

Abstract
A lanthanide‐binding tag site‐specifically attached to a protein presents a tool to probe the protein by multiple spectroscopic techniques, including nuclear magnetic resonance, electron paramagnetic resonance and time‐resolved luminescence spectroscopy. Here a new stable chiral LnIII tag, referred to as C12, is presented for spontaneous and quantitative reaction with a cysteine residue to generate a stable thioether bond. The synthetic protocol of the tag is relatively straightforward, and the tag is stable for storage and shipping. It displays greatly enhanced reactivity towards selenocysteine, opening a route towards selective tagging of selenocysteine in proteins containing cysteine residues. Loaded with TbIII or TmIII ions, the C12 tag readily generates pseudocontact shifts (PCS) in protein NMR spectra. It produces a relatively rigid tether between lanthanide and protein, which is beneficial for interpretation of the PCSs by single magnetic susceptibility anisotropy tensors, and it is suitable for measuring distance distributions in double electron–electron resonance experiments. Upon reaction with cysteine or other thiol compounds, the TbIII complex exhibits a 100‐fold enhancement in luminescence quantum yield, affording a highly sensitive turn‐on luminescence probe for time‐resolved FRET assays and enzyme reaction monitoring.
Keywords: EPR spectroscopy, FRET, lanthanoid tags, luminescence, NMR spectroscopy
Lanthanoid keys to proteins: A stable chiral lanthanide(III) tag reacts spontaneously and quantitatively with a cysteine residue to generate a stable thioether bond. The lanthanide‐binding tag presents a tool to probe proteins using multiple spectroscopic techniques, including NMR, electron paramagnetic resonance and time‐resolved FRET experiments.

Introduction
Site‐specific labelling of proteins with lanthanide complexes offers a powerful tool for a range of spectroscopic techniques, including nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR) and luminescence spectroscopy. Paramagnetic lanthanide ions produce large effects in protein NMR spectra that present long‐range structural restraints. Thus, paramagnetic relaxation enhancement (PRE) effects are observed for protons over 20 Å from a gadolinium(III) ion, [1] pseudocontact shifts (PCS) are observable for nuclear spins over 40 Å from the paramagnetic centre [2] and residual dipolar couplings (RDC) are observed throughout the entire molecule due to molecular alignment in the magnetic field conferred by a bound paramagnetic lanthanide ion (with the exception of GdIII). [3]
PCSs are of particular interest, as they are manifested in easy‐to‐measure changes in chemical shifts, which encode the position of the nuclear spin relative to the coordinate system defined by the magnetic susceptibility anisotropy (Δχ) tensor associated with the paramagnetic centre. The PCS, δ PCS, of a nuclear spin is given by
| (1) |
where δ PCS is measured as the difference in chemical shift measured in the presence and absence of the paramagnetic metal ion, Δχ ax and Δχ rh are the axial and rhombic components of the Δχ tensor, and r, θ and ϕ are the polar coordinates of the nuclear spin relative to the principal axes of the tensor. PCSs thus encode accurate long‐range structural information that can be used for analysing protein conformations in solution and determining the structures of protein‐protein and protein‐ligand complexes.[ 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ] PCSs can even be used as the sole experimental restraints for protein fold determination.[ 12 , 13 , 14 ]
Among the paramagnetic lanthanide(III) ions, terbium(III) and thulium(III) stand out for featuring large Δχ tensors and generating PCSs that tend to shift the NMR signals in opposite directions, assisting their assignment relative to the corresponding diamagnetic reference, which can be prepared with a metal of similar ionic radius such as yttrium(III). (Yttrium is not a lanthanide but together with the lanthanides can be referred to as lanthanoid.) These three lanthanoid ions have thus been used extensively to assess the performance of lanthanide tags for PCS measurements in protein NMR.[ 5 , 7 , 15 ] Stable lanthanoid tags enable PCS measurements in living cells and these data can be sufficient for 3D structure determinations of the tagged proteins.[ 16 , 17 ]
GdIII is the metal ion with the largest paramagnetic moment, which can be exploited not only for PRE measurements in protein structural biology but, even more prominently, in contrast agents for magnetic resonance imaging.[ 18 , 19 , 20 ] Owing to slow electronic relaxation, GdIII ions have also gained an important role in EPR investigations, as double electron–electron resonance (DEER) experiments deliver precise distances between two GdIII ions ranging between about 2.5 and 8 nm.[ 21 , 22 , 23 , 24 ] Such measurements deliver unique data for probing the conformation of proteins following the attachment of GdIII tags at two sites.[ 25 , 26 ] Unlike traditional nitroxide radicals, the EPR signal of GdIII tags is free of orientation selection effects at high (W‐band) EPR frequencies, enabling sensitive DEER measurements, which can be analysed in a straightforward manner. [27] Furthermore, GdIII tags are insensitive to chemical reduction under physiological conditions, which is important for in‐cell DEER measurements.[ 28 , 29 ]
Distance information on the nanometre scale can also be obtained from FRET experiments based on the luminescence of TbIII or EuIII ions. With suitable antennas for efficient photo‐excitation, the luminescence is extremely sensitive and, besides serving applications in structural biology, these lanthanide ions offer outstanding probes in bioassays and for live cell‐imaging.[ 30 , 31 , 32 , 33 , 34 , 35 , 36 ] In particular, luminescent lanthanide tags are integral to commercial time‐resolved (TR)‐FRET assays to study dynamic processes such as protein conformation, protein–protein interactions and receptor‐ligand binding interactions.[ 37 , 38 , 39 , 40 ] LnIII complexes of europium and terbium offer advantages over organic fluorophores for FRET‐based assays, including up to millisecond excited state lifetimes, which enables background autofluorescence to be completely removed through time‐resolved measurements, enhancing sensitivity.[ 41 , 42 ] In addition, they possess narrow emission bands, which enables selective observation of lanthanide emission, and large pseudo‐Stokes shifts, which minimises self‐absorption processes.[ 43 , 44 ]
All these applications of lanthanoid ions demand stable attachments to the targeted biological macromolecule, which must be achieved by linking a suitable lanthanide chelating complex to the molecule, as proteins with natural high‐affinity binding sites for lanthanoids are rare. [45] Different approaches for tagging of proteins have been developed over the past two decades, most of which focus on attachment to single cysteine residues, as thiols are chemically more reactive than any other chemical group of the 20 canonical amino acids.[ 5 , 46 ] Useful tags for single cysteine residues need to fulfil a number of criteria. i) The tether connecting the lanthanide ion with the sulfur of the cysteine should be rigid, with the smallest possible number of rotatable bonds, as precise structural information can be obtained only if the lanthanide ion does not move relative to the protein. [47] At the same time, the tag must not affect the protein structure. ii) The tagged protein should be chemically stable. Lanthanide tags producing thioether bonds are superior to lanthanide tags attached via disulfide bonds, which are sensitive towards reducing agents. Furthermore, a disulfide bond adds more flexibility to the tether than a thioether group. iii) The lanthanide complex should be convenient to use to minimize protein handling. Most users prefer tags already containing the lanthanide ion over tagging approaches that require titration with lanthanides after installation of the tag, as achieving accurate titration ratios can be difficult. Furthermore, tags containing cysteine‐reactive moieties are preferred over tags that require prior chemical activation of the cysteine residue in the protein. iv) The lanthanide complex of the tag needs to be kinetically and thermodynamically stable in order not to dissociate in aqueous or biological media. v) For measurements of PCSs by NMR spectroscopy, the lanthanide complex must form a single stereoisomer to prevent peak doubling as a consequence of diastereomer formation in the chiral environment of the target protein.[ 48 , 49 , 50 ] vi) Synthesis of the lanthanide tag should be straightforward and affordable. Among the many tags published to date, the P4T‐DOTA and Ln‐M7‐Nitro tags recently published by Häussinger and co‐workers[ 51 , 52 ] (Figure 1) fulfil these criteria, except that their synthesis is challenging.
Figure 1.

Chemical structures of existing LnIII tags and the new tag Ln.C12 presented in this work. Ln.C8 is the same as Ln.C7, except for having the opposite chirality in all pendants.
Here, we present a new chiral lanthanide binding tag, C12, which is based on a stable cyclen complex, reacts rapidly with cysteine thiols in quantitative yield, produces a thioether bond and a rigid aromatic tether, and is enantiomerically pure and easier to synthesise than the P4T‐DOTA or Ln‐M7‐Nitro tags. The tag combines the reactive para‐nitropyridyl group of the previously reported Ln.L1 tag, where the nitro group acts as a leaving group in the reaction with cysteine, [53] with the chiral phenylethylamide pendant arms of the C1 tag (Figure 1). [54] We demonstrate the performance of this tag with different proteins for measuring PCSs in NMR experiments, GdIII−GdIII distances in DEER experiments, and luminescence in peptides and proteins labelled with the TbIII complex. We show that cysteine labelling of the TbIII complex of C12 elicits a dramatic 100‐fold enhancement in TbIII luminescence from a dark background. The TbIII complex is resistant to oxygen‐mediated quenching and is suitable for use in homogenous time‐resolved luminescence assays, demonstrated by a FRET experiment with a labelled aurora A protein kinase. Finally, we demonstrate fast and complete reaction with selenocysteine, which raises the prospect of site‐selective tagging in the presence of cysteine residues.
Results
Lanthanide tag synthesis
The synthesis of ligand C12 from 1,4,7,10 tetraazacyclododecane (cyclen) is described in Scheme 1. Briefly, the chiral bromoacetamide arm 3 was prepared by reacting (S)‐1‐phenylethanamine and bromoacetyl bromide, followed by N‐alkylation onto cyclen to give the macrocyclic compound 4. Next, N‐alkylation of 2‐methyl(sulfonyloxymethyl)‐4‐nitropyridine 2 onto the remaining secondary amine of compound 4 gave the ligand C12. Column chromatography was required after each alkylation step to remove impurities; however, the synthesis avoided the need for protecting groups and afforded ligand C12 in good overall yield. Lanthanoid complexes of C12 were readily prepared by the addition of one equivalent of the metal chloride salts, LnCl3 (Ln=TbIII, EuIII, TmIII, GdIII, YIII) in a 1 : 1 mixture of acetonitrile/water.
