

Abstract
Soft rot disease of edible mushrooms leads to rapid degeneration of fungal tissue and thus severely affects farming productivity worldwide. The bacterial mushroom pathogen Burkholderia gladioli pv. agaricicola has been identified as the cause. Yet, little is known about the molecular basis of the infection, the spatial distribution and the biological role of antifungal agents and toxins involved in this infectious disease. We combine genome mining, metabolic profiling, MALDI‐Imaging and UV Raman spectroscopy, to detect, identify and visualize a complex of chemical mediators and toxins produced by the pathogen during the infection process, including toxoflavin, caryoynencin, and sinapigladioside. Furthermore, targeted gene knockouts and in vitro assays link antifungal agents to prevalent symptoms of soft rot, mushroom browning, and impaired mycelium growth. Comparisons of related pathogenic, mutualistic and environmental Burkholderia spp. indicate that the arsenal of antifungal agents may have paved the way for ancestral bacteria to colonize niches where frequent, antagonistic interactions with fungi occur. Our findings not only demonstrate the power of label‐free, in vivo detection of polyyne virulence factors by Raman imaging, but may also inspire new approaches to disease control.
Keywords: antifungal agents, Burkholderia, MALDI, natural products, Raman
A combination of genome mining, metabolic profiling, and imaging techniques (MALDI and UV Raman spectroscopy) provided insight into the arsenal of antifungals produced by the mushroom pathogen Burkholderia gladioli pv. agaricicola. Targeted gene knockouts and bioassays link these antifungals to prevalent symptoms of mushroom soft rot, browning, and impaired mycelium growth.
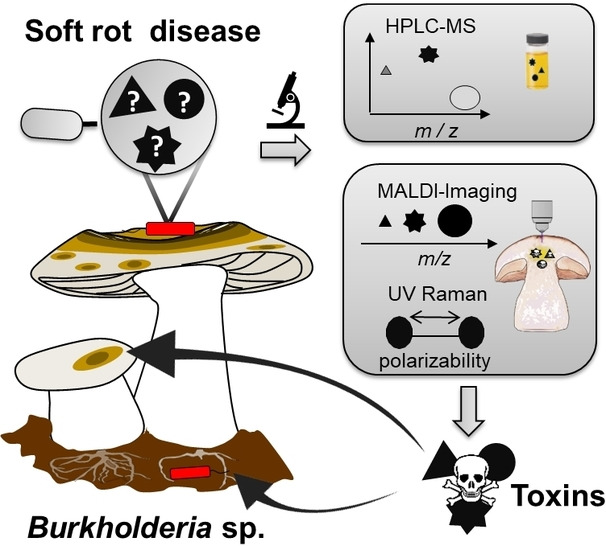
Introduction
The genus Burkholderia comprises versatile bacterial pathogens that cause severe diseases in humans, [1] animals, [2] plants, [3] and fungi. [4] Burkholderia gladioli is a remarkable example, as this species falls into various pathovars (pv.) and strains that inhabit specialized ecological niches. For mushroom farming, B. gladioli pv. agaricicola is of particular interest as it causes soft rot and cavity disease in edible fungi, namely Agaricus bisporus (white button mushroom), Agaricus bitorquis (banded agaric), Hypsizygus marmoreus (brown beech mushroom), Pleurotus ostreatus (oyster mushroom), and Pleurotus eryngii (king oyster mushroom).[ 4 , 5 ] Additionally, B. gladioli strains and pathovars from distinct environmental and clinical sources appear to be potential mushroom pathogens, as they degrade mushroom tissue to different degrees. [6] Rapid decay, soft rot and cavity formation in these mushrooms [7] threaten the global multiple‐billion‐dollar mushroom industry [8] and causes an average annual loss of approximately 25 % of the total production in western countries. [9] Notable outbreaks have been reported in New Zealand, [10] England, [11] Japan and South Korea. [12] Once a mushroom farm is infected, the bacterial pathogens are spread by means of human contact and irrigation. [13] Counter measurements are limited as common methods for crop disease management, such as formalin, sodium hypochlorite solution, or antibiotic treatment, are either forbidden, costly, or affect the indispensable commensal microbial community of mushrooms. [9] Thus, it is important to shed light on the molecular basis of the mushroom infection process, as this knowledge may inspire new ways of controlling the disease, such as strategies to inactivate the toxins. Moreover, most crops fall victim to some kind of bacterial soft rot disease, namely blackleg and tuber soft rot of potato, foot rot of rice, [14] ear soft rot of corn, [15] summer canker of kiwi fruits, [16] bacterial sheath rot of bananas, [17] and bacterial wilt of chrysanthemum. [18] Hence, studying mushroom soft rot and cavity disease might inspire further studies to targeting these diseases. In general, the colonization and invasion of a microbial pathogen is commonly promoted by virulence factors, secondary metabolites, and enzymes, which evolved in an ancient, on‐going arms race for survival. We have previously found that B. gladioli pv. agaricicola attaches, prevails, and disseminates on the mushroom by means of linear lipopeptides. [19] As soon as the bacteria colonize the mushroom, proteases and chitinases degrade the fungal cell wall, [20] enabling as to yet cryptic toxins to damage fungal cell components, eventually leading to cell death (Figure 1A). [21] Furthermore, it is known that B. gladioli pv. agaricicola inhibits mycelium growth by one or more diffusible compounds. [13] In light of the economic importance of the mushroom disease, it is remarkable that the toxins and antifungal agents involved have yet remained elusive.
