

Summary
Deep‐sea hydrothermal vents harbour diverse and abundant animals and their symbiotic microorganisms, which together comprise holobionts. The interplay between bacterial members of holobionts and their viruses (phages) is important for maintaining these symbiotic systems; however, phage–bacterium interactions in deep‐sea vent holobionts are not well understood. Marine sponges serve as good models for such studies and are used to unveil phage–bacterium interplay via metagenomic analysis. In three demosponges from deep‐sea hydrothermal vent fields in the southern Okinawa Trough, the genomes of a diverse array of symbiotic bacteria, including 10 bacterial phyla, were found to lack intact prophages. Genes related to diverse anti‐viral defence systems, for example, the restriction–modification and toxin–antitoxin systems, were abundant in the bacterial communities. We also detected phage genes that could complement or compensate host bacterial metabolism, indicating beneficial roles of phage infection. Our findings provide insight into phage–bacterium interplay in sponges from deep‐sea hydrothermal vents.
Introduction
Since 1977, hundreds of deep‐sea hydrothermal vent ecosystems have been discovered (Corliss et al., 1979). These ecosystems are unique and extreme among marine environments (Staudigel, 2003; He et al., 2017). Studies of these habitats have revealed diverse organisms, including symbiotic animals, bacteria/archaea and viruses, which constitute holobionts (Fujiwara et al., 2000; Kuwahara et al., 2007; Anantharaman et al., 2014; Ikuta et al., 2016) and yielded new views on biology and ecology (Dick, 2019). Symbioses of animals and chemolithoautotrophic bacteria are pervasive because bacteria can provide a primary energy source for their animal host and thereby give rise to a bloom of vent fauna (Dick, 2019). For instance, a symbiosis of chemolithoautotrophic γ‐proteobacteria and tubeworms enables this invertebrate holobiont to thrive in vent fields (Halanych, 2005). Viruses, including bacterial phages, also contribute to holobionts, where they are often found in abundance (5.8 × 107 to 4.5 × 109 particles per gram) (Yoshida‐Takashima et al., 2012). How this viral abundance influences symbiotic bacteria, and how viruses interact with bacteria in vent holobionts, have not been widely examined.
As marine sponge holobionts harbour abundant and diverse microbes, including bacteria and phages (Webster and Taylor, 2012; Pascelli et al., 2018), they have been used as model systems to explore the interplay of different organisms (Jahn et al., 2019). Microbial organisms can make up to 40% of the volume of a sponge (Taylor et al., 2007; Webster and Taylor, 2012), where they serve as a food source and are actively involved in the ecology and biogeochemistry of the symbiotic system. For example, in the deep‐sea glass sponge Lophophysema eversa, bacterial symbionts are important for the cycling of sulfur, carbon and nitrogen (Tian et al., 2016). Viruses are also abundant in sponges (Laffy et al., 2018; Pascelli et al., 2018; Jahn et al., 2019; Zhou et al., 2019); at least 50 morphotypes and 4484 viral populations have been found in shallow‐water sponges (Pascelli et al., 2018; Jahn et al., 2019). Notably, viruses encoding ankyrins that reduce phagocytosis rates are thought to promote the survival of bacterial symbionts by suppressing immune function in the animal host (Jahn et al., 2019). Furthermore, metagenomic profiling of deep‐sea sponge microbiomes revealed the potential involvement of sponge‐associated SUP05 phages in the iron–sulfur cluster assembly, suggesting that the viruses contribute to bacterial symbiont metabolism (Zhou et al., 2019).
The Okinawa Trough is in the south‐eastern East China Sea between the Eurasian Continent and Ryukyu Arc. The ecosystems in the vent fields of the Okinawa Trough are characterized by fauna associated with sediments (Watanabe and Kojima, 2015), such as the squat lobster Shinkaia crosnieri and deep‐sea mussel Bathymodiolus spp. (Makabe et al., 2016; Chen et al., 2017). Unusually, a large sponge population consisting mostly of demosponges from the orders Haplosclerida and Poecilosclerida was recently found at two closely located and hydrothermally influenced vent sites ‐ Swan site, Tarama Knoll; and Crane site, Tarama Hill (Makabe et al., 2016; Chen et al., 2017). In this study, we performed metagenomic sequencing and individual genome bins to detect bacterial and viral signals, and explore phage–bacterium interplay, in three demosponges collected from the Swan and Crane sites.
