

Abstract
Copolymers are the answer to property limitations of homopolymers. In order to use the full variety of monomers available, catalysts active in more than one polymerization mechanism are currently investigated. Iron guanidine catalysts have shown to be extraordinarily active in ROP of lactide and herein prove their versatility by also promoting ATRP of styrene. The presented iron complex is the first polymerizing lactide and styrene simultaneously to a defined block copolymer in a convenient one‐pot synthesis. Both mechanisms work hand in hand with ROP using the dominantly present FeII species on the dormant side of the ATRP equilibrium. This orthogonal copolymerization by a benign iron catalyst opens up new pathways to biocompatible polymerization procedures broadening the scope of ATRP applications.
Keywords: ATRP, copolymerization, iron, orthogonal catalysis, ROP
Orthogonal catalysis combining two polymerization mechanisms in a one‐pot synthesis enables facile production routes to block copolymers. An iron guanidine catalyst is able to copolymerize styrene via ATRP and lactide via ROP simultaneously. By using the FeII species on the dormant side of the ATRP equilibrium for ROP both mechanisms proceed in a controlled manner without interference.

Polymeric materials have revolutionized building, packaging and products in the past century in a way they have become irreplaceable in our everyday life. [1] However, with the innovation massive environmental problems came along. [2] Therefore, the end‐of‐life options should be already considered during the design and, if possible, renewable resources be used. [3] A versatile polymer with extraordinary future prospects is polylactide (PLA) since it comes from a renewable source and covers several end‐of‐life options: mechanical and chemical recycling as well as biodegradation. [4] Next to high‐performing catalysts polymerizing in solution, [5] alternative, biocompatible catalysts for the currently used toxic tin compound in the industrial production of PLA have been developed recently to make the polymers' life cycle even more sustainable. [6] Besides impressive polymeric materials already developed, a treasure yet to be fully understood lies in the combination of different monomers to form copolymers. [7] By adjusting monomer ratios and microstructures materials with tailor‐made physical and mechanical properties as well as a defined durability can be synthesized. [8] Usually, monomers can only be copolymerized if they possess the same functionality and both respond to the same polymerization mechanism, which limits the possible combinations dramatically. Two strategies have been developed to be able to copolymerize orthogonally. In one approach, the first monomer is polymerized and the end group of the produced polymer is functionalized in order to initiate the polymerization of the second monomer. This macroinitiator route enables many combinations, but is quite work‐intensive due to the work‐up between the polymerizations. [9] In a second approach a bifunctional initiator is used which can either promote a sequential or simultaneous polymerization by delivering the initiator for both polymerizations. [10] The main limitations for such an orthogonal one‐pot polymerization are the catalysts, which have to tolerate all components present in the reaction mixture and must not interfere with the other mechanism. The solution to this is one catalyst being active in several polymerization mechanisms. Such catalysts are rare in literature but some examples are found in switch catalysis combining the ring‐opening polymerization of cyclic esters (ROP) with ring‐opening copolymerization of epoxides with CO2 or anhydrides (ROCOP) producing well‐defined block copolymers in a convenient one‐pot procedure. [11] Furthermore, it was shown that redox‐switches can be applied to modify the monomer preference of a catalyst. [12] Attempts have been made to combine ROP with atom transfer radical polymerization (ATRP), yet, to the best of our knowledge no molecular metal catalyst was capable of performing this orthogonal copolymerization in a one‐pot synthesis until today. [13] There are reports about the copolymerization of methyl methacrylate (MMA) with different lactones, however, an uncontrolled polymerization mechanism is followed for the vinyl monomer. [14]
ATRP is an efficient way to produce well defined polymers of unsaturated monomers for numerous applications. [15] Additionally to the synthetically available monomers like styrene or MMA, nature provides interesting substrates containing the potential to replace conventional plastics or conquer new property limitations as polymers. [16] Typically, Cu catalysts are applied due to their good performance, anyhow, their toxic properties in biological systems rule out a number of applications. [17]
Iron complexes on the other hand have received more and more attention in recent years as ATRP catalysts because iron possesses the two oxidation states necessary for the ATRP equilibrium, is inexpensive and abundant while it can be metabolized and subsequently used by many living organisms. [19] Iron catalysts in ATRP allow the exploration of new polymerization techniques in combination with biological systems: Growth in or from cells has been reported as well as naturally occurring iron compounds being active in ATRP. [20] A biologically compatible copolymerization pathway combining ATRP monomers with PLA, a material widely used for implants, generates new options. [21] We recently presented iron guanidine catalysts being highly active in the ROP of lactide—even surpassing the industrially used tin(II) octoate under industrially relevant melt conditions. [18] Until today, only copper guanidine catalysts were applied in ATRP, [22] but herein we show that also iron guanidine catalysts can catalyze ATRP. Its performances in both polymerization mechanisms confer the unique property to the catalyst to copolymerize styrene and lactide from a monomer mixture orthogonal at the same time. The oil‐derived and non‐biodegradable styrene is applied as well‐investigated model monomer to establish a method for orthogonal polymerization using different functional groups. In future, biobased monomers, potentially even containing both functionalities like itaconic acid derivatives[ 16b , 23 ] or α‐methylene‐γ‐butyrolactone (MBL),[ 16a , 24 ] are intended to be polymerized.
