

Abstract
Background
Coagulation factor XI (FXI) contributes to the development of thrombosis but appears to play a minor role in hemostasis and is, therefore, an attractive anticoagulant drug target.
Objectives
To evaluate the safety, pharmacokinetic, and pharmacodynamic properties of BAY 2433334, an orally administered small molecule targeting activated FXI (FXIa), in healthy men.
Patients/Methods
This phase 1 study was conducted in two parts. In part 1, 70 volunteers were randomized 4:1 to receive a single oral dose of BAY 2433334 (5–150 mg as oral solution or immediate‐release tablets) or placebo. In part 2, 16 volunteers received a single oral dose of five BAY 2433334 5‐mg tablets with or without a high‐calorie breakfast in a randomized crossover study design. Adverse events, pharmacokinetic parameters, and pharmacodynamic parameters were assessed up to 72 h after drug administration. Volunteers were followed up after 7 to 14 days.
Results
BAY 2433334 demonstrated favorable safety and tolerability with a dose‐dependent increase in exposure and a terminal half‐life of 14.2 to 17.4 h. A high‐calorie breakfast reduced mean maximum plasma concentration and exposure by 31% and 12.4%, respectively. AY 2433334 was associated with a dose‐dependent inhibition of FXIa activity and an increase in activated partial thromboplastin time. Bleeding times in volunteers who had received BAY 2433334 were similar to those in volunteers who had received placebo.
Conclusions
These data indicate that BAY 2433334 is a promising development candidate for once‐daily oral anticoagulation; it is being evaluated in phase 2 dose‐finding studies in patients at risk of thrombosis.
Keywords: anticoagulant, factor XI, hemostasis, phase 1, thrombosis
Essentials.
BAY 2433334 is an orally administered small molecule targeting activated coagulation factor XI (FXIa).
In this phase 1 study in healthy men, BAY 2433334 exhibited favorable safety and tolerability.
BAY 2433334 dose‐dependently inhibited FXIa activity and increased activated partial thromboplastin time.
BAY 2433334 is a promising development candidate for once‐daily oral anticoagulation.
1. INTRODUCTION
Anticoagulants are cornerstones of the prevention and treatment of thrombotic disorders, but their efficacy may be limited by a dose‐dependent risk of bleeding. 1 In patients with risk factors for bleeding, such as renal impairment or advancing age, anticoagulants may be underdosed or withheld, even if they are indicated. 2 , 3 A substantial need remains for new anticoagulants with improved safety profiles and equivalent or superior efficacies compared with existing therapies.
Data suggest that the inhibition of the activated coagulation factor XI (FXIa) or its zymogen, factor XI (FXI), presents an attractive target mechanism for novel antithrombotic therapies. 4 It has been hypothesized that inhibition of FXI or FXIa may block the amplification or propagation of the intrinsic coagulation pathway to an extent that is sufficient to prevent thrombosis, while leaving the extrinsic (tissue‐factor) pathway and the common pathway intact to ensure an appropriate hemostatic response in case of a vessel injury. 5 , 6 This hypothesis is supported by several lines of preclinical and clinical evidence. 5 , 7 , 8 , 9 , 10 Phase 2 proof‐of‐concept studies in patients undergoing total knee replacement have demonstrated that subcutaneous administration of an antisense oligonucleotide that lowers FXI levels or intravenous administration of an antibody that inhibits FXIa reduced the risk of venous thromboembolism, with a numerically lower rate of bleeding events than enoxaparin. 11 , 12 Further studies with larger sample sizes are required to confirm the efficacy and safety of these approaches.
BAY 2433334 is an orally administered chemically synthesized molecule, which binds directly, potently, and reversibly to the active site of FXIa and thereby inhibits its activity. In in vitro assays, BAY 2433334 inhibited human FXIa in buffer with a half maximal inhibitory concentration of 1.0 (± 0.17) nM (mean ± standard error of the mean). BAY 2433334 is orally bioavailable and has demonstrated potent anticoagulant activity in vitro in human and rabbit plasma and antithrombotic efficacy in vivo in a rabbit arterial thrombosis model. 13 Administration of high and efficacious doses of BAY 2433334 did not increase bleeding time or blood loss in rabbit models, whereas equivalent doses of non‐vitamin K antagonists led to increased bleeding.
The aim of this study (EudraCT number: 2017‐002590‐18) was to evaluate the safety, and pharmacokinetic (PK) and pharmacodynamic (PD) properties of BAY 2433334 in healthy men.
2. METHODS
2.1. Volunteers
Healthy Caucasian men aged 18 to 45 years with a body mass index in the range 18 to 30 kg/m2 were eligible for inclusion in this study. Key exclusion criteria are described in Table S1.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation Good Clinical Practice Guideline and German drug law. All relevant study documents were approved by the local ethics committee and volunteers provided written informed consent.
2.2. Study design
This phase 1, randomized, single‐center study was conducted in two parts (Figure 1).
FIGURE 1.