Results and discussion
Microbial diversity and phage–bacterial connections
Analyses of 16S rRNA genes showed that the tedaniid sponge from the Crane site (TC) (Fig. S1‐A) and haplosclerid sponge from the Crane site (HC) and Swan site (HS) harboured diverse microorganisms (Fig. 1). These microorganisms included 12 phyla of bacteria and archaea, such as Proteobacteria, Actinobacteria, Bacteroidetes and Planctomycetes (Fig. 1A). Proteobacteria was the most abundant phylum, accounting for 88%–96% of the total microbial community in each sponge, within which SUP05 bacteria showed the highest abundance (54%–77%) (Fig. S1‐B and (Zhou et al., 2019)). Previous studies revealed that the community composition of microbial symbionts is typically determined by sponge species regardless of the habitats (Cleary et al., 2013; Sacristan‐Soriano et al., 2020). In our study, the compositional variation in HC‐ and HS‐associated microbiota (HC and HS are the same species) was relatively low compared with that between HC (or HS) and TC, though HC and TC were from the same site, Crane. HC and HS shared the phyla of Proteobacteria, Actinobacteria, Bacteroidetes, Planctomycetes, Firmicutes, Cyanobacteria, Tenericutes, Crenarchaeota and Euryarchaeota. In contrast, TC lacked the phyla of Actinobacteria, Bacteroidetes and Tenericutes but was colonized by Acidobacteria, Chlamydiae and Thermotogae. Ten bacterial genome bins were obtained from the three vent demosponges (Table S1). Each bin was given a short name ‐ such as TC_Bin1_SUP05, HC_Bin1_Evansiales and HS_Bin1_RS24 (Table S1) ‐ based on the host, bin number and NCBI/GTDB/SILVA taxonomy.
Fig. 1.
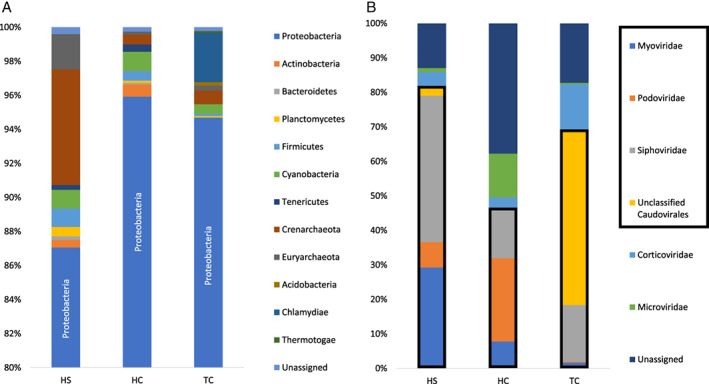
Community structure in three vent sponges.
A. Bacterial and archaeal community structure at the phylum level.
B. Viral community structure at the family level. The dominant Caudovirales is outlined in black. HC, haplosclerid sponge from Crane site; HS, haplosclerid sponge from Swan site; TC, tedaniid sponge from Crane site.
Taxonomic analysis of the viromes of the vent demosponges revealed that the viral community was composed of at least five families, including Siphoviridae, Podoviridae, Myoviridae, Corticoviridae and Microviridae (Fig. 1B). The Caudovirales ‐ comprising the Siphoviridae, Podoviridae and Myoviridae ‐ formed the most abundant viral group, accounting for 46%–81% of the viral community of each sponge (Fig. 1B). We generated 191 viral bins of sponge‐associated viruses (Tables S2 and S3), of which 28 were predicted to infect bacteria (nine bacterial bins) (Table S4). Notably, all vent sponges displayed SUP05–phage connections, suggesting that the previously reported SUP05–phage association in haplosclerid sponges (Zhou et al., 2019) is conserved in a wide range of deep‐sea vent sponges.
Prophages in vent sponge symbionts
As previous studies suggested that phages tend to integrate their genomes into their bacterial hosts in animal‐associated habitats (Knowles et al., 2016), we searched for prophages in 12 binned bacterial genomes (10 bins from this study, two from Zhou et al. (2019)) derived from the three vent sponges using three identifiers, VirSorter (Roux et al., 2015), PHASTER (Arndt et al., 2016) and Prophage Hunter (Song et al., 2019). Unexpectedly, none of the bacterial bins contained intact prophages or attachment sites for phage genome insertion (Table 1 and Table S5). The number of contigs with incomplete prophages was also quite low, ranging from 2 to 11 compared with the thousands of bacterial contigs. Only one contig coding integrases was found in TC_Bin3_UBA1609, which was a low‐abundance bacterial bin (average read coverage <50×, number of reads recruited per kilobase of genome per gigabase of metagenome =19.6, Table S1) in the dominant bins such as TC_Bin1_SUP05 (average read coverage >7000×, reads recruited per kilobase of genome per gigabase of metagenome =4141.5, Table S1). In addition, in the unbinned bacterial metagenomes, only one to three contigs with low abundances (average read coverage <40×) were predicted to contain putative prophage regions (Table 1 and S5). These sequences were incomplete and contained no lysogeny indicators such as attachment sites (att) and integrases.
Table 1.