Scheme 1.

Synthetic scheme for the lanthanide(III) tag Ln.C12.
Photophysical data
To establish the fundamental chemical and photophysical properties of C12, its performance in luminescence applications was explored first. Photophysical data for the TbIII and EuIII complexes of C12, together with their cysteine‐tagged derivatives, are provided in Table 1. The TbIII and EuIII complexes of C12 have similar absorption spectra (Figure 2), characterized by a broad band centred at approximately 300 nm. Upon excitation of the nitropyridine moiety at 300 nm, the TbIII complex of C12 displays weak emission with four characteristic bands in the green region (475–630 nm) of the visible spectrum (Figure 2a). The EuIII complex of C12 emits red light weakly upon excitation at 300 nm, displaying characteristic emission bands in the range 550–720 nm (Figure 2b). The quantum yields of the Ln‐centred luminescence of C12 were determined by indirect excitation via the nitropyridine antenna to be in the range 0.1–0.2 %. The emission lifetimes for Tb.C12 were 1.47 ms in H2O and 2.50 ms in D2O and corresponding values for the EuIII homologue were 0.56 and 2.04 ms. In each case, the change in lifetime in deuterated solvent is consistent with one inner sphere water molecule (q=1) for each Ln complex. [55]
Table 1.
Photophysical data for the TbIII and EuIII complexes of C12 and their cysteine derivatives (10 mM HEPES, pH 7.0).[a]
|
Complex |
λ max [nm] |
ϵ [M−1 cm−1] |
Φ em [%] |
τ [ms] |
τ [ms] |
q |
|---|---|---|---|---|---|---|
|
Tb.C12 |
300 |
1 200 |
0.23 |
1.47 |
2.50 |
1.1 |
|
Tb.C12‐Cys |
278 |
15 500 |
20 |
1.48 |
2.40 |
1.0 |
|
Eu.C12 |
297 |
1 580 |
0.03 |
0.56 |
2.04 |
1.0 |
|
Eu.C12‐Cys |
277 |
11 040 |
0.90 |
0.57 |
2.08 |
1.0 |
Figure 2.
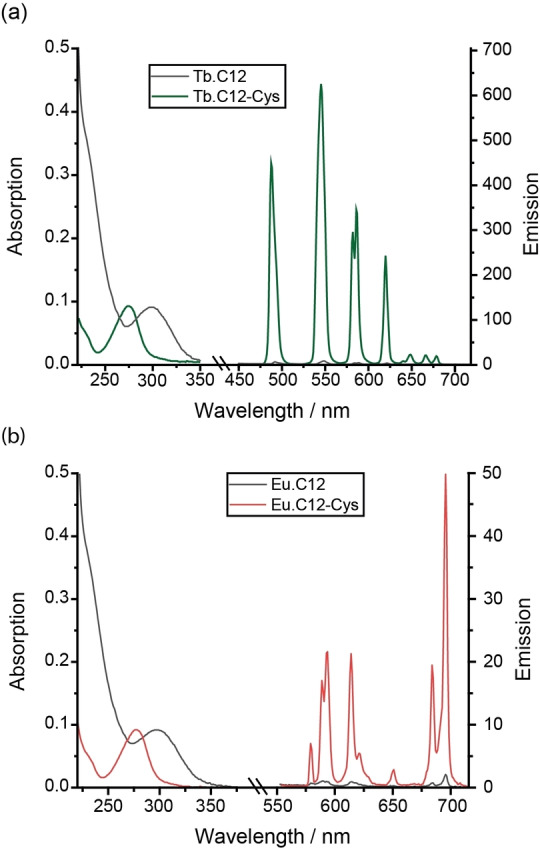
Absorption and emission spectra for a) Tb.C12 and b) Eu.C12 and their cysteine derivatives measured in aqueous buffer (10 mM HEPES, pH 7.0). To emphasise the wavelength shift upon reaction with cysteine, the vertical axes of the absorption spectra for the cysteine derivatives of Tb.C12 and Eu.C12 were scaled down by factors of 13 and 7, respectively. To confirm that the emission intensity increase was due to the successful reaction of Tb.C12, rather than being associated with a noncovalent binding involving displacement of coordinated water, the cysteine derivative was purified by preparative reversed‐phase HPLC.
Cysteine tagging reactions
The ability of Tb.C12 to react with cysteine and other biological thiols was evaluated in water at pH 7.0. Incubation of Tb.C12 (250 μM) with 4 mM cysteine, homocysteine (hCys) and glutathione (GSH) at 37 °C for 16 h resulted in quantitative reaction of the TbIII complex as indicated by LCMS analysis (Figure S1 in the Supporting Information).
Upon ligation of cysteine, the TbIII complex of C12 exhibits a remarkable 100‐fold enhancement in TbIII‐centred luminescence (475–630 nm; Figure 2a). The TbIII emission is effectively ‘switched on’ upon ligation with cysteine. Similar enhancements in emission intensity were observed upon reaction with homocysteine and glutathione (Figure S2). The absorption spectrum of the purified complex, Tb.C12‐Cys, showed a blue‐shifted band centred at 280 nm (Figure 2a), different from the untagged complex Tb.C12 (λ max=300 nm). Notably, the extinction coefficient of Tb.C12‐Cys was measured to be 15 500 M−1 cm−1, approximately 13 times higher than for the untagged complex. The overall quantum yield of the TbIII complex increased from 0.2 to 20 % following ligation with cysteine. The emission lifetimes for Tb.C12‐Cys in H2O and D2O were very similar to those obtained for the unreacted complex Tb.C12 (Table 1), indicating one coordinated water molecule (q=1) and confirming that the observed increase in TbIII luminescence was not associated with displacement of a water molecule by cysteine.
The substantial increase in both the quantum yield and the extinction coefficient upon ligation of Tb.C12 with cysteine means that the overall brightness of the TbIII complex, defined as the product of ϵ and ϕ, increases approximately 1100‐fold. In comparison, the EuIII complex of C12 exhibits a smaller 30‐fold enhancement in EuIII luminescence upon reaction with cysteine (Figure 2b and Table 1) and the overall quantum yield increases only to about 1 %. The ability of Tb.C12 to switch on its emission from a dark background confers significant advantages over other LnIII‐based protein tags that display constant luminescence in their free form, including obviating the need for a washing step to remove the unreacted tag, and providing a luminescence method to report the extent of cysteine tagging under physiological conditions.
The optimum number of equivalents of cysteine required to achieve near‐quantitative ligation of Tb.C12 and Eu.C12 after 16 h was determined using ESI mass spectrometry, which revealed that four equivalents of cysteine were sufficient to achieve greater than 95 % conversion at 37 °C after 16 h (Figures 3a and S3–S5). Real‐time monitoring of the reaction between Tb.C12 and cysteine at 37 °C was achieved using UV/Vis and emission spectroscopy, which revealed complete reaction after 3 h (Figure S6).
Figure 3.
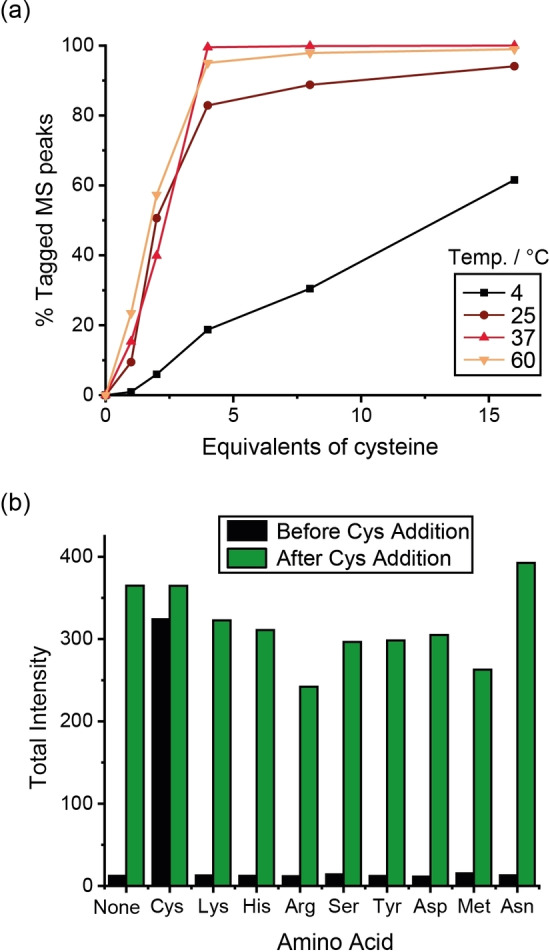
Reaction yields with cysteine. a) Incubation of Tb.C12 (250 μM) with different equivalents of cysteine in water at pH 7.0 and different temperatures for 16 h. Reaction completion was monitored by ESI mass spectrometry (% cysteine‐tagged MS peak). b) Total terbium emission intensity (λ ex=280 nm) after 24 h of incubation of Tb.C12 (250 μM) with various amino acids (4 mM; black), and after a further 24 h of incubation with cysteine (4 mM; green). Incubations were run in water at pH 7.0 and 37 °C. Emission spectra (λ ex=280 nm) were recorded after tenfold dilution into 10 mM HEPES, pH 7.0.
