

Abstract
The xCT antiporter is a cell membrane protein involved in active counter‐transportation of glutamate (outflux) with cystine (influx) over the human cell membrane. This feature makes the xCT antiporter a crucial element of the biosynthesis of the vital free radical scavenger glutathione. The prodrug sulfasalazine, a medication for the treatment of ulcerative colitis, was previously proven to inhibit the xCT antiporter. Starting from sulfasalazine, a molecular scaffold jumping followed by SAR‐assisted design and synthesis provided a series of styryl hydroxy‐benzoic acid analogues that were biologically tested in vitro for their ability to decrease intracellular glutathione levels using four different cancer cell lines: A172 (glioma), A375 (melanoma), U87 (glioma) and MCF7 (breast carcinoma). Depletion of glutathione levels varied among the compounds as well as among the cell lines. Flow cytometry using propidium iodide and the annexin V marker demonstrated minimal toxicity in normal human astrocytes for a promising candidate molecule (E)‐5‐(2‐([1,1′‐biphenyl]‐4‐yl)vinyl)‐2‐hydroxybenzoic acid.
Keywords: Heck cross-coupling, 2-hydroxy-5-styrylbenzoic acid, SAR, xCT antiporter inhibitor, glioblastoma
Glutathione is one of the cells major protective means against oxidative stress. The biosynthesis of glutathione use cystine as substrate that is transported into the cell by the xCT antiporter. Sulfasalazine was known to be an xCT antiporter inhibitor and was used as a lead compound. The team performed scaffold hopping, molecular moiety truncations, and bioisostere alteration to synthesize a library of novel xCT antiport inhibitor candidates that was evaluated in vitro for four various cell lines with various xCT expression.

Introduction
The cystine‐glutamate transporter known as the Xc − or xCT system is an antiporter that is an essential membrane protein involved in active transport of two distinct amino acids across the phospholipid membrane of the cell. [1] The counter‐transporter xCT carries cystine into the cell concomitantly as glutamate is passed out of the cell. The xCT system regulates synaptic activity by stimulating extra‐synaptic receptors and performs non‐vesicular (extracellular) glutamate release. The influx of cystine provides the substrate for the intracellular biosynthesis of L‐cysteine, which in subsequent biosynthetic steps deliver the antioxidant glutathione, Figure 1.
Figure 1.

(a) Biosynthesis of glutathione is dependent on the xCT antiporter, a function that is proven to be inhibited by sulfasalazine. (b) Glutathione operates as an antioxidant for ROS, scavenging free radical generated from oxidative stress as well as for free radical generated by γ‐irradiation during radio therapy.
Glutathione is an essential antioxidant present in mammalian cells and is of paramount importance as a bioorganic free radical scavenger and constitutes a major intracellular defence system against oxidative stress, being induced chemically or by radiation, Figure 1(b). Previous studies have revealed that cancer cells produce the antioxidant glutathione at elevated rates compared to normal cells. [2] This up‐regulated glutathione production enables cancer cells to cope with the elevated free radical activity caused by tumour hypoxia (oxygen deficiency) as a consequence of tumours having grown out of their blood supplies. [3]
Sulfasalazine was previously reported to be an efficient inhibitor of the xCT antiporter, [4] and was therefore investigated as a radiosensitizer on various glioma cells in vitro and in human glioblastomas xenografted into nude rats, demonstrating that sulfasalazine effectively inhibited the biosynthesis of glutathione that protected cancer cells from reactive oxygene species induced by radiation. As such, sulfasalazine potentiated the effect of ionizing radiation leading to increased cancer cell death.[ 5 , 6 ]
Sulfasalazine was originally designed and developed as a prodrug to be orally administrated for the treatment of inflammatory bowel disease, rheumatoid arthritis, ulcerative colitis, and Crohn's disease. [7] When the sulfasalazine prodrug is orally administered, it is activated in the intestine by diazoreductases present in gut bacteria, which cleave the diazo bond present in the sulfasalazine producing sulfapyridine (M&B) and mesalazine (5‐ASA), [8] Scheme 1. Neither of the two metabolites possess xCT antiporter inhibitor activity. Most of the administered sulfasalazine is degraded in the intestines, limiting systemic uptake [9] and its clinical potential as an xCT inhibitor.
Scheme 1.

The prodrug sulfasalazine is ingested orally, whereupon it is metabolized by operation of diazo reductases on the central diazo bond forming sulfapyridine (M&B) and mesalazine (5‐ASA) as metabolites.
Moreover, although sulfasalazine is generally well tolerated, it is associated with toxicity in heavily pre‐treated cancer patients. Side effects bone marrow suppression (myelotoxicity), liver damage, [10] Stevens‐Johnson syndrome, [11] and kidney injury. [12]
Importantly these side effects, have been linked to the sulfapyridne group. [13] Thus, even if sulfasalazine demonstrates good performance as an inhibitor of the system xCT antiporter, several limitations related to properties and undesirable side effects makes sulfasalazine not usable as an xCT inhibitor for clinical use.
To address the limitations of sulfasalazine, we have in the present study devised and synthesised a series of styryl hydroxy‐benzoic acid analogues aided by the SAR approach.
Results and Discussion
SAR assisted design. Our ambition at the outset of this project was to develop an xCT antiporter inhibitor that possesses similar inhibitor characteristics as exhibited by sulfasalazine DC00, but the novel reagent should:
not contain the toxic sulfapyridine moiety
demonstrate low toxicity toward normal cells
possess metabolic stability
possess chemical properties consistent with penetrating ability through the wall of the gastrointestinal tract as well as capacity to penetrate the blood‐brain barrier
Previously, attempts have been made to design and synthesize new xCT antiport inhibitors using sulfasalazine DC00 as a lead compound. Tsukamoto and collaborators [14] synthesized a series of new xCT antiport inhibitor candidates, which unfortunately revealed weaker IC50 than parent xCT antiporter inhibitor sulfasalazine. Nandave and collaborators [15] developed and produced another series of potential xCT antiport inhibitor candidates, which however comprised the unsolicited diazo bond that will be cleaved in‐vivo by diazoreductases. These candidate inhibitor molecules revealed moreover deficient IC50 values compared to the parent inhibitor sulfasalazine DC00. Stockwell and collaborators [16] disclosed recently a study revealing erastin and some analogs to operate as xCT antiporter inhibitors, but were observed to be unstable in‐vivo.
We realized that in order to approach the goals 1–4 listed above, it was necessary to perform a molecular scaffold‐hopping [17] to get rid of the embedded degradable molecular moiety of sulfasalazine, namely the diazo bond. We also deduced that it was essential to retain the overall molecular structure shape. In order to get rid of the serious side effects associated with the systemic use (as with sulfasalazine), a “molecular moiety pruning” was needed, especially with regards to the sulfapyridine moiety. Alternatively, the sulfapyridine moiety could be replaced with a bio‐isostere if it should prove necessary to maintain or boost the xCT antiporter inhibitor activity.
Through these assessments and changes, we arrived at a simple molecular structure (E)‐2‐methoxy‐5‐styryl benzoic acid DC01, that we assumed could represent an early lead compound that we subsequently aimed to optimize with respect to the xCT antiport inhibition ability, Figure 2. For this undertaking, the approach of structure activity relationship (SAR) was used.
Figure 2.

Sketch of the design pathway that includes scaffold hopping •, pruning of molecular moieties •, and interchange of bio‐isosteres •.
The compounds that were synthesized (shown in Figure 3) were successively submitted for in vitro testing versus a panel constituted by four different cancer cell lines.
Figure 3.

Compound library produced by means of SAR assisted design and synthesized. All the compounds were used successively to treat a panel of cell lines composed of: A172, A375, MCF7, and U87.
The glutathione (GSH) levels [%] obtained after treatment of the four cell lines with each of the inhibitor candidates DC00–DC21 at two different concentrations (doses at 250 μM and 500 μM) are graphically portrayed in Figure 4. This graphic shows that the synthesized compounds cause a large variation with respect to the xCT antiport inhibitor ability, which is reflected by the large variation of the glutathione level.
Figure 4.

All the candidate inhibitor compounds of the compound library (DC00–DC21) were successively used to treat a panel of four different cancer cell lines: A172, A375, MCF7, and U87. Each compound (DC00–DC21) was assessed at two different concentrations (doses) 250 μM and 500 μM).
Consistent with the role of glutathione as a principal scavenger of reactive oxygen species in mammalian cells, it is known that reducing its levels increases oxidative stress [5] in cancer cells. However, ROS has a dual role in tumor development, as low ROS are pro‐tumorigenic whereas higher levels are cytotoxic. [18] Thus, elevated ROS does not trigger tumor cell death per se, but rather depends on the degree of increase. Importantly, ROS levels below the threshold for cytotoxicity, may still potentiate the anti‐tumor efficacy of treatments that induce ROS formation, such as radiotherapy
The compounds DC07, DC08, DC10, DC11, DC18, and DC21 shows moderate to high potency. Moreover, DC00 (our benchmarking molecule sulfasalazine) and DC14 show both high efficacies. The difference between these two molecular structures is that the DC00 containing a diazo bond while in DC14, the diazo bond is replaced by an olefin bond and the pyridine moiety of DC00 is replaced by a bioisostere (a phenyl group) in DC14).
Furthermore, the inhibitor candidate molecules DC01–DC05, DC13, and DC17 reveals negligible potency at a dose of 250 μM and low to moderate potency only at a dose of 500 μM.
Chemical Synthesis
The (E)‐stilbene scaffold bearing a hydroxyl group and a carboxylic acid group on one of the aromatic rings and constitutes a mutual structure scaffold for the majority of the devised molecules present in the produced molecular library, Figure 3.
A general synthetic route might involve protection of the phenol group, the formation of the (E)‐stilbene scaffold through C−C bond formation and finally the installation of the carboxylic acid functional group through one of two potential methods: i) by means of a Grignard salt formation and a carboxylation using CO2, [19] or ii) a Pd‐catalysed hydroxyl carbonylation. [20] Both of these methods can also be performed using 11C‐labelled CO2 or CO. [21] This was also an important part of this project, as we intended in a future project to examine one or more of the most promising xCT inhibitor candidates as PET tracers in animal models.
Synthesis of the (E)‐stilbene scaffold
The synthesis of the (E)‐stilbene scaffold was investigated by means of three distinct methods: the Wittig reaction, [22] the Heck cross‐coupling, [23] and the oxidative Heck cross‐coupling reaction. [24] Experimental conditions and other details about the three methods together with obtained results are summarized in Figure 5.
Figure 5.

Comparison of the three methods used for the synthesis of the styryl benzoic acid scaffold.
The Wittig reaction provided a quantitative conversion under mild reaction conditions, but both of the E and Z isomers were formed (in a ratio E : Z≈60 : 40). Silica gel column chromatography appeared as an ineffective method to separate the two isomers, and the I2‐catalyzed isomerisation [25] was fruitless too. Because of the laborious purification that was needed, the method that involved the Wittig reaction was discarded as a feasible method to proceed with the SAR study.
The method based on the Heck cross‐coupling reaction afford an excellent E‐Z selectivity, in fact the Z isomer was never observed. Furthermore, the reagent needed for the coupling is simply either an iodo‐ or a vinyl‐aromatic compound; whereas for the Wittig reaction, the formation of a phosphonium salt was needed beforehand.
The Heck cross‐coupling presented another selectivity issue, namely the formation of both the 1,1 and the 1,2 regioisomers. Nevertheless, the undesired by‐product was provided in yields (GC) of 5–15 %. Given the similar chemical structure, the separation of the two isomers were carried through by means of column chromatography.
Finally, with the aim to overcome the drawbacks of the Heck cross‐coupling method, a new method was developed.
A novel method for oxidative Heck cross‐coupling
An oxidative Heck cross‐coupling is a Pd‐catalysed reaction that involves a terminal olefin and an aromatic boronic acid with an oxidant present. The aromatic boronic acid and the oxidant replaces the aryl halide and the base that is used in the classical Heck cross‐coupling. In the present investigation, the oxidative Heck cross‐coupling was proven to produce, under mild conditions (20 °C), target molecular scaffold in good yields (>70 %). Even though full substrate conversion was achieved for all three methods, only the oxidative Heck cross‐coupling method was successfully performed under mild conditions without formation of any by‐product.
Synthesis of DC01
The synthetic route leading to (E)‐2‐methoxy‐5‐styrylbenzoic acid DC01 start with a bromination of 4‐hydroxybenzaldehyde to obtain 3‐bromo‐4‐hydroxybenzaldehyde 1 that is methylated giving 2 a. A Wittig reaction using 2 a and the Witting salt 3 was then performed. The ultimate reaction step comprised a carbonylation, see Scheme 2.
Scheme 2.

Synthesis of DC01. Method i: a) Mg, 1,2‐dibromoethane. THF. 60 °C, 18 h. b) CO2, 10 min. c) H3O+.
Synthesis of DC02, DC07, DC08, DC13 and DC14
The syntheses of compounds DC02, DC07, DC08, DC13 and DC14 were conducted by means of a Heck cross‐coupling reaction, see Scheme 3. The methodology employs aryl iodides (compounds 7 b and 18) with palladium catalysis and the base potassium carbonate with microwave irradiation to arylate the desired styrene reagents. The corresponding products were submitted for a hydroxyl carbonylation reaction that installed the carboxylic acid group to give the title molecules.
Scheme 3.

Syntheses of ① DC02, DC07, and DC08, ② DC13, and ③ DC14 that involves a Heck cross‐coupling reaction. Method ii: a) Oxalic acid, Ac2O, TEA, Pd(OAc)2, Xantphos, H2O (10 equiv.), DMF, 100 °C, 3 h. b) H3O+.
Synthesis of DC10, DC15, DC16, DC18, DC19 and DC21
Syntheses of DC10, DC15, DC16, DC18, DC19, and DC21, Scheme 4, demanded new method development, namely an adapted oxidative Heck cross‐coupling method. The olefins 12, 21 and 24 were functionalized with desired boronic acid, to afford the corresponding stilbene derivatives, which were submitted for hydroxyl carbonylation, pathways ① and ③ of Scheme 4, or the tetrazole ring formation, target molecule DC15, pathway ② of Scheme 4.
Scheme 4.

Syntheses of ① DC10, DC18, DC19, and DC21 ② DC15, and ③ DC16.
Synthesis of DC03, DC04 and DC05
The syntheses of the derivatives without the olefin bridge, different approaches to join the two aromatic rings were required, see Scheme 5. DC03, DC04 and DC05 were all prepared by using 7 a as substrate. is obtained by bromination and methylation of 4‐iodophenol. When compound 7 a underwent a Sonogashira reaction to obtain compound 8. The latter was either carboxylated to obtain DC03, or first reduced and then carboxylated to achieve DC04.
Scheme 5.

Syntheses of ① DC03‐DC05, ② DC11, ③ DC12, ④ DC17 and ⑤ DC20.
