

Abstract
Background and Aim
Both type 2 diabetes mellitus and non‐alcoholic fatty liver disease are closely associated with elevated levels of low‐density lipoprotein cholesterol and its oxidized form (ox‐LDL). This study aimed to investigate the regulation of sortilin in liver tissue and its potential implications for lipid metabolism.
Methods
Sixty male Wistar rats were randomly divided into four groups: control group (n = 15), ox‐LDL group (n = 15), PD98059 group (n = 15), and ox‐LDL + PD98059 group (n = 15). Liver sinusoidal endothelial cells were extracted from liver tissue of the control group and were identified using an anti‐CD31 antibody. Lipid droplet accumulation was observed by Oil red O and hematoxylin–eosin staining. The protein expression levels were detected by immunohistochemical staining, real‐time reverse transcription‐polymerase chain reaction, and western blot. Histopathologic examinations were performed by Gomori methenamine silver staining.
Results
The ox‐LDL group exhibited increased lipid droplet accumulation. Further, ox‐LDL activated the extracellular signal‐regulated kinase (ERK)‐mediated downregulation of sortilin expression, whereas blocking of ERK signaling by PD98059 increased sortilin protein expression. Consistently, hematoxylin–eosin staining showed that the structure of the hepatocytes was loose and disordered in arrangement, with lipid droplets present in the cytoplasm of the ox‐LDL group. However, PD98059 significantly improved the integration of the scaffold structure. Gomori methenamine silver staining showed that the ox‐LDL group had darker and more obvious fragmented silver nitrate deposits in the basement membrane and sinus space.
Conclusions
Sortilin can protect liver sinusoidal endothelial cells from injury and maintain integration of the liver scaffold structure in ox‐LDL‐induced lipid‐injured liver.
Keywords: Diabetes, ERK, Liver sinusoidal endothelial cells, Non‐alcoholic fatty liver disease, Oxidized low‐density lipoprotein, Sortilin
Introduction
Diabetes and non‐alcoholic fatty liver disease (NAFLD) are both associated with impaired lipid uptake and metabolism in liver tissue; both are characterized by the accumulation of low‐density lipoprotein (LDL) and triglyceride (TG) in the liver. 1 , 2 , 3 Dysfunction of liver sinusoidal endothelial cells (LSECs) is observed in the early stage of NAFLD, caused partly by damage produced by oxidized LDL (ox‐LDL). 4 , 5 Genome‐wide association studies (GWAS) have revealed a strong association between a specific single nucleotide polymorphism (SNP) in the SORT1 gene (SORT1 encodes sortilin) and decreased serum levels of LDL cholesterol (LDL‐C). 6 , 7 Indeed, in a meta‐analysis of >100 000 individuals, this locus was most strongly associated with LDL‐C out of any locus in the genome (P = 1 × 10−170). 8 , 9 Within the locus, sortilin has been established as the causal gene responsible for modulating plasma LDL‐C levels and, by extension, diabetes and NAFLD risk. 10 Further, elevated ox‐LDL causes abnormal activation of protein kinase C (PKC) isoforms and mitogen‐activated protein kinase (MAPK), resulting in inflammation and insulin resistance. 11 , 12
Although it is well known that ox‐LDL is closely related to sortilin expression in LSECs, the downstream mechanisms are still not fully understood. Here, a series of studies was performed to determine the function of sortilin in LSECs.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), phosphate‐buffered saline (PBS), penicillin/streptomycin, trypsin‐ethylenediaminetetraacetic acid, and Hanks Balanced Salt Solution (HBSS) were purchased from Hyclone (Logan, UT, USA). An Oil red O (ORO) stain kit, radio immunoprecipitation assay lysis buffer, 0.1% Tween‐20/Tris‐buffered saline (TBST), bovine serum albumin (BSA), 4′6‐diamidino‐2‐phenylindole, bicinchoninic acid protein quantitative kits, Gomori methenamine silver (GMS), ox‐LDL, and sodium dodecyl sulfate polyacrylamide gel electrophoresis were purchased from SolarBio (Beijing, China). Type II collagenase was obtained from Sigma (St. Louis, MO, USA). A FastKing RT kit was purchased from TIAN‐GEN (Beijing, China). TRIzol® reagent was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Anti‐sortilin, anti‐extracellular signal‐regulated kinase (ERK) antibody (phosphor‐ERK and total ERK), anti‐PKC (PKCα and phosphor‐PKC), anti‐LDL‐C, and β‐actin were purchased from Abcam (Hong Kong, China). Rabbit anti‐CD31 and goat anti‐rabbit horseradish peroxidase (HRP)‐conjugated secondary antibodies were obtained from Bioss (Beijing, China). PD98059 was purchased from Selleck Chemicals (Houston, TX, USA). Polyvinylidene fluoride membranes and enhanced chemiluminescence reagents were purchased from Millipore (Billerica, MA, USA). The primer pairs for β‐actin, sortilin, IL‐6, and TNF‐α were synthesized by Takara Biotechnology Co. Ltd. (Dalian, China). Lentiviral particles (LV‐Sortl and LV‐pERK) were purchased from GeneChem (Shanghai, China). All other chemicals of analytical grade were purchased from commercial suppliers. The cell culture equipment was obtained from Corning Inc. (Corning, NY, USA).