Next, the chemoselectivity of the tagging reaction was examined by incubation of Tb.C12 with a range of amino acids (4 mM) containing nucleophilic functional groups in water at pH 7 and 37 °C. After 24 h incubation, no enhancement in TbIII emission intensity was observed with Lys, His, Arg, Ser, Tyr, Asp, Met, Asn or a combination of all of these amino acids, consistent with no reaction occurring (Figure 3b). This was verified by mass spectrometry (Figures S7 and S8), which showed only the signal for unreacted Tb.C12. The subsequent addition of 4 mM cysteine resulted in a substantial increase in TbIII emission intensity corresponding to formation of the cysteine complex (confirmed by mass spectrometry, Figures S7 and S8), demonstrating excellent selectivity for cysteine over all other nucleophilic amino acid residues.
Finally, dilution studies conducted with Tb.C12‐Cys revealed that nanomolar concentrations of the TbIII tag can be readily detected using standard instrumentation (Figure S9). Further, the Tb.C12‐Cys complex is completely insensitive to oxygen‐mediated quenching (Figure S10).
Tb.C12 was shown to react rapidly and cleanly with the reduced form of GSH but not with the oxidized form (GSSG), which lacks the nucleophilic moiety (similar reactivity of Tb.C12 was observed with cysteine over cystine, Figure S11).
We reasoned that the selective enhancement in TbIII emission intensity upon reaction with GSH could be used for monitoring the enzymatic conversion of GSSG to GSH. An attractive feature of Tb.C12 for this purpose is its long luminescence lifetime (1.47 ms in H2O, Table 1), which permits time‐resolved analysis of the GSH/GSSG ratio, wherein the introduction of a short delay (60 μs) between the excitation and emission measurement enables complete removal of any short‐lived autofluorescence from biomolecules in the sample, enhancing signal‐to‐noise. Additionally, any light scattering in the instrument is also avoided because the light source is off during acquisition of the emitted light. The first TbIII complex to be reported for monitoring glutathione reductase activity [57] operates by Michael addition of GSH to a pendant maleimide arm, forming a thiol‐maleimide conjugate with enhanced TbIII luminescence. However, maleimide–thiol conjugates are known to be susceptible to thiol exchange and ring opening reactions, which can compromise their longer‐term stability.[ 58 , 59 ]
Tb.C12 was added to different concentrations of GSH and the time‐resolved emission intensity recorded as a function of time. The increase in time‐resolved luminescence was immediate (Figure 4a and b) and linearly dependent on GSH concentration even at low μM concentrations and for very short incubation (Figure 4c and d). After a 5‐minute incubation period, the emission intensity increased about threefold. Again, the luminescence of the TbIII complex increased linearly with GSH concentration (Figure S12).
Figure 4.

Monitoring glutathione reductase activity by using Tb.C12. a) and b) Time‐resolved emission intensity over time following the addition of Tb.C12 (25 μM) to different concentrations of glutathione in 50 mM Tris⋅HCl, pH 7.4. c) and d) Time‐resolved emission intensity (λ ex=292–366 nm, λ em=510–500 nm, integration time=60–400 μs) recorded 30 s after addition of Tb.C12 (25 μM) to different concentrations of GSH in 50 mM Tris⋅HCl, pH 7.4. e) Time‐resolved emission intensity (corrected to allow for dilution and background reaction) against initial concentration of oxidised glutathione after adding Tb.C12 (25 μM) to a glutathione reductase reaction mixture containing NADPH (1 mM), glutathione reductase (0.01 U mL−1) and various concentrations of oxidised glutathione (GSSG) in 50 mM Tris⋅HCl at pH 7.4 following a 30‐min incubation. Data fitted to the Michaelis‐Menten equation.
To demonstrate the ability of Tb.C12 to monitor the reduction of GSSG to GSH, a series of enzyme reactions were carried out using glutathione reductase in the presence of the reducing agent NADPH and different concentrations of oxidised glutathione. After a 30‐minute incubation period, Tb.C12 (25 μM) was added to the reaction mixture and the time‐resolved emission intensity was recorded. Figure 4e shows a plot of the time‐resolved emission intensity as a function of initial GSSG concentration (corrected to account for the background of an enzyme‐free reaction). As expected, increasing the GSSG concentration increased the emission intensity, consistent with an increased rate of reaction. The data were fitted to the Michaelis‐Menten equation to give a Michaelis‐Menten constant, K m, of 1.14 mM for GSSG. Thus, the rapid and selective reaction of Tb.C12 with GSH over GSSG enables the enzymatic reduction of glutathione to be monitored in a convenient, luminescence‐based increase‐in‐signal format.
Protein ligation reactions for NMR analysis
To explore the performance of C12 in NMR experiments, we prepared samples of the uniformly 15N‐labelled ubiquitin mutant S57C and reacted with the Tb.C12, Tm.C12 or Y.C12 tags. Quantitative ligation yields were obtained in reactions conducted overnight at room temperature in 20 mM HEPES buffer, pH 7, as indicated by mass spectrometry (Figure S13). Ligation reactions with the proteins IMP‐1N172C, ERp29 S114C and ERp29 G147C similarly resulted in fully tagged protein (Figures S14 and S15). All reactions were performed with 50 μM solutions of protein at a tag/protein molar ratio of 5 : 1.
Observation of PCSs and Δχ tensors
NMR experiments of the ubiquitin mutant S57C ligated with the Y.C12 tag displayed chemical shifts closely similar to the wild‐type protein, indicating little if any structural perturbation introduced by the tag. The Tb.C12 or Tm.C12 tags produced significant PCSs (Figure 5a and Table S1). Observation of single peaks for each backbone amide confirmed that the presence of chiral phenylethylamide pendants resulted in a single diastereomer with the protein, which is a prerequisite for avoiding increased complexity of the NMR spectrum, thus enabling straightforward PCS measurements. As the C12 tag generates fewer rotatable bonds between lanthanide and protein backbone than the C1 [54] or C2 [60] tags, the C12 tag is expected to hold the metal ion more rigidly, thus resulting in less averaging between PCSs of different sign. Indeed, the PCSs measured with the C12 tag tended to be larger than those reported previously with the Tb.C1 tag (Figure S16) [61] and the quality factor associated with the Δχ‐tensor fit was significantly better (Table 2).
Figure 5.

PCSs observed in ubiquitin S57C with C12 tag. a) Superimposition of 15N HSQC spectra. The C12 tag was loaded with YIII (black), TbIII (red) or TmIII ions (blue). The PCSs of selected amide protons are highlighted by lines connecting the corresponding crosspeaks of the protein with paramagnetic and diamagnetic tags. b) Correlation plot of back‐calculated vs. experimental PCSs for the backbone amide protons of ubiquitin S57C with the Tb.C12 (red) or Tm.C12 tag (blue). c) PCS isosurfaces representing the Δχ tensor obtained with the Tb.C12 (left) or Tm.C12 (right) tag. The isosurfaces correspond to PCSs of +1 ppm (blue) and −1 ppm (red) and are plotted on the first conformer of the PDB coordinates 2KOX. [63] The backbone of the protein is drawn in a ribbon representation, and the metal position is shown as a sphere. The Δχ‐tensor fit positioned the paramagnetic centre about 6.3 Å from the Cα atom of residue 57, as anticipated from the covalent structure of the tag.
Table 2.
Δχ‐tensor parameters of ubiquitin S57C tagged with Tb.C12, Tm.C12 or Tb.C1.[a]
|
Tag |
Δχ ax [b] [10−32 m3] |
Δχ rh [b] [10−32 m3] |
x [Å] |
y [Å] |
z [Å] |
α [°] |
β [°] |
γ [°] |
Q [c] |
|---|---|---|---|---|---|---|---|---|---|
|
Tb.C12 |
−13.5(1) |
−8.0(1) |
13.810 |
11.561 |
−3.533 |
65 |
56 |
87 |
0.01 |
|
Tm.C12 |
11.1(2) |
3.2(2) |
13.810 |
11.561 |
−3.533 |
56 |
60 |
67 |
0.06 |
|
Tb.C1 [d] |
−9.06(3) |
−2.5(1) |
17.220 |
8.370 |
−4.000 |
12 |
105 |
151 |
0.06 |
[a] The Δχ‐tensor fits used PCSs measured with TbIII and TmIII, using YIII as the diamagnetic reference and the NMR ensemble structure of ubiquitin (PDB ID: 2KOX). [63] Fits with the Tb.C12 and Tm.C12 tags were to a common set of x, y and z coordinates of the metal position. Euler angles α, β and γ are reported in degrees relative to the structure 2KOX. [b] Uncertainties (in brackets) were determined from fits obtained by randomly omitting 10 % of the PCS data. [c] The quality factor was calculated as the root‐mean‐square deviation between experimental and back‐calculated PCSs divided by the root‐mean‐square of the experimental PCSs. [d] Parameters reproduced from Pearce et al. [61]
To confirm these results, we also tagged the N172C mutant of the Pseudomonas aeruginosa metallo‐β‐lactamase IMP‐1 with C12 and C2 tags. The wild‐type protein already contains a cysteine residue, but this residue coordinates the two zinc ions in the active site [62] and proved to be unreactive towards the tags. As in the case of ubiquitin, conservation of the NMR chemical shifts following ligation with Y.C12 or Y.C2 indicated maintenance of structural integrity. Following ligation with tags loaded with YIII, TmIII or TbIII ions, PCSs were observed both with the C12 and C2 tags (Tables S2 and S3). In this case, the C12 tag produced smaller Δχ tensors than the C2 tag (Figures 6 and S17), but the Q factors of the Δχ‐tensor fits were consistently better (Table 3). In the case of IMP‐1, the Δχ tensor fits identified lanthanoid ion positions that were slightly further from the protein backbone for the C12 than the C2 tag (Figure 6c and d). It is unclear whether this indicates de‐coordination of the pyridine moiety in the C12 tag (despite limited lanthanide hydration indicated by q=1 for the derivative with cysteine; see Table 1), or is an artifact of fitting a single Δχ tensor to PCS data generated by a mobile tag, which places the metal ion at variable positions relative to the protein. [47]
Figure 6.