DC05 was obtained by subjecting compound 7 a for a Suzuki cross‐coupling reaction to obtain intermediate 10, which in the following step was converted into a Grignard reacgent that was carboxylated.
Synthesis of DC11, DC12, DC17 and DC20
DC11, DC17 and DC20 were obtained with the similar synthetic route that comprise a Suzuki cross‐coupling reaction with the respective MOM protected derivatives (7 b or 31) followed by the Pd‐catalysed hydroxyl carbonylation. Finally, DC12 was achieved by a simple Suzuki reaction, Scheme 5.
The compound library DC00–DC21 (Figure 3), which was produced using SAR assisted design and synthesized were in‐vitro tested versus the cancer cell lines A172, A375, U87 and MCF7.
Biology
Assessing depletion of intracellular glutathione levels
Previously, we reported that sulfasalazine DC00 reduces intracellular glutathione levels thereby acting as a radiosensitizer that significantly prolonged survival when administered in combination with radiotherapy in a brain tumour animal model. [5] In this study, we have by means of SAR guided design and synthesis produced a series of potential inhibitors, Figure 3. Each molecule was screened for its ability to inhibit glutathione biosynthesis, using an assay measuring intracellular glutathione levels. This assay was chosen for validation since glutathione biosynthesis is a key element in the events leading to cancer cells radioresistance, [5] and therefore therapeutically relevant.
The biosynthesis of glutathione largely depends upon a rate‐limiting step of interchange of cystine with glutamate through a transmembrane xCT antiporter. Thus, inhibiting cystine uptake by xCT subsequently inhibits glutathione synthesis, which eventually leads to depletion of intracellular glutathione. This again increases oxidative stress and potentiates the effect of radiotherapy. Therefore, we explored xCT expression in a range of cancer cell lines, and representatives from the high, intermediate, and low xCT expressors were chosen for testing the analogues DC00–DC21. The effect of these analogues was compared against each other, as well as between the cell lines, Figure 4.
Notably, cancer cells with high xCT expression exhibited a higher degree of glutathione inhibition compared to cancer cells with lower xCT expression. As previously mentioned, selected four cancer cell line representing different tumour types and with varying degrees of xCT expression, Figure 6. A172 (high xCT expressor), A375 (high xCT expressor), U87 (low expressor) and MCF7 (intermediate xCT expressor). The cells were cultured and treated for 48 h with 250 μM and 500 μM of each analogue. After the incubation, glutathione assay was conducted, and glutathione levels in cancer cells treated with the various analogues were compared to the untreated control cells. Most of the compounds exhibited a dose‐dependent inhibition of glutathione. The effect of glutathione inhibition is shown in Figure 7 where two analogues DC10 and DC14 revealed similar or higher potency of inhibition of glutathione synthesis than sulfasalazine (DC00).
Figure 6.
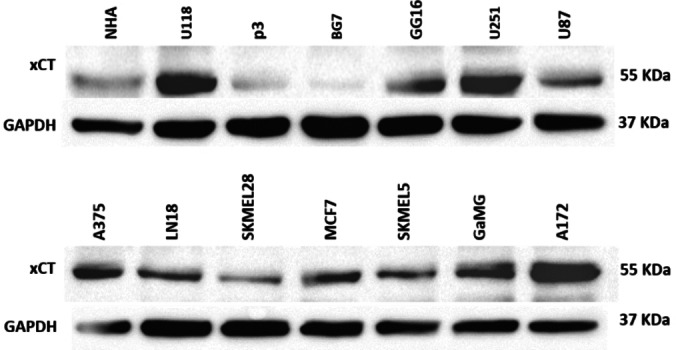
xCT expression in different cancer cell lines assessed by western blot. GAPDH was used as loading control.
Figure 7.
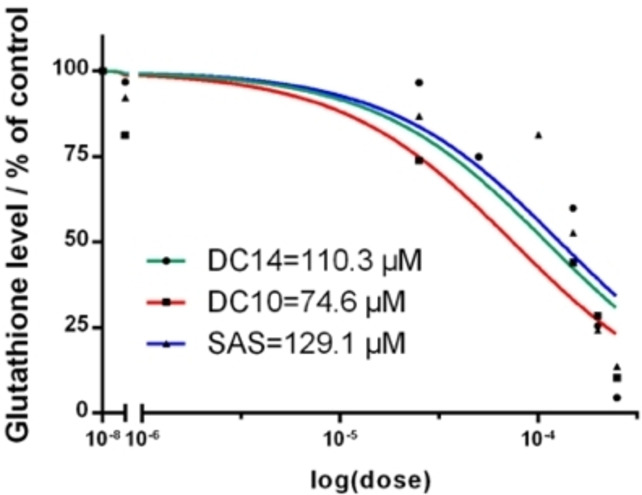
Dose (C compound) ‐ dependent effects (IC50) versus glutathione, DC10 and DC14 on A172 human glioma cell line by glutathione assy.
Particularly, in the U87 cell line with low xCT expression, the effect of the test drugs drug was relatively lower than in the high expressor cell lines. This finding indicates that the effect of glutathione inhibition is mediated by xCT inhibition, consistent with our hypotheses for the project.
These findings provide crucial information about the correlation between structure of the test compounds and their effect on glutathione synthesis, which can inform design of future compounds. Moreover, for the analogues exhibiting a high degree of glutathione inhibition, further validation with additional in vitro assays as well as animal experiments are warranted.
Toxicity versus normal human astrocytes
Among the analogues synthesised, DC10 appeared as one of the more promising xCT inhibitors with a higher potency than sulfasalazine. Thus, as a preliminary screen for toxicity, we performed flow cytometry to assess cell viability in normal human astrocytes (NHA) following treatment with the sulfasalazine DC00 and the lead compound DC10, [26] see Figure 8. As such, NHA were treated with DC00 and DC10 at three various concentrations 50, 100, and 150 μM for a period of 24 h prior to flow cytometry. Untreated NHA cells served as control.
Figure 8.
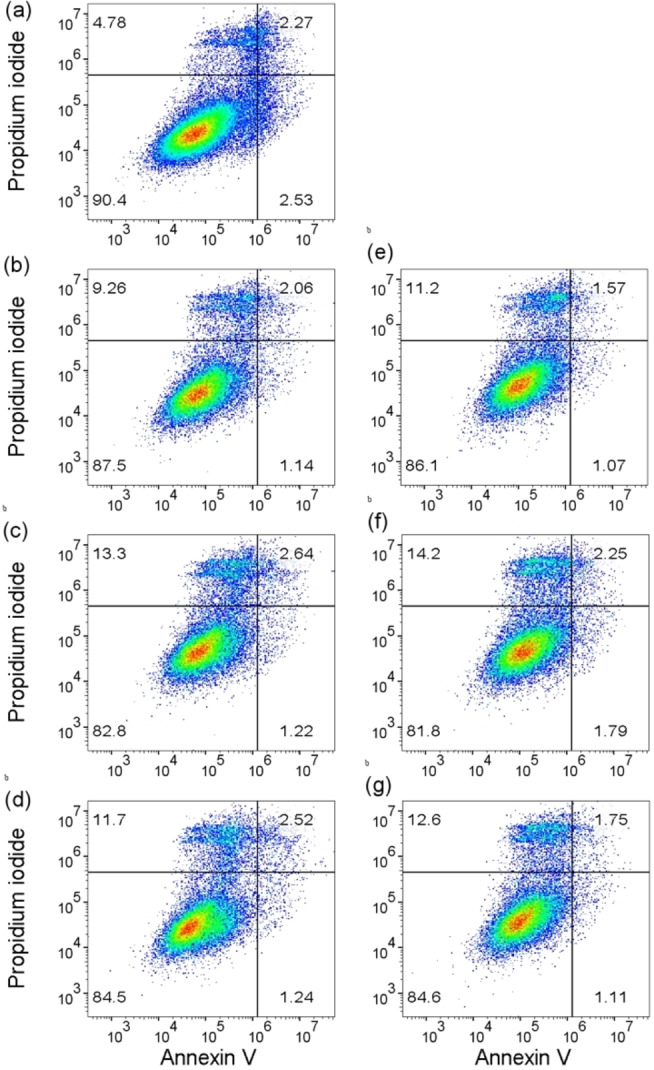
Measurement of cell viability with flow cytometry. Scatter plots from a representative experiment (N=3) comprising the various treatment groups. (a) Control (untreated), (b) DC10 50 μM, (c) DC10 100 μM, (d) DC10 150 μM, (e) DC00 50 μM, (f) DC00 100 μM, (g) DC00 150 μM. The upper left quadrant (Q1) represents propidium iodide positive cells, i. e. dead (necrotic or late apoptosis) cells. The upper right quadrant (Q2) represents Annexin V and propidium iodide positive cells that are dead, lower right quadrant (Q3) represents Annexin V positive cells that are pre‐apoptotic, and lower left quadrant (Q4) contains non‐stained cells representing viable cells.
Flow cytometry revealed no difference in viability between the different treatment groups as well as untreated controls. The viability of the cells was >90 % following treatment with all the applied doses. Collectively, these experiments serve to demonstrate the absence of severe toxicity on a cellular level. Although these experiments do not allow for comparing the toxicity profiles of DC10 with sulfasalazine, our styryl benzoic acid analogues do not contain the sulfapyridine moiety to which the side effects of sulfasalazine have been attributed. Thus, our data suggests further biological validation is warranted to investigate the potential of our most potent compounds for clinical use as xCT inhibitors.
The disrupted cell membranes of apoptotic and dead cells allow the fluorescent dye propidium iodide to enter and stain the cells, whereas the intact cell membrane prevents these dyes from entering and staining viable cells. The fluorescently labelled Annexin V antibody detects a cellular protein associated with apoptotic cell death. The samples were analysed on a flow cytometer, [27] whereupon the acquired data were analysed with the FlowJoTM software. [28] These findings suggest that DC10 exhibit minimal toxicity in normal cells.
SAR of the xCT inhibitor scaffold
The functional groups hydroxyl and carboxylic acid installed on the A‐ring, Figure 9, are both important for the inhibitor activity. Methylated hydroxyl group provides still activity, but significantly reduced. This may indicate that a hydrogen bond is formed between the inhibitor molecule and the hydrogen bonding site found in the xCT antiporter. The hydrogen bonding site is of the HBA type, whereof the methyl group reduces the interactions and therefore acts as a steric hindrance to obtain a strong bond. The carboxylic acid group appears as an essential for the inhibition strength of the xCT inhibitor. It is two possibilities, that it is formed H‐bond interaction between the carboxylic acid group of the A‐ring and the binding region situated in the antiporter. However, this site may as well be a source for ionic bonding interaction as the carboxylic acid group might easily be ionized at blood pH. The carboxylic acid group (DC02) can be substituted with the bioisostere tetrazole (DC15), which afford similar response towards the investigated cell lines. At least two lipophilic regions forming van der Waals interacting bonds between the inhibitor molecule and the antiporter macromolecule site have been identified. Thanks to this interaction, a more active inhibitor was created (DC11) with a rigidified molecular scaffold with additional C−C bonds created in the ring‐B giving ring‐B’. Moreover, prolonging of the ring systems (ring‐C) enhanced further the inhibitor properties, suggesting another more hydrophobic binding site in the xCT antiporter macromolecule.
Figure 9.

Outline for the interaction bonds between the inhibitor molecular scaffold and the xCT antiporter system.
Conclusion
By means of the SAR approach, we have successfully designed, synthesised, and biologically evaluated a molecular library of xCT antiporter inhibitors candidates. From the SAR study, new features about the pharmacophore have been revealed and some novel (DC10, DC14, and DC21) potent xCT antiport inhibitors were revealed.
The toxicity of DC10 was furthermore assessed versus normal human astrocytes. The results from flow cytometry suggest that DC10 exhibit minimal toxicity in normal cells, and thus be a candidate for further biological research.
A binding model from the small molecule inhibitor molecules versus the xCT macro molecule was elaborated. This molecule is not only more active than the parent compound (DC00), but also the pharmacokinetics characteristics have been implemented such as the lipophilicity and the metabolic stability. Further in vitro and in vivo evaluations is currently conducted to test its role as a possible new tool in the fight against cancer.
Experimental Section
Chemistry
GC analyses were performed on a capillary gas chromatograph equipped with a fused silica column (l: 25 m, i.d.: 0.20 mm, film thickness: 0.33 mm) at a helium pressure of 200 kPa, split less/split injector and flame ionization detector. DART‐MS spectra were obtained using PEG as an internal standard under positive ionization mode with a ToF mass analyzer. NMR spectra were recorded on a 500 MHz instrument. Chemical shifts were referenced to the deuterated solvent used in that experiment. All melting points are uncorrected. Synthesis of precursors 2, 3, 4, and 10 were reported previously. The micro‐ wave‐assisted experiments were performed with a Biotage Initiator Sixty EXP Microwave System operating at 0–400 W at 2.45 GHz. The instrument operates in the temper‐ ature range 40–250 °C, a pressure interval of 0–20 bar (2 MPa, 290 psi) with reactor vial volumes of 0.2–20 mL. A SciFinder® database search was performed on 2019–09‐02 to control if the synthesized compounds described below previously have been reported and thus assigned CAS number. In the header of each method description, the CAS number is given as [xxx‐xxx‐xxx] optionally [NEW] if the compound is “NEW”, that is not previously reported.
3‐bromo‐4‐hydroxybenzaldehyde 1 [2973‐78‐6]
A mixture of 4‐hydroxybenzaldehyde (5,0 g, 40.94 mmol) in acetonitrile (25 mL) was cooled at 0 °C. p‐Toluenesulfonic acid monohydrate (1.1 equiv., 8.57 g, 45.04 mmol) and N‐bromosuccinimide (1.1 equiv., 8.02 g, 45.04 mmol) were then added. The reaction mixture was stirred overnight at room temperature using a flask wrapped in aluminum foil. The reaction was monitored by TLC using an eluent composed by hexane : ethyl acetate=8 : 2. The solvent was removed under reduced pressure, whereupon the crude was diluted with DCM and a saturated solution of Na2S2O3 in water and the aqueous phase is extracted two more times with DCM. The organic phases are reunited, dried over Na2SO4 anhydrous and concentrated under reduced pressure to afford the desired compound 1 (7.1 g, 35.32 mmol) as yellow oil and with a yield of 86 %. 1H NMR (400 MHz, DMSO) δ 10.70 (s, 1H), 9.68 (s, 1H), 7.89 (d, J=2.0 Hz, 1H), 7.58 (dd, J=8.3, 2.0 Hz, 1H), 7.00 (d, J=8.3 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 188.84, 159.44, 134.28, 130.16, 129.16, 129.13, 116.00, 110.14.