Figure 1.

The soft rot disease and involved chemical mediators and enzymes produced by B. gladioli pv. agaricicola. A) Schematic representation of different traits of soft rot disease. B) Metabolic potential encoded in the genome of B. gladioli pv. agaricicola. BGC, biosynthetic gene cluster; NRP, nonribosomal peptide; PK, polyketide; T, terpene; Mb, mega base pairs.
Here we report a multimodal and complementary approach that combines metabolic profiling and label‐free imaging techniques to detect, identify, and visualize virulence factors involved in mushroom soft rot disease. Furthermore, we investigate the roles of these chemical mediators in vitro and in vivo using isolated compounds and targeted null mutants, respectively. Finally, we shed light on the distribution of the corresponding gene clusters in other ecological contexts of B. gladioli.
Results and Discussion
Genome mining uncovers biosynthetic potential
To unravel the full metabolic potential of B. gladioli pv. agaricicola, we mined its genome sequence for biosynthetic gene clusters (BGCs) using antiSMASH 5.0 and the basic local alignment search tool (BLAST) (Figure 1B). [22] In addition to the previously characterized BGCs for the non‐ribosomal peptide synthetase (NRPS)‐derived lipopeptides haereogladin (1) and burriogladin (2), mediating swarming and biofilm formation, [19] as well as the siderophore gladiobactin (3), [23] we found a BGC for the swarming inhibiting, diacylated lipopeptide icosalide A1 (4). [24] We also detected two BGCs coding for polyketide synthases (PKS) that could assemble the antifungal gladiofungin A (syn. gladiostatin) (5)[ 23 , 25 ] and the antimicrobial enacyloxins (6). [26] Furthermore, the genome comprises PKS‐ and NRPS‐independent gene loci for the biosynthesis of the fatty acid derived, antifungal polyyne caryoynencin (7), [27] the antifungal isothiocyanate sinapigladioside (8), [28] and the antibiotic toxoflavin (9). [29] Taken together, the soft rot pathogen has a remarkably high and diverse biosynthetic potential.
Metabolic profiling and multimodal imaging of virulence factors
To gain insight into the chemical mediators that are actually produced during the infection process, we employed three complementary approaches; a) metabolic profiling based on high‐performance liquid chromatography coupled to mass spectrometry (HPLC‐MS), b) matrix‐assisted laser desorption ionization (MALDI)‐ based imaging‐MS, and c) ultraviolet (UV) Raman‐based microspectroscopy (Figure 2A). Comparison of HPLC‐MS profiles of crude extracts of infected and healthy mushrooms with authentic reference compounds identified haereogladin A (1, m/z 859.3882 [M+H]+), burriogladin A (2, m/z 961.5040 [M+H]+), icosalide A1 (4, m/z 711.4550 [M−H]−), gladiofungin A (5, m/z 504.2606 [M−H]−), sinapigladioside (8, m/z 468.1342 [M−H]−) and caryoynencin (7, m/z 279.1032 [M−H]−), albeit only in trace amounts (Figure 2B). Gladiobactin (3) and toxoflavin (9) were not detected by this method.
Figure 2.

Multimodal identification of virulence factors involved in the soft rot disease. A) Workflow of B) organic phase extraction and LC–MS analysis (extracted ion chromatograms of crude extract of infected (red) and non‐infected (black) mushroom), and C) imaging of infected mushroom slices. D) MALDI imaging of infected mushroom tissue, visualization of m/z 194 Da [M+H]+(right). E) DFT calculated and measured UV Raman spectra of 7. Measurements carried out on mushrooms slices: non‐infected (black), infected (blue/red). F) Box plots of the area under the curve of the Raman spectral band region associated with 7 (2,120–2,220 cm−1) of infected mushroom tissue at different time points. Each box plot shows the distribution of integrated Raman intensities of three subsequent measurements of mushroom slices at various incubation times.