PCSs of the metallo‐β‐lactamase mutant IMP‐1N172C with the C12 tag. a) Superimposition of 15N HSQC spectra of IMP‐1N172C tagged with the C12 tag loaded with YIII (black), TbIII (red) or TmIII (blue) ions. The PCSs of selected amide protons are identified by lines connecting the corresponding crosspeaks. b) Correlation plot of back‐calculated vs. experimental PCSs for amide protons of IMP‐1N172C tagged with the Tb.C12 (red) or Tm.C12 (blue) tag. c) PCS isosurfaces representing the Δχ tensors obtained with the Tb.C12 (left) or Tm.C12 (right) tag attached at position 172. The isosurfaces correspond to PCSs of +1 ppm (blue) and −1 ppm (red) and are plotted on the PDB structure 4UAM. [64] The distance of the paramagnetic centre from the Cα atom of the tag attachment site is indicated by a dotted line. d) Same as (c), but for the protein with Tb.C2 (left) and Tm.C2 (right) tag.
Table 3.
Δχ‐tensor parameters of IMP‐1N172C tagged with C2 and C12 tags loaded with TbIII or TmIII ions.[a]
|
Tag |
Δχ ax [b] [10−32 m3] |
Δχ rh [b] [10−32 m3] |
x [Å] |
y [Å] |
z [Å] |
α [°] |
β [°] |
γ [°] |
Q [c] |
|---|---|---|---|---|---|---|---|---|---|
|
Tb.C12 |
13.5(2) |
6.4(2) |
43.966 |
85.262 |
25.852 |
61 |
52 |
112 |
0.11 |
|
Tm.C12 |
−10.5(2) |
−5.1(3) |
43.966 |
86.252 |
25.852 |
57 |
54 |
106 |
0.12 |
|
Tb.C2 |
−18.2(3) |
−4.7(2) |
40.869 |
77.901 |
25.202 |
22 |
29 |
172 |
0.33 |
|
Tm.C2 |
13.9(2) |
3.6(3) |
40.869 |
77.901 |
25.202 |
20 |
31 |
172 |
0.35 |
[a] The Δχ‐tensor fits used PCSs measured with TbIII and TmIII, using YIII as the diamagnetic reference. The metal coordinates and tensor parameters for the IMP1 mutant are reported relative to the crystal structure of IMP‐1 (PDB ID: 4UAM). [64] [b] Uncertainties (in brackets) were determined from fits obtained by randomly omitting 10 % of the PCS data and the quality factor was calculated as described in footnote [c] of Table 2.
EPR characterisation of the Gd.C12 tag
To evaluate the performance of Gd.C12 for DEER applications we turned to ERp29. It is a homodimeric protein and its mutants S114C and G147C have been used previously to assess the performance of different gadolinium tags for distance measurements.[ 65 , 66 , 67 ] Figure 7 compares the EPR line shape of the Gd.C12 tag ligated to ERp29 S114C and ERp29 G147C with that of the free tag. The line shape of the central ms |− ⟩→|+ ⟩ transition is similarly narrow as that of the structurally related C1 and C9 tags [66] and undergoes minimal change upon attachment of the tag to the protein, except for about two‐fold narrowing of the central transition. T 1 relaxation (120 μs, 10 K) as determined from three‐pulse inversion recovery did not change significantly between the unbound and bound tag (Figure S18, Table S4). Phase memory times T M (8–10 μs) as determined from spin‐echo decay experiments were also similar (∼15 % longer, Table S6), provided the protein was incubated in D2O over 24 h (Figure S19). As the protein samples were not perdeuterated, the sensitivity of T M to hydrogen exchange must be attributed to amide and hydroxy groups, most likely the amides of the pendant arms of the C12 tag, which are close to the GdIII ion (Figure 1).
Figure 7.

Central transition of echo‐detected EPR spectra of the Gd.C12 tag, free and bound to cysteine mutants of ERp29 recorded at 94 GHz (scaled to the exact frequency). The full width at half maximum (fwhm) of the central sharp |− ⟩→|+ ⟩ transition are annotated on each spectrum. The pump and probe pulse positions applied for the DEER experiments of ERp29 S114C and ERp29 G147C are indicated. The inserts show the full spectra. Line‐shape simulations are shown in Figure S20.
DEER experiments
DEER measurements performed with the mutants S114C and G147C of ERp29 ligated with the Gd.C12 tag are shown in Figure 8. A modulation depth λ of about 6–8 % was obtained, which is very good for DEER with GdIII. [68] For the mutant S114C and G147C, narrow distance distributions centred at 5.7 and 5.6 nm respectively were observed, in close agreement with modelling by tag rotamer libraries generated with the program PyParaTools [69] (Figure S23). These distances are somewhat shorter than obtained with the C1 tag, which were 6.1 and 5.7 nm, respectively. [65] The shorter distance obtained with the C12 tag can be attributed to the shorter linker length to the cysteine residues, which was also reflected in the modelled distance distributions (Figures S22 and S23). Despite a more rigid linker, the experimentally determined distribution widths obtained with C12 were similar to those obtained with the C1 tag.
Figure 8.

DEER distance measurement of ERp29 S114C (left) and ERp29 G147C (right) tagged with Gd.C12. a) Form factor after background subtraction. The vertical axis plots the normalized echo intensity, the red line corresponds to the fitted trace. b) Distance distribution calculated by DeerAnalysis2018. [70] The red lines indicate the maxima of the modelled distance distributions. The full DEER data are shown in Figure S21.
The performance of C12 is also comparable with the C7 and C8 tags, which display distances closer to 6 nm. [67] All three tags have the same number of bonds in the linker between the cyclen ring and cysteine residue. Comparing the distribution widths with those obtained with the C7 and C8 tags is more difficult, because these tags yield remarkably narrow distance distributions for ERp29 S114C but, for unknown reasons, unexpectedly broad distributions for ERp29 G147C. [67] As this is not observed for C12, this difference must be an artefact, which we speculate the C12 tag is less susceptible to.
Time‐resolved FRET using TbIII‐labelled aurora A kinase
Distance information on the nanometre scale can also be obtained from FRET experiments utilising luminescent TbIII or EuIII donors. The selective reaction of Tb.C12 with cysteine‐containing biomolecules, coupled with its bright and long‐lived luminescence signal upon labelling, make the TbIII complex an ideal candidate for use in homogenous time‐resolved FRET assays.[ 39 , 71 , 72 ] Due to the spectral overlap of the emission bands of Tb.C12 and the absorption spectrum of AlexaFluor 633 (AF633; λ ex 633 nm, λ em 650 nm), these two molecules were used as a FRET donor and acceptor, respectively. The suitability of thiol‐tagged Tb.C12 and AF633 as a FRET donor/acceptor pair was established in experiments conducted with GSH‐tagged Tb.C12 donor and freely diffusible AF633 acceptor, which indicated a rate constant for energy transfer of 1.5×109 M−1 s−1 (Figure S24).
To demonstrate FRET experiments in a protein, we used a construct of the protein kinase aurora A engineered to contain exactly two surface exposed cysteine residues (D274 N/S278 C/C290 A/H373 C/C393 A mutant). Aurora A regulates entry into mitosis and other processes integral to cell proliferation and is a target of several cancer drug discovery programmes.[ 73 , 74 , 75 ] Incubating the protein with Tb.C12 at 4 °C and pH 7.4 for 18 h, the maximum enhancement in TbIII emission intensity was observed for a tag/protein molar ratio of 4 : 1 (Figure S25).
FRET measurements of aurora A were performed following stochastic labelling with the Tb.C12/AF633 pair, using 15 μM solutions of aurora A and 120 μM solutions of Tb.C12, whilst varying the concentration of AF633 from 0–60 μM. Background fluorescence from the sample was effectively eliminated by applying a time delay of 60 μs between excitation (λ ex=292–366 nm) and detection (λ em=510–550 nm for Tb.C12 or 660–670 nm for AF633). FRET was demonstrated by the presence of AF633 acceptor decreasing the TbIII emission intensity (Figure 9a), time‐resolved emission intensity of the AF633 acceptor, which increased in a concentration dependent manner (Figure 9b), and a concomitant decrease in the TbIII emission lifetime (Figure 9c). These data are consistent with increasing FRET due to an increase in the number of protein molecules stochastically labelled with both donor and acceptor dyes. The labelling sites on Aurora A are expected to be separated by 19–31 Å (no single distance since one site is on a flexible loop) indicating that high FRET signals can be detected in this distance range. Control experiments involving the protein tagged only with the AF633 acceptor confirmed that both the donor and acceptor must be present for FRET to be observed (Figure 9).
Figure 9.

Time‐resolved emission intensity (λ ex=292–366 nm, integration time=60–400 μs) of TbIII emission (λ em=510–550 nm) and AF633 emission (λ em=660–670 nm). The tagging reaction was performed by incubating 15 μM aurora A with 120 mM Tb.C12 and different amounts of AF633 at 4 °C for 18 h in 50 mM Tris⋅HCl, 50 mM NaCl, 1.8 % DMSO at pH 7.4. Control experiments involved aurora A with no TbIII complex (red) or only AF633 (orange). a) TbIII emission after incubation of aurora A with different equivalents of AF633 (relative to aurora A). b) Same as (a), but monitoring AF633 emission. Observation of some background emission without AF633 is expected from the emission spectrum of the TbIII tag (Figure 2a). c) TbIII emission lifetimes (λ ex=280 nm, λ em=545 nm) of the samples in (b).
We used our measured quantum yield and absorbance/emission spectra to calculate the critical distance (R 0) for the Tb.C12‐AF633 FRET pair. We determined this to be 53±4 Å which is similar to that for many other commonly used FRET pairs. We cannot easily calculate the expected FRET efficiency for specific distances using this value of R 0 since our standard calculation assumes free rotation of both dye molecules (i. e., κ 2=2/3) and our NMR measurements indicate that this is unlikely to be the case for Tb.C12. Nevertheless, our measurements of high FRET in the 19–31 Å range are consistent with our calculation which predicts FRET efficiency >78 % for values of κ 2 for these distances in the physically meaningful range 0.1≤κ 2≤4. Thus, we conclude that Tb.C12 is highly suitable for time‐resolved FRET assays.