Experimental animals and design
Animal experiments were approved by the research ethics committee of the Gansu Provincial Hospital (number: syll20160031, Gansu, China). Sixty male Wistar rats were used in this study. Rats were housed in specific pathogen‐free conditions in the animal house facility under a 12‐h light–dark cycle at 22°C; they had free access to food and water. After 1 week of adaptation, rats were randomly divided into four groups: control group (n = 15), ox‐LDL group (n = 15), PD98059 group (n = 15), and ox‐LDL + PD98059 group (n = 15). The control group was given normal chow feed for 6 weeks, whereas the ox‐LDL and ox‐LDL + PD98059 groups were given high‐fat/high‐cholesterol feed (21% fat and 1.25% cholesterol) for 6 weeks. Additionally, the PD98059 and ox‐LDL + PD98059 groups were given PD98059 by intraperitoneal injection once a day for 6 weeks at a dose of 10 mg/kg of body weight.
Isolation, culture, and identification of liver sinusoidal endothelial cells
For primary cultures, LSECs were isolated from the control group rats. The yield of rat LSECs was, on average, 5 × 106 sinusoidal endothelial cells per 1 g of liver. 13 First, the rats were anesthetized with pentobarbital sodium and then sacrificed by cervical dislocation; the entire liver was carefully resected and repeatedly washed with PBS, cut into 5‐mm3 cubes, and then incubated with 2.5 mL of 0.1% type II collagenase in DMEM at 37°C for 1.5 h. 13 Following this, digestion was terminated with HBSS. The cell suspension was passed through a 100 mesh and washed twice with 5 mL of HBSS containing 10% FBS. Isolated cells were incubated in DMEM supplemented with 10% FBS, 1% penicillin–streptomycin, and 1‐mM l‐glutamine, respectively, 5 , 14 as well as with hepatocyte growth factor and vascular endothelial growth factor (both 10 ng/mL) under standard cell culture conditions (humidified atmosphere with 5% CO2 at 37°C). 15 The cells were identified using an anti‐CD31 antibody via immunofluorescence. 16 , 17
Experimental protocol
First, the cells were treated with ox‐LDL (25, 50, 100, or 200 μg/mL) for 24 h to investigate the effects of ox‐LDL on LSECs. Following this, the cells were divided into four groups (control group, ox‐LDL group, PD98059 group, and ox‐LDL + PD98059) to investigate the effects of ox‐LDL on sortilin expression and the release of inflammatory cytokines in LSECs. Cells in the control group were treated with glucose (5.5 mM) for 24 h; cells in the ox‐LDL group were treated with ox‐LDL (100 μg/mL) for 24 h; cells in the PD98059 group were treated with PD98059 (10 μM) for 24 h; and cells in the ox‐LDL + PD98059 group were treated with ox‐LDL (100 μg/mL) plus PD98059 (10 μM) for 24 h. The levels of sortilin, IL‐6, and TNF‐α were then determined. The cells were treated in the presence or absence of Go6983 for a further 24 h to examine the mechanism underlying the effect of ox‐LDL on sortilin expression.
Hematoxylin–eosin staining, immunohistochemistry, and Gomori methenamine silver
Liver tissue were embedded in paraffin and immunohistochemistry, and hematoxylin–eosin (HE) and GMS staining were performed on consecutive 5‐μm‐thick sections. The sections were deparaffinized in xylene and rehydrated using standardized procedures. Citrate buffer (pH 6.0) was used for antigen retrieval. After washing with distilled water, the sections were blocked for 20 min with peroxidase (0.3% H2O2/methanol). Then, the sections were incubated with 0.05% PBS‐Tween and blocked with 2% BSA‐PBS blocking solution. Following this, they were rinsed with 0.05% PBS‐Tween and incubated with primary antibodies overnight at 4°C, with their respective dilutions, that is, rabbit anti‐ERK at 1:200 and rabbit anti‐sortilin at 1:300. After washing, the sections were incubated for 30 min with HRP‐labeled secondary antibodies. The reaction products were visualized with the diaminobenzidine substrate system, and sections were counterstained with hematoxylin. The images were observed on an Olympus Bx51 fluorescence microscope.
Oil red O staining
Oil red O is a lysochrome diazo dye used for staining neutral lipids and lipid droplets. LSECs were plated in a six‐well plate at a density of 1.0 × 106 cells per well. Then, cells were treated according to the above‐mentioned treatments. The cells were then rinsed three times with PBS, fixed with 10% formalin for 20 min at room temperature, and then stained with fresh‐filtered ORO solution at a ratio of 6:4 (ORO:water) for 15 min. The stain was removed, and the cells were washed gently with distilled water three times and then counterstained with hematoxylin for another 5 min. The dye was then extracted using isopropanol. The lipid droplet content was expressed as the average value of the integrated optical density and was analyzed using Image‐Pro Plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA). 18
Real‐time reverse transcription‐polymerase chain reaction
Liver sinusoidal endothelial cells were seeded in a Petri dish, and after treatment, the culture medium was removed. Then the cells were gently washed twice with chilled PBS and digested with trypsin. Total RNA was extracted from the LSECs using TRIzol reagent. Concentrations were determined by measuring the optical absorbance ratio at 260/280 nm immediately after the sample was dissolved in diethylpyrocarbonate‐treated water. cDNA was synthesized using 2.0‐μg total RNA reverse transcribed in a final volume of 20 μl using a FastKing RT kit (with gDNase), according to the manufacturer's protocol. The cDNA samples were then used for quantitative real‐time reverse transcription‐polymerase chain reaction (RT‐PCR) reactions in a LightCycler thermocycler instrument (Roche Diagnostic) using LC‐FastStart DNA Master SYBY Green 1 (Roche Diagnostics). RT‐PCR amplification was performed using the following primers:
for sortilin, forward 5′‐GACACATGGAGCATGGCACA‐3′,
and reverse 5′‐TGCCTCGGTCATCAGAGGTAAAG‐3′;
for IL‐6, forward 5′‐CAACCACGGCCTTCCCTACT‐3′,
and reverse 5′‐TTGGGAGTGGTATCCTCTGTGA −3′;
for TNF‐α, forward 5′‐ACCGTCAGCCGATTTGCTAT‐3′,
and reverse 5′‐TTGACGGCAGAGAGGAGGTT‐3′;
for β‐actin, forward 5′‐GGAGATTACTGCCCTGGCTCCTA‐3′,
and reverse 5′‐GACTCATCGTACTCCTGCTTGCTG‐3′;
for LV‐Sort1, forward 5′‐GACCAACAATACGCACCAGCAT‐3′,
and reverse 5′‐GAGTTCTCGGGACCAATAGCC‐3′;
for LV‐pERK, forward 5′‐ATGGAGCGCGCCATCAGC‐3′,
and reverse 5′‐ATTGCTTGGCAAAGGGCTATGG‐3′.