Patient flow. Abbreviation: IR, immediate‐release
In part 1, a single‐blind, parallel‐group, placebo‐controlled, dose‐escalation study, eligible volunteers were sequentially assigned by an independent group to a unique randomization number in ascending order and randomized in a 4:1 ratio to receive a single dose of BAY 2433334 or placebo. Seven dose cohorts (oral solution: 5, 12.5, and 25 mg; immediate‐release [IR] tablets [in multiples of 5 or 50 mg]: 25, 50, 100, and 150 mg) were studied. Dose escalation was performed only after the safety and tolerability of the preceding dose steps were confirmed.
In part 2, an open‐label, non‐controlled, two‐group crossover, food‐interaction study, volunteers were randomized 1:1 to receive a single oral dose of BAY 2433334 25 mg (5 × 5 mg) IR tablets following an overnight fast of 10 h (treatment A) or following a high‐fat, high‐calorie breakfast (treatment B), in accordance with European Medicines Agency and US Food and Drug Administration guidance. 14 , 15 After a washout period of at least 7 days, volunteers crossed over to the other treatment. Part 2 was conducted after the safety and tolerability of the 50‐mg dose in part 1 was confirmed to be acceptable.
In parts 1 and 2, the study drug was administered on day 1. Volunteers were admitted to the study site 1 day before administration of the study drug and were discharged on day 4. A follow‐up visit took place 7 to 14 days after BAY 2433334 administration.
2.3. Safety
Adverse events (AEs), including symptomatic bleeding, signs of hepatobiliary dysfunction or pancreatic disorders, were assessed at screening, predose, 1, 2, 3, 4, 6, 8, 12, 15, 24, 36, 48, and 72 h after BAY 2433334 administration, and at the follow‐up visit. AEs were considered to be treatment‐emergent if they started or worsened after the first dose of study medication up to 3 days after the end of treatment. AEs of special interest included fatal bleeding, hemorrhage, symptomatic bleeding in a critical area or organ, bleeding resulting in a clinically significant reduction in hemoglobin level, and AEs related to hepatobiliary dysfunction or disorders of the pancreas. Other safety assessments were clinical laboratory variables, physical examination, vital signs, and electrocardiogram findings.
2.4. Pharmacokinetics
Blood samples were taken predose and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 15, 24, 36, 48, and 72 h after BAY 2433334 administration. In part 2, additional samples were taken 2.5, 3.5, and 5 h after administration. Plasma concentrations of BAY 2433334 were measured using a validated liquid chromatography–tandem mass spectrometry method. 16 , 17 , 18 The calibration range of the procedure was from 5.00 µg/L (lower limit of quantification [LLOQ]) to 5000 µg/L (upper limit of quantification). Quality control samples from 15.0 µg/L to 20000 µg/L were determined with an accuracy of 97% to 101% and a precision of 2.8% to 4.2% (n = 69). All samples were stored at or below −15°C and analyzed within 6 months after sampling.
The evaluated PK parameters included: maximum plasma concentration (Cmax); maximum plasma concentration divided by dose (D; Cmax/D); area under the plasma concentration–time curve (AUC); AUC divided by dose (AUC/D) of BAY 2433334 in plasma; time to Cmax (tmax); terminal elimination half‐life (t½); and apparent oral clearance (CL/F). Cmax and tmax were assessed by inspection of the concentration–time plots. AUC was equal to the sum of AUC(0–tlast) and AUC from the last data point to infinity (AUC[tlast−∞]). AUC(0–tlast) was calculated using the log‐linear trapezoidal rule up to the last time point that had a concentration above the LLOQ (tlast). The extrapolated portion (AUC[tlast−∞]) was calculated using the formula C’(tlast)/λZ, in which C′(tlast) was the estimated concentration at tlast and λZ was the terminal‐phase rate constant (i.e., the slope of the log‐linear regression of the last n data points, where n ≥ 3).
PK parameters were calculated by the noncompartmental method using WinNonlin software (version 5.3, Certara, Princeton, NJ, USA) in conjunction with the Automation Extension (version 2.90, Bayer AG, Berlin, Germany).
2.5. Pharmacodynamics
Blood samples for the analysis of PD parameters were taken predose, 1, 2, 4, 8, 12, 24, 36, 48, and 72 h after dosing, and at the follow‐up visit. Citrated blood samples were collected, centrifuged for plasma preparation within 30 min, and then frozen immediately at −20°C.
Activated partial thromboplastin time (APTT) was evaluated using a validated method in which citrated plasma was recalcified in the presence of a standardized quantity of cephalin (a platelet substitute) and a factor XII activator (kaolin). The analysis was performed using the Stago STA compact coagulation analyzer and STA‐C.K. PREST test kit (Diagnostica Stago, France). The calibration range of the assay was 20 to 240 s and the analytical range determined was 25.4 to 76.6 s. Quality control samples with a mean of 29.8 to 56.2 s were determined with a precision of 2.4% to 4.1% coefficient of variation (CV).
FXIa activity was measured using a proprietary fluorogenic substrate assay. The level of FXIa activity was derived from a calibration curve constructed from native FXI protein. The calibration range of the assay was 0% to 150% and the analytical concentration determined ranged from lower than the LLOQ to 142.2%. Quality control samples with a mean of 5.74% to 117.31% were determined with a precision of 5.06% to 15.84% CV.
