AA817298, and AA142274) were identified in the GenBank EST database (nonredundant database of GenBank non-mouse and non-human EST entries) by using the TBLASTN homology search program with the human XRCC1 amino acid sequence as the query. Two EST clones were obtained for further analysis: AA392201, which was a full-length cDNA from the IMAGE consortium (IMAGE reference no. EST LD11571/AA392201), and AA142274, which was incomplete, from the Berkeley Drosophila Genome Project (BDGP clone CK350). The encoded ORFs were sequenced completely, and the amino acid sequence of the full-length clone was deposited in EMBL (accession code AJ010073). Comparison of the amino acid sequence of the full-length 1.8-kb ORF with that of human XRCC1 by using Multiple Alignment Construction and Analysis Workbench (MACAW) revealed significant homology to human XRCC1 (37.7% identity; 55.2% similarity). The region encoding XRCC1 was PCR amplified from genomic DNA extracted from D. melanogaster (Oregon-R) by using Taq polymerase and primers specific to the 5′- and 3′-untranslated regions of the fruit fly clone, AA392201. The product was sequenced directly.
DNA strand break repair assays.
Strand break repair was assayed by alkaline single-cell agarose gel electrophoresis (24, 31). Cells were harvested (∼105 per pellet), mixed with low-gelling-temperature agarose (Sigma; type VII), and layered onto agarose-coated glass slides. Slides were maintained in the dark at 4°C to gel and for all further steps. Slides were submerged in lysis buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris-Cl [pH 7.0], 1% Triton X-100, 1% dimethyl sulfoxide [DMSO]) for 1 h, washed with distilled water, and incubated for 45 min in alkaline electrophoresis buffer (50 mM NaOH, 1 mM EDTA, 1% DMSO [pH 12.8]). After electrophoresis (25 min, 25 V), air-dried and neutralized slides were stained with SYBR-Green I (FMC BioProducts). Average Comet Tail Moment (24) was scored for duplicate slides (100 cells/slide) by using Comet Assay II software (Perseptive Instruments). Under alkaline conditions, this assay quantifies DNA strand breaks and alkali-labile abasic sites. To quantitate repair, the fraction (percent) of EMS-induced DNA strand breakage remaining after incubation in drug-free medium was calculated by the equation [(mean tail momentafter repair − mean tail momentuntreated cells)/(mean tail momentinitial damage − mean tail momentuntreated cells)] × 100.
Indirect immunofluorescence.
Subconfluent monolayers were resuspended in medium or phosphate-buffered saline (PBS) at 1 × 105 to 5 × 105 cells/ml. Cells (0.1 × 105 to 1 × 105) were deposited on glass slides by cytospin and fixed in 50% methanol–50% acetone. PBS-washed slides were incubated sequentially for 1 h in anti-XRCC1 monoclonal antibody (33-2-5), fluorescein isothiocyanate–goat anti-mouse Fab (DAKO; 1/50 dilution in PBS), anti-Rad51 (FBE-1 [2]), or anti-Pol-β (7545) polyclonal antibody and Cy3-goat anti-rabbit immunoglobulin G (Sigma; 1/300 or greater dilution). Cells were incubated in antifade and 4′,6′-diamidino-2-phenylindole (DAPI) and analyzed with a Zeiss fluorescent microscope at ×400 magnification. Images were photographed and colored or overlaid by using Adobe Photoshop. Frequencies of focus-positive cells were calculated from >100 cells per data point per slide and from multiple independent experiments. A single clone of EM9-X was used for immunostaining to circumvent the heterogeneity of expression levels observed in pooled transfectants.
RESULTS
XRCC1 mediates SSBR by BRCT domain-dependent and -independent mechanisms.
The discovery of an XRCC1 homologue in D. melanogaster (Fig. 1a) identifies a number of domains that are highly conserved with the human protein, including an internal BRCT domain (BRCT I) and the amino terminus that binds DNA polymerase β (8, 18). Surprisingly, however, the BRCT II domain that interacts with DNA ligase III in human XRCC1 (23, 34) was absent from two fruit fly cDNA clones, recovered from independent libraries, and from the genomic sequence of these clones (Fig. 1a) (unpublished observations). Although the presence in fruit flies of an XRCC1 homologue that contains a BRCT II domain cannot be excluded, it was considered more likely that BRCT II is evolutionarily a recent addition to the XRCC1 structure and that the conserved role of this protein does not involve interaction with DNA ligase III.