The gene expression of β‐actin in each sample was detected as an endogenous control. Relative mRNA expression was quantified using the comparative Ct method and expressed as 2−ΔΔCt.
Western blot
Treated cells were grown to confluence, washed three times with ice‐cold PBS, detached using scrapping, and harvested by centrifugation (12 000 rpm for 10 min at 4°C); then, the supernatant was removed. The pellet was resuspended in radio immunoprecipitation assay buffer supplemented with a protease inhibitor cocktail. After 30‐min incubation on ice under vortex, the lysates were centrifuged at 12 000 rpm for 5 min at 4°C, and the clear supernatants containing total protein were collected and transferred to a new tube, where the protein concentrations were evaluated using a bicinchoninic acid assay kit. Equal amounts of protein (50 μg) were electrophoresed through sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (0.45‐mm pore size). The membranes were blocked with 5% skim milk in TBST at room temperature for 2 h and then incubated with primary antibodies (anti‐sortilin antibody, anti‐ERK antibody [phosphor‐ERK and total ERK] and anti‐PKC [PKCα and p‐PKC]) overnight at 4°C. After washing three times with TBST, the membranes were incubated with HRP‐conjugated goat anti‐rabbit IgG secondary antibodies at a ratio of 1:500 for 2 h at room temperature. Washing was repeated and the bands were visualized with enhanced chemiluminescence reagents. Quantification of all samples was performed relative to β‐actin using imagej software (National Institutes of Health, Bethesda, MD, USA).
Lentivirus infection of liver sinusoidal endothelial cells
Liver sinusoidal endothelial cells were grown on Petri dishes to ~70% confluence. LV‐Sort1 and LV‐pERK were infected into the LSECs, according to the manufacturer's instructions. Twenty‐four hours after infection, ox‐LDL was added for cell culture. After 24 h, the cells were harvested and used for subsequent experiments. The cells were selected, pooled, and tested for sortilin expression by western blot.
Statistical analysis
The results are presented as mean ± SD of at least three independent experiments. The significance of the differences among the experimental groups was determined using one‐way analysis of variance. When statistically significant differences were found, a post‐hoc analysis was performed using Tukey's test. P < 0.05 was considered statistically significant.
Results
-
1.
Sortilin is expressed in LSECs, and ox‐LDL affects lipid droplets in LSECs.
Liver sinusoidal endothelial cells were isolated from rat livers and were cultured as described earlier. The morphology of the LSECs was assessed on days 2 and 4 of culturing by immunofluorescence microscopy. The cells grew by static adherence; the cells exhibited a fusiform or polygon shape, resembling cobblestones (Fig. 1a). The cells were identified by immunofluorescence using an anti‐CD31 antibody. Consistent with previous reports, 10 , 19 , 20 the results indicated that CD31 was constitutively expressed in cultured LSECs (Fig. 1b). ORO and HE staining for lipid droplets analysis revealed the presence of many lipid droplets in LSECs and liver tissue when treated with ox‐LDL (Fig. 1c,d). In the control group, most LSECs had no lipid droplets, indicating a low intracellular lipid content. In the ox‐LDL group, many lipid droplets could be observed in the cytoplasm.
Figure 1.
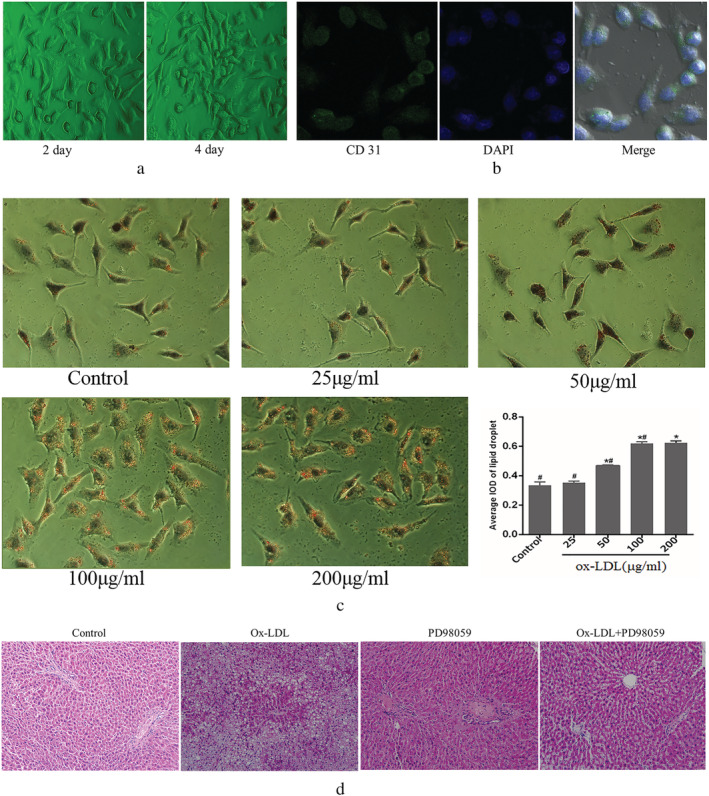
Characterization and identification of liver sinusoidal endothelial cells and oxidized low‐density lipoprotein (ox‐LDL) increased intracellular lipid accumulation. (a,b) Fluorescence images were acquired at an original magnification of 20×; (c) representative lipid droplet staining images were shown as dose dependent, and the average integrated optical density of lipid droplets stained with oil red O from liver sinusoidal endothelial cells was calculated; (d) The liver tissue sections were stained with hematoxylin–eosin. *P < 0.05 versus control. # P < 0.05 versus 200 μg/mL. Data are expressed as the mean ± SD. (green, CD31 expression; blue, nuclei staining with 4′6‐diamidino‐2‐phenylindole). Scale bar: 20 μm. IOD, integrated optical density.