FXI (clotting) activity was measured using a modified APTT assay (using SynthASil APTT reagent) and an ACL TOP coagulation analyzer, both provided by Instrumentation Laboratory Coagulation Systems (Bedford, MA, USA). In brief, plasma samples were diluted and mixed with human plasma that had been immunodepleted of FXI activity. Correction of the clotting time of the FXI‐deficient plasma to that of the mixture was proportional to the residual activity of FXI in the plasma sample, interpolated from a calibration curve. The calibration range of the assay was 3.3% to 150.0% and the analytical range determined was 33.0% to 148.0%. Quality control samples with a mean of 11.2% to 89.3% were determined with a precision of 1.30% to 9.20% CV.
FXI concentration was measured using an enzyme‐linked immunosorbent assay with a VisuLize, FXI antigen kit (Affinity Biologicals, Ontario, Canada) according to the manufacturer's instructions. The calibration range of the assay was 0.015 to 1.0 IU/ml and the analytical concentration determined was 0.54 to 1.57 IU/ml. Quality control samples from 0.27 to 0.92 IU/ml were determined with a precision of 7.75% to 16.25% CV.
Additional PD parameters included rotational thromboelastometry (ROTEM) whole blood clotting time (ROTEM delta analyzer [TEM International GmbH, Munich, Germany]) and bleeding time (Surgicutt adult device [Accriva Diagnostics Inc., San Diego, CA, USA]). Blood samples for whole blood clotting time were taken predose and 1, 4, and 24 h after dosing; measurements were made within 4 h of blood sampling in citrated blood (3.2% or 3.8% citrate), using the manufacturer's reagents for intrinsically activated test measurements. To assess bleeding time, a standard incision (5‐mm length, 1‐mm depth) was made on the forearm, after which blood was absorbed from the incision using filter paper every 30 s until bleeding stopped.
2.6. Statistics
No formal sample‐size calculation was performed for this exploratory study. Data are presented per dose step and placebo data from part 1 were pooled. In part 1, changes from baseline in aPTT (BAY 2433334 vs placebo) were evaluated using an exact Wilcoxon rank‐sum test at the one‐sided α‐level of 0.05 in a sequential order starting with the highest dose. The Hodges‐Lehmann estimate of the change from baseline in APTT was presented with the 90% CI.
For part 1, an exploratory analysis of dose proportionality was performed using analysis of variance (anova), including the factor treatment, on the log‐transformed values of AUC/D and Cmax/D for each BAY 2433334 formulation. For part 2, AUC and Cmax were analyzed assuming log‐normally distributed data; the logarithms of these parameters were analyzed using anova including sequence, subject (sequence), period and treatment effects. The statistical evaluation was performed using the software package SAS release 9.2 (SAS Institute Inc., Cary, NC, USA). No imputation for missing data was performed.
3. RESULTS
3.1. Demographics and disposition
Of the 177 volunteers screened for eligibility, 86 were randomized and completed the study (Figure 1). Key demographic characteristics are shown in Table 1.
TABLE 1.
Demographics
| Part 1 (n = 70) | Part 2 (n = 16) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 14) | BAY 2433334 Oral Solution, mg | BAY 2433334 IR Tablet, mg | BAY 2433334 5 × 5 mg IR Tablet | |||||||
| 5 (n = 8) | 12.5 (n = 8) | 25 (n = 8) | 25 (n = 8) | 50 (n = 8) | 100 (n = 8) | 150 (n = 8) | Fasted–fed (n = 8) | Fed–fasted (n = 8) | ||
| Age, years | ||||||||||
| Mean (SD) | 29.0 (8.1) | 34.4 (7.9) | 25.0 (4.4) | 30.0 (5.0) | 25.1 (5.1) | 27.9 (4.8) | 29.6 (5.5) | 30.1 (5.8) | 32.1 (7.9) | 30.3 (8.5) |
| Range | 19–4 | 24–45 | 19–30 | 22–39 | 20–33 | 21–33 | 21–38 | 22–39 | 24–44 | 20–43 |
| Weight, kg | ||||||||||
| Mean (SD) | 77.1 (11.8) | 81.8 (13.1) | 80.0 (9.9) | 85.1 (9.4) | 80.5 (7.2) | 83.6 (10.1) | 84.2 (19.2) | 77.3 (10.1) | 79.5 (10.9) | 83.8 (15.0) |
| Range | 62.0–102.4 | 58.0–102.0 | 69.0–93.0 | 74.6–104.7 | 73.0–92.3 | 70.8–97.3 | 62.3–125.1 | 66.2–99.0 | 59.9–96.9 | 69.0–115.8 |
| BMI, kg/m2 | ||||||||||
| Mean (SD) | 24.2 (2.8) | 24.8 (3.8) | 23.6 (2.6) | 26.1 (2.7) | 24.2 (2.5) | 26.2 (1.6) | 25.2 (3.3) | 24.3 (3.0) | 24.3 (3.2) | 25.5 (3.0) |
| Range | 19.2–28.7 | 18.9–29.5 | 20.4–28.4 | 21.6–29.9 | 21.1–27.6 | 23.9–28.2 | 20.6–29.2 | 19.6–29.9 | 19.8–29.3 | 21.0–29.5 |
| Smoking status, n (%) | ||||||||||
| Never | 9 (64.3) | 5 (62.5) | 5 (62.5) | 5 (62.5) | 3 (37.5) | 6 (75.0) | 5 (62.5) | 6 (75.0) | 6 (75.0) | 4 (50.0) |
| Former | 1 (7.1) | 0 | 2 (25.0) | 2 (25.0) | 3 (37.5) | 0 | 2 (25.0) | 0 | 1 (12.5) | 1 (12.5) |
| Current | 4 (28.6) | 3 (37.5) | 1 (12.5) | 1 (12.5) | 2 (25.0) | 2 (25.0) | 1 (12.5) | 2 (25.0) | 1 (12.5) | 3 (37.5) |
Abbreviations: BMI, body mass index; IR, immediate‐release; SD, standard deviation.