Selectivity of the C12 tag for selenocysteine
The broad utility of C12 for many different types of experiments prompted us to explore its potential for site‐selective tagging of proteins containing selenocysteine residues. The selenol group of selenocysteine (Sec) is much more nucleophilic than the thiol group of cysteine and recently developed technology for genetic encoding of a photocaged selenocysteine residue, in principle, enables site‐specific installation of a selenocysteine residue, although the protein yields obtainable are still too low for routine use. [76] As solvent‐exposed selenocysteine residues are highly prone to forming Se−Se bonds, free selenol groups can be maintained only in the presence of reducing agents, [77] which are incompatible with tags that contain activated disulfide bonds. In the present work, we therefore tested the compatibility of the C12 tag with reducing agents and its potential for ligation to selenocysteine.
The mutant Q32Sec of the protein GB1 was prepared by cell‐free protein synthesis with the exclusion of cysteine and provision of selenocystine to incorporate selenocysteine in response to the cysteine codon. Tagging reactions with Y.C12 were conducted in parallel with GB1 Q32Sec and GB1 Q32C. The reaction with GB1 Q32Sec was complete within 10 minutes, whereas only a small fraction of GB1 Q32C had reacted even after 5 h at room temperature (Figure 10). After the tagging reaction, GB1 Q32Sec displayed three additional mass peaks, two of which may be attributed to the reduction of the selenocysteine residue to serine (m/z 8275.86 Da) and alanine (m/z 8256.85 Da) caused by the presence of TCEP, which are not amenable to reaction with the C12 tag. The third minor peak (m/z 8292.85 Da) may arise from a minor level of cysteine instead of selenocysteine incorporation. These results indicate that the correctly tagged protein was the main species and illustrate the potential of selenocysteine for site‐specific tagging in the presence of free thiol groups.
Figure 10.

Mass spectra illustrating the much faster reaction of the Y.C12 tag with selenocysteine than cysteine. a) and b) Reaction with the mutant GB1 Q32Sec after 5 and 10 min, respectively. The calculated masses of the untagged and tagged proteins are 8339.98 and 9172.32 Da, respectively. c) and d) Reaction with the uniformly 15N‐labelled mutant GB1 Q32C after 10 min and 5 h, respectively. The masses calculated for 100 % 15N‐enriched untagged and tagged proteins are 8388.38 and 9220.73 Da, respectively.
Discussion
Many lanthanide tags have been developed and compared for PCS and DEER measurements.[ 5 , 7 , 24 ] Most of the tags are designed for attachment to single cysteine residues in the target proteins, either via a disulfide or a thioether bond. Thioether bonds are generally preferrable, as they produce a shorter tether between metal ion and protein, and disulfide bonds tend to be flexible and readily broken by chemical reduction. Attachment via thioethers has been obtained with tags containing phenylsulfonated pyridines,[ 78 , 79 , 80 ] bromo‐ or iodoacetamides[ 81 , 82 , 83 ] or (methylsulfonyl)thiazolo[5,4‐b]pyridines,[ 51 , 78 ] or by a thiol‐ene reaction. [84] Phenylsulfonated tags react only slowly with cysteine residues [78] and require high solvent exposure of the thiol group. Halo‐acetamides entail a relatively long and flexible tether with cysteine and tend to be unstable towards lyophilisation.[ 81 , 82 ] (Methylsulfonyl)thiazolo[5,4‐b]pyridines, such as in the P4T‐DOTA tag (Figure 1), combine high reactivity towards thiol groups with high rigidity of the resulting tether between protein and metal ion, but the synthetic protocol of this group involves a number of steps of modest yield. [51] The methylsulfonyl group on the activated pyridine ring of the Ln‐M7‐Nitro tag (Figure 1) reacts with cysteine thiols within minutes, [78] which may make it difficult to selectively tag selenocysteine in the presence of cysteine.
The 4‐nitropyridyl group of the C12 tag combines good reactivity with a rigid resulting tether, which is shorter than that obtained with (methylsulfonyl)thiazolo[5,4‐b]pyridines. An important advantage of the 4‐nitropyridyl group as the tethering moiety is its ready synthetic accessibility allowing installation on the cyclen ring in just three steps (Scheme 1). In preliminary experiments, we observed that the C12 tag was significantly more reactive towards cysteine thiol groups than the related Ln.L1 tag (Figure 1), [53] suggesting that the electrophilicity of the pyridine ring is enhanced if the positive charge of the lanthanide ion is not compensated by negatively charged acetate pendants. The effect supports the notion that the metal ion is directly coordinated by the nitrogen of the pyridine ring as designed. Finally, the reactivity of the C12 tag is compatible with the presence of TCEP to maintain cysteine and selenocysteine in their reduced forms and it is sufficiently stable for shipping at room temperature. Despite its more modest reactivity compared with the Ln‐M7‐Nitro tag, we readily obtained 100 % ligation yields with different proteins, without any evidence of reaction with amino acids other than cysteine. Notably, however, the large hydrophobic pendants of the C12 tag and its positive net charge carry the potential for undesired interactions with the protein. While this cannot be excluded, the present work found no evidence for such effects.
As in the C1 and C2 tags,[ 54 , 60 ] the chiral phenylethylamide groups on the cyclen ring of the C12 tag shift the equilibrium between different diastereomeric conformations of the lanthanide complex towards a single species, which is maintained in the adduct with a target protein. As expected for a shorter and more rigid tether between protein and metal ion, the present results confirmed our expectations that the C12 tag in general delivers larger PCSs and Δχ‐tensor fits of better quality than the C1 and C2 tags. The Gd.C12 tag also proved to be applicable for the measurement of DEER distance distributions, which displayed comparable short distances and distribution widths.
An outstanding feature of the C12 tag is its capacity to form a stable selenoether bond with selenocysteine. The greater reaction rate of the C12 tag observed with selenocysteine compared with cysteine is underpinned by the greater nucleophilicity of a selenol versus thiol group near neutral pH. This sets the stage for site‐selective tagging of a protein that contains a single selenocysteine residue, regardless of the presence or absence of cysteine residues. We anticipate that this will present an attractive approach once systems become available that incorporate caged selenocysteine residues with greater yield than hitherto achievable,[ 76 , 85 ] as the specificity towards selenocysteine would eliminate the need to mutate native cysteine residues which is a major bottleneck for large proteins that contain several cysteine residues.
Finally, while double‐arm tags attached to two neighbouring cysteine residues immobilise metal ions more easily,[ 81 , 86 , 87 , 88 , 89 , 90 ] suitable sites for double‐cysteine mutations require careful selection and stable attachment of the tag is not guaranteed. [91] Strategies that immobilize the lanthanide ion by simultaneous coordination to two phosphoserine residues [92] or chelating moieties installed on neighbouring cysteine residues[ 93 , 94 ] are similarly restricted in the choice of attachment sites. By allowing attachment to a single amino acid residue, the C12 tag opens a much greater choice of suitable tagging sites.
Among the cyclen tags that react with cysteine by formation of a stable thioether bond,[ 17 , 51 , 68 , 78 , 79 , 81 , 82 ] the most recent designs use a linker with an aromatic ring that is capable of coordinating the lanthanide ion. This design is attractive as it rigidifies the tether to the protein [53] and in this way limits the averaging between positive and negative PCSs that occurs when the lanthanide complex reorientates relative to the protein. This design is also attractive for DEER measurements in order to obtain the narrowest possible distance distributions. Our present results indicate that the different widths in distance distribution obtained for the sites 114 and 157 in ERp29 mostly reflect the conformational space accessible to the tags, as both C1 and C12 yield consistently narrower distribution widths for the site 114 than the site 157 and the latter is more solvent‐exposed. Despite a shorter and more rigid tether in C12, however, the widths of the distance distributions were similar, contrary to our simulations which assumed stable coordination between the pyridine nitrogen atom and GdIII ion. In contrast, our NMR data suggested that the C12 tag restricts translational movements of the lanthanide ion relative to the protein better than the C1 tag, as manifested by better quality factors obtained in the Δχ‐tensor fits of Tb.C12 on the cysteine mutants of ubiquitin and IMP‐1 compared to those obtained with the Tb.C1 and Tb.C2 tags (Tables 2 and 3). [47] Notably, the Gd.C7 and Gd.C8 tags delivered much narrower distance distributions for the ERp29 mutant S114C, but unexpectedly broad distributions for the G147C mutant. [67] A similar discrepancy between these two sites was obtained with the Gd.C9 tag (Figure 1), indicating that the widths of the distance distributions very much depend on the specific tag and its interactions with the protein environment. [66] This may hold in particular for tags with very rigid linkers, as any chemical tag can potentially also affect the protein structure and rigid tags may be more problematic in this regard. In the case of the Gd.C12 tag, the distance distribution widths varied less between different sites than for the previously published Gd.C7 and Gd.C8 tags. [67] In addition, each backbone amide displayed a single peak in the protein NMR spectra, indicating fast exchange between different tag conformations as far as they occur. More examples will need to be evaluated to confirm the consistency in the performance of the C12 tag.