For the catalyst synthesis the ligand methyl 2‐((bis(dimethylamino)methylene)amino)benzoate (TMGasme) was obtained by reported procedures [6b] and crystallized with FeBr2 since it was shown that bromide complexes have better polymerization properties than their chloride analogues in ATRP. [25] The molecular structure of [FeBr2(TMGasme)] (1) is shown in Figure 1, crystallographic details are given in the Supporting Information. 1 crystallizes in a ratio of 1:1 of ligand and iron salt. The ligand has a bite angle of 86.4(1)° and coordinates the iron atom with the guanidine nitrogen atom N1 as well as the carbonyl oxygen atom O1 of the ester function. The fourfold coordination is completed by two bromide ligands which open an angle of 122.1(1)°. The bond lengths and angles of 1 show a high similarity to those of the analogous chloride complex [FeCl2(TMGasme)] leaving the larger Br1‐Fe1‐Br2 angle as a result of the bigger size of bromide the main difference. [18] Since the access to the metal center of the catalyst is a major point of influence for the polymerization activity, the different sizes of the halide ligands become relevant when investigating the polymerization properties. 1 was tested under the same conditions as the analogous chloride complex, bulk polymerization at 150 °C with l‐lactide recrystallized once, in an in situ Raman monitored reactor, to evaluate its features in the ring‐opening polymerization. Table 1 summarizes the details of the polymerizations including the reaction rate constants while Figure S2 shows the corresponding semilogarithmic plot. In comparison with its chloride analogue, the polymerization activity of 1 is found to be in the same order of magnitude, nonetheless slightly lower. The reduction might be the result of the sterically more demanding bromide ligands hampering the access of the monomer to the active chain end. The polymer produced by 1 shows high average molar masses even though the degree of control, expressed by stronger deviations from the theoretically expected molar mass and a higher dispersity, is slightly reduced in comparison with the chloride complex. Due to the very similar polymerization behavior of the two complexes, we imply that 1 performs a living polymerization following the coordination–insertion mechanism as it was shown for the chloride analogue. [18]
Figure 1.
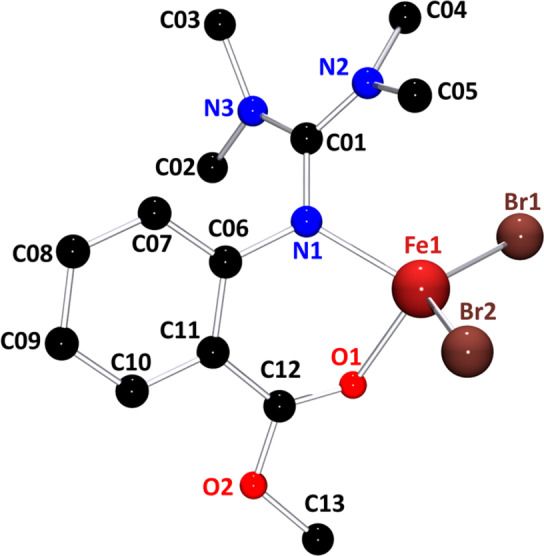
Molecular structure of 1 in the solid state determined by single crystal X‐ray diffraction. Bond lengths: Fe1–Br1 2.393(1) Å, Fe1–Br2 2.406(1) Å, Fe1–N1 2.041(1) Å, Fe1–O1 2.042(2) Å; angles: N1‐Fe1‐O1 86.4(1)°, Br1‐Fe1‐Br2 122.1(1)°. Further bond lengths and angles are listed in Table S2 in comparison with the analogous chloride complex.
Table 1.
ROP of lactide with chloride and bromide complexes.
|
Catalyst[a] |
t [min] |
k app [×10−3 s−1] |
p [b] |
M n,theo [c] [g mol−1] |
M n [d] [g mol−1] |
Đ [d] |
|---|---|---|---|---|---|---|
|
1 |
65 |
0.44 |
0.56 |
80 700 |
42 200 |
1.6 |
|
1 |
45 |
0.36 |
0.55 |
77 900 |
68 800 |
1.6 |
|
[FeCl2(TMGasme)] [18] |
30 |
0.72 |
0.66 |
95 100 |
96 700 |
1.3 |
|
[FeCl2(TMGasme)] [18] |
40 |
0.61 |
0.64 |
92 200 |
82 500 |
1.5 |
[a] Monomer‐to‐initiator (M/I) ratio 1000:1, 150 °C, 260 rpm, solvent free. [b] Calculated from 1H NMR spectrum. [c] Calculated by conversion×molar mass×M/I ratio. [d] Obtained from GPC measurement in THF.