-
2.
ERK inhibitor attenuates ox‐LDL‐induced hepatic scaffold structure damage.
Consistently, compared with the control group, HE staining showed that the structure of hepatocytes in the ox‐LDL group was loose with a disorderly arrangement. Many lipid droplets were observed in the cytoplasm, revealing obvious LSEC injury (non‐alcoholic steatohepatitis), and the scaffold structure was broken (Fig. 1d). Compared with the ox‐LDL group, PD98059 significantly improved liver injury and attenuated non‐alcoholic steatohepatitis, improving the integration of the scaffold structure (Fig. 1d). GMS staining revealed that the ox‐LDL group had a broken scaffold structure. In the control group, liver tissues showed intact scaffold structures with black continuous reticular fibers in parallel at the hepatic cords. The liver sections of the ox‐LDL group exhibited severe broken hepatic scaffolds with fragmented silver nitrate deposits scattered in the hepatic tissue. In contrast, in the PD98059 group, the severe broken hepatic scaffold structure was not evident, and silver nitrate deposits were more regularly arranged (Fig. 2).
Figure 2.
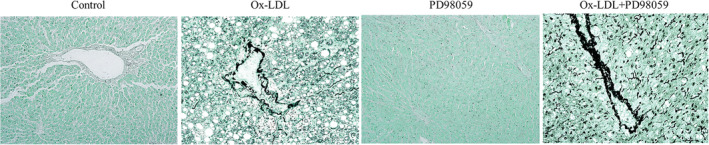
Histopathology staining of liver tissue by Gomori methenamine silver. Scale bar: 20 μm. Ox‐LDL, oxidized low‐density lipoprotein.
-
3.
Sortilin expression levels in LSECs were decreased when cells were cultured with ox‐LDL.
The expression levels of sortilin were examined by western blot, RT‐PCR, and immunohistochemical staining to investigate whether ox‐LDL affected sortilin expression of LSECs. The results revealed dose‐dependent decreases in sortilin protein and mRNA levels in the ox‐LDL group compared with the control group (Fig. 3a,b,c,g). Furthermore, the inflammatory cytokines IL‐6 and TNF‐α were assessed by western blot and RT‐PCR to explore the specific function of ox‐LDL in LSECs. Consistently, the results showed that the expressions of IL‐6 and TNF‐α were elevated after ox‐LDL treatment (Fig. 3d–f). Taken together, these results suggest that ox‐LDL is associated with impaired liver sortilin function and enhanced secretion of inflammatory cytokines.
Figure 3.
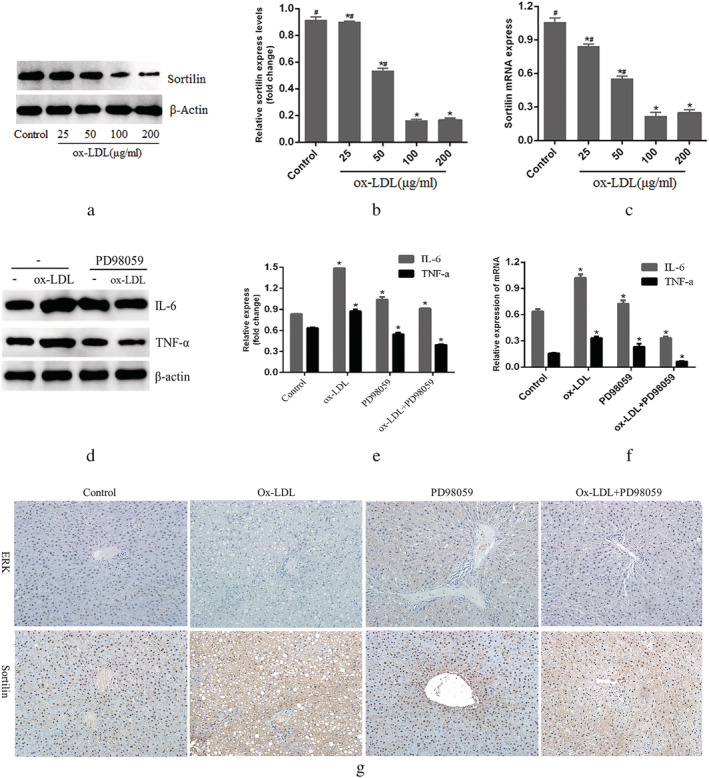
Evaluation of sortilin, IL‐6 and TNF‐α expression in liver sinusoidal endothelial cells. (a,b) Western blot evaluated sortilin expression in a dose‐dependent manner; real‐time reverse transcription‐polymerase chain reaction was performed to measure the mRNA level of sortilin in a dose‐dependent manner; (d,e) western blot evaluated IL‐6 and TNF‐α expression in liver sinusoidal endothelial cells. (f) Real‐time reverse transcription‐polymerase chain reaction was performed to measure the mRNA level of IL‐6 and TNF‐α. (g) Immunohistochemical staining detection sortilin and extracellular signal‐regulated kinase (ERK) in the liver tissue. *P < 0.05 versus control. #
P < 0.05 versus 200 μg/mL. Data are expressed as the mean ± SD. Scale bar: 20 μm. Ox‐LDL, oxidized low‐density lipoprotein. (e,f)  , IL‐6;
, IL‐6;  , TNF‐α.