Composition of sponge‐associated bacterial sequences with complete/incomplete prophages, genes for phage integrases and phage attachment sites.
| Sample | Bacterial metagenome/bin | No. of contigs with complete prophages | No. of contigs with incomplete prophages | No. of contigs with integrases | No. of contigs with attachment sites | No. of total contigs |
|---|---|---|---|---|---|---|
| TC | Bacterial metagenome | ND | 1 | ND | ND | 7826 |
| TC_Bin1_SUP05 | ND | ND | ND | ND | 230 | |
| TC_Bin2_AqS2 | ND | ND | ND | ND | 358 | |
| TC_Bin3_UBA1609 | ND | 1 | 1 | ND | 50 | |
| HC | Bacterial metagenome | ND | 3 | ND | ND | 5896 |
| HC_Bin1_Evansiales | ND | 2 | ND | ND | 221 | |
| HC_Bin2_Endozoicomonadaceae | ND | ND | ND | ND | 143 | |
| HC_Bin3_UBA1609 | ND | ND | ND | ND | 50 | |
| HC_Bin4_RS24 | ND | 4 | ND | ND | 218 | |
| HC_SUP05 | ND | 2 | ND | ND | 161 | |
| HS | Bacterial metagenome | ND | 1 | ND | ND | 12 670 |
| HS_Bin1_RS24 | ND | 1 | ND | ND | 227 | |
| HS_Bin2_Shewanella | ND | ND | ND | ND | 348 | |
| HS_Bin3_Endozoicomonadaceae | ND | ND | ND | ND | 380 | |
| HS_SUP05 | ND | 1 | ND | ND | 192 |
ND: not detected. HC_SUP05 (NCBI Accession VSKZ00000000.1) and HS_SUP05 (NCBI Accession VSKZ00000000.1) were retrieved from Zhou et al. (2019).
The small genome size of sponge symbionts may explain the low detection of prophages in the metagenome. Bacteria with genome size larger than 6 Mb have been predicted to have high prophage frequency (77%) (Touchon et al., 2016). The genome sizes of our dominant symbiotic bacteria are estimated to be much less than 6 Mb. For example, SUP05 bacterial genomes range from 1.07 to 1.18 Mb (88.89% ≤ completeness ≤96%), and Bdellovibrionales was approximately 1.2 Mb with a completeness of 90%–94% (Table S1). Generally, symbionts of deep‐sea animals undergo genome reduction and evolve streamlined genomes (Wolf and Koonin, 2013; Tian et al., 2016; Tian et al., 2017; Rubin‐Blum et al., 2019). According to our analysis, the dominant symbionts with reduced genomes (1.37–2.69 Mb) from other deep‐sea sponges (Hymedesmia (Stylopus) methanophila sp. nov., Lophophysema eversa, and Suberites sp.) also lack complete prophages (Table S5). These sponges are affiliated with different species and inhabit different locations and water depths (Table S5), suggesting that the lack of intact prophages is widespread in deep‐sea sponges.
Diverse antiviral defence systems in symbiotic bacteria
To reveal the antiviral defences of vent sponge bacterial communities, we examined bacterial sequences by querying predicted genes against the restriction enzyme database REBASE (Roberts et al., 2015) with BLASTp and the PFAM database (El‐Gebali et al., 2019) using HMMScan. We found 813–1538 genes associated with 17 antiviral defence systems in the three bacterial communities (Tables S6–S11). The restriction–modification system (RM), bacteriophage exclusion system, DNA phosphorothioation defence, and defence island system associated with restriction–modification (DISARM) of innate immunity are predominant in the bacterial immune systems (290–605 genes accounting for 36%–42% of total defence genes) (Fig. 2A). In contrast, only 19–47 genes were discovered participating in the CRISPR–Cas system. In the defence related to dormancy induction or programmed cell death, toxin–antitoxin (TA)‐related genes were abundant (75–202 genes) compared to abortive infection genes. Genes related to the Zorya, Gabija, Septu, Hachiman, Thoeris, Lamassu, Wadjet, Druantia, Shedu and Kiwa defence systems also formed a relatively large proportion (125–221 genes). Furthermore, the recognition sites of restriction enzymes and CRISPR spacers were compared to phage sequences from three sponges. All restriction enzymes recognize sites in phage sequences (Tables S6, S8 and S10). In contrast, only two (k141_66712 from HC and k141_75926 from HS) of 73 contigs (with 433 spacers, Table S12) contained 15 spacers that matched the phage sequences in our study.
Fig. 2.