In comparison with other robust iron or zinc catalysts applied in bulk polymerization, 1 still performs very well and ranges among the most active robust catalysts for lactide polymerization.[ 6b , 6c , 26 ] 1 is therefore well suited to expand the scope of iron guanidine complexes towards ATRP and orthogonal copolymerizations.
The polymerization activity of 1 as catalyst in ATRP was hence investigated. Styrene was polymerized in bulk with a monomer to initiator to catalyst ratio (M/I/Cat ratio) of 100:1:1 at 110 °C. The bifunctional initiator (4‐(bromomethyl)phenyl)methanol (BrPhOH) was used to rule out effects caused by the change of initiator when performing copolymerizations (vide supra). The polymerization was conducted in a Schlenk tube and kinetic data were derived from 1H NMR spectra from aliquots removed after defined reaction times. Figure 2 shows the semilogarithmic plot of conversion versus time and the development of the molar masses and dispersities during the polymerization.
Figure 2.

Polymerization of styrene. M/I/Cat ratio 100:1:1, 110 °C, 800 rpm, under solvent‐free conditions. Top: semilogarithmic plot of the conversion p vs. time (duplicate measurement, error bars partially invisible due to too small variations. The values are displayed in Table S3). Bottom: theoretical molar masses (black line), experimentally found molar masses (black squares), and dispersities Đ (red circles) in the course of the polymerization.
The polymerization of styrene follows a linear development in the semilogarithmic plot showing a conversion of 25 % after 60 min. Therefore, a pseudo first order kinetic can be assumed. The molar masses grow with the conversion in good approximation to the theoretical values. With dispersities below 1.2 1 performs a highly controlled polymerization producing defined polymer chains typical for a catalyst establishing an ATRP equilibrium. To identify 1 as the active catalytic system, polymerization experiments leaving out one or several components were performed (see Table S4). They confirm that the exact molar masses and narrow dispersities can only be reached when isolated 1 is used. If the catalyst is added in situ, iron bromide promotes an uncontrolled radical polymerization before an equilibrium is established leading to broad dispersities (see Figure S37). Despite the good control crystalline 1 exhibits, it struggles with reaching high conversions since a deactivation can be observed at longer reaction times (see Table S3 and Figure S4). Iron catalysts are known for side reactions promoting terminations like catalytic chain transfer (CCT) or catalytic radical termination (CRT) mostly involving the accumulation of FeIII and leading to a deactivation with time. [27] Applying the principles of initiators for continuous activator regeneration (ICAR) or activators regenerated by electron transfer (ARGET) ATRP sometimes leads to a better performance of iron catalysts, which will be assessed in future.[ 25 , 28 ] For now, 1 is one of the rare examples of iron complexes that are highly active in the ROP of lactide and perform a well‐controlled ATRP of styrene. Hence, 1 is a promising candidate to be the first metal catalyst accomplishing a simultaneous ROP and ATRP in a one‐pot reaction.
Therefore, the polymerization behavior of 1 in a monomer mixture was investigated. A polymerization with a M/I/Cat ratio of 100+100:1:1 (lactide+styrene:BrPhOH:1) was conducted by mixing the components in the reactRaman reactor. Toluene was added to prevent diffusion limitation. Figure 3 shows the collected Raman spectra during the polymerization and the semilogarithmic plot of the integrated monomer bands. For styrene, the band at 1630 cm−1 referring to the double bond stretching is integrated. [29] For lactide, the band at 655 cm−1 reflecting the ring vibration is monitored for the conversion. Furthermore, a band at 873 cm−1 appears during the polymerization which was identified as carbonyl vibration of PLA. The experiment reveals the stunning properties of 1 as catalyst: Both monomers are simultaneously converted to polymer in a controlled manner. The conversion of lactide via ROP proceeds significantly faster than the ATRP of styrene reaching nearly full conversion within the first hour. The polymerization of styrene shows in the beginning a linear growth