FIG. 1.

SSBR independent of XRCC1-DNA ligase III complex. (a) An alignment of human and fruit fly XRCC1 proteins was conducted by MACAW. The ORF is boxed, with gaps for optimal alignment inserted by MACAW. Filled blocks indicate identical residues, and regions of extensive homology are grouped in large blocks. The latter include the N-terminal domain that contains the DNA polymerase β binding site and the internal BRCT I domain. The BRCT II domain in human XRCC1 is hatched, and the positions of the W611D, V584D, and I585D mutations in XRCC1pmBRCT are underlined. (b) Total cell extract (top panel) or protein recovered by metal-chelate affinity chromatography (middle and bottom panels) from the indicated cell lines (see Key) was immunoblotted for DNA ligase III (top and bottom panels) or XRCC1 (middle panel). (c) EM9-X cells, EM9-XpmBRCT cells, or EM9 cells harboring empty vector (EM9-V) were treated with EMS for 1 h and incubated in drug-free medium for 7 to 10 days. The fraction of surviving cells (percent) is shown. Error bars are within 10% of each value and omitted for clarity. (d) Single-strand breakage (expressed as Tail Moment) present in EM9 transfectants before (−EMS) and immediately after (+EMS) EMS treatment (10 mg/ml; 15 min). (e) EMS-induced single-strand breakage (percent) remaining after 3 h in drug-free medium is shown. Values are the mean (± standard deviation) of at least three experiments.
To directly examine the importance of the BRCT II domain to mammalian SSBR, human XRCC1 in which this structure was disrupted was examined for the ability to correct XRCC1 mutant EM9 cells. Two point mutations were chosen (Fig. 1a): one which removes the single most highly conserved amino acid in BRCT domains and is proposed to abolish correct folding of BRCT II (W611D) (40) and a double mutation that abolishes measurable interaction with DNA ligase III in vitro (V584D-I585D) (34). Expression constructs encoding either wild-type human XRCC1 or the mutated derivative (designated XRCC1pmBRCT) were introduced into the XRCC1 mutant CHO cell line, EM9, and G418-resistant transfectants were pooled for analysis. Strikingly, despite the inability of XRCC1pmBRCT protein to interact with DNA ligase III in vitro, the resistance of EM9-XpmBRCT cells expressing this protein to the alkylating agent EMS was similar to that of EM9-X cells expressing wild-type XRCC1 (Fig. 1c). Two observations support the notion that the XRCC1-DNA ligase III complex was absent from EM9-XpmBRCT cells. First, whereas DNA ligase III copurified with XRCC1 from EM9-X cell extract during affinity chromatography, it did not measurably copurify from EM9-XpmBRCT cell extract (Fig. 1b, middle and bottom panels). Second, whereas expression of wild-type XRCC1 increased DNA ligase III levels more than sixfold, expression of XRCC1pmBRCT failed to increase such levels above those present in EM9-V cells harboring empty vector (Fig. 1b, top panel). Staining with Ponceau S confirmed that equal amounts of total protein were present on these immunoblots (unpublished observations).
SSBR proficiency in the CHO cells after EMS treatment was measured by alkaline single-cell agarose gel electrophoresis (SCGE). Strand breakage measured in this assay was expressed as the Tail Moment, which is the product of the fraction of DNA that exited the nucleus during electrophoresis multiplied by the distance migrated (24, 31). The DNA strand breakage measured in these experiments reflects the balance between breaks arising from base excision or alkali-labile sites and breaks rejoined by SSBR. The level of DNA strand breakage present in EM9-X cells immediately after EMS treatment was lower than that present in EM9-XpmBRCT cells or EM9-V cells harboring empty vector (Fig. 1d, +EMS). Given that these cell lines differ only in XRCC1 status, and that this protein has only been observed to affect the rate of strand break repair (36, 39), this difference presumably reflects relative SSBR proficiency during the 15-min incubation with EMS. This, in turn, suggests that the BRCT II domain is required for efficient SSBR at early times after base damage. In contrast, DNA strand breakage decreased by 60 to 65% in both EM9-X and EM9-XpmBRCT cells during a subsequent 3-h repair incubation in drug-free medium, suggesting that the BRCT II domain was largely dispensable for SSBR over longer periods (Fig. 1e). In EM9-V cells, the amount of strand breakage appeared to increase by ∼35% during repair incubation, presumably reflecting continued base excision, or exonuclease activity, in the absence of any XRCC1-dependent SSBR (Fig. 1e).