3.2. Safety
Safety was evaluated in 86 volunteers (Table 2). In part 1, 16 volunteers (22.9%) experienced at least one treatment‐emergent AE (TEAE); the incidence of TEAEs in the active treatment groups ranged from 12.5% (n = 1) to 62.5% (n = 5). TEAEs considered to be related to study drug were reported in six volunteers (8.6%) and included dysgeusia (n = 4), ventricular extrasystoles (n = 1), and nausea (n = 1). In part 2, six volunteers (37.5%) experienced at least one TEAE; of these, two volunteers experienced a TEAE related to BAY 2433334 (palpitations, vertigo, and sensation of a foreign body [n = 1], and paresthesia, dry mouth, and feeling cold [n = 1]). All AEs were of mild intensity. No other signs of potential drug‐related hypersensitivity reactions, spontaneous, prolonged or hidden bleeding, or thrombocytopenia were observed during the study. There were no treatment‐related serious AEs or deaths in any group and no findings of clinical relevance with respect to laboratory tests, vital signs, electrocardiograms, or physical examinations.
TABLE 2.
Safety profile of BAY 2433334 in healthy volunteers
| Part 1 (n = 70) | Part 2 (n = 16) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 14) | BAY 2433334 Oral Solution, mg | BAY 2433334 IR Tablet, mg | BAY 2433334 5 × 5 mg IR Tablet | |||||||
| 5 (n = 8) | 12.5 (n = 8) | 25 (n = 8) | 25 (n = 8) | 50 (n = 8) | 100 (n = 8) | 150 (n = 8) | Fasted (n = 16) | Fed (n = 16) | ||
| TEAEs | 1 (7.1) | 2 (25) | 1 (12.5) | 2 (25) | 1 (12.5) | 1 (12.5) | 3 (37.5) | 5 (62.5) | 1 (6.3) | 6 (37.5) |
| Mild | 1 (7.1) | 2 (25) | 1 (12.5) | 2 (25) | 1 (12.5) | 1 (12.5) | 3 (37.5) | 5 (62.5) | 1 (6.3) | 6 (37.5) |
| Drug‐related TEAEs | 0 | 1 (12.5) | 1 (12.5) | 0 | 0 | 0 | 0 | 4 (50) | 1 (6.3) | 2 (12.5) |
| Serious AEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious TEAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs leading to discontinuation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Data are presented as n (%).
Abbreviations: AE, adverse event; IR, immediate‐release; TEAE, treatment‐emergent adverse event.
3.3. Pharmacokinetics
PK parameters were evaluated in 72 volunteers (Table 3). BAY 2433334 was absorbed with a median tmax of 1.00 h and 2.50 to 3.97 h after administration as oral solution and IR tablets, respectively (fasted state). The geometric mean t½ of BAY 2433334 was 14.2 to 17.4 h and CL/F was 3.19 to 4.21 L/h. The interindividual variability in exposure was low with geometric CV of 16.1% to 25.4%. In part 1, there was no deviation from a dose‐proportional increase in BAY 2433334 exposure administered as oral solution or IR tablets (Figure 2; Table S2). A high bioavailability was observed for the five BAY 2433334 5‐mg IR tablets with 89.5% and 86.0% for AUC and Cmax, respectively, when comparing with the oral solution in an interindividual manner. In part 2, intake of a high‐fat, high‐calorie breakfast before administration of BAY 2433334 had a minimal effect on the bioavailability of the five 5‐mg IR tablets. Mean AUC decreased by 12.4% and Cmax was reduced by 31.4% in the fed versus the fasted state. The median tmax increased from 2.50 h in the fasted state to 5.00 h in the fed state (Table 3).
TABLE 3.