3‐Bromo‐4‐methoxybenzaldehyde 2 a [34841‐06‐0]
3‐Bromo‐4‐hydroxybenzaldehyde 1 (7.0 g, 34.82 mmol) in acetone (25 mL) was added Na2CO3 (2 equiv., 7.38 g, 69.65 mmol) and iodomethane (2 equiv., 9.89 g, 4.34 mL, 69.65 mmol). The reaction mixture was stirred at 20 °C for 18 h. The progress of the reaction was monitored by TLC, using as hexane : ethyl acetate=9 : 1 as eluent. The solvent was removed under reduced pressure and the mixture was extracted with DCM (3×250 mL). The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure to obtain target compound 2 a (7.03 g, 32,69 mmol) as yellow oil in a yield of 94 %. 1H NMR (500 MHz, CDCl3) δ 9.75 (s, 1H), 8.24 (d, J=2.0 Hz, 1H), 7.79 (dd, J=8.5, 2.0 Hz, 1H), 6.86 (d, J=8.5 Hz, 1H), 3.91 (s, 3H). 13C NMR (126 MHz, Acetone) δ 190.34, 161.50, 134.71, 132.12, 132.07, 113.25, 112.76, 57.23.
3‐Bromo‐4‐(methoxymethoxy)benzaldehyde 2 b [162269‐90‐1]
3‐Bromo‐4‐hydroxybenzaldehyde 1 (7.0 g, 34.82 mmol) in DCM (25 mL) was cooled at 0 °C and then added N,N‐Dicyclohexylmethylamine (2 equiv., 13.61 g, 14.92 mL, 69.65 mmol) and chloromethyl methyl ether (1.5 equiv., 4.21 g, 3.97 mL, 52.23 mmol). The reaction mixture was stirred at 20 °C for 18 h. The progress of the reaction was monitored by TLC, using as hexane : ethyl acetate=9 : 1 as eluent. The reaction mixture was poured in HCl (0.1 M) and then extracted with DCM (3×250 mL). The organic phases were combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The white solid obtained is filtered and washed with Et2O. The filtrated was collect and concentrated under reduced pressure to afford the desired compound 2 b (8.15 g, 33.26 mmol) as yellow oil and with a yield of 95 %. 1H NMR (500 MHz, CDCl3) δ 9.86 (s, 1H), 8.09 (d, J=2.0 Hz, 1H), 7.79 (dd, J=8.5, 2.0 Hz, 1H), 7.27 (d, J=8.5 Hz, 1H), 5.35 (s, 2H), 3.53 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 189.55, 158.42, 134.47, 131.45, 130.81, 115.06, 113.33, 94.79, 56.62.
Benzyltriphenylphosphonium chloride 3 [1100‐88‐5]
Benzyl chloride (1.1 equiv., 5.31 g, 1.10 mL, 41.94 mmol) was added to a solution of triphenylphosphine (10.0 g, 38.13 mmol) in toluene (25 mL), that was flushed with N2, heated at reflux, and stirred overnight. A white precipitate was filtrated off using hot toluene and the afforded solid was concentrated under reduced pressure. The desired compound 3 (14.60 g, 37.54 mmol) as white solid and with a yield of 98 %. 1H NMR (500 MHz, CDCl3) δ 7.74–7.64 (m, 9H), 7.59–7.50 (m, 6H), 7.18–7.11 (m, 1H), 7.08–7.00 (m, 4H), 5.46 (d, J=14.5 Hz, 2H).
(E)‐2‐bromo‐1‐methoxy‐4‐styrylbenzene 4 [1018068‐98‐8]
KOH (1.2 equiv., 219 mg, 3.91 mmol) was added to a solution of benzyltriphenylphosphonium chloride 3 (1.2 equiv., 1.52 g, 3.91 mmol) in DCM (10 mL). Then, after a period of 30 min., 3‐bromo‐4‐methoxybenzaldehyde 2 a (700 mg, 3.26 mmol) was added to the mixture. The reaction mixture was stirred for 3 h at room temperature. The reaction was monitored by means of TLC using an eluent composed by hexane : ethyl acetate=9 : 1. The reaction crude obtained is filtered and washed with Et2O. The filtrated was collect and concentrated under reduced pressure. To the crude, containing a mixture of the E and Z isomers, is added I2 (1 %, 8 mg, 32,55 μmol) using as solvent a mixture of hexane : EtOAc=8 : 2 ratio. The reaction is kept under stirring overnight 65 °C. The mixture was extracted with ethyl acetate (3×50 mL) from a sat. Na2S2O3, (aq) (50 mL) dried over anhydrous Na2SO4 , filtrated, solvent evaporated under reduced pressure. The crude was re‐crystalized in hexane. The desired product 4 (456 mg, 1.58 mmol) was obtained as a white solid with a yield of 48 %. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J=2.2 Hz, 1H), 7.39–7.33 (m, 2H), 7.24 (dt, J=9.4, 5.0 Hz, 3H), 7.19–7.10 (m, 1H), 6.84 (s, 2H), 6.73 (d, J=8.5 Hz, 1H), 3.76 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 155.36, 137.18, 131.64, 131.01, 128.71, 127.99, 127.60, 126.87, 126.77, 126.39, 112.10, 111.94, 56.34.
(E)‐2‐bromo‐1‐(methoxymethoxy)‐4‐styrylbenzene 5 [NEW]
Benzyltriphenylphosphonium chloride 3 (1.2 equiv., 1.33 g, 3.43 mmol) was dissolved in DCM (10 mL) using a reaction flask (25 mL). The mixture was then added KOH (1.2 equiv., 192 mg, 3.43 mmol) and after 30 min., 3‐Bromo‐(4‐methoxymethoxy) benzaldehyde (2 b) (700 mg, 2.86 mmol) was also added to the solution. The reaction mixture is stirred for 3 h at room temperature. The reaction is monitored by TLC, using as eluent Hexane : EtOAc=9 : 1. The reaction crude obtained is filtered and washed with Et2O. The filtrated is collect and concentrated under reduced pressure. To the crude, containing a mixture of the E and Z isomers was added I2 (1 %, 7 mg, 28,56 μmol) using as solvent a mixture of hexane and ethyl acatate=8 : 2. The reaction was stirred overnight at 65 °C. The mixture was extracted from a saturated water solution of Na2S2O3 with ethyl acetate (3×250 mL), dried over Na2SO4 and purified through silica gel column chromatography (Hexane : EtOAc=99.5 : 0.5). The desired product 5 (398 mg, 1.25 mmol) is obtained as a yellow oil with a yield of 43 %. 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J=2.1 Hz, 1H), 7.48–7.44 (m, 2H), 7.37–7.30 (m, 3H), 7.27–7.21 (m, 1H), 7.10 (d, J=8.5 Hz, 1H), 6.96 (s, 2H), 5.23 (s, 2H), 3.51 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 153.18, 137.11, 132.89, 131.06, 128.75, 128.47, 127.73, 126.75, 126.70, 126.49, 116.13, 113.26, 95.14, 56.45.
(E)‐2‐methoxy‐5‐styrylbenzoic acid DC01 [NEW]
THF anhydrous (6 mL) was added in a Schlenk flask containing Mg (5 equiv., 63 mg, 2.59 mmol), followed by the addition of dibromoethane (2 equiv., 195 mg, 89 μL, 1.04 mmol). After 10 min., 4 (150 mg, 519 μmol) is added in the mixture. The reaction is kept overnight under stirring at reflux. Afterwards, the CO2 is bubbled for 10 min. through the reaction by means of a gas diffuser. The post‐reaction mixture was acidified and extracted with DCM (3×50 mL). The organic phases was combined, dried over anhydrous Na2SO4, filtrated, and concentrated under reduced pressure. The crude is purified through silica gel column chromatography (DCM/MeOH 95 : 5). Target molecule DC01 (53 mg, 208 μmol) was isolated as a white solid with a yield of 40 %. 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J=2.4 Hz, 1H), 7.69 (dd, J=8.6, 2.4 Hz, 1H), 7.51 (dd, J=7.3, 1.2 Hz, 2H), 7.39–7.34 (m, 2H), 7.30–7.27 (m, 1H), 7.15–7.04 (m, 3H), 4.11 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.14, 157.18, 136.89, 132.82, 132.02, 131.50, 129.32, 128.77, 127.90, 126.53, 126.30, 117.80, 112.03, 56.92. HR‐MS (ESI): [M+H]+: Calcd for C16H15O3 255.10212, found 255.10237 and [M+Na]+: Calcd for C16H14NaO3 277.08406, found 277.08412.
(E)‐2‐hydroxy‐5‐styrylbenzoic acid DC02 [1072937‐49‐5]
THF anhydrous (6 mL) was added in a Schlenk flask containing Mg (5 equiv., 57 mg, 2.35 mmol), followed by the addition of dibromoethane (2 equiv., 177 mg, 81 μL, 940 μmol). After 10 min., 5 (150 mg, 470 μmol) is added in the mixture. The reaction is kept overnight under stirring at reflux. Afterwards, the CO2 is bubbled for 10 min. through the reaction by means of a gas diffuser. The reaction mixture is extracted with DCM (3×50 mL). The organic phases are reunited, dried over Na2SO4 anhydrous and concentrated under reduced pressure. The crude was purified through silica gel column chromatography (DCM/MeOH 9 : 1). The desired product DC02 (51 mg, 212 μmol) is obtained as a white solid with a yield of 45 %. 1H NMR (500 MHz, CD3CN) δ 8.05 (d, J=1.7 Hz, 1H), 7.79 (dd, J=8.6, 2.0 Hz, 1H), 7.58 (d, J=7.6 Hz, 2H), 7.40 (t, J=7.6 Hz, 2H), 7.29 (t, J=7.3 Hz, 1H), 7.16 (dd, J=37.0, 16.5 Hz, 2H), 7.01 (d, J=8.7 Hz, 1H). 13C NMR (126 MHz, CD3CN) δ 171.30, 161.22, 137.19, 132.96, 128.61, 128.39, 128.32, 127.14, 126.92, 126.84, 125.96, 117.39 (one carbon peak under solvent peak). HR‐MS (ESI): [M−H]−: Calcd for C15H11O3 239.07082, found 239.07055.
2‐bromo‐4‐iodophenol 6 [133430‐98‐5]
To a solution of 4‐iodophenol (5.0 g, 22.73 mmol) in acetonitrile (25 mL) cooled at 0 °C was added first p‐toluenesulfonic acid monohydrate (1.1 equiv., 4.76 g, 25.00 mmol) and then N‐Bromosuccinimide (1.1 equiv., 4.45 g, 25.00 mmol).The reaction mixture is stirred overnight at room temperature and the flask is wrapped in aluminum foil. The reaction is monitored by TLC, using as eluent hexane : ethyl acetate=8 : 2. The solvent is removed under reduced pressure. The crude was diluted with DCM and a saturated solution of Na2S2O3 in water and the aqueous phase was extracted two more times with DCM. The organic phases are reunited, dried over Na2SO4 anhydrous and concentrated under reduced pressure to afford the desired compound 1 (6.25 g, 20.91 mmol) as yellow solid and with a yield of 92 %. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J=1.7 Hz, 1H), 7.49 (dd, J=8.6, 1.7 Hz, 1H), 6.78 (d, J=8.8 Hz, 1H), 4.53 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 152.34, 139.64, 138.04, 118.12, 111.34, 82.02.
2‐bromo‐4‐iodo‐1‐methoxybenzene 7 a [182056‐39‐9]
Na2CO3 (2 equiv., 4.26 g, 40.15 mmol) and iodomethane (2 equiv., 5.70 g, 2.50 mL, 69.65 mmol) were added to a solution of 2‐bromo‐4‐iodophenol 6 (6.0 g, 20.07 mmol) in acetone (25 mL). This reaction mixture was stirring 20 h at a temperature 20 °C. The reaction was monitored by TLC using an eluent composed by hexane : ethyl acetate=9 : 1. The solvent was removed under reduced pressure and the mixture was extracted (3×250 mL) with DCM from water. The organic phases were combined and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford target compound 7 a (5.91 g, 18.89 mmol) as yellow oil and with a yield of 94 %. 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J=2.1 Hz, 1H), 7.54 (dd, J=8.7, 2.1 Hz, 1H), 6.65 (d, J=8.6 Hz, 1H), 3.87 (s, 3H).13C NMR (126 MHz, CDCl3) δ 155.98, 140.99, 137.27, 113.84, 112.96, 82.44, 56.31.
2‐bromo‐4‐iodo‐1‐(methoxymethoxy)benzene 7 b [269716‐51‐0]
N,N‐Dicyclohexylmethylamine (2 equiv., 7.84 g, 8.60 mL, 40.15 mmol) and then chloromethyl methyl ether (1.5 equiv., 2.42 g, 2.29 mL, 30.11 mmol) were added to a cold (0 °C) solution of 2‐bromo‐4‐iodophenol 6 (6.0 g, 20.07 mmol) in DCM (25 mL). This reaction mixture was stirred 18 h at a temperature of 20 °C. The reaction mixture was monitored by TLC by using as eluent hexane : ethyl acetate=9 : 1. The reaction mixture was poured in HCl 0.1 M and extracted with DCM (3×250 mL). The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The white solid obtained is filtered and washed with Et2O. The filtrated is collect and concentrated under reduced pressure to afford the desired compound 7 b (6.20 g, 18.08 mmol) as yellow oil and with a yield of 90 %. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J=2.1 Hz, 1H), 7.52 (dd, J=8.7, 2.1 Hz, 1H), 6.91 (d, J=8.7 Hz, 1H), 5.22 (s, 2H), 3.50 (s, 3H).13C NMR (126 MHz, CDCl3) δ 153.85, 141.12, 137.29, 117.87, 114.06, 95.06, 84.21, 56.45.
2‐bromo‐1‐methoxy‐4‐(phenylethynyl)benzene 8 [2253656‐73‐2]
Phenylacetylene (1.1 equiv., 373 mg, 401 μL, 3.66 mmol), 7 (1.04 g, 3.32 mmol) and diethylamine (5 mL) were transferred to a round bottom flask (50 mL) under an argon atmosphere. This mixture was poured into another round bottom flask (50 mL) containing a mixture of Pd(PPh3)4 (3 %, 115 mg, 100 μmol) and CuI (10 %, 63 mg, 332 μmol) in toluene (6 mL). The mixture was heated at 50 °C and stirred for 1 h. The reaction was monitored by TLC, using as eluent hexane : ethyl acetate=9 : 1. The solvent was then removed under reduced pressure and the mixture was extracted with DCM (3×250 mL) from water. The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford target compound 8 (811 mg, 2.82 mmol) as white off solid and with a yield of 85 %. 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J=2.1 Hz, 1H), 7.52–7.49 (m, 2H), 7.45 (dd, J=8.5, 2.0 Hz, 1H), 7.36–7.32 (m, 3H), 6.87 (d, J=8.5 Hz, 1H), 3.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 156.03, 136.29, 132.00, 131.52, 128.37, 128.26, 123.14, 116.89, 111.57, 111.43, 89.07, 87.81, 56.31.