BRCT II-dependent and -independent SSBR processes are cell cycle distinct.
The results described above suggest that the BRCT II domain is important for efficient SSBR at early times after base damage but that XRCC1 can mediate SSBR independently of this structure over longer periods. In an attempt to discriminate these SSBR mechanisms further, we examined whether they exhibited cell cycle specificity. EM9 transfectants were synchronized in late G1 by a combination of serum starvation and mimosine treatment (24), treated with EMS while arrested in G1 or after release into S phase, and subsequently incubated for 3 h in drug-free medium to allow time for repair. Whereas the levels of strand breakage present in G1 and S phase were similar after EMS treatment, that present before EMS treatment was slightly higher in the latter (Fig. 2, middle panels). This may reflect the presence of Okazaki fragments in S-phase cells. As expected, EM9-V cells were deficient, and EM9-X cells were proficient, in SSBR in both cell cycle phases (Fig. 2, right panels). Strikingly, however, EM9-XpmBRCT cells were deficient in SSBR in G1 but were proficient in SSBR in S phase, in the latter case reducing strand breakage by ∼80% (Fig. 2, right panels). To examine whether the inability of XRCC1pmBRCT protein to restore efficient SSBR to EM9 was due solely to its inability to restore normal levels of DNA ligase III, we compared SSBR proficiencies in EM9-X and EM9-XpmBRCT cells in the presence of specific inhibitors of the 20S proteosome. However, despite increases in DNA ligase III levels in EM9-XpmBRCT of three- to fourfold (∼50% of wild-type levels), the proteosome inhibitors did not increase G1 repair (Fig. 3). The inhibitors similarly did not affect repair in EM9-X cells. Similar results were observed when DNA ligase III levels were increased twofold by expression of the human cDNA (unpublished observations). These results suggest that the BRCT II domain is required during G1 SSBR for more than the maintenance of DNA ligase III protein levels, although complete restoration of such levels is required to confirm this.
FIG. 2.

Cell cycle-distinct SSBR. (Left panels) Flow cytometry profile of G1 (A)- or S (B)-phase cells; (middle panels) DNA strand breakage before (−EMS) or immediately after (+EMS) treatment of G1 (a)- or S (b)-phase cells with EMS (10 mg/ml; 15 min); (right panels) EMS-induced strand breakage (percent) remaining in G1 (A)- or S (B)-phase cells after 3 h in drug-free medium.
FIG. 3.

DNA ligase III protein levels and SSBR proficiency. (A) DNA strand breakage in mock-treated (−EMS) or EMS-treated (+EMS) G1-synchronized EM9-X or EM9-XpmBRCT cells (see Key). The proteosome inhibitors (Calbiochem) lactacystin (1 μM) and MG-132 (10 μM) were present (+) or absent (−) for 4 h prior to and during EMS or mock treatment. (b) Fraction of EMS-induced strand breakage remaining in G1-synchronized cells after a 3-h repair incubation in EMS-free medium in the presence (+) or absence (−) of proteosome inhibitors. Strand breakage present immediately after EMS is set at 100% (dotted line). (c) Immunoblotting of cell extract from EM9-X or EM9-XpmBRCT cells (see Key) following synchronization in G1 and incubation for 4 h in the presence (+) or absence (−) of proteosome inhibitors for XRCC1 or DNA ligase III. Strand break data are the mean (± data range) of two determinations.
Since EM9-XpmBRCT cells exhibited wild-type levels of EMS sensitivity over the dose range employed (Fig. 1c), it was considered possible that S-phase SSBR can remove not only strand breaks induced in S phase but also those that persist from G1. To test this prediction, EM9 transfectants were synchronized in G1 and treated with EMS for 15 min and then either maintained in G1 for a further 7 h in drug-free medium or released into S phase for the same period (see Fig. 4a for experimental design). As expected, EM9-XpmBRCT cells failed to reduce EMS-induced DNA strand breakage if maintained in G1 throughout repair incubation (Fig. 4b). In contrast, however, strand breakage fell by ∼70% in EM9-XpmBRCT cells that were released into S phase during repair incubation (Fig. 4b). Control experiments confirmed that EM9-V cells failed to reduce EMS-induced strand breakage in either cell cycle phase (Fig. 4b) and that EM9-X cells did so in both cell cycle phases (unpublished observations). These results confirm that S-phase SSBR can rejoin DNA strand breaks that persist from G1.