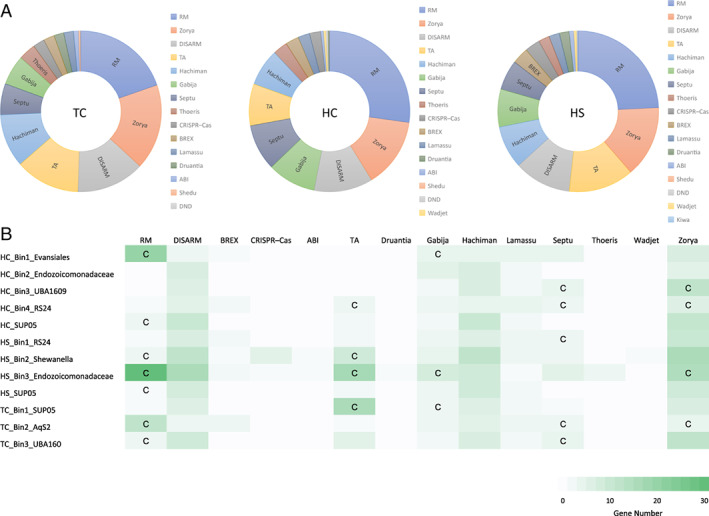
Defence systems of bacterial communities in three vent sponges.
A. Number of defence system genes in sponges TC, HC and HS.
B. Number of defensive genes in each bacterial bin. C represents a complete set of genes for a defence system. HC, haplosclerid sponge from Crane site; HS, haplosclerid sponge from Swan site; TC, tedaniid sponge from Crane site.
All binned bacteria shared the Zorya, Hachiman, Lamassu, DISARM and Gabija systems, among which the complete Gabija and/or Zorya systems were displayed by six bins (Fig. 2B). RM, TA and Septu were found in most bins, 10 of which had a complete set of genes for one or two systems (Fig. 2B). In accordance with the small number of genes encoding Cas enzymes, sequences having putative CRISPR spacers were detected in only seven bacterial bins (Table S13), none of which contained spacers matching the viral sequences obtained in our study (Table S13). On the contrary, RM recognition sequence from symbionts such as HC_Bin1_Evansiales and TC_Bin2_AqS2 were matched to phage sequences obtained in the present study (Table S14), indicating that the RM systems were responsive to infection by phages.
High abundance of SUP05 bacteria and their phages
Our analysis of microbial composition based on 16S rRNA genes revealed that SUP05 populations were dominant in the three vent sponges; phage‐host prediction (Table S4) and analysis of phage abundance showed that SUP05 phages were among the most abundant viruses (Fig. 3; Tables S2 and S3). The coexistence of both abundant SUP05 and their phages is inconsistent with the classical ‘Kill‐the‐Winner’ model (Thingstad, 2000) in which lytic infection suppresses blooms of fast‐growing microbes. A similar case for both abundant SAR11 bacteria and their phages was observed in the open ocean and explained by their life strategy (Zhao et al., 2013). In contrast, the coincident high abundance of SUP05 phages may be explained by the co‐infection of different bacteria, which could increase viral abundance. Indeed, phage Bin9 in sponge HC is predicted to infect both HC_SUP05 and HC_Bin4_RS24 (Table S4).
Fig. 3.
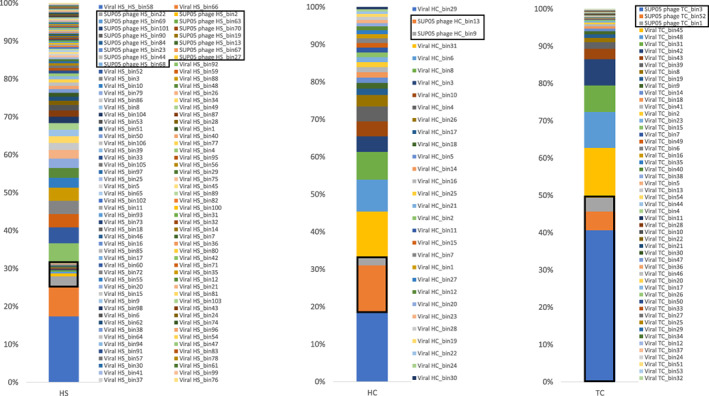
Relative abundance of phage bins (populations) in three vent sponges. HC, haplosclerid sponge from Crane site; HS, haplosclerid sponge from Swan site; TC, tedaniid sponge from Crane site. SUP05 phages are outlined in black.
In addition, the SUP05 bacteria in three vent demosponges were closely related and had undergone genome reduction (Fig. S2‐1–2). Streamlined genomes have been suggested to function as competitive traits (Grote et al., 2012), indicating that the SUP05 are competition strategists. Regardless of their small genomes, SUP05 harboured complete sets of sulfur metabolism‐related genes, which may be utilized to oxidize the abundant reduced sulfur compounds of the vent environment and produce energy for cellular activity.