(Z)‐2‐bromo‐1‐methoxy‐4‐styrylbenzene 9 [1865698‐16‐3]
2‐Bromo‐1‐methoxy‐4‐(phenylethynyl)benzene 8 (350 mg, 1.22 mmol) in ethylacetate (10 mL) was transferred to a round bottom flask (25 mL) wrapped with aluminum foil. Quinoline (0.4 equiv., 63 mg, 58 μL, 487 μmol) and the Lindlar catalyst 5 % Pd w/w (380 mg, 15 % Pd equiv.) were then added. The reaction mixture was stirred under a H2 atmosphere at a temperature of 40 °C for 18 h. The reaction mixture was monitored by TLC using hexane : ethyl acetate=9 : 1 as the eluent. The solvent was then removed under reduced pressure and the mixture was extracted with DCM (3×50 mL). The organic phases were combined, dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified through silica gel column chromatography (hexane : ethyl acetate 99.5 : 0.5). Target compound 9 (288 mg, 996 μmol) was obtained as a yellow solid in a yield of 81 %. 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J=2.0 Hz, 1H), 7.26–7.18 (m, 5H), 7.13 (dd, J=8.6, 2.1 Hz, 1H), 6.72 (d, J=8.5 Hz, 1H), 6.55 (d, J=12.2 Hz, 1H), 6.45 (d, J=12.2 Hz, 1H), 3.85 (s, 3H).13C NMR (126 MHz, CDCl3) δ 154.91, 137.07, 133.78, 131.15, 130.12, 129.09, 128.84, 128.40, 128.37, 127.31, 111.54, 111.31, 56.21.
(Z)‐2‐methoxy‐5‐styrylbenzoic acid DC03 [NEW]
Mg (5 equiv., 63 mg, 2.59 mmol) was transferred to a Schlenk flask (100 mL) together with anhydrous THF (6 mL). Then, dibromoethane (2 equiv., 195 mg, 89 μL, 1.04 mmol) was added to the reaction mixture. After a short period of time (≈10 min.), (Z)‐2‐bromo‐1‐methoxy‐4‐styrylbenzene 9 (150 mg, 519 μmol) was added to the reaction mixture. The reaction mixture was stirred continuously at reflux for another 18 h. Then CO2 was bubbled by means of a gas diffuser (porous glass sinter at the tip of the glass tube) through the reaction mixture for a period of 10 min. The reaction mixture was extracted with DCM (3×50 mL). The organic phases are combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude was purified using a silica gel chromatography column (DCM : MeOH=95 : 5). Targed product DC03 (60 mg, 236 μmol) was obtained as a transparent oil with a yield of 45 %. 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J=2.3 Hz, 1H), 7.39 (dd, J=8.7, 2.3 Hz, 1H), 7.26–7.16 (m, 5H), 6.84 (d, J=8.7 Hz, 1H), 6.63 (d, J=12.1 Hz, 1H), 6.53 (d, J=12.2 Hz, 1H), 4.01 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.54, 156.98, 136.72, 135.05, 134.29, 131.23, 130.81, 128.63, 128.33, 128.02, 127.28, 117.40, 111.42, 56.62. HR‐MS (ESI): [M+H]+: Calcd for C16H15O3 255.10212, found 255.10103.
2‐methoxy‐5‐(phenylethynyl)benzoic acid DC04 [NEW]
Mg (5 equiv., 63 mg, 2.61 mmol) and anhydrous THF (6 mL) were transferred to a Schlenk flask (100 mL) whereupon dibromoethane (2 equiv., 196 mg, 90 μL, 1.04 mmol) was added. Then, after a period of ≈10 min., 2‐bromo‐1‐methoxy‐4‐(phenylethynyl)benzene 8 (150 mg, 522 μmol) was added to the mixture. The reaction mixture was continuously stirred at reflux for a period of 18 h. Then, CO2 was bubbled through the reaction mixture by means of a gas diffuser (porous glass sinter at the tip of the glass tube) through the reaction mixture for a period of 10 min. The reaction mixture is extracted with DCM (3×50 mL). The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified through column chromatography using silica gel using an eluent composed by DCM: methanol=95 : 5. The desired product DC04 (63 mg, 249 μmol) was obtained as a transparent oil in a yield of 48 %. 1H NMR (500 MHz, CDCl3) δ 8.36 (d, J=2.1 Hz, 1H), 7.71 (dd, J=8.6, 2.2 Hz, 1H), 7.57–7.48 (m, 2H), 7.39–7.32 (m, 3H), 7.05 (d, J=8.6 Hz, 1H), 4.10 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 164.68, 157.62, 137.74, 137.09, 131.57, 128.47, 128.39, 122.84, 117.89, 117.72, 111.91, 89.76, 87.36, 56.90. HR‐MS (ESI): [M+H]+: Calcd for C16H13O3 253.08647, found 253.08677.
2‐(3‐bromo‐4‐methoxyphenyl)naphthalene 10 [NEW]
2‐Bromo‐4‐iodo‐1‐methoxybenzene 7 a (1 g, 3.20 mmol), naphthalen‐2‐ylboronic acid (1 equiv., 550 mg, 3.20 mmol), K2CO3 (2 equiv., 883 mg, 6.39 mmol), Pd(PPh3)4 (5 %, 158 mg, 160 μmol), and DMF (10 mL) were transferred to a round bottom flask (50 mL). This reaction mixture was flushed with argon and then continuously stirred for 2 h at 100 °C. The reaction mixture was monitored by TLC using an eluent composed by hexane : ethyl acetate=9 : 1. The solvent was removed from the post reaction mixture under reduced pressure and then extracted with DCM (3×100 mL). The organic phases were combined, dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified through silica gel chromatography column (hexane : ethyl acetate=99.5 : 0.5). Target compound 10 (734 mg, 2.34 mmol) was obtained as a yellow solid in a yield of 73 %. 1H NMR (500 MHz, CDCl3) δ 7.81 (s, 1H), 7.79 (d, J=2.2 Hz, 1H), 7.72 (dd, J=12.9, 7.6 Hz, 3H), 7.51 (dd, J=8.5, 1.8 Hz, 1H), 7.44 (dd, J=8.5, 2.2 Hz, 1H), 7.39–7.32 (m, 2H), 6.80 (d, J=8.5 Hz, 1H), 3.77 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 155.42, 136.73, 135.02, 133.72, 132.57, 132.12, 128.61, 128.19, 127.71, 127.34, 126.46, 126.03, 125.30, 125.13, 112.22, 56.37 (one carbon peak is not visible).
2‐methoxy‐5‐(naphthalen‐2‐yl)benzoic acid DC05 [NEW]
Oxalic acid (3 equiv., 86 mg, 958 μmol), Pd(OAc)2 (5 %, 4 mg, 16 μmol), Xantphos (5 %, 9 mg, 16 μmol), 2‐(3‐bromo‐4‐methoxyphenyl)naphthalene 10 (100 mg, 319 μmo) and DMF (5 mL) were transferred to a glass tube reactor, that was sealed and left under stirring in an oil bath at 100 °C. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production) and TEA (3 equiv., 97 mg, 134 μL, 958 μmol) was added. Then Ac2O (3 equiv., 98 mg, 90 μL, 958 μmol) was drop‐wise added over a period of 15 min. by means of a syringe pump. During the Ac2O addition, CO was formed, though the decomposition of oxalic acid. The reaction mixture was stirred at 100 °C for 3 h. The post reaction mixture was extracted with DCM (3×50 mL). The organic phases are combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The isolated crude product was purified by means of a chromatography column filled with silica gel with an eluent composed of DCM : methanol=95 : 5). Target product DC05 was obtained as a white solid with a yield of 57 % (51 mg, 183 μmol). 1H NMR (500 MHz, CDCl3) δ 10.70 (s, 1H), 8.57 (s, 1H), 8.04 (s, 1H), 7.96–7.82 (m, 4H), 7.73 (d, J=8.5 Hz, 1H), 7.50 (t, J=6.8 Hz, 2H), 7.17 (d, J=8.6 Hz, 1H), 4.13 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.42, 157.45, 136.20, 135.32, 133.64, 133.57, 132.69, 132.43, 128.70, 128.20, 127.66, 126.52, 126.20, 125.59, 124.92, 117.95, 112.28, 56.94. HR‐MS (ESI): [M+H]+: Calcd for C18H13O3 277.08647, found 277.08678.
Methyltriphenylphosphonium iodide 11 [2065‐66‐9]
Iodomethane (1.1 equiv., 11.91 g, 5.22 mL, 83.88 mmol) was added to a solution of triphenylphosphine (20.0 g, 76.25 mmol) in THF (25 mL) was added. The reaction was flushed with N2, heated at reflux and continuously stirred for 18 h. A white precipitate that was formed was filtrated off and the afforded solid is concentrated under reduced pressure. Target methyl‐triphenyl phosphonium iodide 11 was isolated as a white solid in a yield of 93 % (28.60 g, 70.75 mmol). 1H NMR (500 MHz, CDCl3) δ 7.85–7.67 (m, 15H), 3.22 (d, J=13.2 Hz, 3H).
2‐bromo‐1‐(methoxymethoxy)‐4‐vinylbenzene 12 [NEW]
At 0 °C, compound 11 (1.2 equiv., 3.96 g, 9.79 mmol) was slowly added to a suspension of NaH 60 % (3 equiv., 979 mg, 24.48 mmol) in anhydrous DCM (15 mL). The mixture was then left for stirring in 30 min., whereupon compound 2 b (2.0 g, 8.16 mmol) was added to the mixture. The mixture was then stirred for 16 h at 20 °C. The reaction mixture was then quenched with a saturated solution of NaHCO3 in water. The quenched post‐reaction mixture was then extracted with DCM (3×250 mL). The combined organic layers was dried over Na2SO4, and then purified using column chromatography filled with silica gel (eluent hexane : ethyl acetate=9 : 1), which afforded the target compound 12 (1.76 g, 7.24 mmol) as transparent oil in a yield of 89 %. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J=2.1 Hz, 1H), 7.27 (dd, J=8.5, 2.1 Hz, 1H), 7.10 (d, J=8.5 Hz, 1H), 6.60 (dd, J=17.6, 10.9 Hz, 1H), 5.64 (d, J=17.3 Hz, 1H), 5.24 (s, 2H), 5.20 (d, J=10.7 Hz, 1H), 3.52 (s, 3H).13C NMR (126 MHz, CDCl3) δ 153.29, 134.95, 133.10, 130.91, 126.38, 115.97, 113.57, 113.05, 95.11, 56.39.
(E)‐2‐bromo‐4‐(4‐fluorostyryl)‐1‐(methoxymethoxy)benzene 13 [NEW]
Method A: 1‐fluoro‐4‐vinylbenzene (106 mg, 867.84 μmol), Pd(OAc)2 (8 %, 16 mg, 69 μmol), K2CO3 (2 equiv., 240 mg, 21.74 mmol), 7 b (1 equiv., 297 mg, 867.84 μmol) and DMF (6 mL) were transferred to a reactor tube that was sealed. The mixture was flushed with Argon and sonicated for a period of 5 min. The reactor tube was immersed into the reactor cavity of a microwave oven at 100 °C for a period of 4 h. The progress of the reaction was monitored by TLC using an eluent composed of hexane : ethyl acetate=9 : 1. The solvent was removed under reduced pressure. The residue was then extracted with DCM (3×50 mL). The combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. The isolated crude product was purified using a chromatography column packed with silica gel using hexane : ethyl acetate=99 : 1 to achieve target compound 13 as a clear liquid in a yield of 35 % (102 mg, 302.50 μmol).
Method B: 2‐Bromo‐1‐(methoxymethoxy)‐4‐vinylbenzene 12 (210 mg, 867.84 μmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 10 mg, 43 μmol), Pd(OAc)2 (5 %, 9 mg, 43 μmol), (4‐fluorophenyl)boronic acid (1.1 equiv., 133 mg, 950.23 μmol), Cu(OAc)2 .H2O (1.5 equiv., 258 mg, 1.30 mmol), and 7 mL of DMF (7 mL) was transferred to a round bottom flask (50 mL). This mixture was continuously stirred for 18 h at 20 °C. The course of the reaction was monitored by TLC, using hexane : ethyl acetate=9 : 1 as eluent. The post reaction mixture was added a solution EDTA in water (10 mL, 0.2 M) that was extracted with DCM (3×50 mL). The organic phases were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The mixture was purified using column chromatography packed with silica gel with hexane : ethyl acetate=99.6 : 0.4 as eluent. Target compound 13 was achieved as a clear liquid in a yield of 87 % (254 mg, 753.29 μmol).1H NMR (500 MHz, CDCl3) δ 3.36 (s, 3H), 5.07 (s, 2H), 6.65–6.77 (m, 2H), 6.82–6.91 (m, 3H), 6.95 (d, 1H), 7.14 (dd, 1H), 7.21–7.27 (m, 2H), 7.53 (d, 1H).13C NMR (126 MHz, CDCl3) δ 162.38 (d, J=247.1 Hz), 153.26, 133.32 (d, J=3.4 Hz), 132.68, 131.01, 127.97 (d, J=7.8 Hz), 127.17, 126.67, 126.47 (d, J=2.3 Hz), 116.07, 115.67 (d, J=21.8 Hz), 113.28, 95.09, 56.36.
(E)‐5‐(4‐fluorostyryl)‐2‐hydroxybenzoic acid DC07 [NEW]
Oxalic acid (3 equiv., 360 mg, 4.00 mmol), Pd(OAc)2 (5 %, 15 mg, 66.73 μmol), Xantphos (5 %, 39 mg, 66.73 μmol), 13 (450 mg, 1.33 mmol), water (10 equiv., 240 mg, 13.35 mmol) and DMF (10 mL) were transferred to an reaction tube, that was sealed and left stirring in a preheated oil bath at 100 °C. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). TEA (3 equiv., 405 mg, 558 μL, 4.00 mmol) was then added, followed by an addition through syringe pump over 15 min. of Ac2O (3 equiv., 408 mg, 378 μL, 4.00 mmol). During the addition CO will be formed, though the degradation of the oxalic acid. The reaction was then stirred at 100 °C for 3 h. The reaction mixture was extracted with DCM (3×50 mL) from acid water. The organic phases were combined and dried over anhydrous Na2SO4, filtered and the concentrated under reduced pressure. The crude was purified through silica gel column chromatography (DCM/MeOH 95 : 5) to afford DC07 (163 mg, 0.631 mmol) with a yield of 47 %. 1H NMR (500 MHz, DMSO) δ 7.97 (d, J=2.3 Hz, 1H), 7.80 (dd, J=8.6, 2.2 Hz, 1H), 7.66–7.61 (m, 2H), 7.23–7.10 (m, 4H), 6.99 (d, J=8.6 Hz, 1H).13C NMR (126 MHz, DMSO) δ 171.71, 161.45 (d, J=236.6 Hz), 160.84, 133.83, 132.82, 128.42, 128.20, 128.08 (d, J=7.3 Hz), 127.32, 125.48, 117.56, 115.51 (d, J=21.0 Hz), 113.61. HR‐MS (ESI): [M−H]−: Calcd for C15H10FO3 257.06149, found 257.06109.