FIG. 4.

S-phase SSBR can remove breaks induced in G1. (Left and middle panels) Flow cytometry profiles of G1-synchronized cells immediately after treatment with EMS (G1-syn.) or after 7 h in drug-free medium (G1+7hr) in the presence (+mim.) or absence (−mim.) of mimosine to maintain G1 status or allow entry into S phase, respectively. (Right panel) DNA strand breakage (percent) remaining after 7 h in drug-free medium in the presence (G1) or absence (S) of mimosine.
S-phase SSBR correlates with XRCC1 foci that partially colocalize with Rad51.
In an attempt to further characterize the role of XRCC1 in S-phase SSBR, the subcellular localization of XRCC1 was examined. Immunostaining of fixed asynchronous EM9-X cells with anti-XRCC1 antibodies revealed nuclei with discrete foci and/or a less-intense speckled staining (Fig. 5A). Both staining patterns were greatly reduced or absent in untransfected EM9 cells. The behavior of the less-intense staining during the cell cycle was difficult to determine. However, the frequency of focus-positive cells appeared to increase in EM9-X cells during progression through S phase but did not increase if cells were maintained in G1 throughout the experiment (Fig. 5B). These data suggest that the level of XRCC1 foci increases in S phase and are consistent with the participation of these structures in S-phase SSBR. XRCC1 focus-positive cells were also observed in asynchronous populations of EM9-XpmBRCT, the frequency of which was increased ∼3-fold 8 h after EMS treatment (Fig. 5C, top panels). EMS-induced increases in focus-positive cells in asynchronous populations of EM9-X were also observed but were slower and less pronounced, with little or no increase observed 8 h after EMS treatment (Fig. 5c, bottom panels) and ∼2-fold more observed 24 h after treatment (unpublished observations). The elevated or accelerated assembly of foci in EM9-XpmBRCT cells after EMS treatment is also consistent with an involvement of these structures in S-phase SSBR, since these cells are more dependent on the latter process. However, it is also possible that the mutations in XRCC1pmBRCT inhibit focus turnover.
FIG. 5.
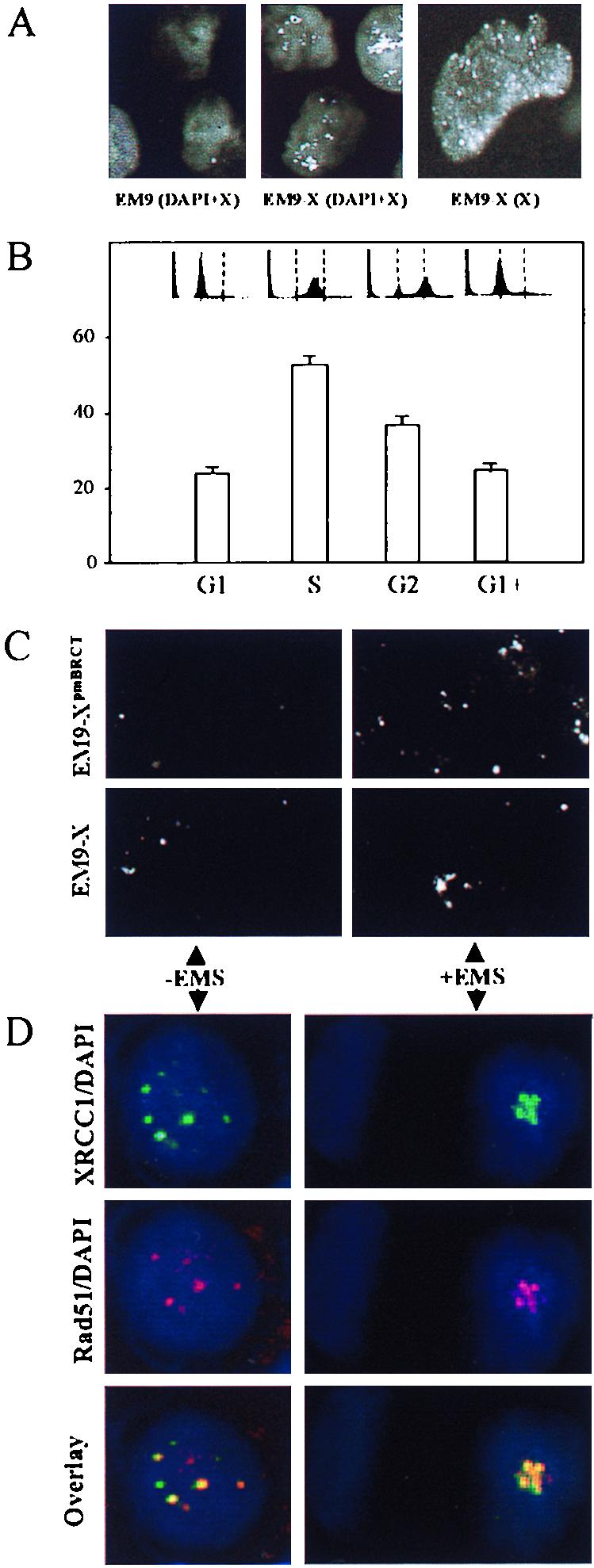
XRCC1 foci. (A) EM9 (left panel) and EM9-X (middle panel) cells immunostained with anti-XRCC1 monoclonal antibody and DAPI. White spots are XRCC1 foci, and grey background is DAPI-stained nuclear DNA. The right panel shows EM9-X cells immunostained with anti-XRCC1 monoclonal antibody. White spots are foci, and grey background is diffuse or speckled staining. (B) Frequency of XRCC1 focus-positive EM9-X cells following synchronization in G1, during transit through S phase or G2, or after maintenance in G1 throughout the experiment (G1+). The flow cytometry profiles are shown with dotted lines indicating G1 and G2. (C) Asynchronous EM9-X or EM9-XpmBRCT cells immunostained