

Abstract
Fungal infections are a major contributor to infectious disease-related deaths across the globe. Candida species are among the most common causes of invasive mycotic disease, with Candida albicans reigning as the leading cause of invasive candidiasis. Given that fungi are eukaryotes like their human host, the number of unique molecular targets that can be exploited for antifungal development remains limited. Currently, there are only three major classes of drugs approved for the treatment of invasive mycoses, and the efficacy of these agents is compromised by the development of drug resistance in pathogen populations. Notably, the emergence of additional drug-resistant species, such as Candida auris and Candida glabrata, further threatens the limited armamentarium of antifungals available to treat these serious infections. Here, we describe our current arsenal of antifungals and elaborate on the resistance mechanisms Candida species possess that render them recalcitrant to therapeutic intervention. Finally, we highlight some of the most promising therapeutic strategies that may help combat antifungal resistance, including combination therapy, targeting fungal-virulence traits, and modulating host immunity. Overall, a thorough understanding of the mechanistic principles governing antifungal drug resistance is fundamental for the development of novel therapeutics to combat current and emerging fungal threats.
Graphical Abstract
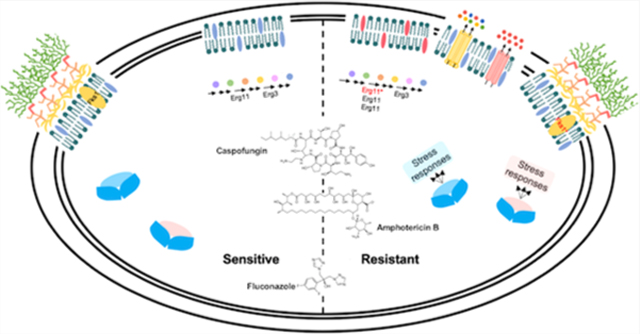
1. INTRODUCTION
Over the past several decades we have witnessed a concerning phenomenon where conventional antimicrobials are becoming increasingly ineffective at combatting infectious disease. With the current situation commonly being described as a postantibiotic era, there remains a great public health need for the development of novel therapeutics to combat pathogenic microbes, as well as a need to predict and prevent the evolution of resistance to our current armamentarium of antimicrobials. This issue of antimicrobial resistance is of particular concern in the context of fungal pathogens given the dearth of distinct classes of antifungals for treatment of invasive infections and the emergence and spread of multidrug-resistant fungal pathogens.
Despite the devastating impact that pathogenic fungi have on human health, they remain a relatively underappreciated contributor to human disease and mortality. Fungi infect billions and kill an estimated 1.5 million individuals annually, which is comparable to the mortality rates of more broadly recognized diseases such as tuberculosis or malaria.1,2 The prevalence of serious diseases caused by fungi has surged in recent decades due to the increasing number of immunocompromised individuals, including cancer patients, organ transplant recipients, individuals infected with HIV, and the increasing elderly population.3,4
The predominant etiological agents of systemic fungal infections include species of Candida, Aspergillus, and Cryptococcus, which collectively account for over 90% of mycotic deaths.1,3 Candida species are the most common cause of invasive mycotic disease in individuals who are severely immunocompromised, have endured invasive clinical procedures, or have experienced major trauma that requires extended treatment in intensive care units.1,3,5 Specifically, Candida species, and in particular Candida albicans, rank as a leading cause of all healthcare-associated bloodstream infections in the United States, with a crude mortality rate of ~40% despite antifungal intervention.1,5–7 In many healthy individuals, C. albicans is present as a harmless commensal in the oral cavity or the gastrointestinal tract; however, in severely immunocompromised patients, this fungus can disseminate into the bloodstream and colonize internal organs, resulting in life-threatening systemic infections.1,8,9 Among non-albicans species, Candida glabrata is the most prevalent cause of invasive candidiasis, with a consistently increasing number of cases reported over the past several years.5 Very recently, the emergence of Candida auris has caused great concern for medical practitioners and researchers across the globe. First isolated in 2009 from the external auditory canal of a patient in Japan,10 C. auris has been reported in over 30 countries on 6 continents in less than a decade.11 Phylogenetic analysis revealed four major clades of C. auris that cluster geographically and are distinguished by thousands of distinct single nucleotide polymorphisms (SNPs).12 Distinguishing features of C. auris include its propensity to colonize the human skin, exceptional nosocomial persistence, and prevalence of resistance to at least one antifungal class.13
Despite the detrimental impact fungi have on human health, only a few classes of antifungal drugs are currently available for treating these life-threatening infections. In general, the development of novel antifungal agents has been slow largely due to the eukaryotic nature of fungal cells, challenges associated with compound permeability across the fungal cell wall and membrane, and limited interest from the pharmaceutical industry in developing novel antifungals.14–16 In fact, a new class of antifungals has not entered clinical practice since the mid-2000s.15,17,18 Further threatening our limited repertoire of clinically relevant antifungals is the ever-increasing prevalence of fungal strains with intrinsic or acquired resistance to one or more drug classes. This Review will summarize our current arsenal of available antifungals, the resistance mechanisms that Candida species have employed to combat these agents, and potential new therapeutic approaches that can be explored to combat these pathogens.
2. CURRENT ARSENAL OF ANTIFUNGAL DRUGS
The polyenes are natural, amphipathic molecules that represent the oldest class of antifungal drugs used to treat systemic fungal infections. Introduced into medical practice in the 1950s, the most commonly used polyene, amphotericin B, exhibits potent activity against a broad spectrum of clinically relevant fungal species, including several species of Candida, Aspergillus, and Cryptococcus.19,20 Traditionally, the polyenes were thought to directly bind to ergosterol, forming drug–lipid complexes that intercalate into fungal cell membranes, causing leakage of intracellular components and, ultimately, cell death.20 However, recently this model was challenged by structural and biophysical studies, which revealed that amphotericin B forms extramembranous aggregates that extract ergosterol from fungal cell membranes, acting as a fungicidal “sterol sponge”21 (Figure 1). Despite its broad-spectrum antifungal activity, the clinical use of amphotericin B is disfavored, primarily due to poor oral bioavailability and dose-dependent toxic effects toward the host, resulting from similarities between ergosterol and cholesterol.20,22 In the mid-1990s, lipid formulations of amphotericin B were introduced that led to vast improvements in nephrotoxicity,18,19 and as such these formulations have served as an alternative treatment option in cases where antifungal availability is restricted or resistance to other more suitable antifungals precludes their utility.23 Fortunately, resistance to amphotericin B rarely occurs in C. albicans likely due to the severe fitness costs associated with the evolution of polyene resistance.24,25 In clinical isolates of C. auris, resistance to amphotericin B can range from 0% to 30%, and preliminary evidence suggests that there is transient and inducible amphotericin B resistance in this species.13
Figure 1.
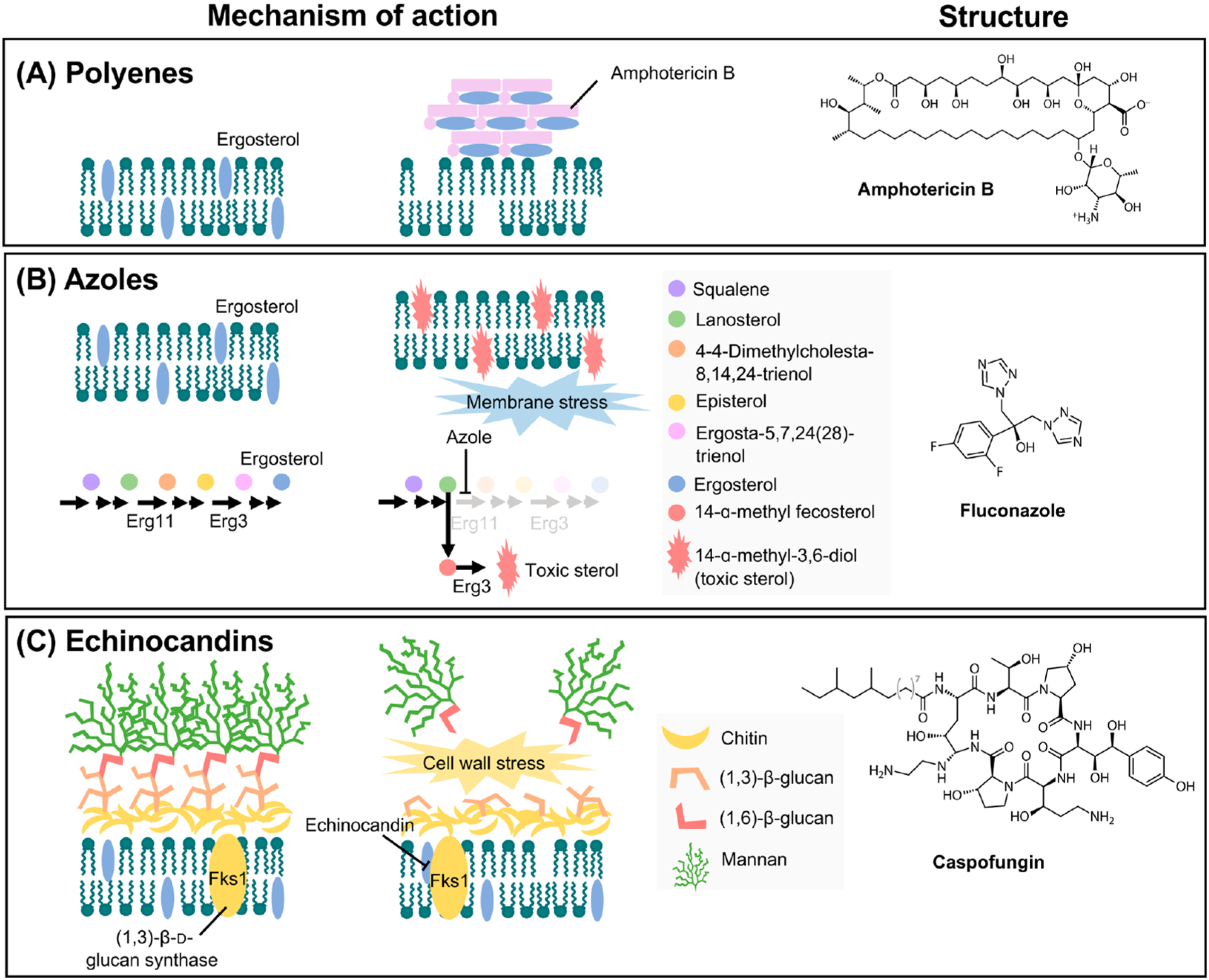
Antifungal mechanisms of action and structures. (A) Polyenes, such as amphotericin B, act as a fungicidal “sterol sponge” by forming extramembranous aggregates that extract ergosterol from lipid bilayers. (B) Azoles exert fungistatic activity by inhibiting lanosterol 14-α-demethylase (encoded by ERG11), which leads to a block in ergosterol synthesis and the accumulation of toxic sterol intermediates, including 14-α-methyl-3,6-diol produced by Erg3. (C) Fungal cell walls are composed of (1,3)-β-d-glucan covalently linked to (1,6)-β-d-glucan, as well as chitin and mannan. Echinocandins prevent the synthesis of (1,3)-β-d-glucan by inhibiting the (1,3)-β-d-glucan synthase (encoded by FKS1 in C. albicans and C. auris and by both FKS1 and FKS2 in C. glabrata); this results in a loss of cell wall integrity and severe cell wall stress. The structure of a representative antifungal from each class is depicted to the right of each mechanism. Adapted with permission from ref 26. Copyright 2008 Springer Nature.
First introduced in the 1980s, the azoles have served as the most frequently deployed drug class in the clinic. These heterocyclic synthetic compounds exert antifungal activity by blocking the synthesis of ergosterol, a key constituent of the fungal cell membrane, which leads to disruptions in membrane stability, permeability, and the function of membrane-bound enzymes.18,19 Azole exposure also causes the accumulation of toxic sterol intermediates, including 14-α-methy-3,6-diol.18,19 Specifically, the azoles inhibit the cytochrome P450 enzyme lanosterol 14-α-demethylase, encoded by ERG11 in Candida species, by binding to the heme group in the active site9 (Figure 1). In contrast to other antifungals, many azole drugs possess exceptional oral bioavailability and are available in both oral and intravenous formulations.27 However, a significant drawback of azoles includes the high potential for interactions with other drugs as they also inhibit mammalian cytochrome P450 enzymes, which are responsible for drug metabolism.14 To overcome this limitation, the development of new azoles (VT-1161, VT-1129, and VT-1598) that possess higher specificity for fungal enzymes is currently underway.28–31 An additional limitation is that azoles only inhibit the growth of Candida and Cryptococcus without killing these pathogens, imposing strong directional selection pressure and promoting the development of antifungal drug resistance. Susceptibility to azoles varies greatly among species of Candida. For the most commonly used azole fluconazole, C. albicans is generally susceptible, Candida krusei is intrinsically resistant, and resistance is steadily increasing for C. glabrata.5,32 Alarmingly, for C. auris, fluconazole resistance is overwhelmingly abundant; for example, in a study of 350 clinical isolates collected from 10 hospitals in India, 90% of isolates exhibited reduced susceptibility to fluconazole.33
The only novel antifungal drug class to enter medical practice in decades is the echinocandins, which are natural product derivatives composed of a cyclic hexapeptide core with an N-linked fatty-acyl side chain. Echinocandins exert antifungal activity by disrupting the cell wall, a structure that is completely absent in mammalian cells. They bind non-competitively to the catalytic subunit of (1,3)-β-d-glucan synthase, which is encoded by FKS1 in C. albicans and by both FKS1 and FKS2 in C. glabrata (Figure 1). A block in the biosynthesis of (1,3)-β-d-glucan culminates in a disruption of cell wall integrity and an imbalance in osmotic pressure, culminating in a fungicidal effect.34 At present, echinocandins are recommended as a first-line treatment for invasive candidiasis given their potent activity against Candida species.23,35 However, this drug class is clinically ineffective against species of Cryptococcus.19 Major advantages of echinocandins include their excellent safety profile, greater fungal selectivity, and low potential for drug–drug interactions.36 However, like polyenes, echinocandins exhibit poor oral absorption, and consequently, clinical use is limited to intravenous administration.37 Recent work has advanced a structurally distinct 1,3-β-d-glucan synthase inhibitor, Ibrexafungerp, which is available in both oral and intravenous formulations and is currently in phase II and phase III clinical trials.38 Given their widespread clinical use, resistance to echinocandins has inevitably emerged in recent years. Resistance has been most prominently recognized in C. glabrata; for example, in one U.S. hospital, the rate of echinocandin resistance in C. glabrata increased from 4.9% to 12.3% over a 10-year period.39 Echinocandin resistance in clinical isolates of C. auris has also been documented.13,33,40 Of utmost concern is the emergence of multidrug-resistant isolates of C. auris, which are impervious to all three antifungal drug classes, deeming these pathogens essentially untreatable.13,33 Fortunately, for other clinically relevant Candida species, including C. albicans, susceptibility to the echinocandins remains high.5
3. MECHANISMS OF ANTIFUNGAL RESISTANCE
Our frequent and prophylactic use of antifungals has led to the development of robust resistance in many Candida species. The term resistance can be defined as a strain that has a minimal inhibitory concentration (MIC) for a particular antifungal above specific clinical breakpoints; resistance can also be used more broadly to indicate a strain with an increase in MIC to an antifungal drug relative to a control or reference strain.41 This is in contrast to the term tolerance, which describes the ability of a drug-susceptible fungal strain to grow in the presence of an antifungal at concentrations above the MIC.41 Numerous adaptive mechanisms of antifungal drug resistance and tolerance have been identified, including drug target alteration or overexpression, upregulation of multidrug transporters, and activation of cellular stress responses (Figures 2 and 3). These mechanisms will be described in additional detail in the sections below.