with time [30] and reaches 38 % conversion after 270 min reflecting the deactivation observed before in the homopolymerization. Different monomer‐to‐monomer ratios were applied and in all cases the conversion of both monomers was recorded (see Table 2). The changing ratios correlate with the found incorporation ratios. The dispersities of the produced polymers are with 1.1 very low, which shows that the polymerization proceeds in a highly controlled manner. The low dispersity is especially impressive with regard to the dispersity of 1.6 of the polymer produced by lactide homopolymerization (see Table 1) and expresses that the conditions chosen for the copolymerization are favorable for a high degree of control in the ROP of lactide. In addition, a sequential approach was tested in which the ATRP as the more sensitive polymerization mechanism was executed first and the ROP of lactide afterwards. The catalyst managed to convert the freshly added lactide, however, much more slowly than expected for 1. Since the deactivation in ATRP is accompanied by the accumulation of the FeIII complex, it is possible that the catalytic system is no longer as active for the ROP of lactide due to the change of oxidation state of the dominantly present iron species. To assess if the FeIII complex of 1 exhibits a lower activity in the ROP of lactide than the FeII complex, lactide was homopolymerized under comparable conditions (volume of styrene replaced by toluene in order to maintain the concentration) with 1 (FeII complex) or with the in situ generated analogous FeIII complex, which could unfortunately not be characterized so far, as catalyst (see Figure S29). The experiment clearly showed that the FeII complex polymerizes lactide significantly faster than the in situ generated FeIII complex, reaching a polymerization activity matching the one of the simultaneous copolymerization. The FeIII complex, on the other hand, hardly converted lactide. Hence, the hindered activity for the ROP of lactide when conducted after the ATRP of styrene is explained. Additionally, the affinity for lactide polymerization of the complexes differing in the oxidation state allows a mechanistical proposal: If only the FeII complex can polymerize lactide with the observed activity, the ROP must take place on the dormant side of the ATRP equilibrium (see Scheme 1). The simultaneous conversion of styrene and lactide becomes possible when ATRP and ROP do not hinder each other but work hand in hand on the different sides of the equilibrium. The ROP of lactide is performed on the mostly “idle” FeII complex, which only has to remain available for the formation of the active ATRP species. Since 1 exhibits living features in ROP as the analogue chloride complex does, an interruption of the ROP does not have consequences.
Figure 3.

Simultaneous copolymerization of lactide and styrene. Top: reactRaman spectra of the polymerization. Bottom: semilogarithmic plot of integrated monomer bands (see also Figure S7).
Table 2.
Details of orthogonal copolymerizations of lactide and styrene via ROP and ATRP.
|
Run |
M/I/Cat ratio[a] |
Monomers[b] |
t [min] |
p(S)[c] |
p(LA)[c] |
Incorp. Ratio (S:LA)[d] |
M n,theo [e] [g mol−1] |
M n [f] [g mol−1] |
Đ [f] |
|---|---|---|---|---|---|---|---|---|---|
|
1 |
100+100:1:1 |
S+LA |
270 |
0.38 |
0.93 |
0.33:1.00 |
27 100 |
31 200 |
1.12 |
|
2 |
50+150:1:1 |
S+LA |
270 |
0.27 |
0.86 |
0.09:1.00 |
24 200 |
33 200 |
1.12 |
|
3 |
150+50:1:1 |
S+LA |
270 |
0.45 |
0.92 |
0.77:1.00 |
27 600 |
22 000 |
1.09 |
|
4 |
100(+100):1:1[g] |
S |
180 |
0.27 |
– |
– |
2800 |
5300 |
1.11 |
|
|
S+LA (seq.) |
180+180 |
0.32 |
0.18 |
1.00:0.22 |
7800 |
7000 |
1.09 |
[a] Styrene+lactide:BrPhOH:1. Polymerizations performed at 110 °C and 260 rpm. [b] S: styrene, LA: lactide. [c] Conversion p calculated from 1H NMR spectrum. [d] Calculated from the 1H NMR spectrum of the isolated polymer. [e] Calculated by M n,theo=p(S)×104.15 g mol−1×S/I ratio+p(LA)×144.13 g mol−1×LA/I ratio/0.56 to obtain the theoretical molar mass in styrene equivalents. [31] [f] Obtained from GPC measurement. [g] Sequential polymerization, styrene was homopolymerized for 180 min, afterwards an aliquot was removed, lactide was added and the polymerization was run for additionally 180 min.
Scheme 1.

Proposed mechanism for combined ROP and ATRP.