To detect the predicted metabolites as well as potentially cryptic mediators in infected tissue, we performed multimodal imaging methods. These approaches could unearth instable metabolites or compounds that are only locally concentrated at the host‐pathogen interface that may evade detection after extraction and workup (Figure 2C). First, we investigated infected mushroom tissue by MALDI imaging, which has been proven to be a valuable method to determine the spatial distribution of metabolites in their biological contexts. [30] Therefore, we infected slices of A. bisporus fruiting bodies and scanned for metabolites using an established MALDI imaging setup.[ 19 , 31 ] In this way we were able to detect toxoflavin (9, m/z 194 [M+H]+) in infected tissue. This toxin seems to be accumulated locally at the infection hotspots and is not ubiquitously distributed throughout the entire mushroom tissue (Figure 2D). This spatial production pattern leads to a low overall concentration when extracting the entire biomass and may explain why 9 was initially not detected by HPLC‐MS. When analyzing a ten‐fold higher amount of infected mushroom we were able to detect 9 by HPLC‐MS, too (Figure S2).
Notably, other antifungal agents namely gladiofungin A (5), caryoynencin (7), and sinapigladioside (8) were not detected by MALDI imaging, likely because these compounds are too reactive or instable. To this end, both HPLC‐MS and MALDI imaging may not adequately represent the involvement of polyyne 7 in soft rot disease.
Thus, we next turned to a complementary optical imaging method. Serendipitously, the four conjugated C−C triple bonds of 7 that confer its instability represent a rare structural motif ideally suited for detection by Raman spectroscopy. This method is based on inelastic light scattering involving the vibrational modes of the chemical bonds of the specimen. Raman spectroscopy can be performed on a sample in situ without the risk of introducing artifacts due to sample preparations. [32] Therefore, it is especially well‐suited to detect highly polarizable units like the four conjugated C−C triple bonds of 7.
The usage of Raman excitation wavelengths in the deep UV at 244 nm offers the advantage to record Raman spectra free of a fluorescence background with a high sensitivity, i. e. detection limit. [33] Furthermore, C−C triple bonds show Raman peaks in the so‐called wavenumber silent region, i. e. are not influenced by Raman signals of the biological matrix (Figure 2E). [34]
Density functional theory (DFT) calculations allowed us to postulate Raman bands originating from 7 (Figure 2E and supplementary information). Raman spectra of samples containing 7 showed characteristic features in the wavenumber region between 2,250 cm−1 and 2,050 cm−1. As these values match the DFT calculations and appear in a wavenumber region where typically C−C triple bonds appear, we considered them as fingerprint marker bands associated with 7. In contrast to healthy mushroom tissue (Figure 2E, black), two zones of infected mushroom tissue, zone I with strongly degraded, yellowish tissue (blue), and zone II with slightly degraded mushroom tissue (red), showed these bands. To corroborate these findings, we monitored the production of 7 in the course of the infection. The calculated area under the curve of the C−C triple fingerprint bands (2,120–2,220 cm−1) of infected mushrooms revealed a signal after 16 h, which is even more prominent at 24 h past infection (Figure 2F). Measurements at later time points were not feasible due to progressive degradation of mushroom tissue. Taken together, these Raman spectroscopic data unequivocally show that the polyyne 7 is produced in the course of soft rot disease.
Evaluation of the antifungal potential in vitro and in vivo
Having established that the antifungals 5, 7, 8, and 9 are produced during infection, we tested whether these compounds can actually damage mushroom tissue. Therefore, we exposed mushroom slices to pure samples of 7, 8 and 9 independently for 48 hours (Figure 3A). Notably, compound 5 could not be assayed in this way as it can only be dissolved in organic solvents that damage the mushroom tissue (Figure S3A). Since 7 cannot be isolated as pure compound as it polymerizes during concentration, [35] we employed an extract with enriched 7, and polymerization was avoided as the samples were not concentrated to complete dryness. [27b] In this assay we optically evaluated damage to mushroom tissue, which is indicated by browning due to the formation of melanin pigments triggered by polyphenol oxidases and tyrosinases. [36] As a positive control, we used tolaasin I, which causes brown spots on mushroom caps. [7] Crude extract containing 7 showed no effect on the mushroom slices, yet rapid degradation of the toxin could be observed. Mild to strong browning of mushroom slices treated with 8 or 9, respectively, indicates that these compounds effectively damage mushroom tissue and thus could promote the bacterial infection.
Figure 3.