Methyl(4‐vinylphenyl)sulfane 14 [18760‐11‐7]
Compound 11 (1.2 equiv., 2.23 g, 5.52 mmol) was slowly added to a suspension of NaH 60 % (3 equiv., 551 mg, 13.80 mmol) in anhydrous DCM (10 mL) and left for stirring in 30 min. at a temperature of at 0 °C. 4‐(Methylthio)benzaldehyde (700 mg, 4.60 mmol) was then added to the mixture and left for stirring for 16 h at 20 °C. The reaction mixture was quenched using a saturated solution of NaHCO3 in water. The crude mixture was extracted with DCM (3×250 mL), dried over Na2SO4, and purified by means of column chromatography packed with silica gel (eluent hexane : ethyl acetate=9 : 1) to achieve target compound 14 (668 mg, 4.39 mmol) as a transparent oil in a yield of 95 %.1H NMR (500 MHz, CDCl3) δ 7.38–7.30 (m, 2H), 7.24–7.17 (m, 2H), 6.67 (dd, J=17.6, 10.9 Hz, 1H), 5.70 (dd, J=17.6, 0.7 Hz, 1H), 5.21 (dd, J=10.9, 0.7 Hz, 1H), 2.48 (s, 3H).13C NMR (126 MHz, CDCl3) δ 138.00, 136.20, 134.59, 126.63 (4 C), 113.23, 15.86.
(E)‐(4‐(3‐bromo‐4‐(methoxymethoxy)styryl)phenyl)(methyl)sulfane 15 [NEW]
Methyl(4‐vinylphenyl)sulfane 14 (150 mg, 998.41 μmol), Pd(OAc)2 (8 %, 18 mg, 80 μmol), K2CO3 (2 equiv., 276 mg, 2.00 mmol), 7 b (1 equiv., 342 mg, 998.41 μmol) and DMF (7 mL) were transferred to a sealed tube reactor. The tube reactor was flushed with Argon, sonicated for 5 min. and then immersed into the reactor cavity of a microwave oven that heated at 100 °C for 4 h. The progress of the reaction was monitored by TLC using hexane : ethyl acetate=9 : 1 as eluent. The solvent was removed under reduced pressure and the mixture was extracted with DCM (3×50 mL). The combined organic phases were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The isolated crude was purified by means of column chromatography using silica gel with hexane : ethyl acetate=99 : 1 as eluent to obtain target compound 15 as a white solid in a yield of 44 % (163 mg, 446.23 μmol). 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J=2.1 Hz, 1H), 7.39–7.34 (m, 2H), 7.31 (dd, J=8.6, 2.1 Hz, 1H), 7.22–7.17 (m, 2H), 7.09 (d, J=8.5 Hz, 1H), 6.89 (s, 2H), 5.22 (s, 2H), 3.50 (s, 3H), 2.46 (s, 3H).13C NMR (126 MHz, CDCl3) δ 153.11, 138.03, 134.01, 132.89, 130.95, 127.79, 126.86, 126.67, 126.64, 126.03, 116.12, 113.26, 95.13, 56.43, 15.77.
(E)‐2‐hydroxy‐5‐(4‐(methylthio)styryl)benzoic acid DC08 [NEW]
Oxalic acid (3 equiv., 59 mg, 0.657 mmol), Pd(OAc)2 (5 %, 3 mg, 10.95 μmol), Xantphos (5 %, 6 mg, 10.95 μmol), 15 (80 mg, 0.219 mmol), water (10 equiv., 40 mg, 2.19 mmol), and DMF (3 mL) were transferred to a reactor tube that was sealed and left for stirring in a preheated oil bath at 100 °C. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). TEA (3 equiv., 67 mg, 91 μL, 657 μmol) was then added. Ac2O (3 equiv., 67 mg, 62 μL, 657 μmol) were added over a period of over 15 min. by means of a syringe pump. During the addition CO was produced due to degradation of oxalic acid. The reaction mixture was stirring at 100 °C for 3 h. The reaction mixture was extracted DCM (3×50 mL) from acid water. The organic phases were combined, dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure. The isolated crude was purified by means of a chromatography column packed with silica gel using DCM : methanol=95 : 5 as eluent to obtain title compound DC08 (37 mg, 0.129 mmol) as a white solid in a yield of 59 %. 1H NMR (500 MHz, DMSO) δ 7.96 (d, J=2.3 Hz, 1H), 7.80 (dd, J=8.7, 2.3 Hz, 1H), 7.56–7.49 (m, 2H), 7.28–7.23 (m, 2H), 7.20 (d, J=16.5 Hz, 1H), 7.09 (d, J=16.5 Hz, 1H), 6.98 (d, J=8.6 Hz, 1H), 2.49 (s, 3H).13C NMR (126 MHz, DMSO) δ 171.72, 160.56, 137.16, 133.90, 132.96, 128.54, 128.32, 126.76, 126.55, 126.27, 126.08, 117.61, 113.18, 30.65. HR‐MS (ESI): [M−H]−: Calcd for C16H13O3S 285.05854, found 285.05887.
(E)‐4‐(3‐bromo‐4‐(methoxymethoxy)styryl)‐1,1′‐biphenyl 16 [NEW].
Compound 12 (500 mg, 2.81 mmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 33 mg, 0,140 mmol), Pd(OAc)2 (5 %, 32 mg, 0,140 mmol), [1,1′‐biphenyl]‐4‐ylboronic acid (1.1 equiv., 611 mg, 3.09 mmol), Cu(OAc)2 monohydrate (1.5 equiv., 764 mg, 4,21 mmol) in 10 mL of DMF were added to a flask (25 mL). The reaction mixture was stirred for 18 h at 20 °C and monitored by means TLC, using hexane : ethyl acetate=9 : 1 as eluent. The mixture was then extracted with DCM (3×100 mL) from a solution EDTA in water (0.2 M). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture was purified by means of column chromatography packed with silica gel (eluent hexane : ethyl acetate=99.6 : 0.4). The target compound 16 (910 mg, 2.30 mmol) was isolated as a white solid in a yield of 82 %. 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J=2.1 Hz, 1H), 7.63–7.52 (m, 6H), 7.44 (dd, J=10.6, 4.8 Hz, 2H), 7.38 (dd, J=8.5, 2.2 Hz, 1H), 7.37–7.32 (m, 1H), 7.14 (d, J=8.5 Hz, 1H), 7.02 (s, 2H), 5.26 (s, 2H), 3.53 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 153.20, 140.64, 140.42, 136.15, 132.89, 131.05, 128.83, 127.96, 127.39, 127.37, 126.93, 126.89, 126.74, 126.73, 116.14, 113.28, 95.14, 56.43.
(E)‐5‐(2‐([1,1′‐biphenyl]‐4‐yl)vinyl)‐2‐hydroxybenzoic acid DC10 [NEW]
Oxalic acid (3 equiv., 63 mg, 759 μmol), Pd(OAc)2 (5 %, 3 mg, 13 μmol), Xantphos (5 %, 7 mg, 13 μmol), 16 (100 mg, 314 μmol), H2O (10 equiv., 45 mg, 45 μL, 2.53 mmol) and DMF (3 mL) were transferred to a reactor tube, that was sealed and stirred at 100 °C whereupon TEA (3 equiv., 77 mg, 106 μL, 759 μmol) was added. Ac2O (3 equiv., 77 mg, 74 μL, 759 μmol) was subsequently added over a period of 15 min by means of a syringe pump. During the addition, CO was produced due to degradation of oxalic acid. The reaction mixture was stirred at 100 °C for 3 h. The reaction mixture was transferred to a separatory funnel (250 mL) that was charged with DCM (50 mL) and water (50 mL). The water phase was adjusted to pH 12 and extracted with DCM (3×50 mL). The water phase was then adjusted to pH 1 and extracted DCM (3×50 mL). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain the desired product DC10 (37 mg, 117 μmol) as a white solid in a yield of 46 %. 1H NMR (500 MHz, DMSO) δ 8.01 (d, J=2.2 Hz, 1H), 7.84 (dd, J=8.7, 2.3 Hz, 1H), 7.73–7.66 (m, 6H), 7.51–7.43 (m, 2H), 7.39–7.34 (m, 1H), 7.31 (d, J=16.5 Hz, 1H), 7.19 (d, J=16.5 Hz, 1H), 7.00 (d, J=8.6 Hz, 1H). 13C NMR (126 MHz, DMSO) δ 171.72, 160.69, 139.64, 138.88, 136.38, 133.08, 128.92, 128.51, 128.50, 127.43, 127.40, 126.86, 126.84, 126.40, 126.31, 117.65, 113.20. HR‐MS (ESI): [M−H]−: Calcd for C21H15O3 315.10212, found 315.10201.
2‐(3‐bromo‐4‐(methoxymethoxy)phenyl)naphthalene 17 [NEW]
Naphthalen‐2‐ylboronic acid (1 equiv., 250 mg, 1.46 mmol), K2CO3 (2 equiv., 402 mg, 2.92 mmol), and Pd(PPh3)4 (5 %, 84 mg, 73 μmol) were added to a solution of compound 7 b (500 mg, 1.46 mmol) in DMF (10 mL). The reaction mixture was flushed with argon and stirred at 100 °C for 2 h. The progress of the reaction was monitored by TLC using hexane : ethyl acetate=9 : 1 as eluent. The solvent was removed under reduced pressure and the mixture was extracted with DCM (3×50 mL) from water. The organic phases were combined, dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure. The product was purified by means of column chromatography packed with silica gel using hexane : ethyl acetate=99.5 : 0.5 as eluent. Target compound 17 (361 mg, 1.05 mmol) was obtained as a yellow solid in a yield of 72 %. 1H NMR (500 MHz, CDCl3) δ 7.93 (d, J=1.4 Hz, 1H), 7.90 (d, J=2.3 Hz, 1H), 7.87–7.79 (m, 3H), 7.63 (dd, J=8.5, 1.9 Hz, 1H), 7.55 (dd, J=3.8, 1.9 Hz, 1H), 7.50–7.42 (m, 2H), 6.86 (d, J=8.6 Hz, 1H), 5.27 (s, 2H), 3.53 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 153.86, 136.67, 136.36, 133.68, 132.62, 132.14, 128.61, 128.20, 127.70, 127.34, 126.46, 126.08, 125.46, 125.14, 117.87, 116.38, 84.30, 56.47.
2‐hydroxy‐5‐(naphthalen‐2‐yl)benzoic acid DC11 [151826‐05‐0]
Oxalic acid (3 equiv., 79 mg, 874 μmol), Pd(OAc)2 (5 %, 3 mg, 15 μmol), Xantphos (5 %, 8 mg, 15 μmol), compound 17 (100 mg, 291 μmol), H2O (10 equiv., 53 mg, 53 μL, 2.91 mmol), and DMF (4 mL) were transferred to a reaction tube that was sealed and left for stirring in a preheated oil bath at 100 °C. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). TEA (3 equiv., 95 mg, 131 μL, 874 μmol) was then added. Ac2O (3 equiv., 96 mg, 89 μL, 874 μmol) was then added over a period of 15 min. by means of a syringe pump. During the addition, CO was produced due to degradation of oxalic acid. The reaction mixture was continuously stirred at 100 °C for 3 h. The reaction mixture was transferred to a separatory funnel (250 mL) that was charged with DCM (50 mL) and water (50 mL). The water phase was adjusted to pH 12 and extracted with DCM (3×50 mL). The water phase was then adjusted to pH 1 and extracted again with DCM (3×50 mL). The organic phases were combined and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the target product DC11 (41 mg, 155 μmol) as a white solid in a yield of 49 %. 1H NMR (500 MHz, DMSO) δ 8.20 (d, J=2.4 Hz, 1H), 8.18 (d, J=1.4 Hz, 1H), 8.02–7.92 (m, 4H), 7.82 (dd, J=8.6, 1.9 Hz, 1H), 7.56–7.48 (m, 2H), 7.12 (d, J=8.6 Hz, 1H). 13C NMR (126 MHz, DMSO) δ 171.73, 160.66, 136.31, 134.11, 133.36, 131.98, 131.04, 128.53, 128.22, 128.08, 127.44, 126.40, 125.96, 124.67, 124.47, 117.92 (one of the carbon peaks is not visible). HR‐MS (ESI): [M−H]−: Calcd for C17H11O3 263.07082, found 263.07098.
(E)‐2‐ammino‐5‐styrylbenzoic acid DC12 [NEW]
(E)‐styrylboronic acid (1 equiv., 56 mg, 0.380 mmol), Pd(PPh3)4 (5 %, 22 mg, 19 μmol) and Na2CO3 (2 equiv., 81 mg, 0.760 mmol) in water (2 mL) and EtOH (1 mL) were added to a solution of 2‐amino‐5‐iodobenzoic acid (100 mg, 0.380 mmol) in toluene (3 mL). The reaction mixture was flushed with argon and stirring and heated at 100 °C for 2 h. The progress of the reaction was monitored by TLC with DCM : MeOH=9 : 1 as eluent. The solvent was then removed under reduced pressure. The obtained crude was purified using chromatography column packed with silica gel using DCM : MeOH=9.5 : 0.5 as eluent with 0.1 % of formic acid present. The target compound DC12 (46 mg, 0.192 mmol was obtained as a yellow solid with a yield of 50 %. 1H NMR (400 MHz, CD3CN) δ 7.93 (d, J=2.1 Hz, 1H), 7.58 (dd, J=2.1, 8.7 Hz, 1H), 7.52 (d, J=7.4 Hz, 2H), 7.34 (t, J=7.6 Hz, 2H), 7.23 (d, J=7.3 Hz, 1H), 7.11 (d, J=16.4 Hz, 1H), 6.98 (d, J=16.5 Hz, 1H), 6.77 (d, J=8.6 Hz, 1H). 13C NMR (126 MHz, CD3CN) δ 169.35, 151.84, 138.51, 132.20, 130.86, 129.22, 128.61, 127.52, 126.56, 125.65, 125.22, 117.61, 109.78. HR‐MS (ESI): [M+H]+: Calcd for C15H14NO2, 240.10245 found 240.10267; [M−CO2−H]+: Calcd for C14H12N, 194.09697 found 194.09691.