, TNF‐α.
-
4.
Activation of the ERK signaling pathway downregulates sortilin expression in LSECs.
The downstream signaling pathways involved were first identified in order to further delineate the mechanisms mediating ox‐LDL‐induced sortilin downregulation. Ox‐LDL has been shown to activate MAPK and PKC signaling. 21 , 22 Consistently, ox‐LDL treatment induced the phosphorylation of ERK and PKC in LSECs (Fig. 4). Interestingly, blocking ERK signaling with PD98059 significantly blocked the repressive effect of ox‐LDL and increased sortilin protein expression (Fig. 4c–e). In contrast, blocking PKC signaling with Go6983 produced no effect on basal sortilin protein levels or ox‐LDL‐induced sortilin expression in LSECs (Fig. 4c). Additionally, the secretion levels of these inflammatory factors were examined (Fig. 3d–f), with PD98059 significantly increasing the levels of IL‐6 and TNF‐α.
Figure 4.
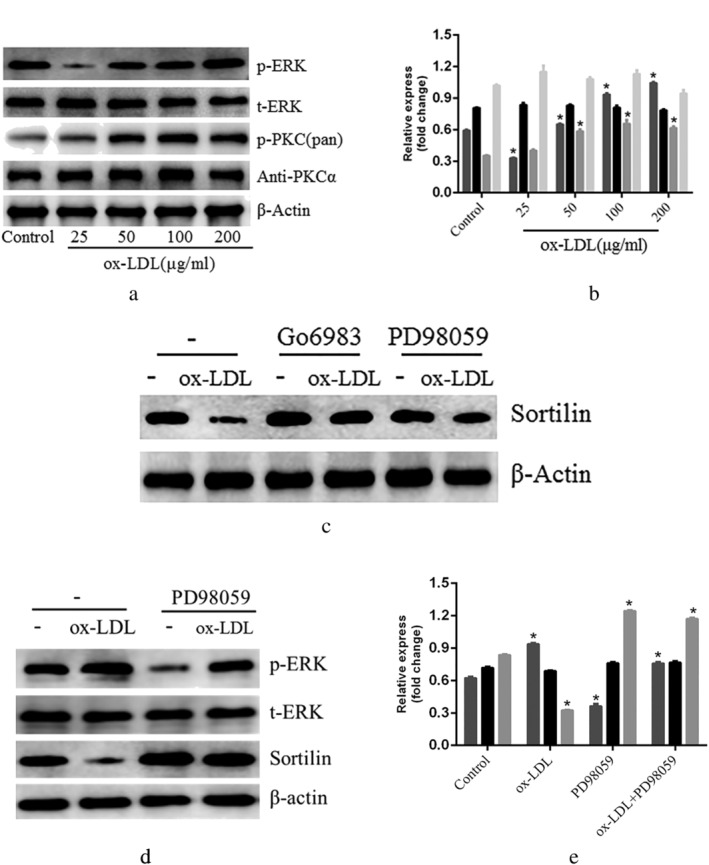
Liver sinusoidal endothelial cells (LSECs) were treated with oxidized low‐density lipoprotein (ox‐LDL) and pathway‐specific inhibitors. (a,b) extracellular signal‐regulated kinase (ERK) phosphorylation and protein kinase C (PKC) protein were measured by western blot in a dose‐dependent manner; (c) LSECs were cultured with ERK inhibitor PD98059 and PKC inhibitor Go6983. Western blot measured sortilin levels; (d,e) the figure shows the evaluation of sortilin, p‐ERK, and t‐ERK expression in LSECs using rabbit anti‐sortilin, rabbit anti‐p‐ERK and rabbit anti‐t‐ERK antibody in the western blot analysis. *P < 0.05 versus control. Data are expressed as the mean ± SD. (b)  , p‐ERK;
, p‐ERK;  , t‐ERK;
, t‐ERK;  , p‐PKC(pan);
, p‐PKC(pan);  , antiPKCα. (e)
, antiPKCα. (e)  , p‐ERK;
, p‐ERK;  , t‐ERK;
, t‐ERK;  , sortilin.
, sortilin.
-
5.
Sortilin expression in LSECs was downregulated, and lentiviral infection increased sortilin expression.
The expression levels of sortilin, t‐ERK, p‐ERK, IL‐6, and TNF‐α proteins induced by ox‐LDL were investigated by lentiviral infection. The results indicated that the t‐ERK protein levels of the western blot gray‐scale band exhibited little variation between the groups. However, band analysis showed that the changes in sortilin, p‐ERK, IL‐6, and TNF‐α protein levels were consistent with the changes in the PD98059 intervention (Figs 5, 6).
Figure 5.