Figure 2.
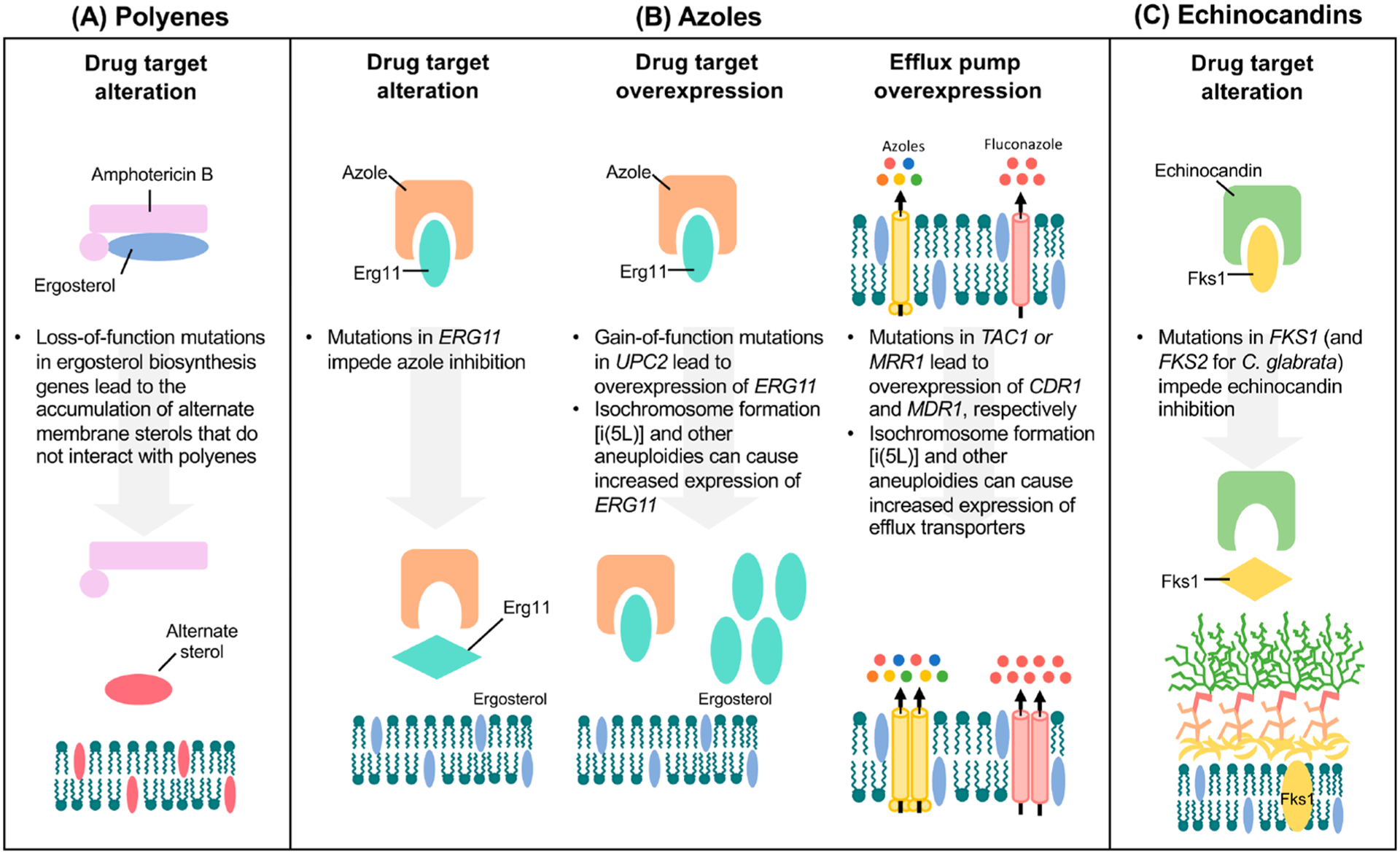
Molecular mechanisms of antifungal drug resistance. (A) Resistance to polyenes is primarily mediated by the depletion of the target ergosterol through loss-of-function mutations in ergosterol biosynthesis genes. This leads to the production of alternate sterols, which do not effectively interact with polyenes and therefore are not extracted from the fungal cell membrane. (B) Resistance to azoles can occur through substitutions to the azole target, Erg11, which leads to lower drug-binding affinity for the lanosterol demethylase enzyme (left panel). Overexpression of the drug target can occur through gain-of-function mutations in the transcriptional activator, UPC2, or through the formation of aneuploidies, such as [i(5L)], which directly increase the copy number of ERG11 (middle panel). Azole resistance is also acquired through the upregulation of ABC transporters (yellow), including Cdr1 and Cdr2, by activating mutations in specific transcription factors (TAC1 in C. albicans and C. auris and PDR1 in C. glabrata). Additionally, overexpression of the MF transporter (pink), Mdr1, through activating mutations in the transcriptional factor, MRR1, confers azole resistance (right panel). Efflux pumps can also be overexpressed through aneuploidy formation. (C) Resistance to echinocandins primarily involves mutations in FKS genes that encode the catalytic subunit of the drug target (1,3)-β-d-glucan synthase. For C. albicans and C. auris, mutations conferring echinocandin resistance occur in FKS1, while for C. glabrata, mutations occur in both FKS1 and its paralogue FKS2.
Figure 3.
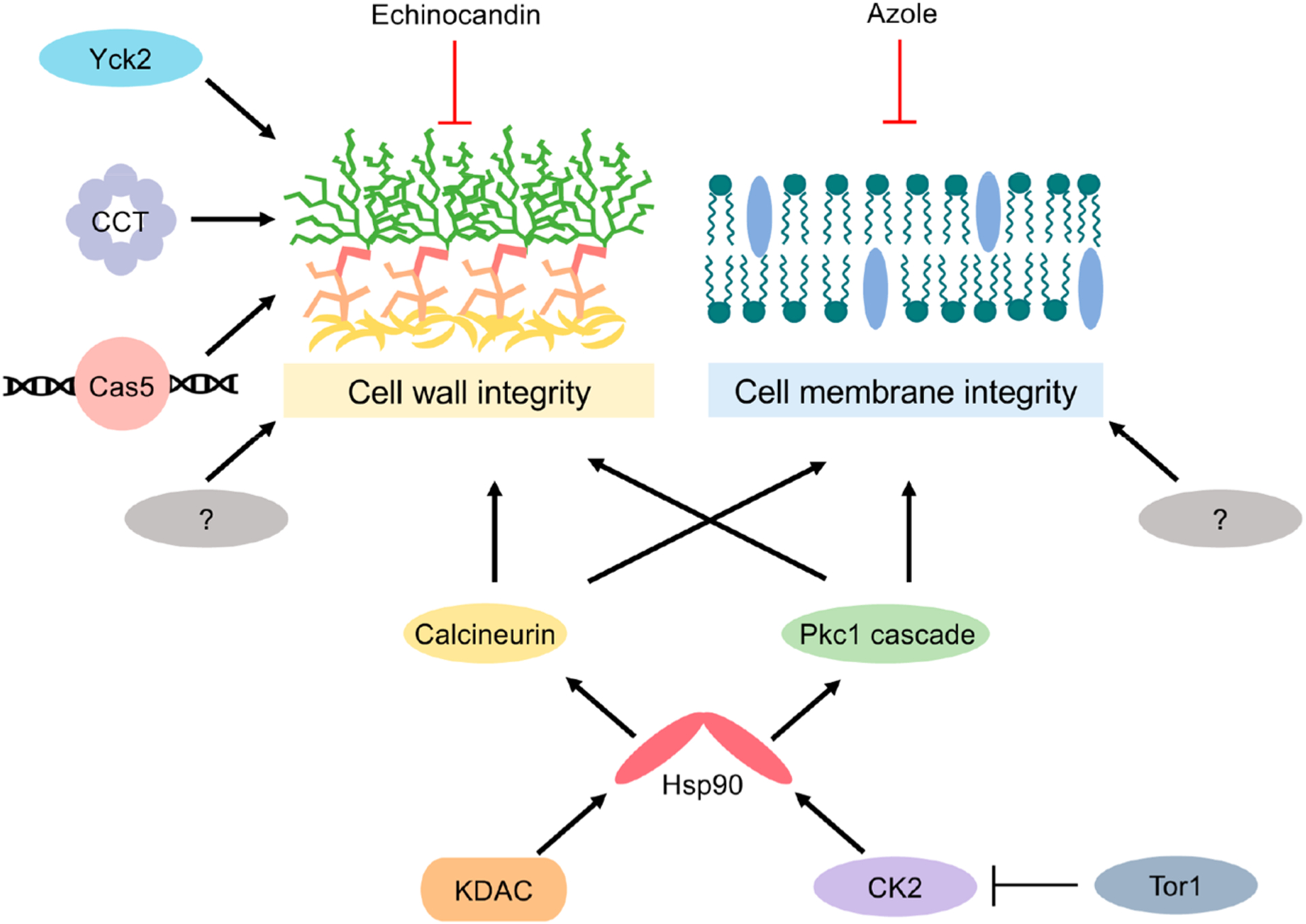
Cellular stress responses important for mediating cell wall and cell membrane integrity. A global cellular regulator governing antifungal tolerance and resistance is the molecular chaperone, Hsp90. In C. albicans, Hsp90 is post-translationally regulated by the protein kinase complex CK2 as well as lysine deacetylases (KDACs). Key client proteins of Hsp90 important for mediating cell wall and cell membrane stress responses include the protein phosphatase calcineurin and several components of the PKC cell wall integrity pathway (Pkc1, Bck1, Mkk2, and Mkc1). Additional cellular factors important for mediating tolerance and resistance to the echinocandins include the transcription factor Cas5, the eukaryotic chaperonin containing TCP-1 (CCT) complex, and the protein kinase Yck2. Notably, many other cellular factors contribute to these cellular stress responses, with many more enigmatic regulators that remain to be identified. Perturbation of any of these signaling pathways in combination with the cell wall or cell membrane stress elicited by the echinocandins and azoles, respectively, culminates in fungal growth inhibition.
3.1. Alterations in Drug Targets
3.1.1. Azoles.
In C. albicans, a common mechanism of azole resistance involves amino acid substitutions in the drug target, Erg11, which leads to lower drug-binding affinity for the lanosterol demethylase enzyme15,42(Figure 2). Over 140 amino acid substitutions in Erg11 have been associated with azole resistance in this species,43 with the majority of these substitutions clustered into hot-spot regions ranging from amino acids 105–165, 266–287, and 405–488.44 Notably, only a few amino acid substitutions have been confirmed experimentally to confer azole resistance.45,46 Molecular mapping of C. albicans ERG11 variant positions revealed that mutations reside in the catalytic site of the enzyme, the fungus-specific external loop, and the proximal surface, as well as between the proximal surface and the heme.47 Preliminary studies have also implicated ERG11 mutations in azole resistance in C. auris. Whole-genome sequencing of 47 C. auris clinical isolates, of which the majority were resistant to fluconazole, identified three hot-spot amino acid substitutions previously implicated in azole resistance in C. albicans: Y132F, K143R, and F126L.13,40 Consistent with these findings, another study of 44 C. auris clinical isolates detected amino acid substitutions at positions Y132 and K143 in Erg11 in 77% of fluconazole-resistant strains.33 Heterologous expression of the C. auris ERG11 allele harboring either the Y132F or K143R substitution into the model yeast Saccharomyces cerevisiae resulted in elevated MIC values to both fluconazole and voriconazole, supporting the causal link between these mutations and C. auris azole resistance.48 Interestingly, ERG11 mutations appear to be strongly associated with specific geographic clades of C. auris, the K143R and Y132F substitutions are linked to isolates in the South Asian and South American clades, whereas the F126L substitution is associated with the African clade.40 In contrast, for C. glabrata, alterations of the drug target have not been reported as a significant mechanism of azole resistance.49–51 Indeed, only a single case of a fluconazole-resistant clinical isolate possessing an amino acid substitution in Erg11 has been documented to date.52
Overexpression of ERG11 is common in azole-resistant clinical isolates of C. albicans and directly contributes to increased target abundance, ultimately lowering drug susceptibility15,17(Figure 2). For C. albicans, the transcriptional activator, Upc2, is a crucial regulator of many ergosterol biosynthesis genes, including ERG11. Indeed, gain-of-function (GOF) mutations in UPC2 cause the constitutive overexpression of ergosterol biosynthesis genes, a higher ergosterol content, and a reduction in fluconazole susceptibility.53–58 Furthermore, disruption of UPC2 in an azole-resistant clinical isolate abrogated ERG11 overexpression and increased fluconazole susceptibility, highlighting the importance of this transcriptional activator in mediating C. albicans azole resistance.59 Similarly, in C. glabrata, disruption of UPC2A in an azole-resistant clinical isolate reduced cellular ergosterol content and increased susceptibility to sterol biosynthesis inhibitors.60
3.1.2. Polyenes.
Resistance to polyenes is extremely uncommon; however, in the rare incidence that it does occur, it is mediated by alterations in enzymes that reduce drug-binding affinity or deplete ergosterol from the membrane61 (Figure 2). In C. albicans, reduced amphotericin B susceptibility can occur through mutations in several ergosterol biosynthesis enzymes, including ERG2,62 ERG3,63 ERG5,64 and ERG11.65 Likewise, for C. glabrata, mutations in ERG2,52 ERG6,66 and ERG1152 have been documented in polyene-resistant clinical isolates. For C. auris, whole-genome sequencing of resistant clinical isolates following treatment with amphotericin B revealed higher expression of several genes in the ergosterol biosynthesis pathway (ERG1, ERG2, ERG6, and ERG13) compared to untreated controls and susceptible isolates.12 Aside from genetic alterations, azole treatment has also been attributed to enhanced polyene resistance in Candida species, as a consequence of azole-mediated reduction in cellular ergosterol levels.67
3.1.3. Echinocandins.
For most Candida species, echinocandin resistance is primarily mediated by mutations in the FKS genes (Figure 2). In C. albicans, mutations that confer echinocandin resistance occur in the essential gene, FKS1. Hot-spot regions correspond to amino acids 641–649 (hot spot 1) and amino acids 1357–1364 (hot spot 2).68 Mutations at these regions decrease the IC50 of the glucan synthase enzyme by several orders of magnitude, elevate MIC values, and result in cross-resistance to diverse echinocandins.69–72 In C. albicans, serine 645 (S645) within hot-spot region 1 exhibits the highest frequency of substitution and is associated with the most prominent resistance phenotype.70–72 For the related FKS2 and FKS3 genes, less is known about their contributions to echinocandin resistance in C. albicans; however, their deletion has been shown to result in higher FKS1 transcript levels and lower echinocandin susceptibility.73 In contrast to C. albicans and other yeasts, mutations conferring increased echinocandin resistance in C. glabrata occur in both FKS1 and FKS2 at regions homologous to those of C. albicans.70,74 In fact, C. glabrata FKS2 is expressed more highly than FKS1,70 and mutations in FKS2 outnumber FKS1 mutations by two-to-one.74 In echinocandin-resistant clinical isolates, the highest frequency of substitutions occurs at residue serine 663 in FKS2, which is equivalent to C. albicans S645.70
For C. auris, sequencing of FKS1 in 38 C. auris clinical isolates revealed a mutation causing a phenylalanine substitution at serine 639 (S369F) (equivalent to S645 in C. albicans), which is associated with resistance to the echinocandins.33 Furthermore, the clinical significance of this mutation was demonstrated using an in vivo murine model of invasive candidiasis. Mice challenged with a S639F mutant failed to respond to treatment with caspofungin in contrast to mice challenged with its susceptible wild-type counterpart.75 Additionally, proline and tyrosine substitutions of the same S639 residue have been detected in C. auris echinocandin-resistant clinical isolates.76