Horizontal gene transfer between phages and hosts
In the vent sponges evaluated in this study, viruses were predicted to harbour transferred genome regions based on their high sequence similarity and maximum likelihood phylogeny (Fig. 4A–C, Fig. S3‐1–11) with the regions encoding 82 protein‐coding genes and five tRNA genes (Table S15). These genes relate to carbohydrate metabolism, amino acid metabolism, metabolism of cofactors and vitamins, lipid biosynthesis, genetic information processing, and signalling and cellular processes. Visualization of the genome structure revealed high similarity between these phage genes and their hosts (Fig. S3‐1–11). For instance, ygfA, dnaA and dnaG homologues in TC_Bin2_AqS2 and phage Bin1 have >96% amino acid similarity (Fig. S3‐1). Among the protein‐coding sequences, nine were classified as putative auxiliary metabolic genes, including genes for the NAD‐dependent epimerase/dehydratase family, phosphoenolpyruvate phosphomutases and the NAD‐binding domain of 6‐phosphogluconate dehydrogenases (Table S15). Based on a maximum likelihood inferred phylogeny and comparison of the genome structure, the auxiliary metabolic gene galE belonging to the NAD‐dependent epimerase/dehydratase family and a gene encoding the NAD‐binding domain of 6‐phosphogluconate dehydrogenases were demonstrated to have been transferred between the γ‐proteobacteria TC_Bin2_AqS2 and the phage Bin1 (Fig. 4D–E and Fig. S4). galE encodes UDP‐glucose 4‐epimerase, which mediates the conversion of UDP‐galactose and UDP‐glucose in galactose metabolism (Thoden and Holden, 1998). With the gain of galE, the phage Bin1 could participate in carbohydrate metabolism and generate energy for replication. In addition, the presence of tRNAs (tRNA‐Thr, tRNA‐Ser, tRNA‐Met and tRNA‐Asp) in phage sequences indicated a complementation/compensation of the host translational machinery for phage reproduction.
Fig. 4.
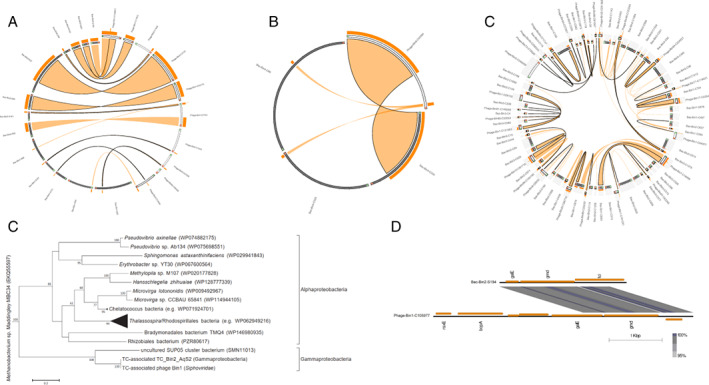
Phage‐host HGT in three vent sponges.
A–C. Circle plots of putatively transferred regions of phages and bacterial hosts in sponge TC (A), sponge HC (B) and sponge HS (C). Viral contigs are shown as protruding grey ideograms; bacterial scaffolds are in black. The direction of orientation is green to red. Orange ribbons show >70% identity. Stacked histograms on top of the ideograms represent the frequency and ratio of the ribbons at each point. Ribbons outlined in black represent the best BLASTn alignment of sequences. Label names have been shortened, for example, Bac‐Bin1‐S36 in (A) represents scaffold_36 of the bacterium TC_Bin1_SUP05; Phage‐Bin52‐C55036 represents contig_55036 of the phage Bin52 in sponge TC.
D. Maximum likelihood‐based phylogeny of galE homologues in TC_Bin2_AqS2 and TC‐associated phage Bin1.
E. Genome structure of contigs containing galE homologues in TC_Bin2_AqS2 and TC associated phage Bin1. Bac‐Bin2‐S194, scaffold_194 of the bacterium TC_Bin2_AqS2; Phage‐Bin1‐C105977, contig_105977 of the phage Bin1. HC, haplosclerid sponge from Crane site; HS, haplosclerid sponge from Swan site; TC, tedaniid sponge from Crane site. HGT, horizontal gene transfer.
Viral complementation/compensation in bacterial metabolic pathways
Viruses in the sediments and seawater of deep‐sea hydrothermal vent fields have been found to complement the metabolic pathways of their hosts and/or compensate for metabolic deficiencies (Anantharaman et al., 2014; He et al., 2017). They can participate in metabolisms of sulfur oxidization, pyrimidine, amino sugar and nucleotide sugar. In our study, metabolic complementation was indicated based on the shared pathways of phages and their hosts, according to a search of the Kyoto Encyclopedia of Genes and Genomes database (Kanehisa and Goto, 2000) using BLASTp (Fig. 5A and B). For instance, TC_Bin1_SUP05 bacteria and their phages Bin1, Bin3 and Bin52 contained homologues of genes encoding UDP‐glucose 4‐epimerases, GDP‐L‐fucose synthases and GDP mannose 4,6‐dehydratases (Fig. S5). Overall, 146 phage genes were involved in 34 metabolic pathways of their hosts (Table S16), in which metabolism of carbohydrates, energy and nucleotides dominated. In certain pathways, we found that genes possibly absent from the host were present in their phages, suggesting that metabolic compensation might occur during infection. For example, in TC_Bin1_SUP05, genes for formate‐tetrahydrofolate ligases were not detected, whereas phages infecting SUP05 contained genes for these enzymes (Fig. 5A). Bacterial symbionts are metabolically important for nutrient cycling in deep‐sea sponges (Tian et al., 2016), as they utilize inorganic materials such as carbon dioxide from the sponge host to synthesize carbohydrates powered by energy from sulfur and/or methane oxidization. The presence of numerous phage genes involved in carbohydrate and energy metabolism indicates that infection contributes to the biogeochemical cycling in the symbiotic system of deep‐sea vent sponges.