Investigation of roles of secondary metabolites in the soft rot disease. A) 10 μL of purified 9 and 8, a crude extract of 7, tolaasin I (all 500 μg mL−1), and water were spotted onto mushroom slices and incubated for 48 hours at 30 °C. Brown spots indicate lesions. B) Exemplary photos of A. bisporus mycelium grown on agar plates with the indicated additives and C) area of mycelium grown; Tebu, tebuconazol; DMSO, dimethyl sulfoxide; PDB, potato dextrose broth extract. An asterisk marks significant results (Statistic standard student t‐test; p‐value <0.05; two‐tail). D) Extracted ion chromatogram of metabolic profiles of B. gladioli pv. agaricicola wild type (WT) and knockout strains; ▵spg, ▵cay, ▵tox, null producers of 8, 7 and 9, respectively. E) Infection assay of mushroom slices with B. gladioli pv. agaricicola wild type and indicated knockout strains as well as a caryoynencin‐KO complemented with 5 μL (2 μg μL−1) of a crude extract enriched with 7.
In addition to destructing mushroom caps, B. gladioli pv. agaricicola was shown to inhibit mycelium growth. [13] To identify the causative agent we evaluated the effect of the antifungals 7–9 on fungal mycelium (Figure 3B). Using tebuconazole as a positive control, [37] DMSO as solvent control, and PDB as media control we measured the effect of these compounds on the growth of A. bisporus mycelium on agar plates.
We observed a significantly reduced mycelium growth (p‐value <0.05) in response to 8, 9, and 7, whereas solvent and media controls did not show any signs of inhibition (Figure 3C). Compound 7 decomposed in the course of this experiment and might be more active than anticipated by these results. However, 8 shows the most potent inhibition of mycelium growth, at the same range as tebuconazole. The amount of 8 from infected mushroom is ∼3.3 μg mL–1 and thus in the same range as the concentration used in the assay. In conclusion, 8 represents a strongly antifungal agent that may be identical with the elusive diffusible agent responsible for the reported mycelium inhibition. [13] Testing of several combinations of bacterial toxins showed no synergistic effect among the antifungals.
To study the roles of the identified metabolites in vivo, we generated targeted null mutants lacking the sinapigladioside (▵spg), toxoflavin (▵tox), caryoynencin (▵cay), icosalide (▵ico), and gladiofungin (▵gla) BGCs. Mutants of B. gladioli pv. agaricicola were constructed by means of chromosomal homologous recombination, using suicide plasmids with a kanamycin resistance cassette flanked by two sequence tags homologous to the target gene. Homologous recombination of the chromosomal allele with the sequence tags of the plasmid resulted in an interruption of the target gene due to the inserted resistance marker. The successful recombination was confirmed by means of colony PCR and metabolic profiling (Figure 3D). We individually infected mushroom slices with the null producer strains (▵spg, ▵tox, ▵ico, ▵gla, ▵cay) and the wild type, and compared the resulting phenotypes. After incubation at 30 °C for two days, the wild type and the ▵spg, ▵tox, ▵ico, and ▵gla mutant strains showed typical signs of tissue degradation (Figure 3E). Only the ▵cay mutant did not show marked disease symptoms. Chemical complementation of the ▵cay mutant with 7, however, did not fully restore the phenotype caused by the wild type. This observation may be rationalized by the instability of the polyyne over time. Furthermore, upon closer inspection of all ▵cay mutants we noted a reduced growth rate and final cell density compared to the wild type and other mutants, which could account for the reduced mushroom tissue degradation. Judging from the mushroom infection assays it seems that the absence of one of the toxins, with the exception of 7, does not impede the progression of the disease. However, as the isolated metabolites 8 and 9 cause damage of the mushroom tissue, a redundant activity is conceivable, yet unexpected as the tox, [29] cay,[ 27b , 35 ] spg [28a] and gla [23] gene clusters have been originally described separately, in plant pathogenic or beetle symbiotic Burkholderia strains. To discover them combined in a fungal pathogen prompted us to investigate the distribution of the BGC in other Burkholderia strains to evaluate whether this combination of metabolites provide advantages in different ecological niches.
Niche‐dependent armory of antifungals and toxins
To learn more about the distribution of these gene clusters in Burkholderia spp. inhabiting diverse niches, we created a 16S rRNA gene‐based phylogenetic tree of Burkholderia spp. complemented with BGCs encoded in the genomes of the respective strains (Figure 4A). We found the tox and cay gene clusters in Burkholderia spp. that are human‐pathogenic (B. mallei and pseudomallei), plant pathogens or plant‐associated (B. glumae BGR1 and B. gladioli pv. gladioli), associated to fungi or mushroom pathogen (B. gladioli pv. cocovenenans and B. gladioli pv. agaricicola, respectively). Only the genome of B. gladioli Lh StG, a beetle symbiont, bears the same identified biosynthetic gene clusters (tox, cay, spg and gla).[ 28a , 38 ]
Figure 4.