3.2. Overexpression of Efflux Pumps
3.2.1. Azoles.
The upregulation of plasma membrane efflux pumps is a major mechanism governing azole resistance in many fungal pathogens. The first main class of efflux pumps implicated in azole drug resistance is the ATP-binding cassette (ABC) superfamily. ABC transporters possess two transmembrane-spanning domains and two cytoplasmic nucleotide-binding domains (NBDs). The NBD drives the movement of substrates across the fungal membrane via ATP hydrolysis.77 The second class of efflux pumps implicated in azole resistance is the major facilitator (MF) superfamily. Like the ABC superfamily, MF transporters also possess transmembrane-spanning helices but use the proton gradient generated across the plasma membrane to drive MF-mediated translocation.77
In C. albicans, overexpression of two homologous ABC transporters, Cdr1 and Cdr2, have been frequently implicated in azole resistance (Figure 2), particularly in patients receiving long-term antifungal therapy.15,17 Of the two transporters, Cdr1 is the major determinant of azole resistance; deletion of CDR1 in a clinical isolate reduces resistance to the azoles by 4-to 8-fold. However, deletion of CDR2 typically results in a much weaker effect.78 The transcription factor Tac1 (transcriptional activator of CDR) binds to cis-acting drug-response elements (DREs) in the promoters of CDR1 and CDR2 to regulate their expression. Indeed, single amino acid substitutions in the activation domain of Tac1 resulting in hyperactive alleles have been linked to azole resistance due to the constitutive overexpression of both CDR1 and CDR2.79 Constitutive upregulation of CDR1 and CDR2 also contributes to azole resistance in clinical isolates of C. glabrata. Genetic disruption of CDR1 caused increases in azole susceptibility, and this effect was further exaggerated by the disruption of CDR2.80,81 In addition to Cdr1 and Cdr2, a third ABC transporter, Snq2, plays an important role in C. glabrata azole resistance.82 In C. glabrata, expression of these ABC transporters is mediated by the zinc cluster transcriptional activator, Pdr1 (pleiotropic drug resistance 1), which directly binds to the pleiotropic drug-response element (PDRE) in the promoter region of target genes.83,84 Indeed, the most prevalent mechanism of clinical azole resistance in C. glabrata involves PDR1 gain-of-function mutations that result in the upregulation of its downstream target transporters.85–87 Specifically, engagement with the KIX domain of the Mediator coactivator subunit Gal11A (also known as Med15A) is essential for the upregulation of C. glabrata Pdr1-target genes.88 More recently, in C. albicans, the Mediator complex was shown to play an important role in Tac1-mediated azole resistance, as deletion of the Mediator tail module in Tac1 gain-of-function mutants reduced CDR1 transcription and increased fluconazole susceptibility.89
Although 95 MF transporters are encoded in the C. albicans genome,90 fluconazole resistance has only been linked to Mdr1 (multidrug resistance 1)91,92 (Figure 2). Expression of this MF pump is regulated by the transcription factor, Mrr1 (multidrug resistance regulator 1), such that deletion of MRR1 abolishes MDR1 expression and increases susceptibility to fluconazole,93 whereas activating point mutations in MRR1 increase azole resistance.94 Interestingly, deleting MRR1 reduces fluconazole resistance to a greater extent than the deletion of the MDR1 efflux pump itself, suggesting that MRR1 may regulate additional determinants of azole resistance.93 More recently, the C. albicans Swi/Snf chromatin remodelling complex was implicated as a major coactivator of Mrr1 as deletion of the catalytic subunit of the Swi/Snf complex, SNF2, in MRR1 gain-of-function strains led to drastic reductions in both Mdr1 activation and fluconazole resistance.95 In contrast to C. albicans, the C. glabrata homologue of MDR1, FLR1, is not significantly associated with azole resistance,96 as is the case with several other non-albicans Candida species.77
In C. auris, the genome encodes for many transporters orthologous to those in C. albicans that primarily belong to the ABC and the MF families.12,97 Phylogenetic analysis revealed that C. auris possesses homologues of MDR1, CDR1/CDR2, and CDR4, as well as two copies of SNQ2.12 Several lines of evidence have indicated the important relationship between efflux pumps and azole resistance in C. auris. Specifically, one study observed that, although CDR1 and MDR1 were both overexpressed in azole-resistant C. auris isolates, only the deletion of CDR1 led to significant reductions in azole MIC values.98 The importance of this efflux pump was confirmed in a separate study in which the deletion of CDR1 was sufficient to confer an 8-fold increase in fluconazole sensitivity.99 Efflux-mediated mechanisms of azole resistance also appear to play an important role in C. auris biofilms. Transcriptomic analysis of drug-resistant C. auris biofilms revealed the upregulation of both ABC and MF transporters, and treatment with efflux pump inhibitors increased fluconazole sensitivity by 4- to 16-fold, highlighting the significance of efflux activity in biofilm-mediated resistance.100 In addition to drug transporters, numerous transcription factors are also present in the C. auris genome, including two copies of both TAC1 and MRR1,12 suggesting that differences in transcriptional regulatory machinery may serve as a mechanism by which efflux pumps are transcribed at a higher constitutive level in C. auris. Recently, a study demonstrated that mutations in TAC1B arise rapidly in vitro upon exposure to fluconazole and that resistance-associated TAC1B mutations are present among several fluconazole-resistant C. auris clinical isolates.101 Interestingly, one of the two copies of MRR1 is truncated at its carboxy terminus, a common regulatory domain, raising the question as to whether C. auris MRR1 has altered regulation relative to homologues in other Candida species.102
3.2.2. Polyenes and Echinocandins.
In contrast to azoles, the upregulation of efflux pumps plays a rather insignificant role in conferring resistance to the echinocandins. This is consistent with the fact that echinocandins function at the outer leaflet of the fungal cell membrane,18,19 as well as the fact that these large cyclic hexapeptide molecules are likely to be poor substrates of fungal efflux pumps due to their size and hydrophobicity.103 Similarly, for polyenes, the lack of efflux-mediated mechanisms of resistance may be attributed to their inability to enter the drug-binding pocket of transporters.103 Interestingly, in C. auris, a recent study highlighted the potential involvement of drug transporters in amphotericin B resistance. Whole-genome sequencing of polyene-resistant clinical isolates led to the identification of four non-synonymous mutations, one of which was in a putative membrane transporter. Notably, none of the SNPs were found in the coding sequences of ergosterol biosynthesis genes, which is the conventional mechanism of polyene resistance in other species of Candida.104 Although C. auris appears to possess a rather unusual mechanism of amphotericin B resistance, the manner by which membrane transporters govern this process requires further analysis.
3.3. Modulation of Stress Responses
The diverse and dynamic niches that fungal pathogens inhabit are subject to a variety of environmental fluctuations, including temperature, pH, and nutrient levels, which are capable of perturbing cellular homeostasis and imposing significant stress on the fungal cell. Antifungal agents represent a chemical stressor these pathogens must recognize, respond to, and adapt to in order to survive.17,105 Consequently, fungal pathogens have evolved broad stress-response circuitry that enable them to thrive in the presence of diverse cellular insults (Figure 3).
3.3.1. Azoles.
A key mechanism through which C. albicans develops resistance to the azoles that is contingent upon stress responses is through alteration of the ergosterol biosynthesis pathway. Loss-of-function mutations in ERG3, which encodes a Δ−5,6-desaturase, block the cellular accumulation of 14-α-methyl-3,6-diol, the toxic sterol intermediate that is otherwise produced as a result of Erg11 inhibition by the azoles.106 Alternatively, 14-α-methyl fecosterol is incorporated into the fungal cell membrane, allowing for continued growth and replication in the presence of azoles. Azole resistance in C. albicans has been associated with five missense mutations in ERG3 (A168V, S191P, G261E, T329S, and A353T) and two further nonsense mutations (Y325* and Y190*), leading to loss of function.51,107
A global cellular regulator governing stress responses in diverse fungal pathogens is the essential molecular chaperone, heat shock protein 90 (Hsp90).108,109 Hsp90 is highly abundant, and its function is tightly coupled to environmental perturbations. It interacts with over 20 cochaperones that facilitate the recognition of specific client proteins, which are enriched in kinases, signal transducers, and transcription factors, many of which serve as hubs in regulatory networks.110–112 These unique attributes enable Hsp90 to serve as both a capacitor for the storage and release of genetic variation and a potentiator to allow for the rapid emergence of new traits.111,112 Specifically, Hsp90 potentiates the rapid evolution of azole resistance in C. albicans, as well as in more evolutionarily distinct fungi, as genetic or pharmacological inhibition of Hsp90 reduces azole tolerance and abrogates resistance.113,114 Primary investigations of Hsp90-mediated resistance in S. cerevisiae revealed that azole resistance acquired via loss-of-function of Erg3 is Hsp90-dependent.114 In C. auris, Hsp90 also governs azole tolerance in azole-susceptible strains, highlighting its role in orchestrating cellular responses upon exposure to membrane stress in this pathogen.99 Notably, Hsp90 does not mediate azole resistance in C. albicans or C. auris isolates where azole resistance is caused by the overexpression of efflux pumps, highlighting this as an Hsp90-independent mode of resistance.99,114
The mechanisms through which Hsp90 confers antifungal resistance are complex given its global impact on cellular signaling. A key Hsp90 client that mediates its effects on antifungal drug tolerance and resistance is the calcium-calmodulin activated protein phosphatase calcineurin.114,115 Azole treatment activates calcineurin-dependent stress responses in C. albicans,115 and genetic or pharmacological impairment of the phosphatase renders C. albicans hypersensitive to the azoles.116 Hsp90 inhibition blocks azole activation of the calcineurin-dependent stress response and phenocopies the effects of calcineurin inhibition,115 highlighting the interconnectedness between calcineurin and Hsp90 in regulating azole tolerance and resistance. The C. albicans protein kinase C, Pkc1, also regulates cellular responses to the azoles through a MAPK cascade consisting of Bck1, Mkk1/2, and Mkc1.117 Compromise of PKC-MAPK signaling phenocopies both Hsp90 and calcineurin inhibition, reducing azole-resistance phenotypes in distinct C. albicans clinical isolates.117 Moreover, genetic depletion of Hsp90 in C. albicans destabilizes Mkc1, Bck1, and Pkc1, ultimately blocking PKC signaling.117 Thus, Hsp90 regulates basal tolerance and resistance to membrane-perturbing azole antifungals through multiple signaling cascades.
In addition to Pkc1, a number of other kinases are involved in stress-response circuity that enables the survival of C. albicans during azole-induced stress. Interestingly, many of these signal transducers also have connections with Hsp90 and other regulators discussed earlier. One promiscuous kinase involved in azole resistance is casein kinase 2 (CK2). Overexpression of the catalytic subunit of the CK2 complex, CKA2, confers fluconazole resistance that is dependent on calcineurin signaling.118 Further, chemical-genetic screens in C. albicans revealed that CK2 subunits genetically interact with Hsp90, and additional biochemical characterization established that CK2 phosphorylates Hsp90 to regulate its chaperoning activity.119,120 Other kinases important for cellular responses to azoles include target of rapamycin (TOR) kinases, which are involved in nutrient sensing and signaling, as well as coupling cellular growth and metabolism to environmental cues. Compromising TOR function with the pharmacological inhibitor rapamycin abrogates erg3-mediated resistance to the azoles.121 Further, the cyclic hexadepsipeptide natural product beauvericin potentiates azoles in C. albicans, C. neoformans, and A. fumigatus.122 Mechanistic studies highlighted a dual mechanism of action of beauvericin in which it both blocks multidrug efflux and inhibits the Tor1 kinase, thereby activating CK2 and inhibiting Hsp90 function.122 Hyper-activation of TOR signaling in fungal pathogens has also been shown to bypass azole toxicity via Hsp90-dependent calcineurin stabilization.123 Thus, fungal kinases play complex and pivotal roles in governing responses to azole-induced cell membrane stress.
Post-translational modifications including methylation, acetylation, and phosphorylation also regulate stress responses, virulence, and antifungal tolerance in Candida species.15,17,19 Specifically, reversible acetylation by various lysine acetyl-transferases (KATs) and lysine deacetylases (KDACs) is important in facilitating chromatin-mediated transcriptional regulation, as well as the post-translational regulation of many cellular proteins. Inhibition of many of these enzymes decreases basal tolerance and resistance to antifungal drugs. For instance, the KDACs Hda1 and Rpd3 deacetylate Hsp90 in S. cerevisiae, and genetic deletion of these factors increases azole sensitivity and impairs the evolution of azole resistance.124 In C. albicans, there is extensive functional redundancy as KDACs Hos2, Hda1, Rpd3, and Rpd31 all contribute to azole resistance.125 Further, a molecule thought to inhibit the KDAC Hos2, MGCD290 (Mirati Therapeutics, San Diego, CA, U.S.A.), displays synergistic activity with azoles against diverse C. albicans drug-resistant clinical isolates.126,127 Furthermore, in vivo studies coupled with preliminary clinical trials have supported the use of MGCD290 in combination with fluconazole to treat C. albicans infections.128 Chromatin remodeling and the activity of the sirtuin-family of KDACs have also been shown to mediate tolerance to the azoles in C. glabrata, where inactivation results in enhanced susceptibility to azoles and other cellular stressors.129 KATs also modulate azole resistance. Genetic impairment of C. albicans ADA2, which encodes a component of the Spt-Ada-Gcn5-acetyl-transferase (SAGA) coactivator complex, confers hyper-sensitivity to fluconazole due to impaired upregulation of efflux pumps Cdr1 and Mdr1.130
Environmental pH plays a role in antifungal tolerance as growth of C. albicans at an acidic pH reduces growth observed at azole concentrations above the MIC.131 In yeast, pH signaling is mediated by the highly conserved Rim pathway, where external pH is detected by the transmembrane proteins Rim21 and Dfg16, which signal through a protein cascade consisting of Rim8, Rim13, Rim20, and the terminal transcription factor Rim101.9 Rim101 regulates the expression of genes involved in cell wall, yeast-to-hyphal transition, adhesion, iron metabolism, and biofilm formation. Phenotypic and transcriptional analyses have implicated multiple components of the Rim pathway in azole tolerance and identified that Hsp90 is a downstream effector of Rim signaling, likely contributing to Rim-mediated effects on antifungal tolerance.132