(E)‐2‐hydroxy‐5‐(4‐(methylsulfonyl)styryl)benzoic acid DC13 [NEW]
Oxone (3 equiv., 120 mg, 0.196 mmol) in water (1 mL) was slowly added to a solution of compound DC08 (15 mg, 52 μmol) in THF (1 mL) and methanol (1 mL). The reaction mixture was stirred for 24 h at 20 °C. Acidic water (50 mL) was added to the post reaction mixture and extracted with DCM (3×50 mL). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified using a chromatography column packed with silica gel (DCM : methanol=95 : 5) to afford target DC13 as an off‐white solid in a yield of 85 % (14 mg, 44 μmol). 1H NMR (500 MHz, DMSO) δ 8.05 (d, J=2.2 Hz, 1H), 7.91–7.80 (m, 5H), 7.48 (d, J=16.5 Hz, 1H), 7.26 (d, J=16.5 Hz, 1H), 7.02 (d, J=8.6 Hz, 1H), 3.22 (s, 3H).13C NMR (126 MHz, DMSO) δ 171.62, 161.25, 142.40, 138.82, 133.38, 131.02, 129.20, 127.79, 127.37, 126.76, 124.99, 117.75, 113.38, 43.58. HR‐MS (ESI): [M−H]−: Calcd for C16H13O5S 317.04837, found 317.04806.
4‐iodo‐N‐phenylbenzenesulfonamide 18 [21226‐28‐8]
4‐Iodobenzenesulfonyl chloride (1.62 g, 5.37 mmol) and TEA (1.1 equiv., 597 mg, 823 mL, 5.91 mmol) were transferred to a flask that contained anhydrous DCM (20 mL). The reaction was cooled at 0 °C whereupon aniline (1 equiv., 500 mg, 5.37 mmol) was added dropwise. The reaction was stirred at 20 °C in 18 h. The mixture was then extracted with DCM (3×250 mL) from solution of HCl (0.1 M). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the target product compound 18 (1.76 g, 4.90 mmol) as a yellow oil in a yield of 91 %. 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J=8.5 Hz, 2H), 7.45 (d, J=8.5 Hz, 2H), 7.27 (t, J=7.6 Hz, 2H), 7.15 (t, J=7.6 Hz, 1H), 7.06 (d, J=7.6 Hz, 2H), 6.59 (s, 1H).13C NMR (126 MHz, CDCl3) δ 138.67, 138.34, 136.02, 129.51, 128.60, 125.84, 121.90, 100.68.
(E)‐4‐(3‐bromo‐4‐(methoxymethoxy)styryl)‐N‐phenylbenzenesulfonamide 19 [NEW]
4‐Iodo‐N‐phenylbenzenesulfonamide 18 (700 mg, 1.95 mmol), Pd(OAc)2 (8 %, 35 mg, 156 μmol), K2CO3 (2 equiv., 539 mg, 3.90 mmol), 7 b (1 equiv., 474 mg, 1.95 mmol) and DMF (10 mL) were transferred to a reactor tube that was sealed. The reactor tube was flushed with Argon and sonicated for 5 min. using an ultrasonic bath. The reactor tube was then immersed into the microwave reactor cavity of a microwave oven for 4 h at 100 °C. The reaction progress was monitored by means of TLC, using hexane : ethyl acetate=9 : 1. The solvent was removed under reduced pressure. The mixture was extracted with DCM (3×100 mL) from water using DCM. The combined organic phases were dried over anhydrous Na2SO4, filtered, and then the solvent was removed under reduced pressure. The crude was purified using column chromatography packed with silica gel and hexane : ethyl acetate=9 : 1 as eluent to obtain target compound 15 (365 mg, 769 μmol) as a white solid with a yield of 39 %. 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J=8.5 Hz, 2H), 7.66 (d, J=2.1 Hz, 1H), 7.62 (s, 1H), 7.42 (d, J=8.5 Hz, 2H), 7.29 (dd, J=2.1, 8.6 Hz, 1H), 7.18–7.23 (m, 2H), 7.12–7.17 (m, 2H), 7.03–7.12 (m, 2H), 6.96 (d, J=16.3 Hz, 1H), 6.84 (d, J=16.3 Hz, 1H), 5.22 (s, 2H), 3.49 (s, 3H). 13C NMR (126 MHz, CDCl3, 27 °C) δ 153.84, 141.78, 137.37, 136.70, 131.80, 131.40, 130.14, 129.37, 127.79, 127.24, 126.72, 126.27, 125.30, 121.53, 116.02, 113.31, 95.04, 56.46.
(E)‐2‐hydroxy‐5‐(4‐(N‐phenylsulfamoyl)styryl)benzoic acid DC14 [NEW]
Oxalic acid (3 equiv., 51 mg, 570 μmol), Pd(OAc)2 (5 %, 2 mg, 9 μmol), Xantphos (5 %, 5 mg, 9 μmol), compound 19 (90 mg, 189 μmol), H2O (10 equiv., 34 mg, 34 μL, 1.90 mmol), and DMF (4 mL) were transferred to a reaction tube that then was sealed. The reaction mixture was heated at 100 °C and stirred and TEA (3 equiv., 57 mg, 79 μL, 570 μmol) was added. Then, Ac2O (3 equiv., 58 mg, 54 μL, 570 μmol) was added over a period of 15 min. by means of a syringe pump. CO was produced due to degradation of oxalic acid. It is advisable to add a balloon to the sealed vial in order to avoid problem with the increasing pressure. The reaction was stirred at 100 °C for 3 h, and then extracted with DCM (3×50 mL) from HCl (0.1 M). The combined organic phases were dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified through chromatography column packed with silica gel using DCM : methanol=9 : 1 and 1 % formic acid as eluent to afford the title compound DC14 (39 mg, 98 μmol) with a yield of 52 %. 1H NMR (500 MHz, DMSO) δ 10.23 (s, 1H), 7.99 (s, 1H), 7.80 (d, J=8.4 Hz, 1H), 7.71 (s, 4H), 7.38 (d, J=16.4 Hz, 1H), 6.96–7.27 (m, 7H). 13C NMR (126 MHz, DMSO, 27 °C) δ 171.58, 160.78, 141.69, 137.71, 137.48, 134.46, 133.22, 130.85, 129.13, 128.69, 127.13, 126.62, 126.00, 124.89, 124.06, 120.09, 117.74. [M−H]−: Calcd for C21H16NO5S 394.07492, found 394.07482.
5‐iodo‐2‐(methoxymethoxy)benzonitrile 20 [2090370‐07‐1]
2‐Hydroxybenzonitrile (2.0 g, 16.79 mmol) in acetonitrile (15 mL) cooled at 0 °C was transferred to a flask (25 mL) that was wrapped in aluminum foil. para‐Toluenesulfonic acid monohydrate (1.1 equiv., 3.51 g, 18.47 mmol) and by N‐iodosuccinimide (1.1 equiv., 4.16 g, 18.47 mmol) were added to the mixture that was stirred at 20 °C for 18 h. The progress of the reaction was monitored by TLC using hexane : ethyl acetate=8 : 2 as eluent. The solvent was removed under reduced pressure. The crude was diluted with DCM and a saturated solution of Na2S2O3 in water. The aqueous phase was extracted further with DCM (2×250 mL). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and then concentrated under reduced pressure to obtain 2‐hydroxy‐5‐benzonitrile that was dissolved in anhydrous DCM (20 mL) to proceed with the following reaction. The same flask was cooled at 0 °C and NaH 60 % (3 equiv., 2.01 g, 50.32 mmol) and chloromethyl methyl ether (1.5 equiv., 2.03 g, 1.91 mL, 25.16 mmol) were added. The reaction was heated to 20 °C for 1 h and then quenched using a sat. NaHCO3 (aq.). The mixture was extracted with DCM (3×250 mL). The organic phases were combined over anhydrous Na2SO, filtered, and concentrated under reduced pressure to obtain compound 21 (4.1 g, 14.18 mmol) as a yellow oil in a yield of 84 %. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J=2.2 Hz, 1H), 7.78 (dd, J=2.2, 8.9 Hz, 1H), 7.03 (d, J=8.9 Hz, 1H), 5.28 (s, 2H), 3.51 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 158.89, 143.03, 141.48, 117.02, 114.73, 105.14, 94.91, 82.89, 56.75.
2‐(methoxymethoxy)‐5‐vinylbenzonitrile 21 [NEW]
Compound 20 (1.00 g, 3.46 mmol), Pd(PPh3)2 (8 %, 319 mg, 276 μmol), tributyl(vinyl)stannane (1.1 equiv., 1.21 g, 3.81 mmol) and DMF (10 mL) were transferred to reactor tube that then was sealed. The mixture was flushed with Argon, and sonicated for 5 min. Then the reactor tube was immersed into the reactor cavity of a microwave oven and heated at 100 °C for 2 h. The progress of the reaction was monitored by TLC with hexane : ethyl acetate=9 : 1 as eluent. The solvent was removed under reduced pressure and the mixture was extracted DCM (3×100 mL) from water. The combined organic phases were dried over anhydrous Na2SO4, filtred, and the solvent was removed under reduced pressure. The crude was purified using a chromatography column packed with silica gel using hexane : ethyl acetate=95 : 5 as eluent to obtain target compound 21 (603 mg, 3.19 mmol) as a yellow oil in a yield of 92 %. 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J=2.3 Hz, 1H), 7.43 (dd, J=2.3, 8.8 Hz, 1H), 7.08 (d, J=8.8 Hz, 1H), 6.50 (dd, J=10.9, 17.6 Hz, 1H), 5.56 (d, J=17.5 Hz, 1H), 5.14–5.18 (m, 4H), 3.40 (s, 3H).13C NMR (126 MHz, CDCl3) δ 158.38, 134.25, 131.87, 131.00, 121.91, 116.16, 115.05, 114.50, 103.01, 94.84, 56.53.
(E)‐2‐(methoxymethoxy)‐5‐styrylbenzonitrile 22 [NEW]
Compound 21 (250 mg, 1.32 mmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 16 mg, 66 μmol), Pd(OAc)2 (5 %, 15 mg, 66 μmol), phenylboronic acid (1.1 equiv., 177 mg, 1.45 mmol), Cu(OAc)2 monohydrate (1.5 equiv., 396 mg, 1.98 mmol) and DMF (6 mL) were transferred to a flask. The reaction mixture was stirred at 20 °C in 18 h. The progress of the reaction was monitored by TLC using hexane : ethyl acetate=9 : 1 as eluent. The mixture was extracted with DCM (3×50 mL) from a solution of EDTA in water (0.2 M). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture is purified using a column chromatography packed with silica gel with using hexane : ethyl acetate=99.4 : 0.6 as eluent. Target compound 22 (288 mg, 1.09 mmol) was obtained as a white solid in a yield of 82 %. 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J=2.2 Hz, 1H), 7.64 (dd, J=2.3, 8.8 Hz, 1H), 7.48–7.51 (m, 2H), 7.32–7.4 (m, 2H), 7.27–7.31 (m, 1H), 7.23 (d, J=8.8 Hz, 1H), 7.01 (d, J=3.5 Hz, 2H), 5.31 (s, 2H), 3.54 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 158.23, 136.69, 132.10, 131.79, 131.21, 129.45, 128.82, 128.08, 126.57, 125.86, 116.28, 115.24, 103.30, 94.90, 56.67.
(E)‐4‐styryl‐2‐(1H‐tetrazol‐5‐yl)phenol DC15 [NEW]
Compound 22 (90 mg, 339 μmol), sodium azide (1.1 equiv., 24 mg, 373 μmol), ammonium chloride (10 %, 2 mg, 34 μmol) and LiCl (1 equiv., 14 mg, 339 μmol) and DMF (3 mL) were transferred to a reactor tube that was sealed and flushed with Ar, whereupon the mixture was heated at reflux for 18 h. The crude was extracted with DCM (3×50 mL) from HCl (0.1 M). The combined organic phases were dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified through chromatography column packed with silica gel using formic acid (0.1 %) in DCM as eluent to obtain target compound DC15 (47 mg, 178 μmol) as an off white solid in a yield of 52 %. 1H NMR (500 MHz, DMSO) δ 8.22 (d, J=2.1 Hz, 1H), 7.71 (dd, J=2.1, 8.7 Hz, 1H), 7.61 (d, J=7.5 Hz, 2H), 7.38 (t, J=7.6 Hz, 2H), 7.24–7.33 (m, 2H), 7.04–7.19 (m, 2H). 13C NMR (126 MHz, DMSO) δ 154.95, 137.18, 130.36, 128.93, 128.67, 127.40, 127.34, 127.21, 126.71, 126.28, 116.77, 110.90. [M−H]−: Calcd for C15H11N4O1 263.09329, found 263.09343.
N‐(2‐bromo‐4‐formylphenyl)acetamide 23 [475150‐63‐1]
N‐(4‐formylphenyl)acetamide (2.0 g, 12.26 mmol) in water (20 mL) were transferred to a aluminum foil wrapped flask. N‐Bromosuccinimide (1.1 equiv., 2.40 g, 13.48 mmol) was then slowly added to the reaction mixture that was stirred at 20 °C for 18 h. The progress of the reaction was monitored by using TLC using hexane : ethyl acetate=8 : 2 as eluent. The solvent was removed under reduced pressure. The crude was diluted with DCM (100 mL) and a saturated solution of Na2S2O3 in water (100 mL). The aqueous phase was extracted further with DCM (2×100 mL). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain target compound 23 (2.76 g, 11.40 mmol) as yellow oil and in a yield of 93 %. 1H NMR (500 MHz, CDCl3) δ 9.88 (s, 1H), 8.63 (d, J=8.5 Hz, 1H), 8.09 (d, J=1.9 Hz, 1H), 7.82 (dd, J=1.8, 8.5 Hz, 1H), 2.77 (s, 1H), 2.30 (s, 3H). Data consistent with literature.
N‐(2‐bromo‐4‐vinylphenyl)acetamide 24 [NEW]
A suspension of NaH 60 % (3 equiv., 2.50 g, 30.98 mmol) in anhydrous DCM (20 mL) was slowly added compound 11 (1.2 equiv., 5.01 g, 12.39 mmol) at 0 °C, which was stirred for 30 min. Then, compound 23 (2.50 g, 10.33 mmol) was added to the mixture, that was stirred at 20 °C for 16 h. The reaction mixture was then quenched using sat. NaHCO3 in water. The crude mixture was extracted with DCM (3×250 mL), dried over anhydrous Na2SO4, filter, and purified using column chromatography packed with silica gel with hexane : ethyl acetate=9 : 1 as eluent to obtain the desired compound 24 (2.01 g, 8.37 mmol) as transparent oil in a yield of 81 %. 1H NMR (500 MHz, CDCl3) δ 8.31 (d, J=8.4 Hz, 1H), 7.58 (d, J=1.8 Hz, 1H), 7.35 (dd, J=1.9, 8.5 Hz, 1H), 6.60 (dd, J=10.9, 17.6 Hz, 1H), 5.69 (d, J=17.5 Hz, 1H), 5.25 (d, J=10.9 Hz, 1H), 2.24 (s, 3H).