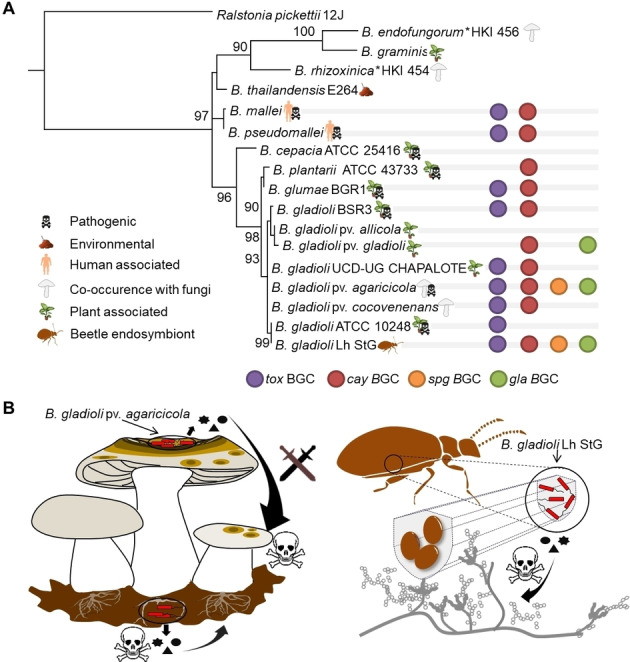
Genetic potential to produce antifungals in related Burkholderia spp. occupying diverse ecological niches. A) 16S rRNA gene‐based phylogenetic tree of Burkholderia spp. from diverse ecological niches. Distribution of gene clusters involved in biosynthesis of 5, 7, 8 and 9 are indicated. Ralstonia picketii 12 J was used as an outgroup; asterisk indicates Burkholderia spp. that have been renamed to Mycetohabitans spp. B) Ecological niches of B. gladioli pv. agaricicola degrading mushroom tissue and inhibiting mycelium growth and B. gladioli Lh StG protecting beetle eggs from fungal pathogens.
B. gladioli Lh StG and B. gladioli pv. agaricicola frequently encounter fungal organisms in their ecological niche. While the toxin mix is used offensively by B. gladioli pv. agaricicola to infect a mushroom host, B. gladioli Lh StG uses the same mediators to defend its habitat, the insect host's eggs (Figure 4B), [28a] in addition to an antifungal (lagriamide) from a yet unculturable symbiont. [39] A plausible explanation of the similar biosynthetic reservoir is the observed dynamic transition between plant pathogenicity and insect‐defensive mutualism of beetle‐associated B. gladioli. [28a]
Conclusion
The aim of this study was to shed light on the bacterial virulence factors involved in mushroom soft rot disease. An important lesson learned is that a multimodal approach, the combination of metabolic profiling and two different imaging techniques, was crucial to identify the blend of chemical mediators involved in the infection process. UV Raman spectroscopy and MALDI‐imaging visualized metabolites that are prone to degradation during work‐up, such as caryoynencin (7), or that are concentrated locally, such as toxoflavin (9), and thus would evade detection by routine HPLC analysis. To avoid overlooking chemical mediators or misjudging their ecological role, it may be beneficial to employ a suite of complementary analytical methods. Raman imaging proved to be particularly suitable for the invasion‐ and label‐free monitoring of instable polyyne virulence factors. This study exemplifies how metabolic profiling and multimodal imaging chemical mediators could be used to investigate the multitude of crops diseases caused by different soft‐rot causing species. [40]
From ecological and evolutionary points of view, it is remarkable that the identified antifungal agents and toxins have previously been reported as players in different host‐microbe interactions. Using pure compounds and genetically engineered null mutants we uncovered new roles of these compounds in the context of soft rot disease. Caryoynencin (7) has first been reported as a metabolite from the bacterial plant‐pathogen B. caryophylli. [27b] Whereas we were able to unequivocally monitor the formation of this potent cytotoxin during the infection process, assays with the pure compound were hampered because of its inherent instability, and the biological effect may be stronger than what we observed in the mushroom browning assay. Toxoflavin (9) has been known as key pathogenicity factor in rice grain rot, [41] we now show a new role of 9 in the context of soft rot. Melanization (mushroom browning), a known response of fungi upon oxidative stresses, [42] may be rationalized by the generation of hydrogen peroxide triggered by 9. [43] Gladiofungin A (5) [24] and sinapigladioside (8) [28a] were originally described as antifungals from beetle symbiotic bacteria with potential egg‐protective functions. Here, we identified 8 as the long sought‐after inhibitor of mycelium growth. [20]
The broad range of antifungals produced by the soft rot pathogen is astonishing. As individual null mutants do not show markedly altered infection phenotypes in mushroom assays, it is possible that the effect is overridden by the excretion of lytic enzymes. Furthermore, it is conceivable that the various toxins complement each other. Even so, using a diverse set of antifungals to attack a host is somewhat reminiscent of combination therapy and prophylaxis approaches. In nature, such a strategy is employed, for example, in the Streptomyces‐beewolf symbiosis where a mix of antibiotic substances produced by the bacterial symbionts enhances the survival probability of the wasp's offspring. [44] The production of a toxin blend could be an evolutionary strategy to prevent the development of host resistance, since a pathogen or host is less likely to develop resistance to multiple metabolites simultaneously.