3.3.2. Echinocandins.
Similar to the azoles, exposure to the cell wall targeting echinocandins initiates a number of interconnected and adaptive responses that modulate fungal susceptibility to this antifungal class. Echinocandin-induced defects in the cell wall are detected by transmembrane proteins that facilitate the activation of the GTPase Rho1. Downstream Rho1 targets include Pkc1 and the β−1,3-glucan synthase subunits.133–135 Cell wall salvage mechanisms can then be activated through these and other signaling networks. The fungal cell wall has dynamic and compensatory capabilities to increase production of one or more components upon inhibition of another. In vitro assessment of echinocandin-mediated β−1,3-glucan inhibition revealed that C. albicans facilitates the compensatory upregulation of chitin synthesis and redistribution of polysaccharides in the cell wall during times of cell wall stress.136,137 This response contributes to the paradoxical growth phenomenon observed at high echinocandin concentrations.138,139 Three signal transduction pathways, the PKC-MAPK pathway, the Ca2+-calcineurin pathway, and the high-osmolarity glycerol (HOG) pathway coordinately regulate chitin synthesis to maintain cell wall integrity.140 PKC-MAPK and calcineurin signaling are also critical in establishing basal echinocandin tolerance through elevated chitin production in C. glabrata.141,142
As discussed, Hsp90 regulates the function of calcineurin, as well as a number of stress-activated protein kinases, which is crucial in mediating responses to the echinocandins.113,143,144 Pharmacological or genetic impairment of Hsp90 function potentiates echinocandin activity in C. albicans, C. glabrata, and the distantly related pathogenic mold A. fumigatus.115,142,145 Furthermore, inhibition of Hsp90 reduces echinocandin resistance in C. glabrata clinical isolates with mutations in the echinocandin target gene FKS1.142 Proof-of-principle experiments established that genetic depletion of C. albicans Hsp90 in vivo enhances the efficacy of the echinocandins in a systemic murine model of infection, and pharmacological inhibition of Hsp90 enhances echinocandin efficacy in a Galleria mellonella model of A. fumigatus infection.113,115 These effects are in part attributed to the role of Hsp90 in stabilizing calcineurin and components of the PKC-MAPK pathway, as deletion of these signal transducers results in decreased chitin synthesis upon echinocandin exposure and increased echinocandin susceptibility.140
An expanded repertoire of cellular factors influencing fungal tolerance to the echinocandins include the C. albicans transcription factor Cas5. Initially identified in a genetic screen for mutants hypersensitive to caspofungin,146 homozygous deletion of CAS5 was shown to reduce Fks1-mediated echinocandin resistance.147 Under basal conditions, Cas5 regulates a distinct set of transcriptional targets under basal physiological conditions compared with those in response to cell wall stress, with a crucial role in transcriptional regulation of cell wall homeostasis and cell cycle dynamics.147 This unveiled novel regulatory circuitry through which stress responses, cell cycle regulation, and antifungal resistance were coupled.147 Further functional genomic screening efforts implicated proteins of the eukaryotic chaperonin containing TCP-1 (CCT) complex in modulating cellular responses to echinocandin-induced stress, enabling both tolerance and resistance.144 The CCT complex is involved in the assembly and folding of cytoskeletal proteins, and its depletion results in actin perturbation and cell wall stress. Hsp90 associates with the CCT complex,144 indicating that it is a member of the Hsp90 interaction network with potential to be targeted to combat echinocandin resistance.
3.3.3. Polyenes.
Resistance to the polyenes in Candida species is a rare phenomenon given the fitness costs associated with the acquisition of resistance. Clinical isolates of C. albicans with loss-of-function mutations in ERG3 have demonstrated cross-resistance to the polyenes and azoles due to their marked reductions in ergosterol content.25,106 The fitness and survival of amphotericin B-resistant Candida isolates are critically dependent upon Hsp90 expression and function. As a consequence, pharmacological inhibition of Hsp90 in resistant C. albicans or C. tropicalis strains abolished amphotericin B resistance.25 These resistant mutants also exhibited constitutive activation of diverse stress responses, with a genetic dependency of resistance on stress-response regulators, including calcineurin, Pkc1, and Hog1.25 It has been postulated that resistance to amphotericin B may enable pathogens to efficiently cope with oxidative stress. C. albicans mounts a compensatory antioxidant response upon exposure to amphotericin B, and studies with amphotericin B-resistant Candida demonstrate significant reductions in reactive oxygen species (ROS) accumulation, reductions in protein carbonylation, reductions in basal mitochondrial respiration, and significant increases in catalase production.148,149
3.4. Genomic Modifications
Fungal pathogens possess remarkable genomic plasticity, allowing them to adapt to environmental perturbations and acquire antifungal resistance. Large-scale genomic alterations including aneuploidies, loss of heterozygosity (LOH), and chromosomal rearrangements can impact the expression of drug targets, efflux pumps, and other factors that contribute to resistance. The emergence of aneuploidy provides a unique opportunity for cells to rapidly develop genotypic and phenotypic diversity without permanently committing to the mutant genotype.150,151 The fungistatic azoles cause alterations in DNA content and cell morphology through an ordered series of aberrant cell cycle events that select for aneuploidy formation.152 Karyotype variability conferring azole resistance has been extensively studied in C. albicans and is commonly observed in both resistant clinical isolates and laboratory strains. A specific aneuploidy on the left arm of chromosome 5 is the most prevalent.153,154 This common segmental aneuploidy consists of an isochromosome composed of two identical left arms of Chr5 flanking a centromere (i(5L)). Gain and loss of i(5L) tightly correlates with the gain and loss of azole resistance. The resistance associated with i(5L)-carrying isolates is mediated by increased copy numbers of ERG11 and TAC1, as well as gain-of-function mutations and LOH of these resistance determinants.155 Variant forms of i(5L) also exist, including those that incorporate a region of chromosome 3 containing MRR1, allowing for further resistance to develop.156
Additionally, aneuploidies of chromosomes 3, 4, and 6 have been implicated in azole resistance in C. albicans.157–159 Experimental evolution in the presence of azoles and the calcineurin inhibitor FK506 revealed extensive aneuploidies in strains that evolved resistance to the drug combination.158 The aneuploidy common in all resistant lineages was an increased copy number of chromosome 4, suggesting that this chromosome may harbor important resistance determinants.158 Further, experimental evolution in a strain sensitized to azoles due to loss of the GTPase activator protein Rgd1 resulted in an amplification of both chromosome 7 and a large segment of chromosome 3, which accompanied suppression of the azole-sensitivity phenotype; overexpression of a transporter gene on the affected region of chromosome 3 was sufficient to confer the suppression phenotype.160 Although most extensively studied in C. albicans, the association between aneuploidy formation and fluconazole resistance in Candida species has also been described in C. glabrata. Similar to C. albicans, an increase in ERG11 copy number has been observed in C. glabrata through the formation of aneuploidies and novel chromosomal configurations.161,162
LOH can also have a profound impact on drug resistance and is common in genomic regions containing determinants of azole susceptibility. LOH events can occur over short regions or over entire chromosomes. In C. albicans, LOH for the transcription factors TAC1 and MRR1 have been reported in azole-resistant isolates.79,94 For both TAC1 and MRR1, LOH can generate two copies of a hyperactive allele, which increases the expression of genes encoding Cdr1 and Cdr2, or Mdr1, respectively.79 Constitutive upregulation of ERG11 has also been reported as a consequence of LOH in the transcription factor UPC2 in C. albicans.54 Interestingly, work has shown that a plethora of stress conditions increase mitotic recombination rates in C. albicans, leading to an increase in LOH rates.163 This suggests a mechanism by which C. albicans can achieve greater genotypic diversity upon exposure to environmental perturbations, which can have important consequences given the lack of a conventional sexual cycle in this organism.
The incidence of echinocandin and polyene resistance arising from chromosomal abnormalities is infrequent compared to azole resistance in C. albicans. Nevertheless, rare genomic alterations have been reported in echinocandin-resistant isolates. Stepwise acquisition of micafungin resistance by homozygous hot-spot mutations in FKS1, followed by LOH, was identified as a mechanism of echinocandin resistance in C. albicans.164 Additionally, while an increase in chromosome 5 copy number confers azole resistance, a chromosome 5 monosomy causes decreases in β−1,3-glucan, increases in chitin, and decreases in echinocandin susceptibility.165 Finally, trisomy of chromosome 2 reduces the susceptibility of C. albicans to caspofungin.166 Adaptation to caspofungin in these strains is mediated by a calcineurin-mediated mechanism that is independent of the downstream transcriptional regulator Crz1.166
Substantial karyotype variability has been observed in C. auris under stress conditions, and preliminary genomic investigations revealed that isolates harbor between 5 and 7 chromosomes of varying sizes.167 Although the relationship between karyotype, drug resistance, and clade affiliation has not been determined, future investigations as to the impact of genomic plasticity on drug resistance for this emerging pathogen will no doubt lead to fascinating insights. C. auris undergoes replicative aging caused by asymmetric cell division. Interestingly, older cells exhibit higher tolerance to fluconazole, micafungin, and amphotericin B compared to younger cells.168 Transient gene duplication in old C. auris cells increases expression of drug-resistance determinants, including ERG11 and CDR1.168 Whether the gene duplications observed resulted from chromosomal duplications remains inconclusive.168 Overall, further mapping of C. auris chromosomes to investigate resistance patterns is required in order to more effectively understand the impact of genomic rearrangements on antifungal resistance in this emerging pathogen.
3.5. Intrinsic Antifungal Resistance
Thus far, we have focused on mechanisms through which fungal pathogens acquire antifungal drug resistance. However, a number of fungi display primary or intrinsic resistance to clinically available antifungals. A classic example of this is the resistance of Cryptococcus species to echinocandins. This has remained enigmatic as C. neoformans not only possesses a β-glucan synthase encoded by FKS1, but echinocandins inhibit the enzyme effectively in vitro.169 Uncovering mechanisms governing intrinsic resistance is an important step to better understand and combat the clinical threat posed by these pathogens.
Intrinsic resistance to the azoles has been reported in several non-albicans Candida species, including C. glabrata, C. krusei, and C. auris. C. glabrata isolates demonstrate striking intrinsic resistance to fluconazole.170 Even without prior azole exposure, C. glabrata isolates develop robust, stable resistance phenotypes within 4 days of in vitro exposure.171 This is largely attributed to enhanced drug efflux through the overexpression of ABC transporters Cdr1, Pdh1, Yor1, and Snq2, which are all regulated by the transcription factor Pdr1.80,82,170 Furthermore, C. glabrata “petite mutants”, or strains with a mitochondrial DNA deficiency, exhibit upregulation of ABC transporters and higher levels of azole resistance.86,172 Finally, C. glabrata can grow with altered membrane sterol composition and can facilitate sterol uptake in both aerobic and anaerobic conditions, as well as in the presence of fluconazole.173 Active transport of exogenous sterols has been attributed to C. glabrata’s capacity to overcome fluconazole-mediated blocks in ergosterol biosynthesis.173 For C. krusei, the mechanisms responsible for innate azole resistance are poorly understood. A number of likely contributors have been suggested, including the variable susceptibility of C. krusei Erg11 to inhibition by azoles, as well as increased efflux pump expression.174,175 For example, C. krusei constitutively expresses the ABC transporter Abc1, which, when pharmacologically inhibited, results in sensitization of the pathogen to the azoles.174 Finally, the remarkable azole resistance exhibited by C. auris, with over 90% of identified isolates displaying MICs above clinical breakpoints, garnered initial suspicions of robust intrinsic azole resistance in this species.11,13,102 Amino acid substitutions in the azole target Erg11 that mediate azole resistance in C. albicans, including Y132F and K143R, were found in resistant C. auris isolates.33 Further endeavors are still crucial to determine the reservoir and evolutionary trajectory of C. auris in order to uncover the mechanism driving the development of such widespread azole resistance in this pathogen.11,13,102
In addition to Candida species displaying intrinsic resistance to azoles, select strains and species also show inherent resistance to the echinocandins. Candida parapsilosis is an example of a species with intrinsically reduced susceptibility to the echinocandins.176 Breakthrough C. parapsilosis infection has been reported during caspofungin therapy in humans, highlighting the clinical significance of this resistance phenotype.177 C. parapsilosis, along with its sibling species Candida orthopsilosis and Candida metapsilosis, possesses a naturally occurring proline-to-alanine amino acid substitution in Fks1, which results in elevated resistance to echinocandins.72 This substitution at position 660 is conserved and distal to hot-spot region 1 of the protein, causing a decreased affinity of the C. parapsilosis glucan synthase to caspofungin.72 Mutations in C. albicans and C. glabrata at the equivalent position cause comparable increases in echinocandin resistance and decreases in enzyme affinity to the echinocandins.72 Interestingly, C. parapsilosis isolates harboring the P660A substitution in Fks1 also demonstrate higher cell wall chitin levels compared to isolates with alternative Fks1 sequences, suggesting that alterations in cell wall composition accompanying this naturally occurring mutation may be important in intrinsic echinocandin resistance.
Antifungal resistance can be intrinsic to fungal species, as well as to distinct growth states adopted by typically nonresistant species. In most natural environments, Candida grows as biofilms, which are organized, three-dimensional microbial communities irreversibly attached to surfaces.178 Candida biofilms are composed of a layer of yeast cells facilitating surface adherence, a heterogeneous layer of filamentous cells, and an outer layer composed of an exopolymeric matrix.178,179 C. albicans is the fungal pathogen most commonly associated with biofilm formation, particularly as medical implants support its colonization. Although planktonic C. albicans cells are not intrinsically resistant to clinically available antifungals, C. albicans biofilms exhibit inherent multidrug resistance.178,180,181 Cells comprising these biofilms overexpress efflux-pump encoding genes and are capable of expressing CDR1 and MDR1 during all developmental phases, which contributes to their robust resistance to azoles.181,182 Glucan synthesis by Fks1 is critical for biofilm-mediated drug resistance, as β−1,3 glucan in the adhesive biofilm matrix sequesters antifungals in a “sponge” to prevent the drugs from reaching their targets.183,184 This mechanism has been implicated in resistance to azoles, echinocandins, and polyenes.15,184,185 Interestingly, Hsp90 and downstream factors of the PKC-MAPK pathway regulate β−1,3 glucan biosynthesis in the biofilm matrix,184–186 and genetic or pharmacological compromise of Hsp90 restores azole efficacy in a rat catheter model of biofilm infection.186 With the ever-growing increase in immunocompromised patients utilizing medical implants, it is particularly critical to develop novel strategies to efficiently combat biofilm-mediated infections.
4. THERAPEUTIC STRATEGIES TO COMBAT RESISTANCE
Although advances are being made to improve our current antifungal arsenal and to identify novel drug targets, the rate at which antimicrobials are being developed is vastly outpaced by the rate at which resistance is emerging. Thus, examining alternative approaches, beyond targeting essential genes or cellular functions, for the development of new antifungal treatments is imperative to bolster the antifungal pipeline. A variety of strategies are in the early stages of being explored, including the use of combination therapy, the development of antivirulence agents, and modulating host immune responses to fungal pathogens to combat or prevent infection.
4.1. Combination Therapy
Combination therapy is routinely implemented for the treatment of diverse infectious diseases, including malaria, tuberculosis, and HIV/AIDS, while its potential in antifungal treatment is only beginning to be explored.19,187 Specific drug combinations can overcome existing resistance and/or delay or prevent further resistance from emerging through numerous modalities (Figure 4). For instance, by combining multiple drugs with distinct mechanisms of action, the number of intracellular processes targeted increases, which makes evolution of resistance more difficult as the pathogen must acquire mutations in multiple genes.17,187 Additionally, combination therapy can increase the fungicidal efficiency of normally fungistatic drugs (e.g. the azoles), reducing pathogen population sizes and, thus, the probability of resistance arising.187,188 Importantly, in rare cases a phenomenon known as selection inversion can occur, reversing drug resistance by neutralizing selective advantages and imposing direct fitness costs on the evolution of drug resistance.189 Finally, combination therapy has the potential to unveil additional efficacious treatments given that fungal biology is controlled by highly interconnected and functionally redundant networks of biological interactions.190–192 The potential for targeting synthetic lethal interactions provides an expanded target space for efficacious combinations of compounds that on their own have little impact on fitness but combined are lethal. Together, this highlights that implementation of combination therapy is a beneficial avenue to address and treat drug-resistant fungal infections.
Figure 4.