for XRCC1 (white dots) before or 8 h after EMS treatment (2 mg/ml, 1 h). Each panel depicts approximately six cells. (D) Asynchronous HeLa cells were harvested and fixed before (−EMS) or 8 h after (+EMS) EMS treatment (2 mg/ml, 1 h) and stained with DAPI plus anti-XRCC1 monoclonal antibody (green; top panels), DAPI plus anti-Rad51 polyclonal antibody (red; middle panels), or DAPI plus anti-XRCC1 and anti-Rad51 (bottom panels, overlapping antibody signals in yellow).
The behavior of XRCC1 foci was reminiscent of Rad51 nuclear foci (14, 33). Rad51 is a homologue of the bacterial RecA recombination protein (1, 4, 22, 30). To examine whether the roles of XRCC1 and Rad51 after base damage might be related, we compared the subnuclear distribution of the two proteins in HeLa cells before and after exposure to EMS. XRCC1 and Rad51 partially colocalized both before and 8 h after EMS treatment (Fig. 5D). Two EMS-treated cells are shown (Fig. 5D, right panels), one of which lacks XRCC1 and Rad51 foci and one of which contains a cluster of both. While the level of colocalization depicted in Fig. 5 is representative, the total number and position of nuclear foci varied. These data raise the possibility that XRCC1 and Rad51 fulfill related roles after DNA base damage.
DISCUSSION
XRCC1-dependent SSBR in G1.
We report here that mammalian SSBR in G1 phase of the cell cycle is abolished by mutation of the XRCC1 BRCT II. The mutant XRCC1 protein employed in this study combines two mutations, W611D and the double mutation V584D-I585D, but similar results were observed with XRCC1 harboring either of these mutations alone (unpublished observations). It seems unlikely that these mutations exert a dominant-negative effect on XRCC1 activities located outside of the BRCT II domain because a number of such activities (e.g., binding to DNA polymerase β) are normal in XRCC1pmBRCT (unpublished observations) and because the mutant protein is still able to mediate SSBR in S phase. Rather, these data suggest that the BRCT II domain is required for SSBR in G1, following DNA base damage. The V584D-I585D double mutation prevents measurable interaction of XRCC1 with DNA ligase III in vitro (34). Since the XRCC1-DNA ligase III interaction maintains normal cellular levels of DNA ligase III (6, 7), reduced levels of which result in decreased SSBR during base excision repair in vitro (11), it seems likely that at least one role for BRCT II is to maintain normal levels of the DNA ligase. However, it is unlikely that this is the only role for this domain because increasing the level of DNA ligase III in EM9-XpmBRCT cells with proteosome inhibitors did not increase SSBR. It is possible that XRCC1 also chaperones DNA ligase III to sites of base damage and/or modifies its activity, or that the BRCT II domain fulfills a role that is independent of the DNA ligase III interaction.