Genomic and metabolic comparisons showed that both the mushroom pathogen and the beetle symbionts are equipped with the same set of antifungals and toxins. The occurrence of these toxin biosynthetic genes in two bacteria from different ecological backgrounds indicates that this biosynthetic potential evolved in an ancestor bacterium in response to antagonistic interactions with fungi. [45] As the descendants were armored with a potent mix of toxins, they could occupy ecological niches that seem distinct at first but share a decisive feature: the bacteria‐fungi interaction. This is an example of secondary metabolites making ecological niches accessible to the producer.
Finally, knowledge on the toxins involved in the infection process further deepens our understanding of soft rot disease and might aid future infection control strategies. It is well conceivable to block the biosynthetic pathways leading to the pathogenicity factors. One could also envisage the use of beneficial microorganisms with the ability to degrade virulence factors. [46] Such approaches might be useful in mushroom farming, especially as the use of conventional pesticides is restricted or affects the indispensable commensal microbial community. [47] Such measures that target the chemical mediators may be promising alternatives to decrease the number of outbreaks of soft rot disease and thus reduce considerable losses in mushroom farming.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank A. Perner for MS/MS measurements. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) SFB 1127/2 ChemBioSys – 239748522 and Leibniz Prize, and InfectoOptics (iTag). M.G.‐A. acknowledges financial support from the European Research Council for a Marie Skłodowska‐Curie Individual Fellowship (IF‐EF) Project reference 700036. Open Access funding enabled and organized by Projekt DEAL.
B. Dose, T. Thongkongkaew, D. Zopf, H. J. Kim, E. V. Bratovanov, M. García-Altares, K. Scherlach, J. Kumpfmüller, C. Ross, R. Hermenau, S. Niehs, A. Silge, J. Hniopek, M. Schmitt, J. Popp, C. Hertweck, ChemBioChem 2021, 22, 2901.
Contributor Information
Prof. Dr. Jürgen Popp, Email: juergen.popp@uni-jena.de, Email: juergen.popp@leibniz-ipht.de.
Prof. Dr. Christian Hertweck, Email: christian.hertweck@leibniz-hki.de, Email: christian.hertweck@uni-jena.de.
References
- 1. Wiersinga W. J., Currie B. J., Peacock S. J., N. Engl. J. Med. 2012, 367, 1035–1044. [DOI] [PubMed] [Google Scholar]
- 2. Mahenthiralingam E., Urban T. A., Goldberg J. B., Nat. Rev. Microbiol. 2005, 3, 144–156. [DOI] [PubMed] [Google Scholar]
- 3. Angus A. A., Agapakis C. M., Fong S., Yerrapragada S., Estrada-de Los Santos P., Yang P., Song N., Kano S., Caballero-Mellado J., De Faria S. M., PLoS One 2014, 9, e83779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Largeteau M. L., Savoie J. M., Appl. Biochem. Biotechnol. 2010, 86, 63–73. [DOI] [PubMed] [Google Scholar]
- 5. Scherlach K., Graupner K., Hertweck C., Annu. Rev. Microbiol. 2013, 67, 375–397. [DOI] [PubMed] [Google Scholar]
- 6. Jones C., Webster G., Mullins A. J., Jenner M., Bull M. J., Dashti Y., Spilker T., Parkhill J., Connor T. R., LiPuma J. J., Microb. Genom. 2021, 7, mgen000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soler-Rivas C., Arpin N., Olivier J. M., Wichers H. J., Physiol. Mol. Plant Pathol. 1999, 55, 21–28. [Google Scholar]
- 8.