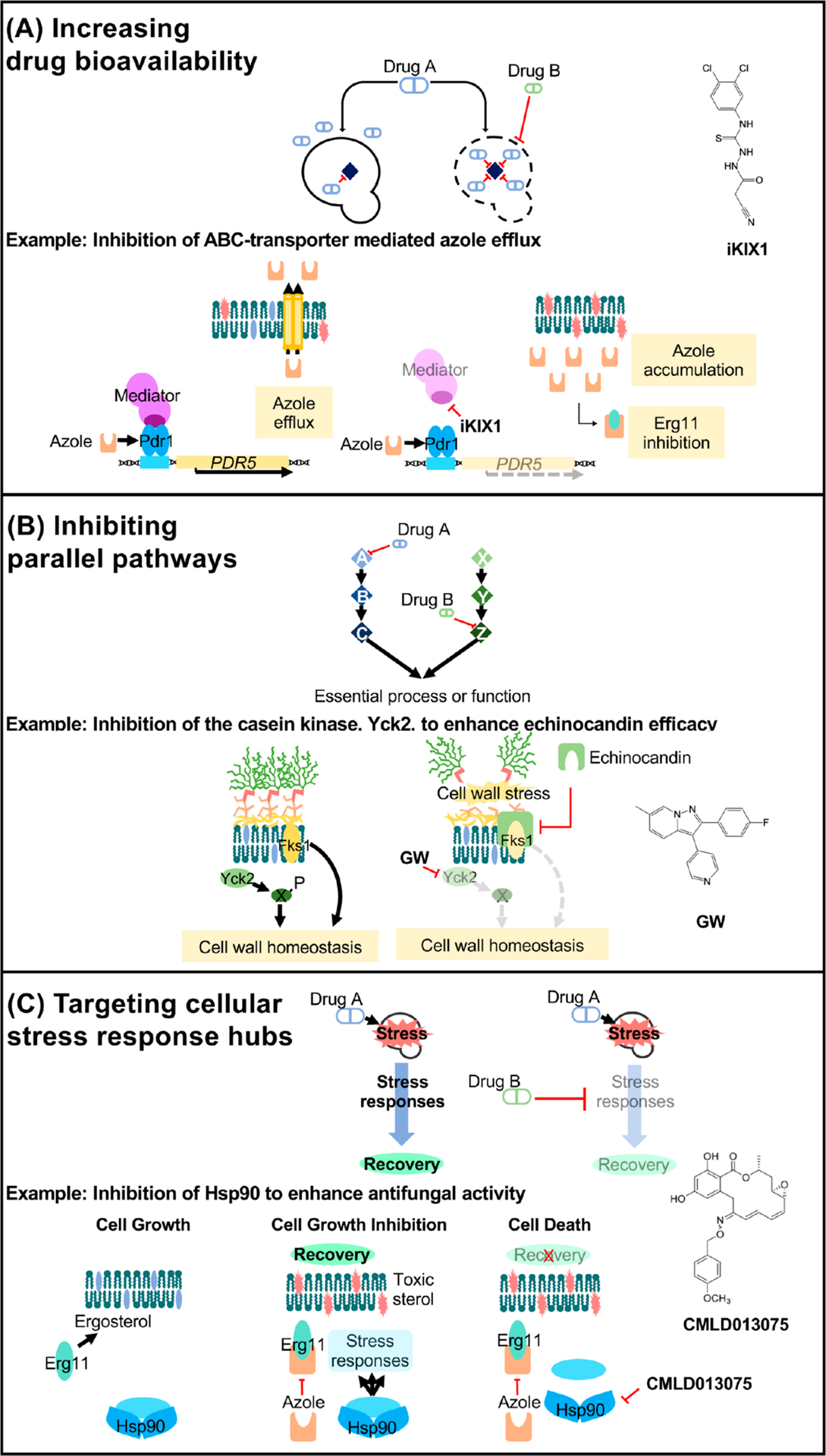
Mechanisms of drug potentiation. (A) Two drugs can potentiate the activity of one another when one drug’s action (drug B) increases the bioavailability of another (drug A) within the target cell. For example, in C. glabrata, the small-molecule iKIX1 (structure shown on right) synergizes with the azoles and resensitizes resistant isolates to fluconazole. iKIX1 disrupts the interaction between the KIX domain within the Gal11/Med15 mediator complex (dark purple) and Pdr1, which prevents upregulation of genes encoding efflux pumps (such as PDR5) in response to the azoles. (B) Two drugs targeting proteins of parallel pathways that converge on a single, essential process or function may also display potentiation activity. Pharmacological inhibition of the nonessential stress kinase Yck2 with the novel 2,3-aryl-pyrazolopyridine compound, GW, potentiates echinocandin efficacy in C. albicans and exacerbates echinocandin-mediated cell wall disruption. (C) Fungal stress responses play a major role in basal antifungal tolerance as well as resistance. Hsp90 is a global regulator of stress-response circuitry, promoting tolerance and resistance to drug-induced stress. Key regulators of cellular stress responses (light blue) are stabilized by Hsp90 in response to antifungal treatment, enabling signal transduction that is necessary to survive in the presence of a drug-induced stress. Pharmacological compromise of Hsp90 using the fungal-selective inhibitor CMLD013075 blocks these signaling networks, thereby preventing the evolution of resistance and abrogating resistance once it has evolved.
Discovery of molecules that target fungal resistance determinants to be used in combination with existing antifungals is an important strategy for the development of antifungal combination therapy. As discussed previously, upregulation of drug efflux is a conserved evolutionary mechanism that fungal pathogens employ to acquire resistance to the azoles, with the consequence that inhibition of efflux should improve azole susceptibility in such isolates. Studies in C. glabrata identified a small molecule termed iKIX1 that is able to bind the C. glabrata Gal11 KIX domain and prevent Pdr1-mediated gene activation, sensitizing previously resistant isolates to the azoles193 (Figure 4). In vivo analysis using both G. mellonella and mouse models of C. glabrata infection demonstrated improved therapeutic efficacy of fluconazole when combined with iKIX1 during treatment.193 Such analyses emphasize the therapeutic potential of small-molecule efflux inhibitors for use in the treatment of azole-resistant fungal infections.
In addition, inhibition of stress-response regulators confers therapeutic benefits in combination with existing antifungal drugs and can overcome preexisting resistance. Although Hsp90 is a global regulator of stress-response circuitry, making it an attractive target in antifungal combination therapy, it is also remarkably conserved across all eukaryotes. Therefore, use of generalist Hsp90 inhibitors, many of which have been explored for use as anticancer agents, causes prohibitive host side effects in the context of systemic fungal infection.113 However, recent work focused on the C. albicans Hsp90 nucleotide-binding domain revealed that, despite Hsp90 sequence conservation across species, C. albicans Hsp90 possesses distinct conformational flexibility.194 This observation enabled the synthesis of the first fungal-selective Hsp90 inhibitor, a resorcylate natural products derivative, as a proof-of-concept that efficiently inhibited C. albicans growth, potentiated azole activity against resistant isolates, and demonstrated low mammalian cell toxicity194 (Figure 4). Building on this effort to produce fungal-selective Hsp90 inhibitors, a series of aminopyrazole-substituted resorcylate amides were synthesized and found to possess potent and fungal-selective Hsp90 inhibitory activity.195 Similar approaches to selectively design inhibitors of fungal stress responses have been applied to the protein phosphatase calcineurin. Crystal structures of calcineurin complexed with the well-characterized inhibitor FK506 and the FK506-binding protein (FKBP12) from human and fungal pathogens revealed conformational differences.196 This, along with additional biochemical characterization, led to the characterization of a less immunosuppressive FK506 analogue, APX879, with an acetohydrazine substitution of the C22-carbonyl of FK506. Intriguingly, APX879 exhibited reduced immunosuppressive activity while maintaining broad-spectrum antifungal activity and efficacy in a murine model of invasive fungal infection.196 Such observations highlight the promise of leveraging chemo-structural approaches to synthesize fungal-selective molecules targeting highly conserved proteins.
In addition to the design of fungal-selective molecules that target known mediators of antifungal tolerance and resistance, a complementary and unbiased approach to identify synergistic drug interactions utilizes high-throughput screening of large compound collections. Molecules that enhance the activity of established antifungals against S. cerevisiae, C. albicans, and C. neoformans have been identified by chemical biology screens197,198 and computational methods.199,200 Many of these studies have not surprisingly observed that compounds that increase azole efficacy often target membrane homeostasis or sphingolipid biosynthesis.201 However, select studies have also used chemical screens to uncover novel cellular targets with previously unappreciated roles in antifungal resistance. For example, recent work described the characterization of a 2,3-aryl-pyrazolopyridine scaffold compound, GW, as an inhibitor of the casein kinase 1 family member Yck2.202 Combination of GW with caspofungin eradicated C. albicans drug resistance in vitro, and genetic depletion of YCK2 caused a significant decline in fungal burden in a mouse model of systemic caspofungin-resistant C. albicans infection. This establishes Yck2 as an important mediator of cell wall integrity and echinocandin resistance202 (Figure 4).
4.2. Targeting Virulence
Targeting fungal virulence provides a complementary approach to the development of fungicidal and fungistatic agents, as in principle these therapeutics simply aim to occlude the ability of a microbe to cause harm to its host. Although the potential utility of targeting nonessential genes has only recently been appreciated, there are already examples of antibiotics that target essential structures rather than essential proteins, as with daptomycin and the Gram-positive cell wall.203 Targeting virulence factors offers many benefits including expanding the repertoire of antifungal targets, minimizing effects on the host mycobiome, and reducing selection pressure for the evolution of drug resistance.204,205 Among the C. albicans virulence factors, its capacity to undergo a morphogenetic transition between yeast and filamentous states is one of the most critical206,207 (Figure 5). While the yeast morphology is important in facilitating early adhesion and dissemination events during C. albicans infection, the filamentous state is responsible for causing physical damage to tissues, systemic invasion, and immune evasion. Filamentation is regulated by numerous, complex signaling pathways, and the identification and analysis of small molecules that target essential components of this developmental transition have been promising in uncovering novel antivirulence agents. For instance, the small-molecule filastatin inhibits filamentous growth, biofilm formation, and pathogenesis in vivo208 (Figure 5). Initial investigations of the clinical utility of this small molecule have suggested that using it to coat medical devices can prevent fungal adherence and disrupt biofilm formation, presenting a new clinical option to prevent acute nosocomial infections resulting from medical implant use.209
Figure 5.
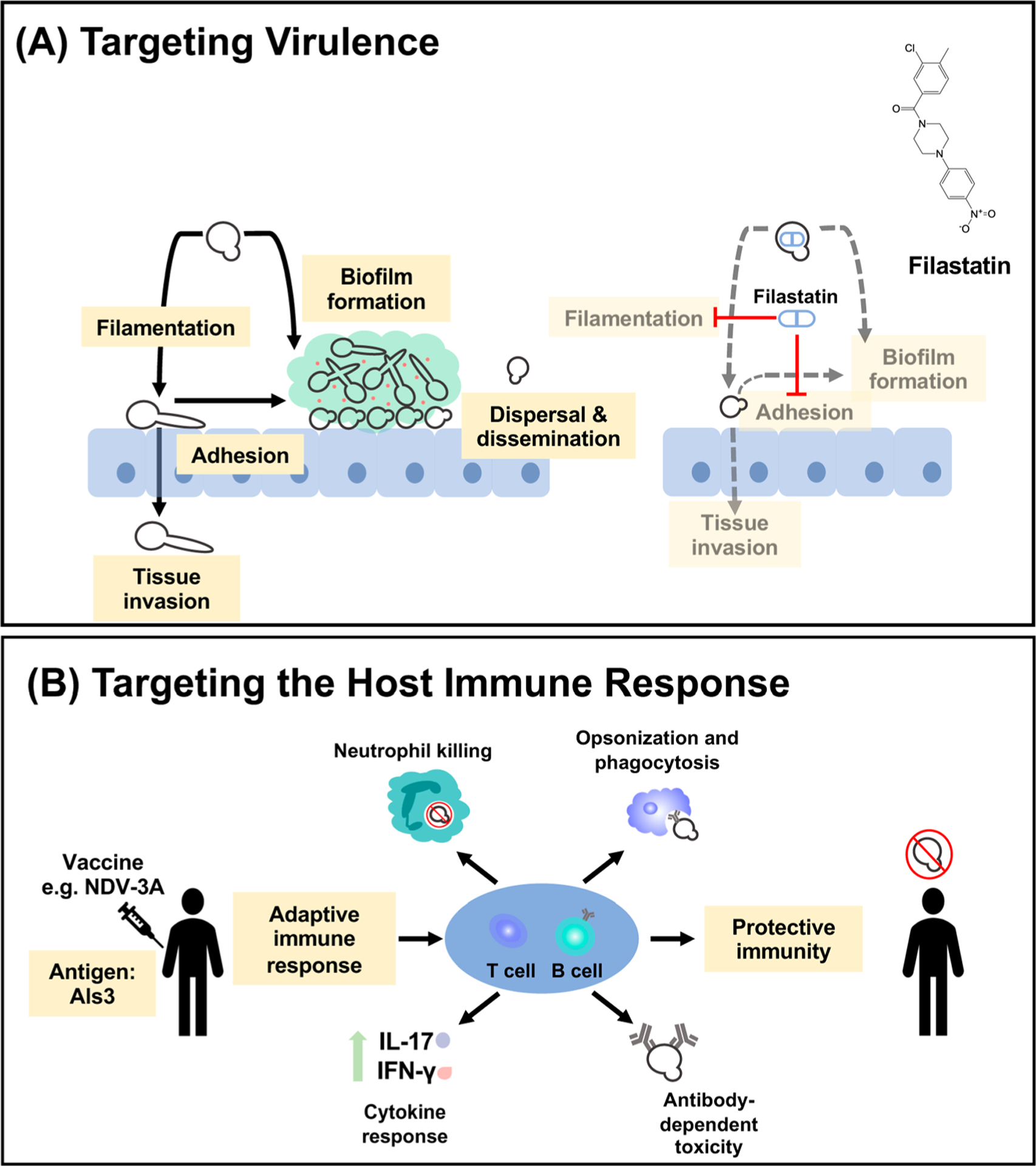
Targeting fungal virulence and the host immune response as alternative antifungal strategies. (A) Thwarting fungal infection can be achieved through manipulation of key fungal virulence traits. The C. albicans morphogenetic transition between yeast and filamentous states enables it to adhere to, damage, invade, and disseminate through host tissues, as well as to form biofilms. The small-molecule filastatin inhibits C. albicans filamentation, surface adhesion, and biofilm formation. (B) Vaccination is a potential therapeutic approach to combat fungal disease, particularly in vulnerable populations. Development of the novel anti-Candida vaccine NDV-3A employs the highly immunogenic adhesin and invasin Als3 to elicit an adaptive immune response mediated by antibody and T-cell responses. Antibody-facilitated opsonization for phagocytosis, T-cell mediated proliferation of proinflammatory cytokines IL-17 and IFN-γ, and enhancement of C. albicans neutrophil killing are characteristic of immunization and facilitate protective immunity in the host.
Intriguingly, many compounds that display bioactivity alone or in combination with conventional antifungals also modulate C. albicans morphogenesis. In fact, even the azoles directly impact C. albicans morphology by preventing the formation of hyphae.9,210 In addition, targeting Hsp90 locks cells in a filamentous state, impairs the dispersal of C. albicans biofilms, and attenuates virulence in mouse models of infection.186,211 Hsp90 has also been shown to regulate virulence programs in Cryptococcus and Aspergillus species.145,212 In addition, the natural product beauvericin, which targets drug efflux and TOR signaling, also represses the expression of many filament-specific genes, blocking the yeast-to-hyphal transition.122 Finally, Pkc1 not only plays pivotal roles in both echinocandin and azole stress responses but is critical for the initiation of filamentous growth.213 Thus, these and other key mediators of fungal signaling networks often play dual roles in both drug resistance and virulence, making them even more promising targets for the development of novel therapeutics.