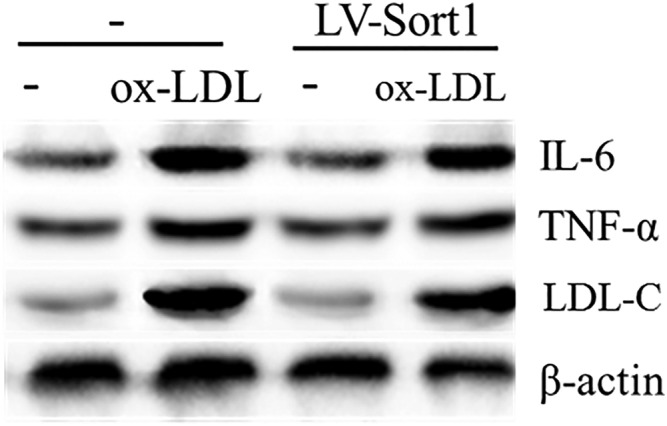
Effect of LV‐Sort1 on low‐density lipoprotein cholesterol (LDL‐C), IL‐6 and TNF‐α protein expression detected by western blot. Ox‐LDL, oxidized low‐density lipoprotein.
Figure 6.
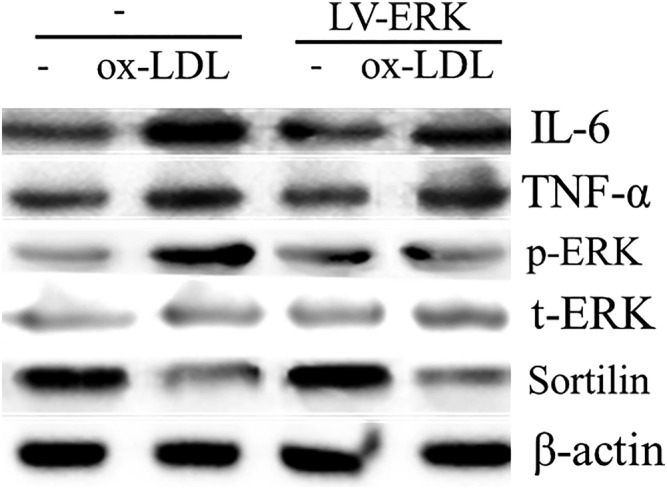
Effect of LV‐pERK on sortilin, t‐ERK, p‐ERK, IL‐6 and TNF‐α protein expression detected by western blot. Ox‐LDL, oxidized low‐density lipoprotein.
Discussion
It is well established that sortilin plays a critical role in the development of dyslipidemia and associated diseases. 6 , 7 Ox‐LDL has been suggested to induce lipid accumulation in LSECs. 4 , 23 LSECs are pivotal regulators of liver microcirculation and play a key role in sinusoidal crosstalk. 24 ERK signaling is one of the critical regulators of cell motility. 25 Our studies are based on detection of sortilin and phosphorylated proteins after activation of relevant signaling pathways with an external stimulus and pathway‐specific inhibitors.
Sortilin is associated with liver lipid accumulation and NAFLD pathogenesis. The current data demonstrated that ox‐LDL promoted lipid accumulation in LSECs. Excessive uptake of ox‐LDL by LSECs seems to be useful for preventing NAFLD; however, it causes over‐accumulation of lipids in LSECs. Our previous studies revealed that ox‐LDL produces cytotoxic effects on liver tissue in a dose‐dependent and time‐dependent manner. 5 This may cause NAFLD and insulin resistance.
Sortilin is involved in dyslipidemia metabolism in the liver. 7 , 26 Obese mice with hepatic steatosis and insulin resistance show markedly decreased liver sortilin proteins. 26 Increasing concentrations of extracellular LDL cause the downregulation of sortilin in LSECs. Because LSECs function to remove potentially dangerous macromolecules from the blood and act as gatekeepers of fibrogenesis by maintaining overall liver homeostasis, 16 , 24 , 27 increased exposure of LSECs to LDL may trigger the transcriptional downregulation of sortilin. This, then, may mediate increased LDL uptake. 28 Evidence also shows that sortilin is a cell surface clearance receptor for LDL and its reduction is anticipated to increase circulating LDL, thereby promoting NAFLD. 6 , 10 , 28 , 29
The ERK regulates multifold proteins via phosphorylation of mainly Ser/Thr‐Pro (S/T‐P) residues. 30 , 31 The current study demonstrated that the ERK signaling pathway was activated in LSECs following their stimulation with ox‐LDL. Interestingly, the results indicated that PD98059 inhibited the ox‐LDL‐mediated activation of the ERK signaling pathway. On the one hand, the ox‐LDL‐mediated activation of MAPKs/ERK and PKC isoforms is associated with substrate phosphorylation and inactivation. Conversely, ERK activation downregulates sortilin in LSECs. These changes presumably result in decreased apoB degradation in both proteasomes and lysosomes and, therefore, apoB overproduction and dyslipidemia. 32 These observations suggest that ERK mediates sortilin downregulation at several levels.
The ox‐LDL induced an inflammatory response in LSECs, including the expression of the inflammatory factors IL‐6 and TNF‐α. Moreover, PD98059 exerted protective effects on cells by inhibiting the ERK pathway. 33 IL‐6 and TNF‐α are both vital inflammatory cytokines in the pathogenesis of NAFLD and type 2 diabetes mellitus, 34 , 35 increasing coagulation and promoting lymphocyte proliferation and differentiation. 36 , 37
However, it should be noted that some studies have reported a positive correlation between sortilin expression and lipid levels and inhibited sortilin function, which may help attenuate lipid accumulation and lipotixicity 38 , 39 , 40 ; this is in contrast to the results of this study. It is possible that the mechanisms involved in sortilin expression, including the mTORC1‐PI3K signaling pathway, are altered under exposure to ox‐LDL. 41 It is also plausible that other lipid‐related proteins involved in liver lipid and cholesterol transport, including ATP‐binding cassette transporter A1 (ABCA1) and G1 (ABCG1), could be affected by sortilin deficiency in NAFLD. 18 , 42 , 43
Our study suggests a novel molecular link between ox‐LDL and ERK activation via downregulation of sortilin in LSECs. Notably, reduction in sortilin in LSECs likely results from chronic metabolic alterations. 44 , 45 However, future large clinical studies should explore the clinical use of sortilin as a diagnostic or therapeutic tool.