Fig. 5.
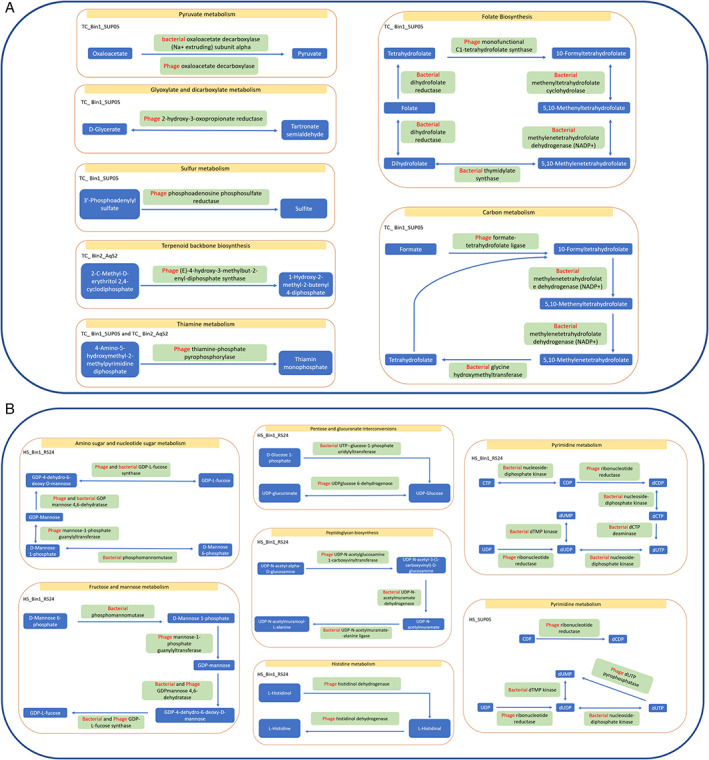
Kyoto Encyclopedia of Genes and Genomes pathways compensated by phage genes in the tedaniid and haplosclerid vent sponges.
A. Pathway compensation in the tedaniid sponge (TC).
B. Pathway compensation in the haplosclerid sponge (HS). HC, haplosclerid sponge from Crane site; HS, haplosclerid sponge from Swan site; TC, tedaniid sponge from Crane site.
Conclusions
Deep‐sea hydrothermal vents are extreme environments that give rise to unique examples of biology and ecology. Our metagenomic analyses of demosponge microbiomes in the deep‐sea hydrothermal vent fields of the Okinawa Trough have unveiled diverse bacteria and phages involved in unique interactions. Prophages in the genomes of bacterial symbionts were less detected in vent sponges. The dominating anti‐viral defence systems of bacterial symbionts may be the restriction‐modification, Zorya, DISARM and toxin–antitoxin systems. SUP05 bacteria and their phages were both abundant, indicating the use of competitive traits and defensive strategies obtained by genetic recombination of SUP05 symbionts. The transfer between Bacterial genes (including potential auxiliary genes) and phage genomes suggests that bacterial genes can be obtained by phages, thereby benefit viral infection and improve viral fitness. Additionally, phage genes may contribute beneficially to bacterial metabolism, including carbohydrate and energy metabolism, thereby influencing nutrient cycling in these symbiotic systems. Collectively, our findings provide a foundation for comprehensively understanding the roles of viruses in deep‐sea vent sponge holobionts. Further studies of larger number of samples, viral activity measurement and isolation of symbiotic bacterium‐phage cultures are required.
Supporting information
Appendix S1. Supplementary Information
Acknowledgements
We thank the captains and crews of the research vessels Kairei and Yokosuka, Kyoko Okino (AORI, University of Tokyo) and the operation team of the ROV KAIKO, for their tireless support during cruises YK14‐16 and KR15‐16. We also thank Dr. Jin Sun (Hong Kong University of Science and Technology), Dr. Chong Chen (Japan Agency for Marine‐Earth Science and Technology), Dr. Ren‐Mao Tian (University of Oklahoma) and Yi Yang (Hong Kong University of Science and Technology) for helping in this program.
Contributor Information
Ying Xu, Email: boxuying@szu.edu.cn.
Rui Zhang, Email: ruizhang@xmu.edu.cn.