A number of other avenues are beginning to be explored to attenuate the virulence of fungal pathogens and circumvent drug resistance. Investigations in C. albicans, C. neoformans, and Aspergillus species have demonstrated the importance of fungal sphingolipids in virulence.214–217 Genetic and pharmacological approaches to deplete these sphingolipids, namely, inositol phosphoryl ceramides or glucosylceramides, cause reduced pathogenicity, making them attractive targets for the development of antivirulence therapeutics.214–217 Furthermore, farnesyltransferases contribute to virulence as well as morphogenesis and cell cycle control in fungal pathogens. C. albicans, C. neoformans, and A. fumigatus farnesyltransferases are essential for both virulence and cellular growth.218–221 Initial analysis of human farnesyltransferase inhibitors developed as anticancer therapeutics, including manumycin A and tipifarnib, has revealed potent antifungal activity.219,222 Efforts targeting other fungal virulence factors including proteinases, phospholipases, and elastases, as well as other cellular pathways that impact C. albicans pathogenicity such as GPI anchor biosynthesis enzymes, are underway to expand the current repertoire of antifungal agents, and they represent auspicious mechanisms to minimize detrimental host effects and the emergence of resistance elicited by conventional antifungals.19,205
4.3. Targeting Host Immunity
Modulating the host immune system by bolstering its capacity to resist fungal infection is another alternative therapeutic avenue to combat fungal infection (Figure 5). Development of preventative immunotherapeutics and vaccines against fungal pathogens has been explored with varying degrees of success. Making use of a shared fungal antigen present in multiple common pathogenic genera led researchers to focus on cell wall β-glucans for vaccine development.223 Although they are poorly immunogenic, conjugation of β-glucans to diphtheria toxoids has shown great promise in mounting strong anti-β-glucan responses in murine models.224 Conjugate β-glucan-based vaccines have demonstrated initial potential in restricting infection and prolonging survival in murine models of Candida, Cryptococcus, and Aspergillus infection, suggesting that they may present a valuable broad-spectrum therapeutic strategy following further development.224–227
The Candida agglutinin-like sequence (ALS) protein Als3, is a hyphal-specific glycophosphatidylinositol cell wall protein with multifunctional adhesin and invasion properties.228,229 Als3 is highly immunogenic, eliciting robust anti-Als3 B-cell responses and T-cell responses in mice, as well as anti-Als3 antibody responses in humans.230,231 Early analysis of a recombinant N-terminal Als3 vaccine, NDV-3A, demonstrated protective efficacy in vaginal, oral, and systemic murine models of C. albicans infection230,232 (Figure 5). Subsequent progression through a phase II randomized clinical trial revealed the safety and efficacy of this vaccine in treating patients with recurrent vulvovaginal candidiasis.233 Further interrogation of the vaccine’s beneficial properties also demonstrated that NDV-3A induces C. auris cross-reactive antibody and T-cell responses and that vaccination with Als3 protects mice from disseminated C. glabrata, C. parapsilosis, and C. tropicalis infection.229 The success of NDV-3A to date highlights that targeting host immune responses through vaccination to combat fungal infection is a viable therapeutic strategy and should encourage further investigation of additional fungal vaccines.
5. CONCLUSION AND FUTURE OUTLOOK
A thorough understanding of the mechanistic principles governing antifungal drug resistance is fundamental for the development of more effective treatment strategies to combat current and emerging fungal threats. Despite the fact that we are still learning of new ways that Candida can become resistant to therapeutic treatments, advances in chemical genomic approaches in diverse fungal species have enabled powerful strategies to understand how antifungal agents exert their activity, as well as how resistance can emerge to these chemical insults.234,235 Continued focus on the identification of new efficacious compounds will also be critical in the future in order to combat these pathogens. While there has been limited success in discovering novel antifungal classes, this is at least in part due to the fact that large-scale chemical libraries are typically biased toward targeting human biology. For example, synthetic compounds that dominate pharmaceutical company libraries are enriched in molecules with chemical properties that adopt the Lipinski rule of five,236 guidelines that may not be ideal for antimicrobials. Consequently, compounds identified in antifungal screens often overlap with compounds that have bioactivity against human cellular targets. Expanding screens of synthetic molecules to libraries that typically have not yielded bioactivity in human targets (so-called chemical dark matter) can offer an entry point into a selective antifungal target space.237 Focusing on scientific methods that adopt clever screens for new targets coupled with mining of natural products and synthetic molecule libraries will provide opportunities to yield new antifungal leads. Collectively, advanced functional genomics resources, promising new screening platforms, and renewed interest from governments and the pharmaceutical industry in combatting drug-resistant infectious disease provide great hope for the accelerated development of novel antifungal therapeutics.
ACKNOWLEDGMENTS
We thank all members of the Cowen lab for helpful discussions. L.E.C. is supported by the Canadian Institutes of Health Research Foundation (Grant FDN-154288) and a National Institutes of Health NIAID R01 (R01AI120958-01A1); L.E.C. is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and co-Director of the CIFAR Fungal Kingdom: Threats & Opportunities program.
Biographies
Yunjin (Rachel) Lee, B.Sc., completed her undergraduate degree in Honours Biology at McMaster University. For her senior thesis project with Dr. Ana Campos’ lab, she screened a library of protein kinase inhibitors to identify novel regulators and signaling pathways underlying Hydra regeneration. This piqued her interest in the use of chemical biology strategies to unravel complex biological mechanisms. After learning about the devastating impact of fungal pathogens, she wanted to be actively involved in researching new therapeutic strategies and understanding the mechanisms of antifungal drug resistance. Rachel joined Dr. Leah Cowen’s lab in December 2018 and is currently investigating the mechanisms of novel antifungal compounds that possess activity against Candida albicans, as well as characterizing the roles of protein kinases in C. albicans pathobiology.
Emily Puumala, M.Sc., completed her integrated Master’s degree at Durham University’s Department of Biosciences in Durham, U.K. Her research focused on the structural and functional properties of Rap, a bacteriophage lambda Holliday junction resolvase, as a member of Dr. Gary Sharples’ research group. Emily has been a Ph.D student in Dr. Cowen’s lab at the University of Toronto since December 2018. She is currently focused on identifying and investigating novel antifungal compounds to combat drug resistance in the emerging pathogen Candida auris.
Nicole Robbins, Ph.D, has been a senior research associate in Dr. Cowen’s lab since July 2016. She received her undergraduate degree from McMaster University and her Ph.D. from the University of Toronto. As a doctorate student with Dr. Cowen, she focused on mechanisms by which the molecular chaperone Hsp90 governed fungal drug resistance in Candida albicans. Nicole then went on to pursue a postdoctoral fellowship with Dr. Gerry Wright at McMaster University, where she used high-throughput screening and chemical biology approaches to elucidate antifungal mode-of-action. As a research associate with Dr. Cowen, she is involved in many projects that aim to provide a global view of the circuitry that govern drug resistance, morphogenesis, and virulence in emerging fungal pathogens.
Leah E. Cowen, Ph.D., is Professor and Chair of the Department of Molecular Genetics at the University of Toronto and Co-founder and Chief Scientific Officer of Bright Angel Therapeutics, a company that leverages state-of-the-art technologies for development of novel antifungal therapeutics. She received her undergraduate degree from the University of British Columbia and a Ph.D. from the University of Toronto, and she pursued postdoctoral studies at the Whitehead Institute, Massachusetts Institute of Technology. Her laboratory takes an interdisciplinary approach to understand what allows some microbes to exploit the host and cause disease and to develop new strategies to treat life-threatening infectious disease. Dr. Cowen has an outstanding track record of excellence in research, scholarship, and education. She has published over 85 high-impact research articles and delivered over 90 invited lectures. She has been recognized with a myriad of awards including a Burroughs Wellcome Fund Career Award, Grand Challenges Canada Star in Global Health Award, Merck Irving S. Sigal Memorial Award, E.W.R. Steacie Award, Canada Research Chair in Microbial Genomics & Infectious Disease, and Fellowship of the American Academy of Microbiology. Dr. Cowen is Co-Director of the CIFAR program The Fungal Kingdom: Threats & Opportunities. She has cultivated a network of international excellence through her extensive team of over 70 collaborators and is advancing knowledge translation as Chief Scientific Officer of Bright Angel Therapeutics.
Footnotes
The authors declare the following competing financial interest(s): L.E.C. is a co-founder and shareholder in Bright Angel Therapeutics, a platform company for development of novel antifungal therapeutics. L.E.C. is a consultant for Boragen, a small-molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.0c00199
REFERENCES
- (1).Brown GD; Denning DW; Gow NA; Levitz SM; Netea MG; White TC Hidden Killers: Human Fungal Infections. Sci. Transl. Med 2012, 4, 165rv13. [DOI] [PubMed] [Google Scholar]
- (2).Fisher MC; Hawkins NJ; Sanglard D; Gurr SJ Worldwide Emergence Of Resistance To Antifungal Drugs Challenges Human Health And Food Security. Science 2018, 360, 739–742. [DOI] [PubMed] [Google Scholar]
- (3).Pfaller MA; Diekema DJ Epidemiology of Invasive Mycoses in North America. Crit. Rev. Microbiol 2010, 36, 1–53. [DOI] [PubMed] [Google Scholar]
- (4).Enoch DA; Yang H; Aliyu SH; Micallef C The Changing Epidemiology of Invasive Fungal Infections. Methods Mol. Biol 2017, 1508, 17–65. [DOI] [PubMed] [Google Scholar]
- (5).Pfaller MA; Diekema DJ; Turnidge JD; Castanheira M; Jones RN Twenty Years of the SENTRY Antifungal Surveillance Program: Results for Candida Species From 1997–2016. Open Forum Infect. Dis 2019, 6, S79–S94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pfaller MA; Diekema DJ Epidemiology Of Invasive Candidiasis: A Persistent Public Health Problem. Clin. Microbiol. Rev 2007, 20, 133–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wisplinghoff H; Bischoff T; Tallent SM; Seifert H; Wenzel RP; Edmond MB Nosocomial Bloodstream Infections in US Hospitals: Analysis of 24,179 Cases From a Prospective Nationwide Surveillance Study. Clin. Infect. Dis 2004, 39, 309–17. [DOI] [PubMed] [Google Scholar]
- (8).Brown GD; Denning DW; Levitz SM Tackling Human Fungal Infections. Science 2012, 336, 647. [DOI] [PubMed] [Google Scholar]
- (9).Shapiro RS; Robbins N; Cowen LE Regulatory Circuitry Governing Fungal Development, Drug Resistance, and Disease. Microbiol. Mol. Biol. Rev 2011, 75, 213–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Satoh K; Makimura K; Hasumi Y; Nishiyama Y; Uchida K; Yamaguchi H Candida auris sp. nov. A Novel Ascomycetous Yeast Isolated from the External Ear Canal of an Inpatient in a Japanese Hospital. Microbiol. Immunol 2009, 53, 41–4. [DOI] [PubMed] [Google Scholar]
- (11).Rhodes J; Fisher MC Global Epidemiology Of Emerging Candida auris. Curr. Opin. Microbiol 2019, 52, 84–89. [DOI] [PubMed] [Google Scholar]
- (12).Munoz JF; Gade L; Chow NA; Loparev VN; Juieng P; Berkow EL; Farrer RA; Litvintseva AP; Cuomo CA Genomic Insights Into Multidrug-Resistance, Mating and Virulence in Candida Auris and Related Emerging Species. Nat. Commun 2018, 9, 5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lockhart SR Candida auris and multidrug resistance: Defining the New Normal. Fungal Genet. Biol 2019, 131, 103243. [DOI] [PubMed] [Google Scholar]
- (14).Roemer T; Krysan DJ Antifungal Drug Development: Challenges, Unmet Clinical Needs, and New Approaches. Cold Spring Harbor Perspect. Med 2014, 4, No. a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Revie NM; Iyer KR; Robbins N; Cowen LE Antifungal Drug Resistance: Evolution, Mechanisms and Impact. Curr. Opin. Microbiol 2018, 45, 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Perlin DS; Rautemaa-Richardson R; Alastruey-Izquierdo A The Global Problem of Antifungal Resistance: Prevalence, Mechanisms, and Management. Lancet Infect. Dis 2017, 17, E383–E392. [DOI] [PubMed] [Google Scholar]
- (17).Robbins N; Caplan T; Cowen LE Molecular Evolution of Antifungal Drug Resistance. Annu. Rev. Microbiol 2017, 71, 753–775. [DOI] [PubMed] [Google Scholar]
- (18).Perfect JR The Antifungal Pipeline: A Reality Check. Nat. Rev. Drug Discovery 2017, 16, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Robbins N; Wright GD; Cowen LE Antifungal Drugs: The Current Armamentarium and Development of New Agents. Microbiol. Spectrum 2016, 4, No. FUNK-0002–2016. [DOI] [PubMed] [Google Scholar]
- (20).Ostrosky-Zeichner L; Casadevall A; Galgiani JN; Odds FC; Rex JH An Insight Into The Antifungal Pipeline: Selected New Molecules and Beyond. Nat. Rev. Drug Discovery 2010, 9, 719–27. [DOI] [PubMed] [Google Scholar]
- (21).Anderson TM; Clay MC; Cioffi AG; Diaz KA; Hisao GS; Tuttle MD; Nieuwkoop AJ; Comellas G; Maryum N; Wang S; et al. Amphotericin Forms an Extramembranous and Fungicidal Sterol Sponge. Nat. Chem. Biol 2014, 10, 400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Agarwal AK; Rogers PD; Baerson SR; Jacob MR; Barker KS; Cleary JD; Walker LA; Nagle DG; Clark AM Genome-Wide Expression Profiling of the Response to Polyene, Pyrimidine, Azole, and Echinocandin Antifungal Agents in Saccharomyces cerevisiae. J. Biol. Chem 2003, 278, 34998–5015. [DOI] [PubMed] [Google Scholar]