(E)‐N‐(2‐bromo‐4‐styrylphenyl)acetamide 25 [NEW]
Compound 24 (500 mg, 2.08 mmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 24 mg, 104 μmol), Pd(OAc)2 (5 %, 23 mg, 104 μmol), phenylboronic acid (1.1 equiv., 279 mg, 2.29 mmol), Cu(OAc)2 monohydrate (1.5 equiv., 623 mg, 3.12 mmol) were dissolved in DMF (8 mL). The reaction mixture was stirred at 20 °C for 18 h. The reaction progress was monitored by TLC using hexane : ethyl acetate=9 : 1 as eluent. The mixture was extracted with DCM (3×50 mL) from a solution EDTA in water (0.2 M). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture was purified column chromatography packed with silica gel using hexane : ethyl acetate=95 : 5 to obtain target product 25 (241 mg, 0,762 mmol) as a white solid in a yield of 36 %. 1H NMR (500 MHz, CDCl3) δ 8.27 (d, J=8.5 Hz, 1H), 7.59–7.63 (m, 1H), 7.55 (s, 1H), 7.41 (d, J=7.4 Hz, 2H), 7.36 (dd, J=1.8, 8.6 Hz, 1H), 7.28 (t, J=7.6 Hz, 2H), 7.15–7.23 (m, 1H), 6.86–6.99 (m, 2H), 2.16 (s, 3H).13C NMR (126 MHz, CDCl3, 27 °C) δ 168.10, 136.91, 134.83, 134.71, 131.45, 129.81, 129.26, 128.75, 127.91, 126.56, 126.52, 121.68, 113.42, 22.72.
(E)‐2‐acetamido‐5‐styrylbenzoic acid DC16 [380365‐20‐8]
Oxalic acid (3 equiv., 85 mg, 948 μmol), Pd(OAc)2 (5 %, 4 mg, 16 μmol), Xantphos (5 %, 9 mg, 16 μmol), compound 25 (100 mg, 316 μmol), H2O (10 equiv., 57 mg, 57 μL, 3.16 mmol) and DMF (5 mL) were transferred to a reaction tube, which was sealed and stirred and heated at 100 °C. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). TEA (3 equiv., 96 mg, 132 μL, 948 μmol) was then added. Ac2O (3 equiv., 97 mg, 89 μL, 948 μmol) was added over a period of 15 min. by means of a syringe pump. During the addition CO was formed through degradation of the oxalic acid. The reaction mixture was stirred and heated at 100 °C for 3 h. Then, the reaction mixture was extracted with DCM (3×50 mL) from HCl (0.1 M). The combined organic phases were dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified with a chromatography column packet with silica gel using DCM : methanol=9 : 1 and 1 % formic acid as eluent to obtain title compound DC16 (38 mg, 135 μmol) in a yield of 42 %. 1H NMR (500 MHz, DMSO) δ 11.26 (s, 1H), 8.49 (d, J=8.7 Hz, 1H), 8.17 (d, J=2.0 Hz, 1H), 7.85 (dd, J=2.0, 8.7 Hz, 1H), 7.62 (d, J=7.4 Hz, 2H), 7.38 (t, J=7.7 Hz, 2H), 7.19–7.32 (m, 4H), 2.15 (s, 3H). 13C NMR (126 MHz, DMSO) δ 169.33, 168.36, 140.04, 136.98, 131.32, 131.03, 129.26, 128.68, 128.36, 127.96, 127.59, 127.17, 126.44, 120.09, 25.01.
3‐bromo‐4‐(methoxymethoxy)‐1,1′ : 4′,1′′‐terphenyl 26 [NEW]
Compound 7 b (500 mg, 1.46 mmol) in DMF (10 mL) was added [1,1′‐byphenyl]‐4‐ylboronic acid (1 equiv., 288 mg, 1.46 mmol), K2CO3 (2 equiv., 402 mg, 2.92 mmol), and Pd(PPh3)4 (5 %, 84 mg, 73 μmol). The reaction mixture was flushed with argon and stirred for 2 h at 100 °C. The progress of the reaction was monitored by TLC using hexane : ethyl acetate=9 : 1 as eluent. The solvent was then removed under reduced pressure and the mixture was extracted with DCM (3×50 mL) from water. The organic phases were combined and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified using column chromatography packed with silica gel using hexane : ethyl acetate=99.5 : 0.5. Target compound 26 (234 mg, 0.633 mmol) was obtained as a yellow solid in a yield of 43 %. 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J=2.2 Hz, 1H), 7.55–7.67 (m, 6H), 7.49 (dd, J=2.3, 8.5 Hz, 1H), 7.44 (t, J=7.6 Hz, 2H), 7.31–7.38 (m, 1H), 7.21 (d, J=8.6 Hz, 1H), 5.27 (s, 2H), 3.54 (s, 3H). 13C NMR (126 MHz, CDCl3, 27 °C) δ 153.25, 140.59, 140.24, 138.29, 135.99, 131.82, 128.88, 127.61, 127.46, 127.18, 127.07, 126.98, 116.35, 113.36, 95.19, 56.47.
(E)‐3‐(3‐bromo‐4‐(methoxymethoxy)styryl)‐1,1′‐biphenyl 27 [NEW]
Compound 12 (500 mg, 2.06 mmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 24 mg, 102 μmol), Pd(OAc)2 (5 %, 23 mg, 102 μmol), [1,1′‐biphenyl‐3‐ylboronic acid (1.1 equiv., 448 mg, 2.26 mmol), and Cu(OAc)2 monohydrate (1.5 equiv., 615 mg, 3.09 mmol) were transferred to a reaction flask (25 mL) and dissolved in DMF (8 mL). The reaction mixture was stirred at 20 °C for 18 h. The progress of the reaction was monitored by TLC with hexane : ethyl acetate=9 : 1 as eluent. The mixture was extracted with DCM (3×50 mL) from a solution EDTA in water (0.2 M). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture was purified using column chromatography packed with silica gel with hexane : ethyl acetate=99.5 : 0.5 as eluent to obtain title compound 27 (698 mg, 1.77 mmol) as a white solid in a yield of 86 %. 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J=2.2 Hz, 1H), 7.71 (t, J=1.6 Hz, 1H), 7.61–7.67 (m, 2H), 7.37–7.51 (m, 7H), 7.16 (d, J=8.5 Hz, 1H), 7.07 (s, 2H), 5.28 (s, 2H), 3.55 (s, 3H).13C NMR (126 MHz, CDCl3, 27 °C) δ 153.27, 141.79, 141.09, 137.59, 132.83, 131.11, 129.16, 128.82, 128.38, 127.46, 127.22, 127.07, 126.79, 126.61, 125.39, 125.34, 116.14, 113.29, 95.15, 56.44.
(E)‐5‐(2‐([1,1′‐biphenyl]‐3‐yl)vinyl)‐2‐hydroxybenzoic acid DC18 [NEW]
Oxalic acid (3 equiv., 102 mg, 1.14 mmol), Pd(OAc)2 (5 %, 4 mg, 19 μmol), Xantphos (5 %, 11 mg, 19 μmol), compound 27 (150 mg, 379 μmol), H2O (10 equiv., 68 mg, 68 μL, 3.79 mmol), and DMF (7 mL) were transferred to a reactor tube that was sealed. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). The mixture was stirred and heated at 100 °C whereupon TEA (3 equiv., 115 mg, 158 μL, 1.14 mmol) was added. Ac2O (3 equiv., 116 mg, 107 μL, 1.14 mol) was then added over a period of 15 min. by means of a syringe pump. During the addition, CO was formed through the degradation of the oxalic acid. The reaction mixture was then stirred at 100 °C for 3 h. The reaction mixture was extracted with DCM (3×50 mL) from HCl (0.1 M). The combined organic phases were dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified using a chromatography column packed with silica gel with 1 % formic acid in DCM as eluent to obtain target compound DC18 (52 mg, 164 μmol) in a yield of 43 %. 1H NMR (500 MHz, DMSO) δ 7.96 (d, J=2.2 Hz, 1H), 7.84 (s, 1H), 7.78 (dd, J=2.3, 8.7 Hz, 1H), 7.66 (dd, J=1.0, 8.1 Hz, 2H), 7.52 (d, J=7.7 Hz, 1H), 7.48 (d, J=7.9 Hz, 1H), 7.37–7.44 (m, 3H), 7.3–7.35 (m, 2H), 7.16 (d, J=16.5 Hz, 1H), 6.94 (d, J=8.6 Hz, 1H).13C NMR (126 MHz, DMSO, 27 °C) δ 171.77, 160.69, 140.55, 140.06, 137.83, 133.11, 129.24, 128.88, 128.58, 128.53, 127.75, 127.52, 126.75, 126.71, 125.68, 125.38, 124.63, 117.67, 113.14. [M−H]−: Calcd for C21H15O3 315.10212, found 315.10192.
(E)‐2‐(3‐bromo‐4‐(methoxymethoxy)styryl)‐1,1′‐biphenyl 28 [NEW]
Compound 12 (500 mg, 2.06 mmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 24 mg, 102 μmol), Pd(OAc)2 (5 %, 23 mg, 102 μmol), [1,1′‐biphenyl‐2‐ylboronic acid (1.1 equiv., 448 mg, 2.26 mmol), and Cu(OAc)2 monohydrate (1.5 equiv., 615 mg, 3.09 mmol) were transferred to a flask and dissolved with DMF (8 mL) that was stirred at 20 °C for 18 h. The progress of the reaction was monitored by TLC using hexane : ethyl acetate=9 : 1 as eluent. The mixture was extracted with DCM (3×50 mL) from a solution EDTA in water (0.2 M). The organic phases were combined and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture was purified using column chromatography packed with silica gel using hexane : ethyl acetate=99.5 : 0.5 as eluent to obtain title compound 28 (654 mg, 1.65 mmol) as a white solid in a yield of 80 %. 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J=7.4 Hz, 1H), 7.46 (d, J=2.1 Hz, 1H), 7.23–7.39 (m, 9H), 7.13–7.17 (m, 1H), 6.99 (d, J=8.5 Hz, 1H), 6.79–6.94 (m, 2H), 5.14 (s, 2H), 3.42 (s, 3H).13C NMR (126 MHz, CDCl3) δ 153.04, 141.15, 140.82, 135.22, 133.22, 131.32, 130.37, 129.89, 128.20, 127.69, 127.64, 127.61, 127.58, 127.22, 126.49, 125.84, 116.15, 113.16, 95.11, 56.41.
(E)‐5‐(2‐([1,1′‐biphenyl]‐2‐yl)vinyl)‐2‐hydroxybenzoic acid DC19 [NEW]
Oxalic acid (3 equiv., 102 mg, 1.14 mmol), Pd(OAc)2 (5 %, 4 mg, 19 μmol), Xantphos (5 %, 11 mg, 19 μmol), 28 (150 mg, 379 μmol), H2O (10 equiv., 68 mg, 68 μL, 3.79 mmol) and DMF (7 mL) were transferred to a reactor tube that was sealed. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). The reaction mixture was stirred at 100 °C, whereupon TEA (3 equiv., 115 mg, 158 μL, 1.14 mmol) was then added. Ac2O (3 equiv., 116 mg, 107 μL, 1.14 mol) was added over a period of 15 min. using a syringe pump resulting in production of CO through degradation of the oxalic acid. The reaction was stirred at 100 °C for 3 h. The mixture was extracted with DCM (3×50 mL) from HCl (0.1 M). The combined organic phases were dried over anhydrous Na2SO4, filtered, and the solvent was then removed under reduced pressure. The crude was purified by column chromatography packed with silica gel using formic acid (1 %) in DCM as eluent to obtain the title compound DC19 (56 mg, 177 μmol) in a yield of 46 %.1H NMR (500 MHz, DMSO) δ 7.89 (d, J=7.5 Hz, 1H), 7.86 (d, J=2.2 Hz, 1H), 7.51–7.6 (m, 3H), 7.44–7.5 (m, 2H), 7.39–7.44 (m, 3H), 7.36 (dd, J=1.4, 7.6 Hz, 1H), 7.24 (d, J=16.4 Hz, 1H), 6.92–7.01 (m, 2H). 13C NMR (126 MHz, DMSO) δ 171.61, 160.68, 140.36, 134.64, 132.76, 131.58, 130.10, 129.52, 128.34, 128.31, 127.71, 127.52, 127.26, 125.67, 125.12, 117.74, 113.20. [M−H]−: Calcd for C21H15O3 315.10212, found 315.10188.
2‐bromo‐4‐iodo‐3,5‐dimethylphenol (29) [854672‐24‐5]
p‐Toluenesulfonic acid monohydrate (1.1 equiv., 844 mg, 4.43 mmol) and N‐Bromosuccinimide (1.1 equiv., 789 mg, 4.43 mmol), 4‐Iodo‐3,5‐dimethylphenol (1 g, 4.03 mmol), and acetonitrile (15 mL) werer transferred to a flask (25 mL) that was cooled at 0 °C. The reaction flask was wrapped in aluminum foil and the mixture was stirred at 20 °C for 18 h. The reaction mixture was monitored by means of TLC using as hexane : ethyl acetate as eluent. The solvent was then removed under reduced pressure, whereupon the isolated crude was diluted with DCM (50 mL) and a sat. Na2S2O3 (aq.) (50 mL). The aqueous phase was extracted with DCM (2×50 mL). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain the title compound 29 (1.23 g, 3.76 mmol) as yellow solid and in a yield of 93 %. 1H NMR (500 MHz, CDCl3) δ 6.87 (s, 2H), 5.68 (s, 2H), 2.72 (s, 6H), 2.42 (s, 7H).13C NMR (126 MHz, CDCl3) δ 151.98, 142.36, 140.50, 114.18, 109.05, 96.65, 30.74, 29.86.
2‐bromo‐4‐iodo‐1‐(methoxymethoxy)‐3,5‐dimethylbenzene (30) [NEW]
NaH 60 % (3 equiv., 451 mg, 11.29 mmol) and DCM (15 mL) were transferred to a flask under inert and anhydrous conditions. The suspension was cooled at 0 °C whereupon compound 29 (1.23 g, 3.76 mmol) was slowly added and the mixture was stirred for 10 min. Chloromethyl methyl ether (1.5 equiv., 454 mg, 0.428 mL, 5.64 mmol) was then added dropwise. The reaction mixture was then heated at 20 °C and stirred for 1 h. The reaction mixture was then quenched with a sat. NaHCO3 (aq.) and extracted with DCM (3×50 mL). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain compound 30 (1.27 g, 3.42 mmol) as a yellow oil in a yield of 91 %. 1H NMR (500 MHz, CDCl3) δ 6.95 (s, 1H), 5.22 (s, 2H), 3.51 (s, 3H), 2.75 (s, 3H), 2.45 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 153.42, 141.78, 141.67, 114.31, 111.50, 99.27, 95.08, 56.43, 30.85, 30.32.