Interaction with DNA ligase III may be evolutionarily a relatively recent addition to XRCC1 function, since a homologue of XRCC1 identified in fruit flies lacks the BRCT II domain that in human XRCC1 binds the DNA ligase. It is possible that XRCC1-DNA ligase III complex has arisen in higher eukaryotes to supplement SSBR because the larger genomes of these organisms acquire more spontaneous base damage. Another DNA ligase apparently involved in SSBR following base damage is DNA ligase I (26, 27). DNA ligase I activity was not detected in this study, however, as indicated by the apparent absence of residual SSBR in EM9-V cells (see Fig. 1 to 3). This may indicate that the half-lives of strand breaks generated and repaired by the DNA ligase I base excision repair complex (26) are too short to be detected.
XRCC1-dependent SSBR in S phase.
Based on the extensive conservation between human and fruit fly XRCC1 outside of the BRCT II domain (Fig. 1a), it seems likely that it is S-phase SSBR that is the evolutionarily conserved role of this protein. Although the BRCT II domain is largely dispensable for mammalian SSBR during S phase, this structure may contribute to the process, since the efficiency of repair in EM9-XpmBRCT cells was slightly less than in EM9-X cells (Fig. 2b, right panel). Nevertheless, SSBR mediated by XRCC1 in S phase was sufficient to maintain cellular resistance to an alkylating agent (0 to 1 mg of EMS per ml) in the absence of repair in G1. It is likely that the ability of S-phase SSBR to remove strand breaks that persist from G1 is important in this respect. It is noteworthy that the substrate specificity of SSBR mediated by XRCC1 during S phase may not be restricted to breaks arising from base damage. This is suggested by the observation that XRCC1 mutant cells are hypersensitive to the antitumor agent camptothecin, which breaks DNA in S phase independently of base damage (3, 9, 15, 16, 28).
The conclusion that S-phase SSBR does not require XRCC1-DNA ligase III complex is based on three observations. First, the mutations in XRCC1pmBRCT prevent measurable interaction with DNA ligase III in vitro (34) and copurification of the two proteins from cell extract (Fig. 1b, bottom panel). Second, XRCC1pmBRCT failed to increase levels of DNA ligase III above those present in EM9 cells transfected with empty vector (Fig. 1b, top panel, compare lanes 1 and 2). This observation is particularly significant because it reflects the amount of complex present in whole cells before lysis. Finally, an XRCC1 deletion mutant lacking the entire DNA ligase III-binding BRCT II domain also confers resistance to EMS (unpublished observations). This observation renders unlikely the caveat that S-phase SSBR mediated by XRCC1pmBRCT results from leaky point mutations that allow assembly of a small but undetectable amount of complex. Since S-phase SSBR does not appear to require XRCC1-DNA ligase III complex, what role does XRCC1 play in this process? It is possible that XRCC1 is required during S phase for its interaction with DNA polymerase β (8, 18) or poly(ADP-ribose) polymerase (8, 21), perhaps to chaperone these proteins to sites of damage or to modify their activity.
XRCC1 nuclear foci.
Immunostaining with anti-XRCC1 monoclonal antibody revealed a speckled distribution of XRCC1 in CHO cell nuclei and also the presence of less frequent, but more intensely staining, foci. Several observations are consistent with the foci being associated with S-phase SSBR. First, the frequency of focus-positive EM9-X cells appeared to increase two- to threefold during progression through S phase. Second, EM9-XpmBRCT cells that are more reliant on S-phase SSBR exhibited elevated and accelerated assembly of XRCC1 foci relative to EM9-X cells. Third, the XRCC1 foci partially colocalized with Rad51 protein, which in somatic cells appears to mediate strand break repair specifically in S/G2 (32). The colocalization of XRCC1 with Rad51 raises the possibility that XRCC1-dependent S-phase SSBR is coordinated or involved with recombination events initiated by strand breaks during replication. Given the frequency at which base damage arises spontaneously (20), XRCC1 and Rad51 might be required routinely during S phase in this scenario, perhaps explaining the similar embryonic-lethal phenotype of XRCC1−/− and Rad51−/− nullizygous mice (19, 35, 38). Irrespective of the relationship between XRCC1 and Rad51, it is tempting to speculate that XRCC1 foci reflect the sequestration of this protein into base excision repair and/or strand break repair “factories” through which replicating DNA can translocate.