- (23).Pappas PG; Kauffman CA; Andes DR; Clancy CJ; Marr KA; Ostrosky-Zeichner L; Reboli AC; Schuster MG; Vazquez JA; Walsh TJ; et al. Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis 2016, 62, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pfaller MA; Espinel-Ingroff A; Canton E; Castanheira M; Cuenca-Estrella M; Diekema DJ; Fothergill A; Fuller J; Ghannoum M; Jones RN; et al. Wild-type MIC Distributions and Epidemiological Cutoff Values for Amphotericin B, Flucytosine, and Itraconazole and Candida spp. as Determined by CLSI Broth Microdilution. J. Clin. Microbiol 2012, 50, 2040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Vincent BM; Lancaster AK; Scherz-Shouval R; Whitesell L; Lindquist S Fitness Trade-Offs Restrict the Evolution of Resistance To Amphotericin B. PLoS Biol 2013, 11, No. e1001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cowen LE The Evolution of Fungal Drug Resistance: Modulating the Trajectory From Genotype to Phenotype. Nat. Rev. Microbiol 2008, 6, 187–98. [DOI] [PubMed] [Google Scholar]
- (27).Allen D; Wilson D; Drew R; Perfect J Azole Antifungals: 35 Years of Invasive Fungal Infection Management. Expert Rev. Anti-Infect. Ther 2015, 13, 787–98. [DOI] [PubMed] [Google Scholar]
- (28).Wiederhold NP; Patterson HP; Tran BH; Yates CM; Schotzinger RJ; Garvey EP Fungal-Specific Cyp51 Inhibitor VT-1598 Demonstrates In Vitro Activity Against Candida and Cryptococcus Species, Endemic Fungi, Including Coccidioides Species, Aspergillus Species and Rhizopus arrhizus. J. Antimicrob. Chemother 2018, 73, 404–408. [DOI] [PubMed] [Google Scholar]
- (29).Warrilow AG; Parker JE; Price CL; Nes WD; Garvey EP; Hoekstra WJ; Schotzinger RJ; Kelly DE; Kelly SL The Investigational Drug VT-1129 Is a Highly Potent Inhibitor of Cryptococcus Species CYP51 but Only Weakly Inhibits the Human Enzyme. Antimicrob. Agents Chemother 2016, 60, 4530–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Warrilow AG; Hull CM; Parker JE; Garvey EP; Hoekstra WJ; Moore WR; Schotzinger RJ; Kelly DE; Kelly SL The Clinical Candidate VT-1161 is a Highly Potent Inhibitor of Candida albicans CYP51 but Fails to Bind the Human Enzyme. Antimicrob. Agents Chemother 2014, 58, 7121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hoekstra WJ; Garvey EP; Moore WR; Rafferty SW; Yates CM; Schotzinger RJ Design and Optimization of Highly-Selective Fungal CYP51 Inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 3455–8. [DOI] [PubMed] [Google Scholar]
- (32).Berkow EL; Lockhart SR Fluconazole Resistance in Candida Species: A Current Perspective. Infect. Drug Resist 2017, 10, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chowdhary A; Prakash A; Sharma C; Kordalewska M; Kumar A; Sarma S; Tarai B; Singh A; Upadhyaya G; Upadhyay S; Yadav P; Singh PK; Khillan V; Sachdeva N; Perlin DS; Meis JF A Multicentre Study of Antifungal Susceptibility Patterns Among 350 Candida auris Isolates (2009–17) in India: Role of the ERG11 and FKS1 Genes in Azole and Echinocandin Resistance. J. Antimicrob. Chemother 2018, 73, 891–899. [DOI] [PubMed] [Google Scholar]
- (34).Letscher-Bru V; Herbrecht R Caspofungin: The First Representative of a New Antifungal Class. J. Antimicrob. Chemother 2003, 51, 513–21. [DOI] [PubMed] [Google Scholar]
- (35).Cornely OA; Bassetti M; Calandra T; Garbino J; Kullberg BJ; Lortholary O; Meersseman W; Akova M; Arendrup MC; Arikan-Akdagli S; et al. ESCMID* Guideline for the Diagnosis and Management of Candida Diseases 2012: Non-Neutropenic Adult Patients. Clin. Microbiol. Infect 2012, 18, 19–37. [DOI] [PubMed] [Google Scholar]
- (36).Patil A; Majumdar S Echinocandins in Antifungal Pharmacotherapy. J. Pharm. Pharmacol 2017, 69, 1635–1660. [DOI] [PubMed] [Google Scholar]
- (37).Denning DW Echinocandin Antifungal Drugs. Lancet 2003, 362, 1142–51. [DOI] [PubMed] [Google Scholar]
- (38).Davis MR; Donnelley MA; Thompson GR Ibrexafungerp: A Novel Oral Glucan Synthase Inhibitor. Med. Mycol 2019, No. myz083. [DOI] [PubMed] [Google Scholar]
- (39).Alexander BD; Johnson MD; Pfeiffer CD; Jimenez-Ortigosa C; Catania J; Booker R; Castanheira M; Messer SA; Perlin DS; Pfaller MA Increasing Echinocandin Resistance in Candida glabrata: Clinical Failure Correlates with Presence of FKS Mutations and Elevated Minimum Inhibitory Concentrations. Clin. Infect. Dis 2013, 56, 1724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lockhart SR; Etienne KA; Vallabhaneni S; Farooqi J; Chowdhary A; Govender NP; Colombo AL; Calvo B; Cuomo CA; Desjardins CA; et al. Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis 2017, 64, 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Berman J; Krysan DJ Drug Resistance and Tolerance in Fungi. Nat. Rev. Microbiol 2020, DOI: 10.1038/s41579-019-0322-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lamb DC; Kelly DE; Schunck WH; Shyadehi AZ; Akhtar M; Lowe DJ; Baldwin BC; Kelly SL The Mutation T315A in Candida albicans Sterol 14alpha-Demethylase Causes Reduced Enzyme Activity and Fluconazole Resistance Through Reduced Affinity. J. Biol. Chem 1997, 272, 5682–8. [DOI] [PubMed] [Google Scholar]
- (43).Morio F; Loge C; Besse B; Hennequin C; Le Pape P Screening for Amino Acid Substitutions in the Candida albicans Erg11 Protein of Azole-Susceptible and Azole-Resistant Clinical Isolates: New Substitutions and a Review of the Literature. Diagn. Microbiol. Infect. Dis 2010, 66, 373–84. [DOI] [PubMed] [Google Scholar]
- (44).Marichal P; Koymans L; Willemsens S; Bellens D; Verhasselt P; Luyten W; Borgers M; Ramaekers FC; Odds FC; Vanden Bossche H Contribution of Mutations in the Cytochrome P450 14alpha-Demethylase (Erg11p, Cyp51p) to Azole Resistance in Candida albicans. Microbiology 1999, 145, 2701–2713. [DOI] [PubMed] [Google Scholar]
- (45).Xiang MJ; Liu JY; Ni PH; Wang S; Shi C; Wei B; Ni YX; Ge HL Erg11 Mutations Associated with Azole Resistance in Clinical Isolates of Candida albicans. FEMS Yeast Res 2013, 13, 386–93. [DOI] [PubMed] [Google Scholar]
- (46).Wu Y; Gao N; Li C; Gao J; Ying C A Newly Identified Amino Acid Substitution T123I in the 14alpha-Demethylase (Erg11p) Of Candida albicans Confers Azole Resistance. FEMS Yeast Res 2017, 17, No. fox012. [DOI] [PubMed] [Google Scholar]
- (47).Flowers SA; Colon B; Whaley SG; Schuler MA; Rogers PD Contribution of Clinically Derived Mutations in ERG11 To Azole Resistance in Candida albicans. Antimicrob. Agents Chemother 2015, 59, 450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Healey KR; Kordalewska M; Jimenez Ortigosa C; Singh A; Berrio I; Chowdhary A; Perlin DS Limited ERG11 Mutations Identified in Isolates of Candida auris Directly Contribute to Reduced Azole Susceptibility. Antimicrob. Agents Chemother 2018, 62, No. e01427–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Brun S; Berges T; Poupard P; Vauzelle-Moreau C; Renier G; Chabasse D; Bouchara JP Mechanisms of Azole Resistance in Petite Mutants of Candida glabrata. Antimicrob. Agents Chemother 2004, 48, 1788–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Salazar SB; Wang C; Munsterkotter M; Okamoto M; Takahashi-Nakaguchi A; Chibana H; Lopes MM; Guldener U; Butler G; Mira NP Comparative Genomic and Transcriptomic Analyses Unveil Novel Features of Azole Resistance and Adaptation to the Human Host in Candida glabrata. FEMS Yeast Res 2018, 18, No. fox079. [DOI] [PubMed] [Google Scholar]
- (51).Spettel K; Barousch W; Makristathis A; Zeller I; Nehr M; Selitsch B; Lackner M; Rath PM; Steinmann J; Willinger B Analysis of Antifungal Resistance Genes in Candida albicans and Candida glabrata Using Next Generation Sequencing. PLoS One 2019, 14, No. e0210397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hull CM; Parker JE; Bader O; Weig M; Gross U; Warrilow AG; Kelly DE; Kelly SL Facultative Sterol Uptake in an Ergosterol-Deficient Clinical Isolate of Candida glabrata Harboring a Missense Mutation in ERG11 and Exhibiting Cross-Resistance to Azoles and Amphotericin B. Antimicrob. Agents Chemother 2012, 56, 4223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hoot SJ; Smith AR; Brown RP; White TC An A643V Amino Acid Substitution in Upc2p Contributes to Azole Resistance in Well-Characterized Clinical Isolates of Candida albicans. Antimicrob. Agents Chemother 2011, 55, 940–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Heilmann CJ; Schneider S; Barker KS; Rogers PD; Morschhauser J An A643T Mutation in the Transcription Factor Upc2p Causes Constitutive ERG11 Upregulation and Increased Fluconazole Resistance in Candida albicans. Antimicrob. Agents Chemother 2010, 54, 353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Dunkel N; Liu TT; Barker KS; Homayouni R; Morschhauser J; Rogers PD A Gain-of-Function Mutation in the Transcription Factor Upc2p Causes Upregulation of Ergosterol Biosynthesis Genes and Increased Fluconazole Resistance in a Clinical Candida albicans Isolate. Eukaryotic Cell 2008, 7, 1180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).MacPherson S; Akache B; Weber S; De Deken X; Raymond M; Turcotte B Candida albicans Zinc Cluster Protein Upc2p Confers Resistance to Antifungal Drugs and is an Activator of Ergosterol Biosynthetic Genes. Antimicrob. Agents Chemother 2005, 49, 1745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Silver PM; Oliver BG; White TC Role of Candida albicans Transcription Factor Upc2p in Drug Resistance and Sterol Metabolism. Eukaryotic Cell 2004, 3, 1391–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Flowers SA; Barker KS; Berkow EL; Toner G; Chadwick SG; Gygax SE; Morschhauser J; Rogers PD Gain-of-Function Mutations in UPC2 are a Frequent Cause of ERG11 Upregulation in Azole-Resistant Clinical Isolates of Candida albicans. Eukaryotic Cell 2012, 11, 1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Vasicek EM; Berkow EL; Flowers SA; Barker KS; Rogers PD UPC2 is Universally Essential for Azole Antifungal Resistance in Candida albicans. Eukaryotic Cell 2014, 13, 933–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Whaley SG; Caudle KE; Vermitsky JP; Chadwick SG; Toner G; Barker KS; Gygax SE; Rogers PD UPC2A is Required for High-Level Azole Antifungal Resistance in Candida glabrata. Antimicrob. Agents Chemother 2014, 58, 4543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ellis D Amphotericin B: Spectrum and Resistance. J. Antimicrob. Chemother 2002, 49, 7–10. [DOI] [PubMed] [Google Scholar]
- (62).Jensen RH; Astvad KM; Silva LV; Sanglard D; Jorgensen R; Nielsen KF; Mathiasen EG; Doroudian G; Perlin DS; Arendrup MC Stepwise Emergence of Azole, Echinocandin and Amphotericin B Multidrug Resistance In Vivo in Candida albicans Orchestrated by Multiple Genetic Alterations. J. Antimicrob. Chemother 2015, 70, 2551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Martel CM; Parker JE; Bader O; Weig M; Gross U; Warrilow AG; Rolley N; Kelly DE; Kelly SL Identification and Characterization of Four Azole-Resistant erg3 Mutants of Candida albicans. Antimicrob. Agents Chemother 2010, 54, 4527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Martel CM; Parker JE; Bader O; Weig M; Gross U; Warrilow AG; Kelly DE; Kelly SL A Clinical Isolate of Candida albicans with Mutations in ERG11 (Encoding Sterol 14alpha-Demethylase) and ERG5 (Encoding C22 Desaturase) is Cross Resistant to Azoles and Amphotericin B. Antimicrob. Agents Chemother 2010, 54, 3578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Sanglard D; Ischer F; Parkinson T; Falconer D; Bille J Candida albicans Mutations in the Ergosterol Biosynthetic Pathway and Resistance to Several Antifungal Agents. Antimicrob. Agents Chemother 2003, 47, 2404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Vandeputte P; Tronchin G; Larcher G; Ernoult E; Berges T; Chabasse D; Bouchara JP A Nonsense Mutation in the ERG6 Gene Leads to Reduced Susceptibility to Polyenes in a Clinical Isolate of Candida glabrata. Antimicrob. Agents Chemother 2008, 52, 3701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Vazquez JA; Arganoza MT; Boikov D; Yoon S; Sobel JD; Akins RA Stable Phenotypic Resistance of Candida Species to Amphotericin B Conferred by Preexposure to Subinhibitory Levels of Azoles. J. Clin. Microbiol 1998, 36, 2690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Perlin DS Resistance to Echinocandin-Class Antifungal Drugs. Drug Resist. Updates 2007, 10, 121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Park S; Kelly R; Kahn JN; Robles J; Hsu MJ; Register E; Li W; Vyas V; Fan H; Abruzzo G; et al. Specific Substitutions in the Echinocandin Target Fks1p Account For Reduced Susceptibility of Rare Laboratory and Clinical Candida Sp. Isolates. Antimicrob. Agents Chemother 2005, 49, 3264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Garcia-Effron G; Lee S; Park S; Cleary JD; Perlin DS Effect of Candida glabrata FKS1 and FKS2 Mutations on Echinocandin Sensitivity and Kinetics of 1,3-Beta-D-Glucan Synthase: Implication for the Existing Susceptibility Breakpoint. Antimicrob. Agents Chemother 2009, 53, 3690–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Garcia-Effron G; Park S; Perlin DS Correlating Echinocandin MIC and Kinetic Inhibition Of Fks1Mutant Glucan Synthases for Candida albicans: Implications for Interpretive Breakpoints. Antimicrob. Agents Chemother 2009, 53, 112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Garcia-Effron G; Katiyar SK; Park S; Edlind TD; Perlin DS A Naturally Occurring Proline-To-Alanine Amino Acid Change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis Accounts for Reduced Echinocandin Susceptibility. Antimicrob. Agents Chemother 2008, 52, 2305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Suwunnakorn S; Wakabayashi H; Kordalewska M; Perlin DS; Rustchenko E FKS2 and FKS3 Genes of Opportunistic Human Pathogen Candida albicans Influence Echinocandin Susceptibility. Antimicrob. Agents Chemother 2018, 62, No. e02299–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Katiyar S; Pfaller M; Edlind T Candida albicans and Candida glabrata Clinical Isolates Exhibiting Reduced Echinocandin Susceptibility. Antimicrob. Agents Chemother 2006, 50, 2892–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kordalewska M; Lee A; Park S; Berrio I; Chowdhary A; Zhao Y; Perlin DS Understanding Echinocandin Resistance in the Emerging Pathogen Candida auris. Antimicrob. Agents Chemother 2018, 62, No. e00238–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Rhodes J; Abdolrasouli A; Farrer RA; Cuomo CA; Aanensen DM; Armstrong-James D; Fisher MC; Schelenz S Genomic Epidemiology of the UK Outbreak of the Emerging Human Fungal Pathogen Candida auris. Emerging Microbes Infect 2018, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Coleman JJ; Mylonakis E Efflux in Fungi: La Piece de resistance. PLoS Pathog 2009, 5, No. e1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Tsao S; Rahkhoodaee F; Raymond M Relative Contributions of the Candida albicans ABC Transporters Cdr1p and Cdr2p to Clinical Azole Resistance. Antimicrob. Agents Chemother 2009, 53, 1344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Coste A; Turner V; Ischer F; Morschhauser J; Forche A; Selmecki A; Berman J; Bille J; Sanglard D A Mutation in Tac1p, A Transcription Factor Regulating CDR1 and CDR2, is Coupled with Loss of Heterozygosity at Chromosome 5 to Mediate Antifungal Resistance in Candida albicans. Genetics 2006, 172, 2139–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Sanglard D; Ischer F; Bille J Role of ATP-Binding-Cassette Transporter Genes in High-Frequency Acquisition of Resistance to Azole Antifungals in Candida glabrata. Antimicrob. Agents Chemother 2001, 45, 1174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Sanglard D; Ischer F; Calabrese D; Majcherczyk PA; Bille J The ATP Binding Cassette Transporter Gene CgCDR1 from Candida glabrata is Involved in the Resistance of Clinical Isolates to Azole Antifungal Agents. Antimicrob. Agents Chemother 1999, 43, 2753–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Torelli R; Posteraro B; Ferrari S; La Sorda M; Fadda G; Sanglard D; Sanguinetti M The ATP-Binding Cassette Transporter-Encoding Gene CgSNQ2 is Contributing to the CgPDR1-Dependent Azole Resistance of Candida glabrata. Mol. Microbiol 2008, 68, 186–201. [DOI] [PubMed] [Google Scholar]