2‐(3‐bromo‐4‐(methoxymethoxy)‐2,6‐dimethylphenyl)naphthalene (31) [NEW]
Compound 30 (1.27 g, 3.42 mmol) in DMF (15 mL) was added naphthalen‐2‐ylboronic acid (1 equiv., 589 mg, 3.42 mmol), K2CO3 (2 equiv., 946 mg, 6.85 mmol) and Pd(PPh3)4 (5 %, 198 mg, 171 μmol). The reaction mixture was then flushed with argon and stirred at 100 °C for 2 h. The reaction mixture was monitored by means of TLC using hexane : ethyl acetate=9 : 1 as eluent. The solvent was then removed under reduced pressure and the mixture was extracted with DCM (3×100 mL) from water. The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified using column chromatography packed with silica gel and hexane : ethyl acetate=99.5 : 0.5 as eluent. Target compound 31 (585 mg, 1.58 mmol) was obtained as a yellow solid in a yield of 46 %. 1H NMR (500 MHz, CDCl3) δ 7.87–7.80 (m, 1H), 7.59 (s, 1H), 7.56–7.49 (m, 2H), 7.24 (dd, J=8.3, 1.5 Hz, 1H), 7.00–6.97 (m, 1H), 5.31 (s, 2H), 3.60 (s, 3H), 2.17 (s, 3H), 2.01 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 152.64, 138.46, 137.62, 136.91, 136.30, 133.53, 132.37, 128.22, 128.09, 127.94, 127.82, 127.78, 126.20, 125.95, 114.75, 113.35, 95.19, 56.47, 21.85, 21.27.
6‐hydroxy‐2,4‐dimethyl‐3‐(naphthalen‐2‐yl)benzoic acid (DC20) [NEW]
Oxalic acid (3 equiv., 291 mg, 3.23 mmol), Pd(OAc)2 (5 %, 12 mg, 54 μmol), Xantphos (5 %, 32 mg, 54 μmol), 31 (400 mg, 1.08 mmol), H2O (10 equiv., 194 mg, 194 μL, 10.8 mmol) and DMF (7 mL) was transferred to a reaction tube that was sealed. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). The reaction mixture was then stirred and heated at 100 °C whereupon TEA (3 equiv., 327 mg, 450 μL, 3.23 mmol) was added. Ac2O (3 equiv., 329 mg, 305 μL, 2.23 mol) was added over a period of 15 min. by means of syringe pump. The reaction was stirring at 100 °C for 3 h. The mixture was extracted with DCM (3×50 mL) from HCl 0.1 M. The combined organic phases were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified using a chromatography column packed with silica gel with 1 % formic acid in DCM as eluent to obtain target compound DC20 (65 mg, 222 μmol) in a yield of 20 %. 1H NMR (600 MHz, DMSO) δ 7.99–7.90 (m, 3H), 7.65 (d, J=0.9 Hz, 1H), 7.56–7.52 (m, 2H), 7.25 (dd, J=8.3, 1.7 Hz, 1H), 6.69 (s, 1H), 1.98 (s, 3H), 1.91 (s, 3H). 13C NMR (151 MHz, DMSO) δ 171.09, 155.69 138.80, 138.37, 134.71, 133.64, 133.10, 132.26, 128.66, 128.55, 128.44, 128.26, 128.04, 126.61, 126.35, 119.71, 115.14, 21.50, 19.01. [M−H]−: Calcd for C19H15O3 291.10212, found 291.10244. [M−CO2−H]−: Calcd for C18H15O 247.11229 found 247.11603.
(E)‐2‐(3‐bromo‐4‐(methoxymethoxy)styryl)‐9,9‐dimethyl‐9H‐fluorene 32 [NEW]
Compound 12 (500 mg, 2.06 mmol), 2,3‐di(pyridin‐2‐yl)pyrazine (5 %, 24 mg, 102 μmol), Pd(OAc)2 (5 %, 23 mg, 102 μmol), (9,9‐dimethyl‐9H‐fluorene‐2‐yl)boronic acid (1.1 equiv., 538 mg, 2.26 mmol), Cu(OAc)2 monohydrate (1.5 equiv., 615 mg, 3.09 mmol) were transferred to a reactor tube and dissolved in DMF (8 mL) and then stirred for 18 h at 20 °C. The progress of the reaction was monitored by means of TLC using hexane : ethyl acetate=9 : 1 as eluent. The mixture was extracted with DCM (3×50 mL) from a solution EDTA in water (0.2 M). The organic phases were combined, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture was purified using a chromatography column packed with silica gel with hexane : ethyl acetate=99.5 : 0.5 as eluent to obtain title compound 32 (803 mg, 1.84 mmol) as a white solid in a yield of 89 %. 1H NMR (500 MHz, CDCl3) δ 7.64 (d, J=2.1 Hz, 1H), 7.55–7.6 (m, 1H), 7.54 (d, J=7.8 Hz, 1H), 7.43–7.46 (m, 1H), 7.31 (s, 2H), 7.24 (dd, J=2.1, 8.5 Hz, 1H), 7.17–7.21 (m, 2H), 7.00 (d, J=8.5 Hz, 1H), 6.93 (d, J=7.7 Hz, 2H), 5.11 (s, 2H), 3.39 (s, 3H), 1.39 (s, 6H).13C NMR (126 MHz, CDCl3, 27 °C) δ 154.22, 153.97, 153.18, 139.14, 138.95, 136.37, 133.15, 131.07, 128.96, 127.39, 127.15, 126.71, 126.14, 125.97, 122.68, 120.47, 120.35, 120.12, 116.20, 113.36, 95.18, 56.45, 46.85, 27.29.
(E)‐5‐(2‐(9,9‐dimethyl‐9H‐fluoren‐2‐yl)vinyl)‐2‐hydroxybenzoic acid DC21 [NEW]
Oxalic acid (3 equiv., 102 mg, 1.14 mmol), Pd(OAc)2 (5 %, 4 mg, 19 μmol), Xantphos (5 %, 11 mg, 19 μmol), compound 32 (165 mg, 379 μmol), H2O (10 equiv., 68 mg, 68 μL, 3.79 mmol), and DMF (7 mL) were transferred to a reactor tube that was sealed and stirred at 100 °C. The septum was penetrated with a syringe needle that was attached to a balloon (for pressure equalization during the in‐situ CO production). TEA (3 equiv., 115 mg, 158 μL, 1.14 mmol) was then added. Ac2O (3 equiv., 116 mg, 107 μL, 1.14 mol) was added over a period of 15 min. by means of a syringe pump. The reaction mixture was then stirred at 100 °C for 3 h. The reaction mixture was extracted with DCM (3×50 mL) from HCl 0.1 M. The organic phases were combined and dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by means of a chromatography column packed with silica gel with 1 % formic acid in DCM as eluent to obtain title compound DC21 (61 mg, 171 μmol) with a yield of 45 % 1H NMR (500 MHz, DMSO) δ 8.05 (d, J=2.2 Hz, 1H), 7.82–7.86 (m, 2H), 7.77–7.82 (m, 2H), 7.51–7.56 (m, 2H), 7.28–7.39 (m, 3H), 7.22 (d, J=16.4 Hz, 1H), 7.02 (d, J=8.6 Hz, 1H), 1.47 (s, 6H). 13C NMR (126 MHz, DMSO) δ 171.82, 160.63, 153.80, 153.52, 138.34, 137.95, 136.52, 132.96, 128.71, 128.45, 127.24, 127.21, 127.04, 126.84, 125.94, 122.73, 120.29, 120.27, 120.04, 117.69, 113.17, 26.86. [M−H]−: Calcd for C24H19O3 355.13342, found 355.13312.
Biology
Determination of glutathione level
The cancer cell lines A172, A375, U87 and MCF7 were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10 % fetal bovine serum, 2 % v/v L‐glutamine, 3.2 % v/v nonessential amino acids, and antibiotics 2 % v/v (penicillin‐streptomycin), 0.02 % v/v Plasmocin at 37 °C and 5 % CO2. xCT expression of cancer cell lines was evaluated by western blotting from protein samples of cell lysates as described before. [5] In brief, protein concentrations were determined by BCA assay (Thermo Fisher Scientific, USA) followed by SDS PAGE and immunoblotting using rabbit anti‐xCT (Thermo Fisher Scientific) and rabbit anti‐GAPDH (Abcam, Cambridge, UK) as a loading control. The analogues were dissolved in dimethylsulfoxide (DMSO) and stock concentrations of 100 mM were stored at −20 °C in aliquots. Final concentrations were made in growth medium before adding to the cell monolayers. The test compounds were evaluated based on the measurement of intracellular Glutathione that was performed by the glutathione‐Glo assay (Promega, Madison, WI, USA, cat#V6912) according to the manufacturer's recommended instruction. A flat bottomed 96‐well plate was seeded with 5000 cells per well. After attachment to the well surface, cells were exposed to 250 μM, 500 μM of each analogue for 48 h. After that, glutathione‐Glo reagent was added followed by short incubation and addition of luciferin detection reagent. Next, bioluminescence was measured using a luminometer. [29] All measurements were normalized to the untreated control and were performed three times.
For the cell viability studies using normal human astrocytes (NHA), the cells were obtained from Applied Biological Materials Inc., Vancouver, Canada.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
D.C. acknowledges for his ESR grant through the PET3D project that was funded by the European Commission under the H2020 – MSCA‐ITN‐2015 programme grant agreement no 675417. S. S. acknowledges for his PhD grant funded by Department of Biomedicine at the University of Bergen.
D. Cirillo, S. Sarowar, P. Øyvind Enger, H.-R. Bjørsvik, ChemMedChem 2021, 16, 2650.
References
- 1. Bröer S., Wagner C., Cell Biochem. Biophys. 2002, 36, 155–168. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Hayes J. D., McMahon M., Trends Biochem. Sci. 2009, 34, 176–188; [DOI] [PubMed] [Google Scholar]
- 2b. Diehn M., Cho R. W., Lobo N. A., Kalisky T., Dorie M. J., Kulp A. N., Qian D., Lam J. S., Ailles L. E., Wong M., Joshua B., Kaplan M. J., Wapnir I., Dirbas F. M., Somlo G., Garberoglio C., Paz B., Shen J., Lau S. K., Quake S. R., Brown J. M., Weissman I. L., Clarke M. F., Nature 2009, 458, 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lluis J. M., Buricchi F., Chiarugi P., Morales A., Fernandez-Checa J. C., Cancer Res. 2007, 67, 7368–7377. [DOI] [PubMed] [Google Scholar]
- 4. Gout P. W., Buckley A. R., Simms C. R, Bruchovsky N., Leukemia 2001, 15, 1633–1640. [DOI] [PubMed] [Google Scholar]
- 5. Sleire L., Skeie B., Netland I. A., Førde H. E., Dodoo E., Selheim F., Leiss L., Heggdal J. I., Pedersen P.-H., Wang J., Enger P. Ø., Oncogene 2015, 34, 5951–5959. [DOI] [PubMed] [Google Scholar]
- 6. Ganz J. C., Gamma Knife Neurosurgery, Springer-Verlag: Wien; 2011, pp 1–376. [Google Scholar]
- 7. Neumann V. C., Grindulis K. A., Sulphasalazine in Rheumatoid Arthritis: An old drug revived; SAGE Publications; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schröder H., Campbell D. E., Clin. Pharmacol. Ther. 1972, 13, 539–331. [DOI] [PubMed] [Google Scholar]
- 9. Peppercorn M. A., Ann. Intern. Med. 1984, 101, 377–386. [DOI] [PubMed] [Google Scholar]
- 10.LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. Sulfasalazine. [Updated 2017 Sep 25]. https://www.ncbi.nlm.nih.gov/books/NBK548792/ (downloaded 2020–03-02).
- 11. Teraki Y., Shibuya M., Izaki S., Clin. Exp. Dermatol. 2009, 35, 723–728. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a.M. C. Stuart, M. Kouimtzi, S. R. Hill (eds.), World Health Organization model formulary 2008, pp 40–41, pp 45–46;
- 12b. Alanazi A. S., J. Med. Cases 2014, 5(5), 289–291; [Google Scholar]
- 12c. Marsia S., Mahmood S., Raza M., Nusrat K., Saleem A., Cureus 2019, 11(4), DOI 10.7759/cureus.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das K. M., Eastwood M. A., McManus J. P., Sircus W., N. Engl. J. Med. 1973, 289, 491–495. [DOI] [PubMed] [Google Scholar]
- 14. Shukla K., Thomas A. G., Ferraris D. V., Hin N., Sattler R., Alt J., Rojas C., Slusher B. S., Tsukamoto T., Bioorg. Med. Chem. Lett. 2011, 21, 6184–61847. [DOI] [PubMed] [Google Scholar]
- 15. Patel D., Kharkar P. S., Gandhi N. S., Kaur E., Dutt S., Nandave M., Drug Dev. Res. 2019, 80, 758–777. [DOI] [PubMed] [Google Scholar]
- 16. Dixon S. J., Patel D. N., Welsch M., Skouta R., Lee E. D., Hayano M., Thomas A. G., Gleason C. E., Tatonetti N. P., Slusher B. S., Stockwell B. R., eLife 2014, 3, e02523. DOI:10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Böhm H.-J., Flohr A., Stahl M., Drug Discovery Today Technol. 2004, 1(3), 217–224; [DOI] [PubMed] [Google Scholar]
- 17b. Sun H., Tawa G., Wallqvist A., Drug Discovery Today 2012, 17(7/8), 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayes J. D., Dinkova-Kostova A. T., Tew K. D., Cancer Cell 2020, 38, 167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li M., O'Doherty G. A., Org. Lett. 2006, 8(18), 3987–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shao C., Lu A., Wang X., Zhou B., Guan X., Zhang Y., Org. Biomol. Chem. 2017, 15(23), 5033–5040. [DOI] [PubMed] [Google Scholar]
- 21. Taddei C., Gee A., J. Labelled Compd. Radiopharm. 2018, 61(3), 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maryanoff B. E., Reitz A. B., Chem. Rev. 1989, 89(4), 863–927. [Google Scholar]
- 23.
- 23a. Heck R. F., J. Am. Chem. Soc. 1968, 90 (20), 5518–5526; [Google Scholar]
- 23b. Crisp G. T., Chem. Soc. Rev. 1998, 27 (6), 427–436. [Google Scholar]
- 24.
- 24a. Yoo K. S., Yoon C. H., Jung K. W., J. Am. Chem. Soc. 2006, 128 (50), 16384–16393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Karimi B., Behzadnia H., Elhamifar D., Akhavan P. F., Esfahani F. K., Zamani A., Synthesis 2010, 2010 (09), 1399–1427; [Google Scholar]
- 24c. Su Y., Jiao N., Curr. Org. Chem. 2011, 15 (18), 3362–3388. [Google Scholar]
- 25. Lee H. J., Seo J. W., Lee B. H., Chung K. H., Chi D. Y., Bioorg. Med. Chem. Lett. 2014, 14, 463–466. [DOI] [PubMed] [Google Scholar]
- 26.NHA, McGill University Health Center, Montreal, QC, Canada.
- 27.Flow cytometer Accuri C6 fby Accuri Cytometers Ltd., Ann Arbor, MI, USA.
- 28.FlowJo™software v10.7 by FlowJo LLC, OR, USA.
- 29.Victor 3 1420 multi-label counter, Perkins Elmer, Waltham, MA, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information