On a technical note, the DEER measurements with Gd.C12 delivered a maximal modulation depth of almost 8 %, which is similar to the record modulation depth of 9 % reported for the [Gd.sTPATCN] spin label. [68] For DEER measurements using nitroxide spin labels, it is well‐known that the phase memory time can be greatly extended by perdeuteration of the protein.[ 95 , 96 ] As deuteration of exchangeable hydrogens was sufficient to extend the phase memory time of the Gd.C12 tag, it may possibly be extended further by synthesizing the tag and/or protein in perdeuterated form. In past experiments of ERp29 S114C tagged with Gd.C1, however, perdeuteration did not extend the phase memory time very much. [65]
In our luminescence experiments, cysteine labelling of the TbIII complex of C12 elicited a remarkably large enhancement in both the extinction coefficient (ϵ increases from 1200 to 15 500 M−1 cm−1) and emission quantum yield (Φ increases from 0.02 to 20 %). The ability to ‘switch on’ TbIII emission upon labelling is highly advantageous for time‐resolved luminescence assays, as it avoids cumbersome washing/purification steps and permits dynamic imaging of biochemical processes in high‐throughput format. Other protein‐labelling methods which display fluorogenic behaviour, such as SNAP‐tag technology [97] require the attachment of the fluorophore to a quencher group, which is released when the tag reacts with the target biomolecule. In the present work, we exemplified the performance of the Tb.C12 tag in straightforward microplate reader‐based bioassays, monitoring the enzymatic reduction of glutathione and observing time‐resolved FRET within a model protein kinase, Aurora A. The quantum yield of the thiol‐tagged Tb.C12 is approximately 10 times larger than that of the europium(III) cryptate (ϕ=0.02) [98] used in several commercial homogeneous time‐resolved FRET assays, indicating that a tenfold increase in sensitivity is achievable with the Tb.C12 tag in such assays. The commercially available Lumi4‐Tb tag, which exhibits unequalled brightness (15 800 M−1 cm−1 at 340 nm in water vs. 3100 M−1 cm−1 for Tb.C12‐Cys at 280 nm), is not silent in its untagged form. [33]
Conclusion
In summary, the C12 tag combines many favourable features, including stable lanthanide binding in a complex forming a single conformation, a relatively short tether favouring high‐quality Δχ‐tensor fits, high reactivity towards cysteine and selenocysteine, attachment to a single solvent‐exposed thiol or selenol group, and, last but not least, relative ease of synthesis and convenience of use in the tagging reaction which, important for tagging of selenocysteine, can be conducted in the presence of reducing agents such as TCEP. The Δχ tensors measured by NMR are of useful magnitude, the EPR properties of the Gd.C12 tag make it suitable for DEER distance measurements and the outstanding luminescent properties of the Tb.C12 tag open a host of attractive applications in structural biology, enzymology and, potentially, even live‐cell imaging applications. We hope to improve these further by modification of the antenna of the Tb.C12 tag to increase its excitation wavelength towards the visible region. Overall, we anticipate that the broad applicability and convenience of the C12 tag will make it an exceptionally popular tool.
Experimental Section
The protocols for synthesis and characterisation of complexes Tb.C12, Eu.C12, Tm.C12, Gd.C12 and Y.C12 are provided in the Supporting Information, as are the protocols for protein production, purification and tagging. The Supporting Information also reports the general procedures for the reaction of Ln.C12 with low‐molecular‐weight thiols, glutathione reductase reactions and tagging of aurora A with Tb.C12 and AlexaFluor 633.
Extinction coefficients and quantum yields: The extinction coefficients of Ln.C12 or Ln.C12‐Cys (Ln=Tb or Eu; 1 mg mL−1) were determined in 10 mM HEPES pH 7.0. Quantum yields were measured using quinine sulfate in 0.05 M H2SO4 as a standard (Φ em=0.52, λex=350 nm). [56]
Luminescence experiments: Luminescence spectra were recorded on a Camlin Photonics luminescence spectrometer with FluoroSENS version 3.4.7.2024 software. Emission spectra were obtained using a 40 μL Hellma Analytics quartz cuvette (Art no. 111‐10‐K‐40). Excitation light was set at 280 nm (or 300 nm for untagged LnIII complexes), and emission read in the range 400–720 using an integration time of 0.5 seconds, increment of 1.0 nm and excitation slit of 0.2 nm and emission slits of 0.5 nm. Plate reader data were obtained on a BMG Labtech CLARIOstar microplate reader in black Fisherbrand™ 384‐well plates, using a total volume of 40 μL per well.
Emission lifetime measurements were performed on the same instrument. Measurements were taken of 1 mL of 0.1 absorbance samples of Ln complexes in 10 mM HEPES, pH 7.0. Measurements were obtained by indirect excitation of the Ln ion via the pyridine antennae using a short pulse of light at λ max (300 nm for untagged complexes, 280 nm for thiol‐tagged complexes), followed by monitoring the integrated intensity of the light emitted at 546 nm (Tb complexes) or 615 nm (Eu complexes), with 500 data points collected over a 10 ms time period. The decay curves were plotted in Origin Labs 2019 version 9.6.0.172, and fitted to the equation:
| (1) |
where I is the intensity at time, t, following excitation, A 0 is the intensity when decay has ceased, A 1 is the pre‐exponential factor and k is the rate constant for the depopulation of the excited state.
The hydration state, q, of the EuIII and TbIII complex of C12 was determined using the modified Horrocks equation: [55]
| (2) |
| (3) |
where τ and τ are the emission lifetime times in water and D2O, respectively, and n is the number of carbonyl‐bound amide NH groups.
Study of specificity for selenocysteine versus cysteine : The tagging reactions of the proteins GB1 Q32Sec and GB1 Q32C were carried out in parallel, using 50 μM GB1 mutant, 250 μM Tb.C12 tag and 1 mM TCEP. Progress of the reaction was monitored by sampling 10 μL aliquots from the reaction mixture after 5, 10, 15, 30, 45 and 60 min, as well as after 5 h. The reaction was quenched by the addition of 0.1 % TFA and snap freezing for storage. The final samples were analysed by intact protein mass spectrometry.
Intact protein mass spectrometry: Intact protein analysis was carried out on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) connected to a Thermo Fisher Scientific UltiMate 3000 HPLC system equipped with ZORBAX 300SB−C3, 3.5μm, 4.6×50mm HPLC column (Agilent Technologies). Each HPLC run was performed with 500 μL/min linear gradient of solvent A (0.1 % (v/v) formic acid in water) and solvent B (0.1 % (v/v) formic acid in acetonitrile), ramping solvent B from 5 % solvent B at the start to 80 % after 12min. Data were collected using an electrospray ionization (ESI) source in positive ion mode. Protein intact mass was determined by deconvolution using the Xtract function in the Qual Browser software tool of the program Xcalibur 3.0.63 (Thermo Fisher Scientific).
NMR measurements : All NMR data were acquired at 35 °C on a Bruker 600 MHz Avance NMR spectrometer equipped with a TCI cryoprobe. The PCSs of amide protons were measured in 15N HSQC spectra recorded with acquisition times of t 1max=90 ms and t 2max=122 ms. The PCSs were measured in ppm as the chemical shifts in the paramagnetic sample minus the chemical shift in the diamagnetic sample.
Δχ‐tensor fits: Δχ‐tensor parameters were determined using the program Paramagpy, [99] using the PCSs of backbone amide protons and atomic coordinates from the Protein Data Bank (PDB ID: 2KOX [63] for the structure of ubiquitin and 4UAM [64] for the structure of IMP‐1). The quality of the fit was assessed by the Q factor, which was calculated as the ratio of the root‐mean‐square deviation between experimental and back‐calculated PCSs and the root‐mean‐square of the experimental PCSs.
EPR characterisation: All EPR measurements were performed at 10 K on a modified Bruker EPR spectrometer operating at W‐band (94 GHz). [100] The line shape of both the protein‐bound and free Gd.C12 tag was measured by an electron spin‐echo field‐sweep sequence π/2–τ–π–echo, using π/2=40 ns, π=80 ns and τ=500 ns. T 1 relaxation was measured by the inversion recovery sequency π–t+dt–π/2–τ–π–echo. The integrated echo intensity was recorded as a function of time t incremented in intervals dt (1000 ns), using the same pulse lengths as above. T 2 relaxation was measured by recording the decay of the integrated echo intensity with time, using the pulse sequence π/2–t+dt–π–t+2dt–echo (dt=20 ns).
DEER measurements: The standard four‐pulse DEER sequence (π/2(νobs)–t 1–π(νobs)–(t 1+dt)–π(Δνpump)–(t 2–dt)–π(νobs)–t 2–echo) was used with averaging over the initial time delay to remove nuclear modulation artefacts; each scan was acquired by averaging four different t 1 values from 400–562 ns. The DEER echo was observed at 93.94 GHz with π/2 and π pulses of 16 and 32 ns, respectively, and an ELDOR pulse of π=16 ns at 120–130 MHz above the probe frequency. Other parameters used were a repetition rate of 255 μs, dt=20 ns and t 2=7.6 μs. The field position for detection was set at the peak of the GdIII spectrum (Figure 7), applying the pump pulse at the centre of the Gd spectrum and the probe pulse on the edge of the ms |− ⟩→|+ ⟩ transition.
The data were analysed using DeerAnalysis2018 [70] and distance distributions were obtained using Tikhonov regularization. The regularization parameter was chosen by the L‐curve criterion. Estimation of uncertainties in distance distributions due to background correction were obtained using the validation option in DeerAnalysis.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Dr. Elwy H. Abdelkader for simulations with the program PyParaTools. S.J.B. gratefully acknowledges financial support by the EPSRC (EP/S032339/1) and the Wellcome Trust (204500/Z/16/Z). Financial support by the Australian Research Council for a Laureate Fellowship to G.O. (FL170100019), project funding (DP170100162, DP210100088) and through the Centre of Excellence for Innovations in Peptide & Protein Science (CE200100012) is also gratefully acknowledged. C.A.D. was supported by an Imperial College Research Fellowship.
I. D. Herath, C. Breen, S. H. Hewitt, T. R. Berki, A. F. Kassir, C. Dodson, M. Judd, S. Jabar, N. Cox, G. Otting, S. J. Butler, Chem. Eur. J. 2021, 27, 13009.
Contributor Information
Prof. Gottfried Otting, Email: gottfried.otting@anu.edu.au.