Pharmacokinetic parameters of BAY 2433334 in plasma (summary statistics)
| Part 1 (n = 56) | Part 2 (n = 16) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BAY 2433334 Oral Solution, mg | BAY 2433334 IR Tablet, a mg | BAY 2433334 5 x 5 mg IR Tablets | |||||||
| 5 (n = 8) | 12.5 (n = 8) | 25 (n = 8) | 25 (n = 8) | 50 (n = 8) | 100 (n = 8) | 150 (n = 8) | Fasted (n = 16) | Fed (n = 16) | |
| Cmax, μg/L | 91.4 (15.9) | 205 (19.1) | 372 (13.5) | 320 (18.3) | 617 (15.6) | 1230 (25.9) | 1720 (22.7) | 374 (20.7) | 257 (15.9) |
| Cmax/D, 10−3/L | 18.3 (15.9) | 16.4 (19.1) | 14.9 (13.5) | 12.8 (18.3) | 12.3 (15.6) | 12.3 (25.9) | 11.5 (22.7) | 15.0 (20.7) | 10.3 (15.9) |
| AUC, μg/h/L | 1570 (18.8) | 3770 (24.9) | 6630 (25.4) | 5940 (19.9) | 13 200 (16.1) | 27 600 (23.5) | 38 700 (24.7) | 6690 (19.9) | 5860 (17.8) |
| AUC/D, h/L | 0.313 (18.8) | 0.302 (24.9) | 0.265 (25.4) | 0.237 (19.9) | 0.264 (16.1) | 0.276 (23.5) | 0.258 (24.7) | 0.268 (19.9) | 0.234 (17.8) |
| tmax, h b | 1.00 (0.75–1.50) | 1.00 (0.75–1.50) | 1.00 (0.75–1.50) | 2.50 (1.00–5.95) | 3.00 (0.983–4.00) | 3.00 (0.750–6.00) | 3.97 (0.983–4.00) | 2.50 (0.750–4.98) | 5.00 (3.00–8.00) |
| t½, h | 14.5 (15.7) | 16.0 (18.1) | 15.2 (21.6) | 14.2 (20.0) | 17.4 (12.4) | 16.3 (13.7) | 14.8 (9.53) | 15.7 (13.3) | 16.1 (13.9) |
Data are presented as geometric mean (% coefficient of variation) unless stated otherwise.
Abbreviations: AUC, area under the plasma concentration–time curve; AUC/D, area under the plasma concentration–time curve divided by dose; Cmax, maximum plasma drug concentration; Cmax/D, maximum plasma drug concentration divided by dose; IR, immediate‐release; t½, terminal elimination half‐life; tmax, time to reach maximum plasma concentration.
Doses administered as 5 mg or 50 mg IR tablets.
Median (range).
FIGURE 2.
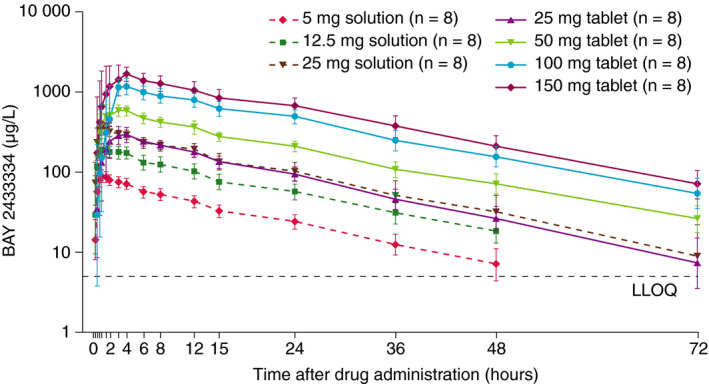
Plasma concentration–time profile of BAY 2433334 in healthy volunteers in part 1. Data are presented as geometric mean. The lower and upper limits of the error bars are geometric mean/geometric SD and mean × geometric SD, respectively. Abbreviations: LLOQ, lower limit of quantification; SD, standard deviation
3.4. Pharmacodynamics
PD parameters were evaluated in 86 volunteers.
3.4.1. APTT
In part 1, a rapid, dose‐dependent increase in APTT was observed (Figure 3a). The increase from baseline was significantly greater than that for placebo (p < .0001) for all BAY 2433334 dose steps (Table S3). The geometric mean maximal ratio to baseline in aPTT was 2.12, corresponding to an absolute value of 36.6 s. Interindividual variability was low, with geometric CVs of 1.24% to 5.52% for all dose steps. aPTT values returned to baseline within 72 h. In part 2, the increase in aPTT was more rapid and the maximum ratio to baseline was higher in the fasted than in the fed state (Figure S1). Maximal effects were observed 4 and 8 h after BAY 2433334 administration in the fasted and fed states, respectively.
FIGURE 3.