Conclusion
In summary, ox‐LDL mediated downregulation of sortilin expression by activating the ERK signaling pathway; this may underlie the increased microangiopathy risk in diabetes and NAFLD. Furthermore, sortilin protected LSECs from injury and maintained integration of the liver scaffold structure in the ox‐LDL‐induced lipid‐injured liver. These findings provide a new insight into the molecular mechanism of sortilin and its therapeutic potential in NAFLD.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 81660148, 81560146, 81960173, and 81760151), Gansu Province Health Industry Research Project (grant number GSWSKY‐2015‐10), and Lanzhou Chengguan District Science and Technology Project (grant number 2018SHFZ0068).
Zhang, Q. , Lin, W. , Tian, L. , Di, B. , Yu, J. , Niu, X. , and Liu, J. (2021) Oxidized low‐density lipoprotein activates extracellular signal‐regulated kinase signaling to downregulate sortilin expression in liver sinusoidal endothelial cells. Journal of Gastroenterology and Hepatology, 36: 2610–2618. 10.1111/jgh.15486.
Wenyan Lin and Qi Zhang contributed equally to this work
Declaration of conflict of interest: The authors declare no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Financial support: This work was supported by the National Natural Science Foundation of China (grant numbers 81660148, 81560146, 81960173, and 81760151), Gansu Province Health Industry Research Project (grant number GSWSKY‐2015‐10), and Lanzhou Chengguan District Science and Technology Project (grant number 2018SHFZ0068).
References
- 1. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014; 59: 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rabinowich L, Fishman S, Hubel E et al. Sortilin deficiency improves the metabolic phenotype and reduces hepatic steatosis of mice subjected to diet‐induced obesity. J. Hepatol. 2015; 62: 175–181. [DOI] [PubMed] [Google Scholar]
- 3. Engin A. Non‐alcoholic fatty liver disease. Adv. Exp. Med. Biol. 2017; 960: 443–467. [DOI] [PubMed] [Google Scholar]
- 4. Liu J, Quan J, Feng J et al. High glucose regulates LN expression in human liver sinusoidal endothelial cells through ROS/integrin αvβ3 pathway. Environ. Toxicol. Pharmacol. 2016; 42: 231–236. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Q, Liu J, Liu J et al. oxLDL induces injury and defenestration of human liver sinusoidal endothelial cells via LOX1. J. Mol. Endocrinol. 2014; 53: 281–293. [DOI] [PubMed] [Google Scholar]
- 6. Goettsch C, Kjolby M, Aikawa E. Sortilin and its multiple roles in cardiovascular and metabolic diseases. Arterioscler. Thromb. Vasc. Biol. 2018; 38: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao A, Cayabyab FS, Chen X et al. Implications of sortilin in lipid metabolism and lipid disorder diseases. DNA Cell Biol. 2017; 36: 1050–1061. [DOI] [PubMed] [Google Scholar]
- 8. Musunuru K, Strong A, Frank‐Kamenetsky M et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010; 466: 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teslovich TM, Musunuru K, Smith AV et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466: 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang X, Raghavan A, Peters DT, Pashos EE, Rader DJ, Musunuru K. Interrogation of the atherosclerosis‐associated SORT1 (sortilin 1) locus with primary human hepatocytes, induced pluripotent stem cell‐hepatocytes, and locus‐humanized mice. Arterioscler. Thromb. Vasc. Biol. 2018; 38: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiwari RL, Singh V, Singh A et al. PKCδ‐IRAK1 axis regulates oxidized LDL‐induced IL‐1β production in monocytes. J. Lipid Res. 2014; 55: 1226–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun JJ, Yin XW, Liu HH et al. Rapamycin inhibits ox‐LDL‐induced inflammation in human endothelial cells in vitro by inhibiting the mTORC2/PKC/c‐Fos pathway. Acta Pharmacol. Sin. 2018; 39: 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chou CH, Lai SL, Ho CM et al. Lysophosphatidic acid alters the expression profiles of angiogenic factors, cytokines, and chemokines in mouse liver sinusoidal endothelial cells. PloS one. 2015; 10: e0122060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silva‐Cote I, Cardier JE. Liver sinusoidal endothelial cells promote lymphohematopoietic differentiation from murine embryonic stem cells: role of soluble factors. Immunol. Lett. 2014; 161: 106–112. [DOI] [PubMed] [Google Scholar]
- 15. Zhao X, Zhao Q, Luo Z et al. Spontaneous immortalization of mouse liver sinusoidal endothelial cells. Int. J. Mol. Med. 2015; 35: 617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poisson J, Lemoinne S, Boulanger C et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J. Hepatol. 2017; 66: 212–227. [DOI] [PubMed] [Google Scholar]
- 17. Sorensen KK, Simon‐Santamaria J, McCuskey RS, Smedsrod B. Liver sinusoidal endothelial cells. Compr. Physiol. 2015; 5: 1751–1774. [DOI] [PubMed] [Google Scholar]
- 18. Zhang BC, Zhang CW, Wang C, Pan DF, Xu TD, Li DY. Luteolin attenuates foam cell formation and apoptosis in ox‐LDL‐stimulated macrophages by enhancing autophagy. Cell. Physiol. Biochem. 2016; 39: 2065–2076. [DOI] [PubMed] [Google Scholar]