Dr. Stephen J. Butler, Email: S.J.Butler@lboro.ac.uk.
References
- 1. Abdelkader E. H., Qianzhu H., Tan Y. J., Adams L. A., Huber T., Otting G., J. Am. Chem. Soc. 2021, 143, 1133–1143. [DOI] [PubMed] [Google Scholar]
- 2. Allegrozzi M., Bertini I., Janik M. B. L., Lee Y.-M., Liu G., Luchinat C., J. Am. Chem. Soc. 2000, 122, 4154–4161. [Google Scholar]
- 3. Pintacuda G., John M., Su X. C., Otting G., Acc. Chem. Res. 2007, 40, 206–212. [DOI] [PubMed] [Google Scholar]
- 4. Hass M. A. S., Ubbink M., Curr. Opin. Struct. Biol. 2014, 24, 45–53. [DOI] [PubMed] [Google Scholar]
- 5. Nitsche C., Otting G., Prog. NMR Spectrosc. 2017, 98–99, 20–49. [DOI] [PubMed] [Google Scholar]
- 6.C. Luchinat, G. Parigi, E. Ravera, Paramagnetism in Experimental Biomolecular NMR, Royal Society of Chemistry, 2018.
- 7. Joss D., Häussinger D., Prog. NMR Spectrosc. 2019, 114–115, 284–312. [DOI] [PubMed] [Google Scholar]
- 8. Softley C. A., Bostock M. J., Popowicz G. M., Sattler M., J. Biomol. NMR 2020, 74, 287–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saio T., Ishimori K., Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129332. [DOI] [PubMed] [Google Scholar]
- 10. Häussinger D., Huang J. R., Grzesiek S., J. Am. Chem. Soc. 2009, 131, 14761–14767. [DOI] [PubMed] [Google Scholar]
- 11. Vlasie M. D., Comuzzi C., van den Nieuwendijk A. M., Prudêncio M., Overhand M., Ubbink M., Chem. Eur. J. 2007, 13, 1715–1723. [DOI] [PubMed] [Google Scholar]
- 12. Yagi H., Pilla K. B., Maleckis A., Graham B., Huber T., Otting G., Structure 2013, 21, 883–890. [DOI] [PubMed] [Google Scholar]
- 13. Pilla K. B., Otting G., Huber T., Structure 2017, 25, 559–568. [DOI] [PubMed] [Google Scholar]
- 14. Pilla K. B., Otting G., Huber T., J. Mol. Biol. 2016, 428, 522–532. [DOI] [PubMed] [Google Scholar]
- 15. Su X.-C., Chen J. L., Acc. Chem. Res. 2019, 52, 1675–1686. [DOI] [PubMed] [Google Scholar]
- 16. Pan B. B., Yang F., Ye Y., Wu Q., Li C., Huber T., Su X. C., Chem. Commun. 2016, 52, 10237–10240. [DOI] [PubMed] [Google Scholar]
- 17. Müntener T., Häussinger D., Selenko P., Theillet F. X., J. Phys. Chem. Lett. 2016, 7, 2821–2825. [DOI] [PubMed] [Google Scholar]
- 18. Caravan P., Ellison J. J., McMurry T. J., Lauffer R. B., Chem. Rev. 1999, 99, 2293–2352. [DOI] [PubMed] [Google Scholar]
- 19. Li H., Meade T. J., J. Am. Chem. Soc. 2019, 141, 17025–17041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aime S., Botta M., Terreno E., Adv. Inorg. Chem. 2005, 57, 173–237. [Google Scholar]
- 21. Qi M., Groß A., Jeschke G., Godt A., Drescher M., J. Am. Chem. Soc. 2014, 136, 15366–15378. [DOI] [PubMed] [Google Scholar]
- 22. Razzaghi S., Qi M., Nalepa A. I., Godt A., Jeschke G., Savitsky A., Yulikov M., J. Phys. Chem. Lett. 2014, 5, 3970–3975. [DOI] [PubMed] [Google Scholar]
- 23.Y. Yang, F. Yang, Y. J. Gong, J. L. Chen, D. Goldfarb, X. C. Su, Angew. Chem. Int. Ed. 2017, 56, 2914–2918; Angew. Chem. 2017, 129, 2960–2964. [DOI] [PubMed]
- 24. Yang Y., Yang F., Gong Y. J., Bahrenberg T., Feintuch A., Su X. C., Goldfarb D., J. Phys. Chem. Lett. 2018, 9, 6119–6123. [DOI] [PubMed] [Google Scholar]
- 25. Kaczmarski J. A., Mahawaththa M. C., Feintuch A., Clifton B. E., Adams L. A., Goldfarb D., Otting G., Jackson C. J., Nat. Commun. 2020, 11, 5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thonon D., Jacques V., Desreux J. F., Contrast Media Mol. Imaging 2007, 2, 24–34. [DOI] [PubMed] [Google Scholar]
- 27. Potapov A., Yagi H., Huber T., Jergic S., Dixon N. E., Otting G., Goldfarb D., J. Am. Chem. Soc. 2010, 132, 9040–9048. [DOI] [PubMed] [Google Scholar]
- 28. Theillet F.-X., Binolfi A., Bekei B., Martorana A., Rose H. M., Stuiver M., Verzini S., Lorenz D., van Rossum M., Goldfarb D., Selenko P., Nature 2016, 530, 45–50. [DOI] [PubMed] [Google Scholar]
- 29.A. Feintuch, G. Otting, D. Goldfarb, Methods Enzymol., eds. P. Z. Qin, K. Warncke, Academic Press, 2015, 563, 415–457. [DOI] [PubMed]
- 30. Bünzli J.-C. G., J. Lumin. 2016, 170, 866–878. [Google Scholar]
- 31. Moore E. G., Samuel A. P. S., Raymond K. N., Acc. Chem. Res. 2009, 42, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. New E. J., Parker D., Smith D. G., Walton J. W., Curr. Opin. Chem. Biol. 2010, 14, 238–246. [DOI] [PubMed] [Google Scholar]
- 33. Xu J., Corneillie T. M., Moore E. G., Law G.-L., Butlin N. G., Raymond K. N., J. Am. Chem. Soc. 2011, 133, 19900–19910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hewitt S. H., Ali R., Mailhot R., Antonen C. R., Dodson C. A., Butler S. J., Chem. Sci. 2019, 10, 5373–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bodman S. E., Butler S. J., Chem. Sci. 2021, 12, 2716–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bünzli J. C. G., Chem. Rev. 2010, 110, 2729–2755. [DOI] [PubMed] [Google Scholar]
- 37. Mathis G., Clin. Chem. 1993, 39, 1953–1959. [PubMed] [Google Scholar]
- 38. Selvin P. R., Annu. Rev. Bioph. Biom. 2002, 31, 275–302. [DOI] [PubMed] [Google Scholar]
- 39. Zwier J. M., Bazin H., Lamarque L., Mathis G., Inorg. Chem. 2014, 53, 1854–1866. [DOI] [PubMed] [Google Scholar]
- 40. Hewitt S. H., Butler S. J., Chem. Commun. 2018, 54, 6635–6647. [DOI] [PubMed] [Google Scholar]
- 41. Bünzli J.-C. G., Coord. Chem. Rev. 2015, 293–294, 19–47. [Google Scholar]
- 42. Heffern M. C., Matosziuk L. M., Meade T. J., Chem. Rev. 2014, 114, 4496–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bünzli J.-C. G., Piguet C., Chem. Soc. Rev. 2005, 34, 1048–1077. [DOI] [PubMed] [Google Scholar]
- 44. Butler S. J., Delbianco M., Lamarque L., McMahon B. K., Neil E. R., Pal R., Parker D., Walton J. W., Zwier J. M., Dalton Trans. 2015, 44, 4791–4803. [DOI] [PubMed] [Google Scholar]
- 45. Cotruvo J. A., Featherston E. R., Mattocks J. A., Ho J. V., Laremore T. N., J. Am. Chem. Soc. 2018, 140, 15056–15061. [DOI] [PubMed] [Google Scholar]
- 46. Chalker J. M., Bernardes G. J. L., Davis B. G., Acc. Chem. Res. 2011, 44, 730–741. [DOI] [PubMed] [Google Scholar]
- 47. Shishmarev D., Otting G., J. Biomol. NMR 2013, 56, 203–216. [DOI] [PubMed] [Google Scholar]
- 48. Ikegami T., Verdier L., Sakhaii P., Grimme S., Pescatore B., Saxena K., Fiebig K. M., Griesinger C., J. Biomol. NMR 2004, 29, 339–349. [DOI] [PubMed] [Google Scholar]
- 49. Pintacuda G., Moshref A., Leonchiks A., Sharipo A., Otting G., J. Biomol. NMR 2004, 29, 351–361. [DOI] [PubMed] [Google Scholar]
- 50. Opina A. C. L., Strickland M., Lee Y. S., Tjandra N., Byrd R. A., Swenson R. E., Vasalatiy O., Dalton Trans. 2016, 45, 4673–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joss D., Häussinger D., Chem. Commun. 2019, 55, 10543–10546. [DOI] [PubMed] [Google Scholar]
- 52. Joss D., Winter F., Häussinger D., Chem. Commun. 2020, 56, 12861–12864. [DOI] [PubMed] [Google Scholar]
- 53. Gempf K. L., Butler S. J., Funk A. M., Parker D., Chem. Commun. 2013, 49, 9104–9106. [DOI] [PubMed] [Google Scholar]
- 54. Graham B., Loh C. T., Swarbrick J. D., Ung P., Shin J., Yagi H., Jia X., Chhabra S., Barlow N., Pintacuda G., Huber T., Otting G., Bioconjugate Chem. 2011, 22, 2118–2125. [DOI] [PubMed] [Google Scholar]
- 55. Beeby A., Clarkson I. M., Dickins R. S., Faulkner S., Parker D., Royle L., de Sousa A. S., Williams J. A. G., Woods M., J. Chem. Soc. Perkin Trans. 2 1999, 493–504. [Google Scholar]