Both polymerization mechanisms do not rely on a constant connection to the active chain end. Therefore, a bifunctional co‐initiator can lead to the formation of a PLA–PS block copolymer. All performed polymerizations containing lactide and styrene (run 1–4, Table 2) show a monomodal molar mass distribution when analyzed with gel permeation chromatography (GPC, see Figure S30). The molar masses of the simultaneous polymerizations match the theoretical ones well, independently of the monomer‐to‐monomer ratio applied. If two unconnected homopolymers had formed, significant deviations from the theoretical molar masses should be found. Furthermore, the sequential polymerization shows a significant growth in molar mass between the aliquot taken before the lactide addition and after completion of the full reaction time, which cannot be caused by the slight growth of styrene conversion only (see Table 2, run 4). 1H NMR spectra confirmed that both functional groups of the co‐initiator are used in the simultaneous copolymerization (see Figure S28). Diffusion‐ordered NMR spectroscopy (DOSY) revealed a single diffusion coefficient for the polymer product produced by simultaneous copolymerization as well as by sequential addition (see Figure S11–S26) supporting the formation of block copolymers further. The mixture of the two homopolymers shows in contrast two diffusion coefficients (see Figure S27). The absence of non‐blocky segments in the copolymer is validated by a defined single peak in the carbonyl region of the 13C NMR spectrum corresponding to the carbonyl carbon atom of lactide which is very sensitive to varying neighboring units. [32] In addition to the spectroscopic investigations, the copolymers as well as the homopolymers and a mixture of the latter were analyzed with differential scanning calorimetry (DSC, see Table S5 and Figure S31–S36). The results show the difference of PLA as a semicrystalline polymer and PS as an amorphous polymer. The melting of the PLA homopolymer is recorded with 174 °C in a typical range, [33] while no glass transition was observed. The degree of crystallinity is determined with 61 %. PS on the other hand only shows a glass transition at 79 °C. The produced copolymers as well as the pestled homopolymer mixture exhibit the features of both polymers: a measurable glass transition as well as a melting event which vary around the values of the homopolymers. The occurrence of melting events in all polymers suggests that the copolymers undergo microphase separation with crystalline domains surrounded by amorphous ones. As expected, the degree of crystallinity rises with the PLA content of the polymer.
The herein presented iron guanidine catalyst is the first to polymerize lactide and styrene via ROP and ATRP simultaneously. It represents a breakthrough for orthogonal catalysts, revolutionizing the synthesis of block copolymers of lactones and vinylic monomers. By using the dormant side of ATRP for ROP both mechanisms go hand in hand and a high degree of control is maintained. This novel working principle will pave the way to further orthogonal polymerization applications. The here reported one‐pot synthesis approach allows copolymerization in a facile way making extensive copolymerization procedures obsolete. Due to the biocompatible character of iron, the catalyst fulfills the criteria for applications with biological contact and opens up new polymerization procedures combining biodegradable lactones with vinylic monomers. The details of the polymerization mechanisms as well as further monomer combinations are currently under investigation.
Details on the molecular structure of 1 can be found at the Cambridge Crystallographic Data Centre under deposition number CCDC 2073861.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
R.D.R. thanks Deutsche Bundesstiftung Umwelt (DBU) for funding. The authors thank Total Corbion PLA for lactide donations, Dr. G. Fink for NMR measurements, and S. Buschmann for DSC measurements. Furthermore, the authors thank the referees for valuable suggestions for the improvement of this manuscript. S. H.‐P. thanks the PrepFund (PFKA007) of RWTH Aachen University. Open access funding enabled and organized by Projekt DEAL.
R. D. Rittinghaus, A. Karabulut, A. Hoffmann, S. Herres-Pawlis, Angew. Chem. Int. Ed. 2021, 60, 21795.
References
- 1. Geyer R., Jambeck J. R., Law K. L., Sci. Adv. 2017, 3, e1700782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Lebreton L. C., Van der Zwet J., Damsteeg J.-W., Slat B., Andrady A., Reisser J., Nat. Commun. 2017, 8, 15611; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Schmidt C., Krauth T., Wagner S., Environ. Sci. Technol. 2017, 51, 12246–12253. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Fagnani D. E., Tami J. L., Copley G., Clemons M. N., Getzler Y. D., McNeil A. J., ACS Macro Lett. 2021, 10, 41–53; [DOI] [PubMed] [Google Scholar]
- 3b. Payne J., McKeown P., Jones M. D., Polym. Degrad. Stab. 2019, 165, 170–181. [Google Scholar]
- 4.