- 19. Strong A, Ding Q, Edmondson AC et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Inves. 2012; 122: 2807–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alkhouri N, Poordad F, Lawitz E. Management of nonalcoholic fatty liver disease: lessons learned from type 2 diabetes. Hepatol Commun. 2018; 2: 778–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang H, Liang B, Li T, Zhou Y, Shang D, Du Z. Orexin A suppresses oxidized LDL induced endothelial cell inflammation via MAPK p38 and NF‐κB signaling pathway. IUBMB Life 2018; 70: 961–768. [DOI] [PubMed] [Google Scholar]
- 22. Chang H, Yuan W, Wu H, Yin X, Xuan H. Bioactive components and mechanisms of Chinese poplar propolis alleviates oxidized low‐density lipoprotein‐induced endothelial cells injury. BMC Complement. Altern. Med. 2018; 18: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yao S, Zong C, Zhang Y et al. Activating transcription factor 6 mediates oxidized LDL‐induced cholesterol accumulation and apoptosis in macrophages by up‐regulating CHOP expression. J. Atheroscler. Thromb. 2013; 20: 94–107. [DOI] [PubMed] [Google Scholar]
- 24. Marrone G, Shah VH, Gracia‐Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016; 65: 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanimura S, Takeda K. ERK signalling as a regulator of cell motility. J. Biochem. 2017; 162: 145–154. [DOI] [PubMed] [Google Scholar]
- 26. Li J, Matye DJ, Li T. Insulin resistance induces posttranslational hepatic sortilin 1 degradation in mice. J. Biol. Chem. 2015; 290: 11526–11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells—gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018; 15: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel KM, Strong A, Tohyama J et al. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ. Res. 2015; 116: 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strong A. Revisiting old friends: sortilin‐1, low‐density lipoprotein receptor, and prorenin receptor as modulators of lipoprotein and energy metabolism. Circ. Res. 2018; 122: 652–654. [DOI] [PubMed] [Google Scholar]
- 30. Unal EB, Uhlitz F, Bluthgen N. A compendium of ERK targets. FEBS Lett. 2017; 591: 2607–2615. [DOI] [PubMed] [Google Scholar]
- 31. Kidger AM, Sipthorp J, Cook SJ. ERK1/2 inhibitors: new weapons to inhibit the RAS‐regulated RAF‐MEK1/2‐ERK1/2 pathway. Pharmacol. Ther. 2018; 187: 45–60. [DOI] [PubMed] [Google Scholar]
- 32. Bi L, Chiang JY, Ding WX, Dunn W, Roberts B, Li T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J. Lipid Res. 2013; 54: 2754–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu B, Zhang B, Guo R, Li S, Xu Y. Enhancement in efferocytosis of oxidized low‐density lipoprotein‐induced apoptotic RAW264.7 cells through Sirt1‐mediated autophagy. Int. J. Mol. Med. 2014; 33: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feldman A, Eder SK, Felder TK et al. Clinical and metabolic characterization of obese subjects without non‐alcoholic fatty liver: a targeted metabolomics approach. Diabetes Metab. 2019; 45: 132–139. [DOI] [PubMed] [Google Scholar]
- 35. Lei L, Zhou C, Yang X, Li L. Down‐regulation of microRNA‐375 regulates adipokines and inhibits inflammatory cytokines by targeting AdipoR2 in non‐alcoholic fatty liver disease. Clin. Exp. Pharmacol. Physiol. 2018; 45: 819–831. [DOI] [PubMed] [Google Scholar]
- 36. Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017; 14: 32–42. [DOI] [PubMed] [Google Scholar]
- 37. Barchetta I, Cimini FA, De Gioannis R et al. Procollagen‐III peptide identifies adipose tissue‐associated inflammation in type 2 diabetes with or without nonalcoholic liver disease. Diabetes Metab. Res. Rev. 2018; 34: e2998. [DOI] [PubMed] [Google Scholar]
- 38. Ogawa K, Ueno T, Iwasaki T et al. Soluble sortilin is released by activated platelets and its circulating levels are associated with cardiovascular risk factors. Atherosclerosis 2016; 249: 110–115. [DOI] [PubMed] [Google Scholar]
- 39. Oh TJ, Ahn CH, Kim BR et al. Circulating sortilin level as a potential biomarker for coronary atherosclerosis and diabetes mellitus. Cardiovasc. Diabetol. 2017; 16: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J, Wang Y, Matye DJ et al. Sortilin 1 modulates hepatic cholesterol lipotoxicity in mice via functional interaction with liver carboxylesterase 1. J. Biol. Chem. 2017; 292: 146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinn WJ 3rd, Wan M, Shewale SV et al. mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J. Clin. Invest. 2017; 127: 4207–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Demina EP, Miroshnikova VV, Schwarzman AL. Role of the ABC transporters A1 and G1, key reverse cholesterol transport proteins, in atherosclerosis. Mol Biol (Mosk) 2016; 50: 223–230. [DOI] [PubMed] [Google Scholar]
- 43. Jiang T, Ren K, Chen Q et al. Leonurine prevents atherosclerosis via promoting the expression of ABCA1 and ABCG1 in a Ppargamma/Lxrα signaling pathway‐dependent manner. Cell. Physiol. Biochem. 2017; 43: 1703–1717. [DOI] [PubMed] [Google Scholar]
- 44. Hivelin C, Mazella J, Coppola T. Sortilin derived propeptide regulation during adipocyte differentiation and inflammation. Biochem. Biophys. Res. Commun. 2017; 482: 87–92. [DOI] [PubMed] [Google Scholar]
- 45. Pirault J, Polyzos KA, Petri MH, Ketelhuth DFJ, Back M, Hansson GK. The inflammatory cytokine interferon‐gamma inhibits sortilin‐1 expression in hepatocytes via the JAK/STAT pathway. Eur. J. Immunol. 2017; 47: 1918–1924. [DOI] [PubMed] [Google Scholar]