ACKNOWLEDGMENTS
R. M. Taylor and D. J. Moore contributed equally to this work.
We thank Grant Haynes and Ian Morris for help with SCGE and Fiona Benson and Steve West for Rad51 antibodies.
This work was funded by the Medical Research Council (grants G9603130 and G9809326).
REFERENCES
- 1.Aboussekhra A, Chanet A, Adjiri A, Fabre F. Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to prokaryotic RecA proteins. Mol Cell Biol. 1992;12:3224–3234. doi: 10.1128/mcb.12.7.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barlow A L, Benson F E, West S C, Hulten M A. Distribution of the Rad51 recombinase in human and mouse spermatocytes. EMBO J. 1997;16:5207–5215. doi: 10.1093/emboj/16.17.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrows L R, Holden J A, Anderson M, D'Arpa P. The CHO XRCC1 mutant, EM9, deficient in DNA ligase III activity, exhibits hypersensitivity to camptothecin independent of DNA replication. Mutat Res. 1998;408:103–110. doi: 10.1016/s0921-8777(98)00022-6. [DOI] [PubMed] [Google Scholar]
- 4.Basile G, Aker M, Mortimer R K. Nucleotide sequencing and transcriptional regulation of the yeast recombinational repair gene, RAD51. Mol Cell Biol. 1992;12:3235–3246. doi: 10.1128/mcb.12.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bork P, Hofmann K, Bucher P, Neuwald A F, Altschul S F, Koonin E V. A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- 6.Caldecott K W, McKeown C K, Tucker J D, Ljungquist S, Thompson L H. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol. 1994;14:68–76. doi: 10.1128/mcb.14.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldecott K W, Tucker J D, Stanker L H, Thompson L H. Characterisation of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res. 1995;23:4836–4843. doi: 10.1093/nar/23.23.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caldecott K W, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase β and possibly poly(ADP-ribose) polymerase, and DNA ligase III is a novel ‘nick sensor’ in vitro. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caldecott K, Jeggo P. Cross-sensitivity of γ-ray-sensitive hamster mutants to cross-linking agents. Mutat Res. 1991;255:111–121. doi: 10.1016/0921-8777(91)90046-r. [DOI] [PubMed] [Google Scholar]
- 10.Callebaut I, Mornon J P. From BRCA1 to RAP1. A widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- 11.Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K, Frosina G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J Biol Chem. 1997;272:23970–23975. doi: 10.1074/jbc.272.38.23970. [DOI] [PubMed] [Google Scholar]
- 12.Carrano A V, Minkler J L, Dillehay L H, Thompson L H. Incorporated bromodeoxyuridine enhances the sister-chromatid exchange and chromosomal aberration frequencies in an EMS-sensitive Chinese hamster cell line. Mutat Res. 1986;162:233–239. doi: 10.1016/0027-5107(86)90090-4. [DOI] [PubMed] [Google Scholar]
- 13.Friedberg E C, Walker G C, Siede W, editors. DNA repair and mutagenesis. Washington, D.C.: American Society for Microbiology; 1995. [Google Scholar]
- 14.Haaf T, Golub E I, Reddy G, Radding C, Ward D C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its colocalisation in synaptonemal complexes. Proc Natl Acad Sci USA. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm C, Covey J M, Kerrigan D, Pommier Y. Differential requirement of DNA replication for the cytotoxicity of CAN topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989;49:6365–6368. [PubMed] [Google Scholar]
- 16.Hsiang Y-H, Lihou M G, Liu L F. Arrest of replication forks by drug-stabilised topoisomerase I-cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 17.Kadkhodayan S, Salazar E P, Lamerdin J E, Weber C A. Construction of a functional cDNA clone of the hamster ERCC2 DNA repair and transcription gene. Somat Cell Mol Genet. 1996;22:453–460. doi: 10.1007/BF02369437. [DOI] [PubMed] [Google Scholar]