- 4a. McKeown P., Jones M. D., Sustain. Chem. 2020, 1, 1–22; [Google Scholar]
- 4b. Drumright R. E., Gruber P. R., Henton D. E., Adv. Mater. 2000, 12, 1841–1846; [Google Scholar]
- 4c. Auras R., Harte B., Selke S., Macromol. Biosci. 2004, 4, 835–864; [DOI] [PubMed] [Google Scholar]
- 4d. Sintim H. Y., Bary A. I., Hayes D. G., Wadsworth L. C., Anunciado M. B., English M. E., Bandopadhyay S., Schaeffer S. M., DeBruyn J. M., Miles C. A., Sci. Total Environ. 2020, 727, 138668; [DOI] [PubMed] [Google Scholar]
- 4e. Román-Ramírez L. A., McKeown P., Shah C., Abraham J., Jones M. D., Wood J., Ind. Eng. Chem. Res. 2020, 59, 11149–11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Pilone A., Press K., Goldberg I., Kol M., Mazzeo M., Lamberti M., J. Am. Chem. Soc. 2014, 136, 2940–2943; [DOI] [PubMed] [Google Scholar]
- 5b. Stopper A., Okuda J., Kol M., Macromolecules 2012, 45, 698–704; [Google Scholar]
- 5c. Pilone A., De Maio N., Press K., Venditto V., Pappalardo D., Mazzeo M., Pellecchia C., Kol M., Lamberti M., Dalton Trans. 2015, 44, 2157–2165; [DOI] [PubMed] [Google Scholar]
- 5d. Thevenon A., Romain C., Bennington M. S., White A. J., Davidson H. J., Brooker S., Williams C. K., Angew. Chem. Int. Ed. 2016, 55, 8680–8685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8822–8827; [Google Scholar]
- 5e. Rosen T., Popowski Y., Goldberg I., Kol M., Chem. Eur. J. 2016, 22, 11533–11536; [DOI] [PubMed] [Google Scholar]
- 5f. Hador R., Lipstman S., Rescigno R., Venditto V., Kol M., Chem. Commun. 2020, 56, 13528–13531; [DOI] [PubMed] [Google Scholar]
- 5g. Driscoll O. J., Leung C. K., Mahon M. F., McKeown P., Jones M. D., Eur. J. Inorg. Chem. 2018, 5129–5135; [Google Scholar]
- 5h. Jones M. D., Brady L., McKeown P., Buchard A., Schäfer P. M., Thomas L. H., Mahon M. F., Woodman T. J., Lowe J. P., Chem. Sci. 2015, 6, 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Jones M. D., Wu X., Chaudhuri J., Davidson M. G., Ellis M. J., Mater. Sci. Eng. C 2017, 80, 69–74; [DOI] [PubMed] [Google Scholar]
- 6b. Schäfer P., Fuchs M., Ohligschläger A., Rittinghaus R., McKeown P., Akin E., Schmidt M., Hoffmann A., Liauw M., Herres-Pawlis S., ChemSusChem 2017, 10, 3547–3556; [DOI] [PubMed] [Google Scholar]
- 6c. Schäfer P. M., McKeown P., Fuchs M., Rittinghaus R. D., Hermann A., Henkel J., Seidel S., Roitzheim C., Ksiazkiewicz A. N., Hoffmann A., Pich A., Jones M. D., Herres-Pawlis S., Dalton Trans. 2019, 48, 6071–6082; [DOI] [PubMed] [Google Scholar]
- 6d. Rittinghaus R. D., Tremmel J., Růžička A., Conrads C., Albrecht P., Hoffmann A., Ksiazkiewicz A. N., Pich A., Jambor R., Herres-Pawlis S., Chem. Eur. J. 2020, 26, 212–221; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Hermann A., Hill S., Metz A., Heck J., Hoffmann A., Hartmann L., Herres-Pawlis S., Angew. Chem. Int. Ed. 2020, 59, 21778–21784; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21962–21968; [Google Scholar]
- 6f. Lackmann C., Brendt J., Seiler T.-B., Hermann A., Metz A., Schäfer P. M., Herres-Pawlis S., Hollert H., J. Hazard. Mater. 2021, 416, 125889. [DOI] [PubMed] [Google Scholar]
- 7. Diaz C., Mehrkhodavandi P., Polym. Chem. 2021, 12, 783–806. [Google Scholar]
- 8.
- 8a. Rosen T., Goldberg I., Navarra W., Venditto V., Kol M., Angew. Chem. Int. Ed. 2018, 57, 7191–7195; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7309–7313; [Google Scholar]
- 8b. Cohn D., Hotovely-Salomon A., Polymer 2005, 46, 2068–2075; [Google Scholar]
- 8c. Diaz C., Tomković T., Goonesinghe C., Hatzikiriakos S. G., Mehrkhodavandi P., Macromolecules 2020, 53, 8819–8828; [Google Scholar]
- 8d. Diaz C., Ebrahimi T., Mehrkhodavandi P., Chem. Commun. 2019, 55, 3347–3350. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Schappacher M., Fur N., Guillaume S. M., Macromolecules 2007, 40, 8887–8896; [Google Scholar]
- 9b. Jakubowski W., Matyjaszewski K., Macromol. Symp. 2006, 240, 213–223; [Google Scholar]
- 9c. Messman J. M., Scheuer A. D., Storey R. F., Polymer 2005, 46, 3628–3638. [Google Scholar]
- 10.