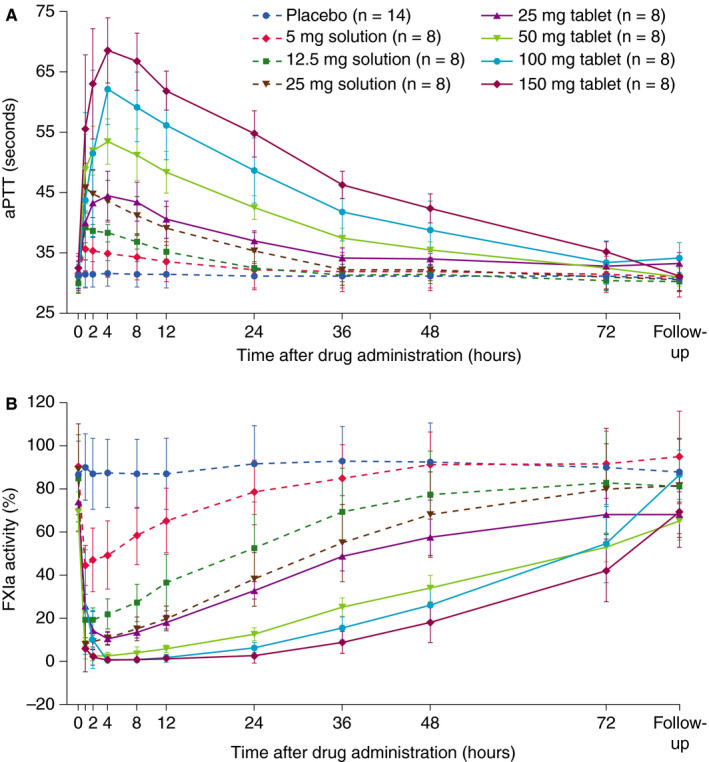
Effects of increasing doses of BAY 2433334 on (A) APTT and (B) FXIa activity in healthy volunteers in part 1. Data are presented as mean and standard deviation. Abbreviations: APTT, activated partial thromboplastin time; FXIa, activated factor XI
3.4.2. FXIa and FXI activity
In part 1, there was a dose‐dependent reduction in FXIa activity (Figure 3b) and in FXI activity (Figure S2). The geometric means of maximal decrease in FXIa and FXI activity relative to baseline were 0.01 and 0.52, respectively. Interindividual variability was low to moderate with geometric CVs of 5.54% to 68.1% (FXIa activity) and 3.48% to 12.42% (FXI activity). The maximal effects of BAY 2433334 on FXIa and FXI inhibition were observed approximately 1 h after administration of oral solution and 2–4 h after administration of tablets. An FXIa activity geometric mean ratio to baseline of 0.20 or less, corresponding to an FXIa activity reduction of 80% or more, was seen for at least 24 h after administration of BAY 2433334 50‐, 100‐, and 150‐mg tablets (Figure 3b). In part 2, the inhibition of FXIa activity was more rapid after administration of BAY 2433334 25 mg in the fasted state than in the fed state, with maximal effects observed 4 and 8 h after administration, respectively (Figure S3).
3.4.3. FXI concentration
No differences in FXI concentrations were observed after administration of BAY 2433334 (Figure S4). The geometric mean maximum ratio to baseline was 1.12 after administration of placebo and 1.17 after administration of BAY 2433334.
3.4.4. ROTEM whole blood clotting time
Following BAY 2433334 administration, there was a trend toward a dose‐dependent increase in clotting time (Figure 4a).
FIGURE 4.
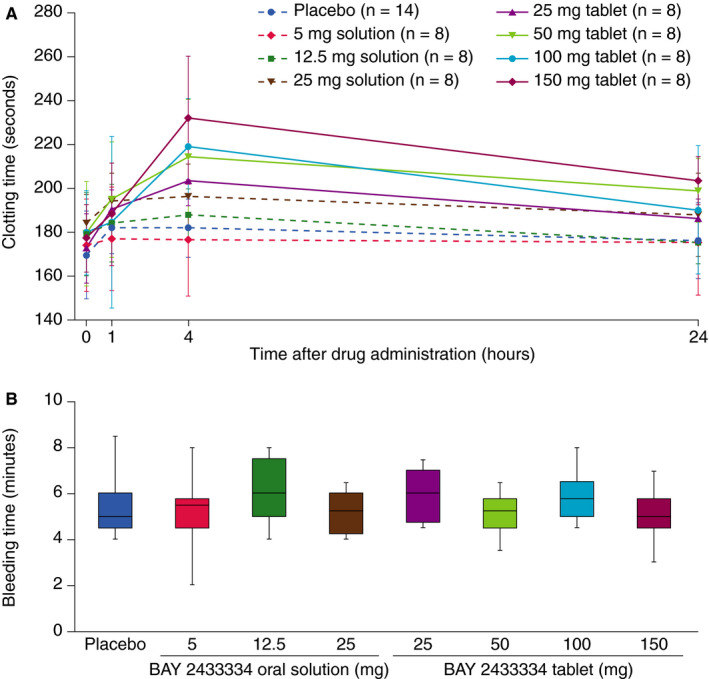
Effect of increasing doses of BAY 2433334 on (A) clotting time measured by rotational thromboelastometry and (B) bleeding time. Bleeding time data were obtained 4 h after drug administration and are presented as box‐and‐whisker plots; upper and lower lines of the box denote the upper and lower quartiles, respectively, midlines denote the medians, and upper and lower lines denote the maximum and minimum values, respectively
3.4.5. Bleeding time
Bleeding times were consistent across all dose cohorts and were similar to those for placebo during the 24 h after drug administration (Figure 4b).