Data Availability Statement
All data are available in NCBI database (www.ncbi.nlm.nih.gov). The accession numbers of raw reads of two DNA libraries of TC metagenome are SRR11301536 for the 500 bp insert and SRR11301537 for the 300 bp insert. The accession number of the assembled TC metagenome is JAAGOD000000000. The genome accession numbers of all bacterial genome bins from TC, HC and HS are presented in Table S1.
References
- Anantharaman, K. , Duhaime, M.B. , Breier, J.A. , Wendt, K.A. , Toner, B.M. , and Dick, G.J. (2014) Sulfur oxidation genes in diverse deep‐sea viruses. Science 344: 757–760. [DOI] [PubMed] [Google Scholar]
- Arndt, D. , Grant, J.R. , Marcu, A. , Sajed, T. , Pon, A. , Liang, Y. , and Wishart, D.S. (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44: W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Watanabe, H.K. , Miyazaki, J. , and Kawagucci, S. (2017) Unanticipated discovery of two rare gastropod molluscs from recently located hydrothermally influenced areas in the Okinawa Trough. PeerJ 5: e4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary, D.F.R. , Becking, L.E. , de Voogd, N.J. , Pires, A.C.C. , Polonia, A.R.M. , Egas, C. , and Gomes, N.C.M. (2013) Habitat‐ and host‐related variation in sponge bacterial symbiont communities in Indonesian waters. FEMS Microbiol Ecol 85: 465–482. [DOI] [PubMed] [Google Scholar]
- Corliss, J.B. , Dymond, J. , Gordon, L.I. , Edmond, J.M. , von Herzen, R.P. , Ballard, R.D. , et al. (1979) Submarine thermal springs on the Galapagos Rift . Science 203: 1073–1083. [DOI] [PubMed] [Google Scholar]
- Dick, G.J. (2019) The microbiomes of deep‐sea hydrothermal vents: distributed globally, shaped locally. Nat Rev Microbiol 17: 271–283. [DOI] [PubMed] [Google Scholar]
- El‐Gebali, S. , Mistry, J. , Bateman, A. , Eddy, S.R. , Luciani, A. , Potter, S.C. , et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res 47: D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara, Y. , Takai, K. , Uematsu, K. , Tsuchida, S. , Hunt, J.C. , and Hashimoto, J. (2000) Phylogenetic characterization of endosymbionts in three hydrothermal vent mussels: influence on host distributions. Mar Ecol Prog Ser 208: 147–155. [Google Scholar]
- Grote, J. , Thrash, J.C. , Huggett, M.J. , Landry, Z.C. , Carini, P. , Giovannoni, S.J. , and Rappé, M.S. (2012) Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3: e00252‐00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halanych, K.M. (2005) Molecular phylogeny of siboglinid annelids (aka pogonophorans): a review. Hydrobiologia 535: 297. [Google Scholar]
- He, T. , Li, H. , and Zhang, X. (2017) Deep‐sea hydrothermal vent viruses compensate for microbial metabolism in virus‐host interactions. mBio 8: e00893‐00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta, T. , Takaki, Y. , Nagai, Y. , Shimamura, S. , Tsuda, M. , Kawagucci, S. , et al. (2016) Heterogeneous composition of key metabolic gene clusters in a vent mussel symbiont population. ISME J 10: 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, M.T. , Arkhipova, K. , Markert, S.M. , Stigloher, C. , Lachnit, T. , Pita, L. , et al. (2019) A phage protein aids bacterial symbionts in eukaryote immune evasion. Cell Host Microbe 26: 542–550. [DOI] [PubMed] [Google Scholar]
- Kanehisa, M. , and Goto, S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, B. , Silveira, C.B. , Bailey, B.A. , Barott, K. , Cantu, V.A. , Cobian‐Guemes, A.G. , et al. (2016) Lytic to temperate switching of viral communities. Nature 531: 466–470. [DOI] [PubMed] [Google Scholar]
- Kuwahara, H. , Yoshida, T. , Takaki, Y. , Shimamura, S. , Nishi, S. , Harada, M. , et al. (2007) Reduced genome of the thioautotrophic intracellular symbiont in a deep‐sea clam, Calyptogena okutanii . Curr Biol 17: 881–886. [DOI] [PubMed] [Google Scholar]
- Laffy, P.W. , Wood‐Charlson, E.M. , Turaev, D. , Jutz, S. , Pascelli, C. , Botte, E.S. , et al. (2018) Reef invertebrate viromics: diversity, host specificity and functional capacity. Environ Microbiol 20: 2125–2141. [DOI] [PubMed] [Google Scholar]
- Makabe, A. , Tsutsumi, S. , Chen, C. , Torimoto, J. , Matsui, Y. , Shibuya, T. , Miyazaki, J. , Kitada, K. , and Kawagucci, S. (2016) Discovery of new hydrothermal vent fields in the mid‐ and southern‐Okinawa Trough. In Goldschmidt Conference Abstracts 2016; 1945.