- 10a. Wolf F. F., Friedemann N., Frey H., Macromolecules 2009, 42, 5622–5628; [Google Scholar]
- 10b. Jakubowski W., Lutz J. F., Slomkowski S., Matyjaszewski K., J. Polym. Sci. Part A 2005, 43, 1498–1510; [Google Scholar]
- 10c. Mecerreyes D., Moineau G., Dubois P., Jérôme R., Hedrick J. L., Hawker C. J., Malmström E. E., Trollsas M., Angew. Chem. Int. Ed. 1998, 37, 1274–1276; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1306–1309. [Google Scholar]
- 11.
- 11a. Romain C., Williams C. K., Angew. Chem. Int. Ed. 2014, 53, 1607–1610; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1633–1636; [Google Scholar]
- 11b. Kernbichl S., Reiter M., Adams F., Vagin S., Rieger B., J. Am. Chem. Soc. 2017, 139, 6787–6790; [DOI] [PubMed] [Google Scholar]
- 11c. Stößer T., Williams C. K., Angew. Chem. Int. Ed. 2018, 57, 6337–6341; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6445–6450; [Google Scholar]
- 11d. Chen T. T., Zhu Y., Williams C. K., Macromolecules 2018, 51, 5346–5351; [Google Scholar]
- 11e. Sulley G. S., Gregory G. L., Chen T. T., Peña Carrodeguas L., Trott G., Santmarti A., Lee K.-Y., Terrill N. J., Williams C. K., J. Am. Chem. Soc. 2020, 142, 4367–4378; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Liu Y., Guo J.-Z., Lu H.-W., Wang H.-B., Lu X.-B., Macromolecules 2018, 51, 771–778; [Google Scholar]
- 11g. Saini P. K., Fiorani G., Mathers R. T., Williams C. K., Chem. Eur. J. 2017, 23, 4260–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Wang X., Thevenon A., Brosmer J. L., Yu I., Khan S. I., Mehrkhodavandi P., Diaconescu P. L., J. Am. Chem. Soc. 2014, 136, 11264–11267; [DOI] [PubMed] [Google Scholar]
- 12b. Wei J., Diaconescu P. L., Acc. Chem. Res. 2019, 52, 415–424; [DOI] [PubMed] [Google Scholar]
- 12c. Lai A., Hern Z. C., Diaconescu P. L., ChemCatChem 2019, 11, 4210–4218; [Google Scholar]
- 12d. Quan S. M., Wei J., Diaconescu P. L., Organometallics 2017, 36, 4451–4457; [Google Scholar]
- 12e. Xu X., Luo G., Hou Z., Diaconescu P. L., Luo Y., Inorg. Chem. Front. 2020, 7, 961–971. [Google Scholar]
- 13.
- 13a. Fazekas E., Nichol G. S., Garden J. A., Shaver M. P., ACS omega 2018, 3, 16945–16953; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Li J., Yang C., Cheng C., IOP Conf. Ser.: Mater. Sci. Eng. 2016, 137, 012058; [Google Scholar]
- 13c. Li M., Wang S., Li F., Zhou L., Lei L., Polym. Chem. 2020, 11, 6591–6598. [Google Scholar]
- 14.
- 14a. Kostakis K., Mourmouris S., Karanikolopoulos G., Pitsikalis M., Hadjichristidis N., J. Polym. Sci. Part A 2007, 45, 3524–3537; [Google Scholar]
- 14b. Yang H., Xu J., Pispas S., Zhang G., Macromolecules 2012, 45, 3312–3317; [Google Scholar]
- 14c. Jung H.-J., Yu I., Nyamayaro K., Mehrkhodavandi P., ACS Catal. 2020, 10, 6488–6496. [Google Scholar]
- 15.
- 15a. Matyjaszewski K., Davis T. P., Handbook of radical polymerization, Wiley, Hoboken, 2003; [Google Scholar]
- 15b. Kato M., Kamigaito M., Sawamoto M., Higashimura T., Macromolecules 1995, 28, 1721–1723; [Google Scholar]
- 15c. Wang J.-S., Matyjaszewski K., Macromolecules 1995, 28, 7901–7910; [Google Scholar]
- 15d. Percec V., Barboiu B., Macromolecules 1995, 28, 7970–7972. [Google Scholar]
- 16.
- 16a. Mosnácek J., Matyjaszewski K., Macromolecules 2008, 41, 5509–5511; [Google Scholar]
- 16b. Okada S., Matyjaszewski K., J. Polym. Sci. Part A 2015, 53, 822–827. [Google Scholar]
- 17.
- 17a. Wang J.-S., Matyjaszewski K., Macromolecules 1995, 28, 7572–7573; [Google Scholar]
- 17b. Flemming C., Trevors J., Water Air Soil Pollut. 1989, 44, 143–158. [Google Scholar]
- 18. Rittinghaus R. D., Schäfer P. M., Albrecht P., Conrads C., Hoffmann A., Ksiazkiewicz A. N., Bienemann O., Pich A., Herres-Pawlis S., ChemSusChem 2019, 12, 2161–2165. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Dadashi-Silab S., Matyjaszewski K., Molecules 2020, 25, 1648; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Yuan M., Cui X., Zhu W., Tang H., Polymer 2020, 12, 1987; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Dunford H. B., Dolphin D., Raymond K., Sieker L., The Biological Chemistry of Iron, Reidel Publishing, Dordrecht, 1981. [Google Scholar]
- 20.