4. DISCUSSION
This first‐in‐human, phase 1, randomized, single‐center study demonstrated a favorable safety and tolerability profile of BAY 2433334 following single oral doses. Dose‐dependent increases in exposure of BAY 2433334 and APTT, as well as decreases in FXIa and FXI activity, were observed. Bleeding times were similar in volunteers who received BAY 2433334 and placebo.
The low CL/F of 3.19–4.21 L/h (corresponding to a blood clearance after oral administration of 4.54–5.99 L/h), assuming high absolute bioavailability, suggests that BAY 2433334 is a low‐clearance drug. High absolute bioavailability was expected because low‐to‐moderate interindividual variability in exposure, high relative bioavailability of the tablet compared with the oral solution and a minor effect of food on BAY 2433334 exposure were observed in this study. The effect of the high‐fat, high‐calorie breakfast on the rate of absorption (decrease in Cmax with prolonged tmax) could mostly be explained by delayed gastric emptying. An exploratory anova did not show any deviation from dose proportionality. Moreover, the observed t½ of 14.2–17.4 h, together with the direct PK/PD relationship, supports a once‐daily dosing regimen, which would improve patient adherence to medication in chronic diseases. 19 In addition, the relatively short half‐life of BAY 2433334 may permit convenient, temporary cessation of anticoagulation before elective surgical procedures.
Generally, the effects of BAY 2433334 on PD parameters are consistent with its mechanism of action. Preclinical data have demonstrated that BAY 2433334 inhibited FXIa activity and prolonged APTT in human plasma, with no significant effect on prothrombin time, highlighting the specific inhibitory effects of BAY 2433334 on the intrinsic coagulation pathway. 13 In this study, APTT was evaluated using a kaolin‐triggered assay, which affords greater sensitivity than conventional silica‐based assays. 20 Data from the Surgicutt test demonstrated that no relevant or dose‐dependent changes in bleeding time occurred in the first 4 h following BAY 2433334 administration; these data are in agreement with preclinical observations. 13 Of note, the clinical predictivity of preclinical bleeding models is limited and the finding that BAY 2433334 is not associated with increased bleeding is yet to be proven in the clinical setting. Preclinical and clinical studies of other pharmacological inhibitors of FXI or FXIa also provide supportive evidence that selective inhibition of FXI or FXIa is not associated with an increased risk of bleeding. 5 , 8 , 9 , 10 , 11 , 12 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27
The limitations of this study are inherent to phase 1 studies. To minimize the risk to volunteers at this early stage of clinical development, women were excluded because data from reproductive studies were not available at the time of study conception. Data from this study of a small number of healthy Caucasian men might not necessarily apply to other populations, including women and patients requiring thromboprophylaxis or those with acute thrombosis, although there are no reports of significant differences in the properties of FXI between populations. In addition, the association between the results of APTT, FXIa, and FXI activity assays and clinical outcomes in patients receiving BAY 2433334 are unknown. Further studies of the safety, PD and PK of BAY 2433334 in heterogenous patient populations are needed.
Based on the safety, PK, and PD results obtained in the current study, BAY 2433334 is a promising development candidate for once‐daily oral anticoagulation. Further evaluation of the PK, PD, safety, and tolerability of BAY 2433334 is ongoing in an extended phase 1 program; data will be published separately. Further, safety and efficacy are under investigation in the PACIFIC program consisting of three phase 2, dose‐finding studies in patients with atrial fibrillation (PACIFIC‐AF, NCT04218266), acute myocardial infarction (PACIFIC‐AMI; NCT04304534), and noncardioembolic ischemic stroke (PACIFIC‐STROKE; NCT04304508).
CONFLICT OF INTEREST
Drs. Thomas, Kanefendt, Schwers, Unger, and Yassen are employees of Bayer AG, which provided funding for this study. Dr. Boxnick is an employee of CRS Clinical Research Services, which received funding from Bayer AG.
AUTHOR CONTRIBUTIONS
Dirk Thomas, Friederike Kanefendt, Stephan Schwers, Sigrun Unger, and Ashraf Yassen contributed to the study design and the analysis and interpretation of data. Stefanie Boxnick was the principal investigator and was responsible for the design and conduct of the study, and the reporting of data. All authors were involved in critically revising the manuscript for important intellectual content and approved the final version.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Bayer AG, Berlin, Germany. We thank Tobias Marquardt for support with PD measurements and Lukas Fiebig for bioanalysis and Dorina van der Mey for support with PK analysis. Editorial support was provided by Emma Bolton, DPhil, of Oxford PharmaGenesis, Oxford, UK, with funding from Bayer AG.