- 18.Kubota Y, Nash R A, Klungland A, Schar P, Barnes D, Lindahl T. Reconstitution of DNA base excision repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 19.Lim D-S, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–714. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 21.Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de-Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita T, Yoshimura Y, Yamamoto A, Murata M, Mori H, Yamamoto H, Matsushiro A. A mouse homolog of the Escherichia coli recA and Saccharomyces cerevisiae RAD51 genes. Proc Natl Acad Sci USA. 1993;90:6577–6580. doi: 10.1073/pnas.90.14.6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nash R A, Caldecott K W, Barnes D E, Lindahl T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry. 1997;36:5207–5211. doi: 10.1021/bi962281m. [DOI] [PubMed] [Google Scholar]
- 24.Olive P L, Banath J P, Durand R E. Heterogeneity in radiation-induced DNA damage and repair in tumour and normal cells measured using the ‘comet’ assay. Radiat Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 25.Orren D K, Petersen L N, Bohr V A. A UV-responsive G2 checkpoint in rodent cells. Mol Cell Biol. 1995;15:3722–3730. doi: 10.1128/mcb.15.7.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prasad R, Singhal R K, Srivastava D K, Molina J T, Tomkinson A, Wilson S H. Specific interaction of DNA polymerase beta and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J Biol Chem. 1996;271:16000–16007. doi: 10.1074/jbc.271.27.16000. [DOI] [PubMed] [Google Scholar]
- 27.Prigent C, Satoh M S, Daly G, Barnes D E, Lindahl T. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol Cell Biol. 1994;14:310–317. doi: 10.1128/mcb.14.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan A J, Squires S, Strutt S, Johnson R T. Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 1991;19:3295–3300. doi: 10.1093/nar/19.12.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen R, Zdzienicka M Z, Mohrenweiser H, Thompson L H, Thelen M P. Mutations in hamster single-strand break repair gene XRCC1 causing defective DNA repair. Nucleic Acids Res. 1998;26:1032–1037. doi: 10.1093/nar/26.4.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 31.Singh N P, McCoy M T, Tice R R, Schneider E I. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 32.Sonoda E, Sasaki M S, Buerstedde J-M, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;14:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tashiro S, Kotomura N, Shinohara A, Tanaka K, Ueda K, Kamada N. S phase specific formation of human Rad51 protein nuclear foci in lymphocytes. Oncogene. 1996;12:2165–2170. [PubMed] [Google Scholar]
- 34.Taylor R M, Wickstead B, Cronin S, Caldecott K W. Role of a BRCT domain in the interaction of DNA ligase III-α with the DNA repair protein XRCC1. Curr Biol. 1998;8:877–880. doi: 10.1016/s0960-9822(07)00350-8. [DOI] [PubMed] [Google Scholar]
- 35.Tebbs R S, Flannery M L, Meneses J J, Hartmann A, Tucker J D, Thompson L H, Cleaver J E, Pederson R A. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 36.Thompson L H, Brookman K W, Dillehav L E, Carrano A V, Mazrimas C L, Mooney C L, Minkler J L. A CHO-cell strain having hypersensitivity to mutagens, a defect in strand-break repair, and an extraordinary baseline frequency of sister chromatid exchange. Mutat Res. 1982;95:427–440. doi: 10.1016/0027-5107(82)90276-7. [DOI] [PubMed] [Google Scholar]
- 37.Thompson L H, Brookman K W, Jones N J, Allen S A, Carrano A V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol Cell Biol. 1990;10:6160–6171. doi: 10.1128/mcb.10.12.6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zdzienicka M Z, van der Schans G P, Natarajan A T, Thompson L H, Neuteboom I, Simons J W I M. A Chinese hamster ovary cell mutant (EM-C11) with sensitivity to simple alkylating agents and a very high level of sister chromatid exchanges. Mutagenesis. 1992;7:265–269. doi: 10.1093/mutage/7.4.265. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Moréra S, Bates P A, Whitehead P C, Coffer A I, Hainbucher K, Nash R A, Sternberg M J E, Lindahl T, Freemont P S. Structure of an XRCC1 BRCT domain, a new protein-protein interaction module. EMBO J. 1998;176:6404–6411. doi: 10.1093/emboj/17.21.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]