- 20a. Layadi A., Kessel B., Yan W., Romio M., Spencer N. D., Zenobi-Wong M., Matyjaszewski K., Benetti E. M., J. Am. Chem. Soc. 2020, 142, 3158–3164; [DOI] [PubMed] [Google Scholar]
- 20b. Ramu A., Kumar K. R., Polym. Chem. 2020, 11, 687; [Google Scholar]
- 20c. Bennett M. R., Gurnani P., Hill P. J., Alexander C., Rawson F. J., Angew. Chem. Int. Ed. 2020, 59, 4750–4755; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 4780–4785. [Google Scholar]
- 21. Da Silva D., Kaduri M., Poley M., Adir O., Krinsky N., Shainsky-Roitman J., Schroeder A., Chem. Eng. J. 2018, 340, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Bienemann O., Froin A. K., dos Santos Vieira I., Wortmann R., Hoffmann A., Herres-Pawlis S., Z. Anorg. Allg. Chem. 2012, 638, 1683–1690; [Google Scholar]
- 22b. Hoffmann A., Bienemann O., Dos Santos Vieira I., Herres-Pawlis S., Polymer 2014, 6, 995–1007; [Google Scholar]
- 22c. Rösener T., Bienemann O., Sigl K., Schopp N., Schnitter F., Flörke U., Hoffmann A., Döring A., Kuckling D., Herres-Pawlis S., Chem. Eur. J. 2016, 22, 13550–13562; [DOI] [PubMed] [Google Scholar]
- 22d. Rösener T., Hoffmann A., Herres-Pawlis S., Eur. J. Inorg. Chem. 2018, 3164–3175; [Google Scholar]
- 22e. Herres-Pawlis S., Kröckert K., Mannsperger J., Rösener T., Hoffmann A., Z. Anorg. Allg. Chem. 2021, 647, 832–842. [Google Scholar]
- 23. Liu B., Chen J., Liu N., Ding H., Wu X., Dai B., Kim I., Green Chem. 2020, 22, 5742–5750. [Google Scholar]
- 24. Hong M., Chen E. Y.-X., Nat. Chem. 2016, 8, 42. [DOI] [PubMed] [Google Scholar]
- 25. Okada S., Park S., Matyjaszewski K., ACS Macro Lett. 2014, 3, 944–947. [DOI] [PubMed] [Google Scholar]
- 26. Herber U., Hegner K., Wolters D., Siris R., Wrobel K., Hoffmann A., Lochenie C., Weber B., Kuckling D., Herres-Pawlis S., Eur. J. Inorg. Chem. 2017, 1341–1354. [Google Scholar]
- 27.
- 27a. Shaver M. P., Allan L. E., Rzepa H. S., Gibson V. C., Angew. Chem. Int. Ed. 2006, 45, 1241–1244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1263–1266; [Google Scholar]
- 27b. Lake B. R., Shaver M. P., Controlled Radical Polymerization: Mechanisms, ACS Publications, Washington, 2015, pp. 311–326. [Google Scholar]
- 28.
- 28a. Jakubowski W., Matyjaszewski K., Angew. Chem. Int. Ed. 2006, 45, 4482–4486; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4594–4598; [Google Scholar]
- 28b. Jakubowski W., Min K., Matyjaszewski K., Macromolecules 2006, 39, 39–45; [Google Scholar]
- 28c. Zhang L., Miao J., Cheng Z., Zhu X., Macromol. Rapid Commun. 2010, 31, 275–280; [DOI] [PubMed] [Google Scholar]
- 28d. Mukumoto K., Wang Y., Matyjaszewski K., ACS Macro Lett. 2012, 1, 599–602. [DOI] [PubMed] [Google Scholar]
- 29. Sears W., Hunt J., Stevens J., J. Chem. Phys. 1981, 75, 1589–1598. [Google Scholar]
- 30.Small deviations from linearity occur due to the fast conversion of lactide in the first 30 min changing the viscosity of the polymerization mixture enormously.
- 31. Kowalski A., Duda A., Penczek S., Macromolecules 1998, 31, 2114–2122. [Google Scholar]
- 32. Kan C., Ma H., RSC Adv. 2016, 6, 47402–47409. [Google Scholar]
- 33. Farah S., Anderson D. G., Langer R., Adv. Drug Delivery Rev. 2016, 107, 367–392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information