- 56. Brouwer A. M., Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar]
- 57. McMahon B. K., Gunnlaugsson T., J. Am. Chem. Soc. 2012, 134, 10725–10728. [DOI] [PubMed] [Google Scholar]
- 58. Starck M., Fradgley J. D., Di Vita S., Mosely J. A., Pal R., Parker D., Bioconjugate Chem. 2020, 31, 229–240. [DOI] [PubMed] [Google Scholar]
- 59. Fontaine S. D., Reid R., Robinson L., Ashley G. W., Santi D. V., Bioconjugate Chem. 2015, 26, 145–152. [DOI] [PubMed] [Google Scholar]
- 60. de la Cruz L., Nguyen T. H. D., Ozawa K., Shin J., Graham B., Huber T., Otting G., J. Am. Chem. Soc. 2011, 133, 19205–19215. [DOI] [PubMed] [Google Scholar]
- 61. Pearce B. J. G., Jabar S., Loh C. T., Szabo M., Graham B., Otting G., J. Biomol. NMR 2017, 68, 19–32. [DOI] [PubMed] [Google Scholar]
- 62. Concha N. O., Janson C. A., Rowling P., Pearson S., Cheever C. A., Clarke B. P., Lewis C., Galleni M., Frère J.-M., Payne D. J., Bateson J. H., Abdel-Meguid S. S., Biochemistry 2000, 39, 4288–4298. [DOI] [PubMed] [Google Scholar]
- 63. Fenwick R. B., Esteban-Martín S., Richter B., Lee D., Walter K. F. A., Milovanovic D., Becker S., Lakomek N. A., Griesinger C., Salvatella X., J. Am. Chem. Soc. 2011, 133, 10336–10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carruthers T. J., Carr P. D., Loh C.-T., Jackson C. J., Otting G., Angew. Chem. Int. Ed. 2014, 53, 14269–14272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14494–14497. [Google Scholar]
- 65. Yagi H., Banerjee D., Graham B., Huber T., Goldfarb D., Otting G., J. Am. Chem. Soc. 2011, 133, 10418–10421. [DOI] [PubMed] [Google Scholar]
- 66. Abdelkader E. H., Lee M. D., Feintuch A., Cohen M. R., Swarbrick J. D., Otting G., Graham B., Goldfarb D., J. Phys. Chem. Lett. 2015, 6, 5016–5021. [DOI] [PubMed] [Google Scholar]
- 67. Prokopiou G., Lee M. D., Collauto A., Abdelkader E. H., Bahrenberg T., Feintuch A., Ramirez-Cohen M., Clayton J., Swarbrick J. D., Graham B., Otting G., Goldfarb D., Inorg. Chem. 2018, 57, 5048–5059. [DOI] [PubMed] [Google Scholar]
- 68. Shah A., Roux A., Starck M., Mosely J. A., Stevens M., Norman D. G., Hunter R. I., El Mkami H., Smith G. M., Parker D., Lovett J. E., Inorg. Chem. 2019, 58, 3015–3025. [DOI] [PubMed] [Google Scholar]
- 69.X.-C. S. M. Stanton-Cook, G. Otting, T. Huber, http://comp-bio.anu.edu.au/mscook/PPT, accessed 18 January 2021.
- 70. Jeschke G., Chechik V., Ionita P., Godt A., Zimmermann H., Banham J., Timmel C. R., Hilger D., Jung H., Appl. Magn. Reson. 2006, 30, 473–498. [Google Scholar]
- 71. Selvin P. R., Nat. Struct. Biol. 2000, 7, 730–734. [DOI] [PubMed] [Google Scholar]
- 72. Rajapakse H. E., Gahlaut N., Mohandessi S., Yu D., Turner J. R., Miller L. W., Proc. Nat. Acad. Sci. 2010, 107, 13582–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lin X., Xiang X., Hao L., Wang T., Lai Y., Abudoureyimu M., Zhou H., Feng B., Chu X., Wang R., Am. J. Cancer Res. 2020, 10, 2705–2729. [PMC free article] [PubMed] [Google Scholar]
- 74. Damodaran A. P., Vaufrey L., Gavard O., Prigent C., Trends Pharmacol. Sci. 2017, 38, 687–700. [DOI] [PubMed] [Google Scholar]
- 75. D'Assoro A. B., Haddad T., Galanis E., Front. Oncol. 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Welegedara A. P., Adams L. A., Huber T., Graham B., Otting G., Bioconjugate Chem. 2018, 29, 2257–2264. [DOI] [PubMed] [Google Scholar]
- 77. Welegedara A. P., Maleckis A., Bandara R., Mahawaththa M. C., Herath I. D., Tan Y. J., Giannoulis A., Goldfarb D., Otting G., Huber T., ChemBioChem 2020, 22, 1840–1848. [DOI] [PubMed] [Google Scholar]
- 78. Müntener T., Kottelat J., Huber A., Häussinger D., Bioconjugate Chem. 2018, 29, 3344–3351. [DOI] [PubMed] [Google Scholar]
- 79. Yang F., Wang X., Pan B.-B., Su X.-C., Chem. Commun. 2016, 52, 11535–11538. [DOI] [PubMed] [Google Scholar]
- 80. Chen J.-L., Wang X., Yang F., Cao C., Otting G., Su X.-C., Angew. Chem. Int. Ed. 2016, 55, 13744–13748; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13948–13952. [Google Scholar]
- 81. Liu W.-M., Skinner S. P., Timmer M., Blok A., Hass M. A. S., Filippov D. V., Overhand M., Ubbink M., Chem. Eur. J. 2014, 20, 6256–6258. [DOI] [PubMed] [Google Scholar]
- 82. Wu Z., Lee M. D., Carruthers T. J., Szabo M., Dennis M. L., Swarbrick J. D., Graham B., Otting G., Bioconjugate Chem. 2017, 28, 1741–1748. [DOI] [PubMed] [Google Scholar]
- 83. Hikone Y., Hirai G., Mishima M., Inomata K., Ikeya T., Arai S., Shirakawa M., Sodeoka M., Ito Y., J. Biomol. NMR 2016, 66, 99–110. [DOI] [PubMed] [Google Scholar]
- 84. Li Q.-F., Yang Y., Maleckis A., Otting G., Su X.-C., Chem. Commun. 2012, 48, 2704–2706. [DOI] [PubMed] [Google Scholar]
- 85. Rakauskaitė R., Urbanavičiūtė G., Rukšėnaitė A., Liutkevičiūtė Z., Juškėnas R., Masevičius V., Klimašauskas S., Chem. Commun. 2015, 51, 8245–8248. [DOI] [PubMed] [Google Scholar]
- 86. Prudêncio M., Rohovec J., Peters J. A., Tocheva E., Boulanger M. J., Murphy M. E. P., Hupkes H.-J., Kosters W., Impagliazzo A., Ubbink M., Chem. Eur. J. 2004, 10, 3252–3260. [DOI] [PubMed] [Google Scholar]
- 87. Keizers P. H. J., Desreux J. F., Overhand M., Ubbink M., J. Am. Chem. Soc. 2007, 129, 9292–9293. [DOI] [PubMed] [Google Scholar]
- 88. Liu W.-M., Keizers P. H. J., Hass M. A. S., Blok A., Timmer M., Sarris A. J. C., Overhand M., Ubbink M., J. Am. Chem. Soc. 2012, 134, 17306–17313. [DOI] [PubMed] [Google Scholar]
- 89. Lee M. D., Dennis M. L., Swarbrick J. D., Graham B., Chem. Commun. 2016, 52, 7954–7957. [DOI] [PubMed] [Google Scholar]
- 90. Welegedara A. P., Yang Y., Lee M. D., Swarbrick J. D., Huber T., Graham B., Goldfarb D., Otting G., Chem. Eur. J. 2017, 23, 11694–11702. [DOI] [PubMed] [Google Scholar]
- 91. Guan J.-Y., Keizers P. H. J., Liu W.-M., Löhr F., Skinner S. P., Heeneman E. A., Schwalbe H., Ubbink M., Siegal G., J. Am. Chem. Soc. 2013, 135, 5859–5868. [DOI] [PubMed] [Google Scholar]
- 92. Mekkattu Tharayil S., Mahawaththa M. C., Loh C. T., Adekoya I., Otting G., Magn. Reson. 2021, 2, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Swarbrick J. D., Ung P., Su X.-C., Maleckis A., Chhabra S., Huber T., Otting G., Graham B., Chem. Commun. 2011, 47, 7368–7370. [DOI] [PubMed] [Google Scholar]
- 94. Swarbrick J. D., Ung P., Chhabra S., Graham B., Angew. Chem. Int. Ed. 2011, 50, 4403–4406; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4495–4498. [Google Scholar]
- 95. El Mkami H., Ward R., Bowman A., Owen-Hughes T., Norman D. G., J. Magn. Reson. 2014, 248, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ward R., Bowman A., Sozudogru E., El-Mkami H., Owen-Hughes T., Norman D. G., J. Magn. Reson. 2010, 207, 164–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Maurel D., Comps-Agrar L., Brock C., Rives M.-L., Bourrier E., Ayoub M. A., Bazin H., Tinel N., Durroux T., Prézeau L., Trinquet E., Pin J.-P., Nat. Methods 2008, 5, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Alpha B., Balzani V., Lehn J.-M., Perathoner S., Sabbatini N., Angew. Chem. Int. Ed. 1987, 26, 1266–1267; [Google Scholar]; Angew. Chem. 1987, 99, 1310–1311. [Google Scholar]
- 99. Orton H. W., Huber T., Otting G., Magn. Reson. 2020, 1, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nalep A., Möbius K., Lubitz W., Savitsky A., J. Magn. Reson. 2014, 242, 203–213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information