Thomas D, Kanefendt F, Schwers S, Unger S, Yassen A, Boxnick S. First evaluation of the safety, pharmacokinetics, and pharmacodynamics of BAY 2433334, a small molecule targeting coagulation factor XIa. J Thromb Haemost. 2021;19:2407–2416. 10.1111/jth.15439
Manuscript handled by: Sabine Eichinger
REFERENCES
- 1. Shoeb M, Fang MC. Assessing bleeding risk in patients taking anticoagulants. J Thromb Thrombolysis. 2013;35:312‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Camm AJ, Accetta G, Ambrosio G, et al. Evolving antithrombotic treatment patterns for patients with newly diagnosed atrial fibrillation. Heart. 2017;103:307‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. García Rodríguez LA, Martín‐Pérez M, Vora P, et al. Appropriateness of initial dose of non‐vitamin K antagonist oral anticoagulants in patients with non‐valvular atrial fibrillation in the UK. BMJ Open. 2019;9:e031341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mackman N, Bergmeier W, Stouffer GA, Weitz JI. Therapeutic strategies for thrombosis: new targets and approaches. Nat Rev Drug Discov. 2020;19:333‐352. [DOI] [PubMed] [Google Scholar]
- 5. Zhang H, Lowenberg EC, Crosby JR, et al. Inhibition of the intrinsic coagulation pathway factor XI by antisense oligonucleotides: a novel antithrombotic strategy with lowered bleeding risk. Blood. 2010;116:4684‐4692. [DOI] [PubMed] [Google Scholar]
- 6. Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36:1316‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep‐vein thrombosis. Thromb Haemost. 2011;105:269‐273. [DOI] [PubMed] [Google Scholar]
- 8. Crosby JR, Marzec U, Revenko AS, et al. Antithrombotic effect of antisense factor XI oligonucleotide treatment in primates. Arterioscler Thromb Vasc Biol. 2013;33:1670‐1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride‐induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695‐702. [DOI] [PubMed] [Google Scholar]
- 10. Yau JW, Liao P, Fredenburgh JC, et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood. 2014;123:2102‐2107. [DOI] [PubMed] [Google Scholar]
- 11. Buller HR, Bethune C, Bhanot S, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372:232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weitz JI, Bauersachs R, Becker B, et al. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA. 2020;323:130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heitmeier S, Visser M, Gäfke A‐G, Harwardt M, Griessbach C, Mueller V, Tersteegen A, Strassburger J, Gerdes C, Laux V, Jimenez‐Nunez E, Ackerstaff J, Roehrig S. Preclinical pharmacology of BAY 2433334, a small molecule inhibitor of coagulation factor XIa [abstract]. https://academy.isth.org/isth/2020/milan/295338/stefan.heitmeier.preclinical.pharmacology.of.bay.2433334.a.small.molecule.html. Accessed May 2021.
- 14. European Medicines Agency . Guideline on the investigation of drug interactions. 2012. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf. Accessed May 2021.
- 15. U.S. Department of Health and Human Services FaDA, Center for Drug Evaluation and Research (CDER) Guidance for industry. Food‐effect bioavailability and fed bioequivalence studies. 2002. https://www.fda.gov/media/70945/download. Accessed May 2021.
- 16. US Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM) . Bioanalytical method validation guidance for industry. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical‐Method‐Validation‐Guidance‐for‐Industry.pdf. Accessed May 2021.
- 17. European Medicines Agency . Guideline on bioanalytical method validation. 2011. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐bioanalytical‐method‐validation_en.pdf. Accessed May 2021. [DOI] [PubMed]
- 18. European Medicines Agency . Reflection paper for laboratories that perform the analysis or evaluation of clinical trial samples. 2012. https://www.ema.europa.eu/en/documents/regulatory‐procedural‐guideline/reflection‐paper‐laboratories‐perform‐analysis‐evaluation‐clinical‐trial‐samples_en.pdf. Accessed May 2021.
- 19. Laliberté F, Bookhart BK, Nelson WW, et al. Impact of once‐daily versus twice‐daily dosing frequency on adherence to chronic medications among patients with venous thromboembolism. Patient. 2013;6:213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomas D, Thelen K, Kraff S, et al. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: first evaluation of safety, pharmacodynamics, and pharmacokinetics. Res Pract Thromb Haemost. 2019;3:242‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa‐mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981‐3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. David T, Kim YC, Ely LK, et al. Factor XIa‐specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci Transl Med. 2016;8:353ra112. [DOI] [PubMed] [Google Scholar]
- 23. Tucker EI, Marzec UM, White TC, et al. Prevention of vascular graft occlusion and thrombus‐associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Montfoort ML, Knaup VL, Marquart JA, et al. Two novel inhibitory anti‐human factor XI antibodies prevent cessation of blood flow in a murine venous thrombosis model. Thromb Haemost. 2013;110:1065‐1073. [DOI] [PubMed] [Google Scholar]
- 25. Yamashita A, Nishihira K, Kitazawa T, et al. Factor XI contributes to thrombus propagation on injured neointima of the rabbit iliac artery. J Thromb Haemost. 2006;4:1496‐1501. [DOI] [PubMed] [Google Scholar]
- 26. Younis HS, Crosby J, Huh JI, et al. Antisense inhibition of coagulation factor XI prolongs APTT without increased bleeding risk in cynomolgus monkeys. Blood. 2012;119:2401‐2408. [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Smith PL, Hsu MY, et al. Effects of factor XI deficiency on ferric chloride‐induced vena cava thrombosis in mice. J Thromb Haemost. 2006;4:1982‐1988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material