- Pascelli, C. , Laffy, P.W. , Kupresanin, M. , Ravasi, T. , and Webster, N.S. (2018) Morphological characterization of virus‐like particles in coral reef sponges. PeerJ 6: e5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, R.J. , Vincze, T. , Posfai, J. , and Macelis, D. (2015) REBASE ‐ a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43: D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, S. , Enault, F. , Hurwitz, B.L. , and Sullivan, M.B. (2015) VirSorter: mining viral signal from microbial genomic data. PeerJ 3: e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin‐Blum, M. , Antony, C.P. , Sayavedra, L. , Martinez‐Perez, C. , Birgel, D. , Peckmann, J. , et al. (2019) Fueled by methane: deep‐sea sponges from asphalt seeps gain their nutrition from methane‐oxidizing symbionts. ISME J 13: 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacristan‐Soriano, O. , Criado, N.P. , and Avila, C. (2020) Host species determines symbiotic community composition in Antarctic sponges (Porifera: Demospongiae). Front Mar Sci 7: 474. [Google Scholar]
- Song, W. , Sun, H.X. , Zhang, C. , Cheng, L. , Peng, Y. , Deng, Z. , et al. (2019) Prophage Hunter: an integrative hunting tool for active prophages. Nucleic Acids Res 47: W74–W80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudigel, H. (2003) Hydrothermal alteration processes in the oceanic crust. Treatise Geochem 3: 659. [Google Scholar]
- Taylor, M.W. , Hill, R.T. , Piel, J. , Thacker, R.W. , and Hentschel, U. (2007) Soaking it up: the complex lives of marine sponges and their microbial associates. ISME J 1: 187–190. [DOI] [PubMed] [Google Scholar]
- Thingstad, T.F. (2000) Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr 45: 1320–1328. [Google Scholar]
- Thoden, J.B. , and Holden, H.M. (1998) Dramatic differences in the binding of UDP‐galactose and UDP‐glucose to UDP‐galactose 4‐epimerase from Escherichia coli . Biochemistry 37: 11469–11477. [DOI] [PubMed] [Google Scholar]
- Tian, R.M. , Sun, J. , Cai, L. , Zhang, W.P. , Zhou, G.W. , Qiu, J.W. , and Qian, P.Y. (2016) The deep‐sea glass sponge Lophophysema eversa harbours potential symbionts responsible for the nutrient conversions of carbon, nitrogen and sulfur. Environ Microbiol 18: 2481–2494. [DOI] [PubMed] [Google Scholar]
- Tian, R.M. , Zhang, W. , Cai, L. , Wong, Y.H. , Ding, W. , and Qian, P.Y. (2017) Genome reduction and microbe‐host interactions drive adaptation of a sulfur‐oxidizing bacterium associated with a cold seep sponge. mSystems 2: e00184‐00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon, M. , Bernheim, A. , and Rocha, E.P. (2016) Genetic and life‐history traits associated with the distribution of prophages in bacteria. ISME J 10: 2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, H. , and Kojima, S. (2015) Vent fauna in the Okinawa trough. In Subseafloor Biosphere Linked to Hydrothermal Systems: TAIGA Concept. Ishibashi, J.‐I. , Okino, K. , and Sunamura, M. (eds). Tokyo: Springer Japan, pp. 449–459. [Google Scholar]
- Webster, N.S. , and Taylor, M.W. (2012) Marine sponges and their microbial symbionts: love and other relationships. Environ Microbiol 14: 335–346. [DOI] [PubMed] [Google Scholar]
- Wolf, Y.I. , and Koonin, E.V. (2013) Genome reduction as the dominant mode of evolution. Bioessays 35: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida‐Takashima, Y. , Nunoura, T. , Kazama, H. , Noguchi, T. , Inoue, K. , Akashi, H. , et al. (2012) Spatial distribution of viruses associated with planktonic and attached microbial communities in hydrothermal environments. Appl Environ Microbiol 78: 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Temperton, B. , Thrash, J.C. , Schwalbach, M.S. , Vergin, K.L. , Landry, Z.C. , et al. (2013) Abundant SAR11 viruses in the ocean. Nature 494: 357–360. [DOI] [PubMed] [Google Scholar]
- Zhou, K. , Zhang, R. , Sun, J. , Zhang, W. , Tian, R.‐M. , Chen, C. , et al. (2019) Potential Interactions between Clade SUP05 sulfur‐oxidizing bacteria and phages in hydrothermal vent sponges. Appl Environ Microbiol 85: e00992‐00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary Information
Data Availability Statement
All data are available in NCBI database (www.ncbi.nlm.nih.gov). The accession numbers of raw reads of two DNA libraries of TC metagenome are SRR11301536 for the 500 bp insert and SRR11301537 for the 300 bp insert. The accession number of the assembled TC metagenome is JAAGOD000000000. The genome accession numbers of all bacterial genome bins from TC, HC and HS are presented in Table S1.