- 8a. Royse D. J., Baars J., Tan Q., in Edible and Medicinal Mushrooms, Wiley, 2017, pp. 5–13; [Google Scholar]
- 8b. Leveau J. H. J., Preston G. M., New Phytol. 2008, 177, 859–876; [DOI] [PubMed] [Google Scholar]
- 8c. Zhang Y., Geng W., Shen Y., Wang Y., Dai Y. C., Sustainability 2014, 6, 2961–2973. [Google Scholar]
- 9. Soković M., van Griensven L. J. L. D., Eur. J. Plant Pathol. 2006, 116, 211–224. [Google Scholar]
- 10. Gill W. M., Cole A. L. J., Can. J. Microbiol. 1992, 38, 394–397. [Google Scholar]
- 11. Lincoln S. P., Fermor T. R., Stead D. E., Sellwood J. E., Plant Pathol. 1991, 40, 136–144. [Google Scholar]
- 12. Lee C. J., Yun H. S., Jhune C. S., Cheong J. C., Yoo Y. B., J. Plant Pathol. 2010, 92, 235–240. [Google Scholar]
- 13. Gill W. M., Tsuneda A., Can. J. Microbiol. 1997, 43, 639–648. [Google Scholar]
- 14. Pu X. M., Zhou J. N., Lin B. R., Shen H. F., Plant Dis. 2012, 96, 1818–1818. [DOI] [PubMed] [Google Scholar]
- 15. Lu S. E., Henn R. A., Nagel D. H., Plant Dis. 2007, 91, 1514–1514. [DOI] [PubMed] [Google Scholar]
- 16. Wu W. X., Liu Y., Huang X. Q., Zhang L., Plant Dis. 2017, 101, 1540. [Google Scholar]
- 17. Liu Q., Xiao W., Wu Z., Li S., Yuan Y., Li H., J. Plant Pathol. 2016, 98, 503–510. [Google Scholar]
- 18. Végh A., Némethy Z., Salamon P., Mándoki Z., Palkovics L., Plant Dis. 2014, 98, 988–988. [DOI] [PubMed] [Google Scholar]
- 19. Thongkongkaew T., Ding W., Bratovanov E., Oueis E., García-Altares M., Zaburannyi N., Harmrolfs K., Zhang Y., Scherlach K., Müller R., Hertweck C., ACS Chem. Biol. 2018, 13, 1370–1379. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Chowdhury P. R., Heinemann J. A., Appl. Environ. Microbiol. 2006, 72, 3558–3565; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Vial L., Groleau M. C., Dekimpe V., Déziel E., J. Microbiol. Biotechnol. 2007, 17, 1407–1429. [PubMed] [Google Scholar]
- 21.
- 21a. Fogliano V., Ballio A., Gallo M., Woo S., Scala F., Lorito M., Mol. Plant-Microbe Interact. 2002, 15, 323–333; [DOI] [PubMed] [Google Scholar]
- 21b. Someya N., Tsuchiya K., Yoshida T., Noguchi M. T., Akutsu K., Sawada H., Biocontrol Sci. 2007, 12, 1–6. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S. Y., Medema M. H., Weber T., Nucleic Acids Res. 2019, 47, W81-W87; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., J. Mol. Biol. 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 23. Hermenau R., Mehl J. L., Ishida K., Dose B., Pidot S. J., Stinear T. P., Hertweck C., Angew. Chem. Int. Ed. 2019, 58, 13024–13029; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13158–13163. [Google Scholar]
- 24. Dose B., Niehs S. P., Scherlach K., Flórez L. V., Kaltenpoth M., Hertweck C., ACS Chem. Biol. 2018, 13, 2414–2420. [DOI] [PubMed] [Google Scholar]
- 25. Challis G., Nakou I. T., Jenner M., Dashti Y., Romero-Canelón I., Masschelein J., Mahenthiralingam E., Angew. Chem. Int. Ed. 2020, 51, 23345–23353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Mahenthiralingam E., Song L., Sass A., White J., Wilmot C., Marchbank A., Boaisha O., Paine J., Knight D., Challis G. L., Chem. Biol. 2011, 18, 665–677; [DOI] [PubMed] [Google Scholar]
- 26b. Ross C., Opel V., Scherlach K., Hertweck C., Mycoses 2014, 57, 48–55. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Yamaguchi M., Park H. J., Ishizuka S., Omata K., Hirama M., J. Med. Chem. 1995, 38, 5015–5022; [DOI] [PubMed] [Google Scholar]
- 27b. Kusumi T., Ohtani I., Nishiyama K., Kakisawa H., Tetrahedron Lett. 1987, 28, 3981–3984. [Google Scholar]
- 28.
- 28a. Flórez L. V., Scherlach K., Gaube P., Ross C., Sitte E., Hermes C., Rodrigues A., Hertweck C., Kaltenpoth M., Nat. Commun. 2017, 8, 15172; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Dose B., Niehs S. P., Scherlach K., Shahda S., Flórez L. V., Kaltenpoth M., Hertweck C., ChemBioChem 2021, 22, 1920–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Nagamatsu T., Yamasaki H., Hirota T., Yamato M., Kido Y., Shibata M., Yoneda F., Chem. Pharm. Bull. 1993, 41, 362–368; [DOI] [PubMed] [Google Scholar]
- 29b. Latuasan H. E., Berends W., Biochim. Biophys. Acta 1961, 52, 502–508. [DOI] [PubMed] [Google Scholar]
- 30. Esquenazi E., Yang Y. L., Watrous J., Gerwick W. H., Dorrestein P. C., Nat. Prod. Rep. 2009, 26, 1521–1534. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Graupner K., Scherlach K., Bretschneider T., Lackner G., Roth M., Gross H., Hertweck C., Angew. Chem. Int. Ed. 2012, 51, 13173–13177; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13350–13354; [Google Scholar]
- 31b. Scherlach K., Lackner G., Graupner K., Pidot S., Bretschneider T., Hertweck C., ChemBioChem. 2013, 14, 2439–2443. [DOI] [PubMed] [Google Scholar]
- 32. Krafft C., Schmitt M., Schie I. W., Cialla-May D., Matthäus C., Bocklitz T., Popp J., Angew. Chem. Int. Ed. 2017, 56, 4392–4430; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4458–4500. [Google Scholar]
- 33. Zukovskaja O., Kloss S., Blango M. G., Ryabchykov O., Kniemeyer O., Brakhage A. A., Bocklitz T. W., Cialla-May D., Weber K., Popp J., Anal. Chem. 2018, 90, 8912–8918. [DOI] [PubMed] [Google Scholar]
- 34. Azemtsop Matanfack G., Rüger J., Stiebing C., Schmitt M., Popp J., J. Biophotonics 2020, 13, e202000129. [DOI] [PubMed] [Google Scholar]
- 35. Ross C., Scherlach K., Kloss F., Hertweck C., Angew. Chem. Int. Ed. 2014, 53, 7794–7798; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7928–7932. [Google Scholar]
- 36. Jolivet S., Arpin N., Wichers H. J., Pellon G., Mycol. Res. 1998, 102, 1459–1483. [Google Scholar]
- 37. Chalaux N., Savoie J. M., Olivier J. M., Agronomie 1993, 13, 407–412. [Google Scholar]
- 38. Waterworth S. C., Flórez L. V., Rees E. R., Hertweck C., Kaltenpoth M., Kwan J. C., mBio 2020, 11, e02430–02419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Flórez L. V., Scherlach K., Miller I. J., Rodrigues A., Kwan J. C., Hertweck C., Kaltenpoth M., Nat. Commun. 2018, 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Charkowski A. O., Annu. Rev. Phytopathol. 2018, 56, 269–288. [DOI] [PubMed] [Google Scholar]
- 41. Jeong Y., Kim J., Kim S., Kang Y., Nagamatsu T., Hwang I., Plant Dis. 2003, 87, 890–895. [DOI] [PubMed] [Google Scholar]
- 42. Cordero R. J. B., Casadevall A., Fungal Biol. Rev. 2017, 31, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J., Park J., Kim S., Park I., Seo Y. S., Mol. Plant Pathol. 2016, 17, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.
- 44a. Kaltenpoth M., Göttler W., Herzner G., Strohm E., Curr. Biol. 2005, 15, 475–479; [DOI] [PubMed] [Google Scholar]
- 44b. Koehler S., Doubský J., Kaltenpoth M., Front. Zool. 2013, 10, 1–13; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44c. Kroiss J., Kaltenpoth M., Schneider B., Schwinger M.-G., Hertweck C., Maddula R. K., Strohm E., Svatoš A., Nat. Chem. Biol. 2010, 6, 261–263. [DOI] [PubMed] [Google Scholar]
- 45. Scherlach K., Hertweck C., Annu. Rev. Microbiol. 2020, 74, 267–290. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Tsukamoto T., Murata H., Shirata A., Biosci. Biotechnol. Biochem. 2002, 66, 2201–2208; [DOI] [PubMed] [Google Scholar]
- 46b. Hermenau R., Kugel S., Komor A. J., Hertweck C., Proc. Natl. Acad. Sci. USA 2020, 117, 23802–23806; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46c. Jung W. S., Lee J., Kim M. I., Ma J., Nagamatsu T., Goo E., Kim H., Hwang I., Han J., Rhee S., PLoS One 2011, 6, e22443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tarkka M. T., Sarniguet A., Frey-Klett P., Curr. Genet. 2009, 55, 233–243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information