- (83).Paul S; Bair TB; Moye-Rowley WS Identification of Genomic Binding Sites for Candida glabrata Pdr1 Transcription Factor in Wild-Type and rho0 Cells. Antimicrob. Agents Chemother 2014, 58, 6904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Paul S; Schmidt JA; Moye-Rowley WS Regulation of the CgPdr1 Transcription Factor from the Pathogen Candida glabrata. Eukaryotic Cell 2011, 10, 187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Vermitsky JP; Earhart KD; Smith WL; Homayouni R; Edlind TD; Rogers PD Pdr1 Regulates Multidrug Resistance in Candida glabrata: Gene Disruption and Genome-Wide Expression Studies. Mol. Microbiol 2006, 61, 704–22. [DOI] [PubMed] [Google Scholar]
- (86).Tsai HF; Krol AA; Sarti KE; Bennett JE Candida glabrata PDR1, a Transcriptional Regulator of a Pleiotropic Drug Resistance Network, Mediates Azole Resistance in Clinical Isolates and Petite Mutants. Antimicrob. Agents Chemother 2006, 50, 1384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Caudle KE; Barker KS; Wiederhold NP; Xu L; Homayouni R; Rogers PD Genomewide Expression Profile Analysis of the Candida glabrata Pdr1 Regulon. Eukaryotic Cell 2011, 10, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Thakur JK; Arthanari H; Yang F; Pan SJ; Fan X; Breger J; Frueh DP; Gulshan K; Li DK; Mylonakis E; et al. A Nuclear Receptor-Like Pathway Regulating Multidrug Resistance in Fungi. Nature 2008, 452, 604–9. [DOI] [PubMed] [Google Scholar]
- (89).Liu Z; Myers LC Mediator Tail Module Is Required for Tac1-Activated CDR1 Expression and Azole Resistance in Candida albicans. Antimicrob. Agents Chemother 2017, 61, No. e01342–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Gaur M; Puri N; Manoharlal R; Rai V; Mukhopadhayay G; Choudhury D; Prasad R MFS Transportome of the Human Pathogenic Yeast Candida albicans. BMC Genomics 2008, 9, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).White TC Increased mRNA Levels of ERG16, CDR, and MDR1 Correlate with Increases in Azole Resistance in Candida albicans Isolates from a Patient Infected with Human Immunodeficiency Virus. Antimicrob. Agents Chemother 1997, 41, 1482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Wirsching S; Michel S; Morschhauser J Targeted Gene Disruption in Candida albicans Wild-Type Strains: The Role of the MDR1 Gene in Fluconazole Resistance of Clinical Candida albicans Isolates. Mol. Microbiol 2000, 36, 856–65. [DOI] [PubMed] [Google Scholar]
- (93).Morschhauser J; Barker KS; Liu TT; Blass-Warmuth J; Homayouni R; Rogers PD The Transcription Factor Mrr1p Controls Expression of the MDR1 Efflux Pump and Mediates Multidrug Resistance in Candida albicans. PLoS Pathog 2007, 3, No. e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Dunkel N; Blass J; Rogers PD; Morschhauser J Mutations in the Multi-Drug Resistance Regulator MRR1, Followed by Loss of Heterozygosity, are the Main Cause of MDR1 Overexpression in Fluconazole-Resistant Candida albicans Strains. Mol. Microbiol 2008, 69, 827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Liu Z; Myers LC Candida albicans Swi/Snf and Mediator Complexes Differentially Regulate Mrr1-Induced MDR1 Expression and Fluconazole Resistance. Antimicrob. Agents Chemother 2017, 61, No. e01344–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Chen KH; Miyazaki T; Tsai HF; Bennett JE The bZip Transcription Factor CgAp1p is Involved in Multidrug Resistance and Required for Activation of Multidrug Transporter Gene CgFLR1 in Candida glabrata. Gene 2007, 386, 63–72. [DOI] [PubMed] [Google Scholar]
- (97).Chatterjee S; Alampalli SV; Nageshan RK; Chettiar ST; Joshi S; Tatu US Draft Genome of a Commonly Misdiagnosed Multidrug Resistant Pathogen. BMC Genomics 2015, 16, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Rybak JM; Doorley LA; Nishimoto AT; Barker KS; Palmer GE; Rogers PD Abrogation of Triazole Resistance upon Deletion of CDR1 in a Clinical Isolate of Candida auris. Antimicrob. Agents Chemother 2019, 63, No. e00057–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Kim SH; Iyer KR; Pardeshi L; Munoz JF; Robbins N; Cuomo CA; Wong KH; Cowen LE Genetic Analysis of Candida auris Implicates Hsp90 in Morphogenesis and Azole Tolerance and Cdr1 in Azole Resistance. mBio 2019, 10, No. e02529–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Kean R; Delaney C; Sherry L; Borman A; Johnson EM; Richardson MD; Rautemaa-Richardson R; Williams C; Ramage G Transcriptome Assembly and Profiling of Candida auris Reveals Novel Insights into Biofilm-Mediated Resistance. mSphere 2018, 3, No. e00334–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Rybak JM; Munoz JF; Barker KS; Parker JE; Esquivel BD; Berkow EL; Lockhart SR; Gade L; Palmer GE; White TC Mutations in TAC1B: a Novel Genetic Determinant of Clinical Fluconazole Resistance in C. auris. mBio 2020, 11, e00365–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Montoya MC; Moye-Rowley WS; Krysan DJ Candida auris: The Canary in the Mine of Antifungal Drug Resistance. ACS Infect. Dis 2019, 5, 1487–1492. [DOI] [PubMed] [Google Scholar]
- (103).Cannon RD; Lamping E; Holmes AR; Niimi K; Baret PV; Keniya MV; Tanabe K; Niimi M; Goffeau A; Monk BC Efflux-Mediated Antifungal Drug Resistance. Clin. Microbiol. Rev 2009, 22, 291–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Escandon P; Chow NA; Caceres DH; Gade L; Berkow EL; Armstrong P; Rivera S; Misas E; Duarte C; Moulton-Meissner H; et al. Molecular Epidemiology of Candida auris in Colombia Reveals a Highly Related, Countrywide Colonization with Regional Patterns in Amphotericin B Resistance. Clin. Infect. Dis 2018, 68, 15–21. [DOI] [PubMed] [Google Scholar]
- (105).Cowen LE; Steinbach WJ Stress, Drugs, and Evolution: The Role of Cellular Signaling in Fungal Drug Resistance. Eukaryotic Cell 2008, 7, 747–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Kelly SL; Lamb DC; Kelly DE; Manning NJ; Loeffler J; Hebart H; Schumacher U; Einsele H Resistance to Fluconazole and Cross-Resistance to Amphotericin B in Candida albicans from AIDS Patients Caused by Defective Sterol Delta5,6-Desaturation. FEBS Lett 1997, 400, 80–2. [DOI] [PubMed] [Google Scholar]
- (107).Morio F; Pagniez F; Lacroix C; Miegeville M; Le Pape P Amino Acid Substitutions in the Candida albicans Sterol Delta5,6-Desaturase (Erg3p) Confer Azole Resistance: Characterization of Two Novel Mutants With Impaired Virulence. J. Antimicrob. Chemother 2012, 67, 2131–8. [DOI] [PubMed] [Google Scholar]
- (108).O’Meara TR; Robbins N; Cowen LE The Hsp90 Chaperone Network Modulates Candida Virulence Traits. Trends Microbiol 2017, 25, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Cowen LE The Fungal Achilles’ Heel: Targeting Hsp90 to Cripple Fungal Pathogens. Curr. Opin. Microbiol 2013, 16, 377–84. [DOI] [PubMed] [Google Scholar]
- (110).Taipale M; Krykbaeva I; Koeva M; Kayatekin C; Westover KD; Karras GI; Lindquist S Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Jarosz DF; Taipale M; Lindquist S Protein Homeostasis and the Phenotypic Manifestation of Genetic Diversity: Principles and Mechanisms. Annu. Rev. Genet 2010, 44, 189–216. [DOI] [PubMed] [Google Scholar]
- (112).Taipale M; Jarosz DF; Lindquist S HSP90 at the Hub of Protein Homeostasis: Emerging Mechanistic Insights. Nat. Rev. Mol. Cell Biol 2010, 11, 515–28. [DOI] [PubMed] [Google Scholar]
- (113).Cowen LE; Singh SD; Kohler JR; Collins C; Zaas AK; Schell WA; Aziz H; Mylonakis E; Perfect JR; Whitesell L; et al. Harnessing Hsp90 Function as a Powerful, Broadly Effective Therapeutic Strategy for Fungal Infectious Disease. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 2818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Cowen LE; Lindquist S Hsp90 Potentiates the Rapid Evolution of New Traits: Drug Resistance in Diverse Fungi. Science 2005, 309, 2185–9. [DOI] [PubMed] [Google Scholar]
- (115).Singh SD; Robbins N; Zaas AK; Schell WA; Perfect JR; Cowen LE Hsp90 Governs Echinocandin Resistance in the Pathogenic Yeast Candida albicans via Calcineurin. PLoS Pathog 2009, 5, No. e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Cruz MC; Goldstein AL; Blankenship JR; Del Poeta M; Davis D; Cardenas ME; Perfect JR; McCusker JH; Heitman J Calcineurin is Essential for Survival During Membrane Stress in Candida albicans. EMBO J 2002, 21, 546–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).LaFayette SL; Collins C; Zaas AK; Schell WA; Betancourt-Quiroz M; Gunatilaka AA; Perfect JR; Cowen LE PKC Signaling Regulates Drug Resistance of the Fungal Pathogen Candida albicans via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90. PLoS Pathog 2010, 6, No. e1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Bruno VM; Mitchell AP Regulation of Azole Drug Susceptibility by Candida albicans Protein Kinase CK2. Mol. Microbiol 2005, 56, 559–73. [DOI] [PubMed] [Google Scholar]
- (119).Diezmann S; Michaut M; Shapiro RS; Bader GD; Cowen LE Mapping the Hsp90 Genetic Interaction Network in Candida albicans Reveals Environmental Contingency and Rewired Circuitry. PLoS Genet 2012, 8, No. e1002562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Mollapour M; Tsutsumi S; Truman AW; Xu W; Vaughan CK; Beebe K; Konstantinova A; Vourganti S; Panaretou B; Piper PW; et al. Threonine 22 Phosphorylation Attenuates Hsp90 Interaction with Cochaperones and Affects its Chaperone Activity. Mol. Cell 2011, 41, 672–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Robbins N; Collins C; Morhayim J; Cowen LE Metabolic Control of Antifungal Drug Resistance. Fungal Genet. Biol 2010, 47, 81–93. [DOI] [PubMed] [Google Scholar]
- (122).Shekhar-Guturja T; Tebung WA; Mount H; Liu N; Kohler JR; Whiteway M; Cowen LE Beauvericin Potentiates Azole Activity via Inhibition of Multidrug Efflux, Blocks Candida albicans Morphogenesis, and Is Effluxed via Yor1 and Circuitry Controlled by Zcf29. Antimicrob. Agents Chemother 2016, 60, 7468–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Khandelwal NK; Chauhan N; Sarkar P; Esquivel BD; Coccetti P; Singh A; Coste AT; Gupta M; Sanglard D; White TC; et al. Azole Resistance in a Candida albicans Mutant Lacking the ABC Transporter CDR6/ROA1 Depends on TOR Signaling. J. Biol. Chem 2018, 293, 412–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Robbins N; Leach MD; Cowen LE Lysine Deacetylases Hda1 and Rpd3 Regulate Hsp90 Function Thereby Governing Fungal Drug Resistance. Cell Rep 2012, 2, 878–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Li X; Robbins N; O’Meara TR; Cowen LE Extensive Functional Redundancy in the Regulation of Candida albicans Drug Resistance and Morphogenesis by Lysine Deacetylases Hos2, Hda1, Rpd3 and Rpd31. Mol. Microbiol 2017, 103, 635–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Pfaller MA; Messer SA; Georgopapadakou N; Martell LA; Besterman JM; Diekema DJ Activity of MGCD290, a Hos2 Histone Deacetylase Inhibitor, in Combination with Azole Antifungals Against Opportunistic Fungal Pathogens. J. Clin. Microbiol 2009, 47, 3797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Pfaller MA; Rhomberg PR; Messer SA; Castanheira M In Vitro Activity of a Hos2 Deacetylase Inhibitor, MGCD290, in Combination with Echinocandins Against Echinocandin-Resistant Candida Species. Diagn. Microbiol. Infect. Dis 2015, 81, 259–63. [DOI] [PubMed] [Google Scholar]
- (128).Besterman J; Nguyen DT; Ste-Croix H MGCD290, an Oral Fungal Hos2 Inhibitor, Enhances the Antifungal Properties of Fluconazole Following Multiple-or Single-Dose Oral Administration in Pre-and Post-Infection Settings. In Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); MethylGene, Inc.: San Francisco, CA, 2012; Vol. Poster M-1711. [Google Scholar]
- (129).Orta-Zavalza E; Guerrero-Serrano G; Gutierrez-Escobedo G; Canas-Villamar I; Juarez-Cepeda J; Castano I; De Las Penas A Local Silencing Controls the Oxidative Stress Response and the Multidrug Resistance in Candida glabrata. Mol. Microbiol 2013, 88, 1135–48. [DOI] [PubMed] [Google Scholar]
- (130).Sellam A; Askew C; Epp E; Lavoie H; Whiteway M; Nantel A Genome-Wide Mapping of the Coactivator Ada2p Yields Insight Into the Functional Roles of SAGA/ADA Complex in Candida albicans. Mol. Biol. Cell 2009, 20, 2389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Cornet M; Gaillardin C pH Signaling in Human Fungal Pathogens: A New Target for Antifungal Strategies. Eukaryotic Cell 2014, 13, 342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Garnaud C; Garcia-Oliver E; Wang Y; Maubon D; Bailly S; Despinasse Q; Champleboux M; Govin J; Cornet M The Rim Pathway Mediates Antifungal Tolerance in Candida albicans through Newly Identified Rim101 Transcriptional Targets, Including Hsp90 and Ipt1. Antimicrob. Agents Chemother 2018, 62, No. e01785–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Levin DE Cell Wall Integrity Signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev 2005, 69, 262–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Kamada Y; Qadota H; Python CP; Anraku Y; Ohya Y; Levin DE Activation of Yeast Protein Kinase C by Rho1 GTPase. J. Biol. Chem 1996, 271, 9193–6. [DOI] [PubMed] [Google Scholar]
- (135).Qadota H; Python CP; Inoue SB; Arisawa M; Anraku Y; Zheng Y; Watanabe T; Levin DE; Ohya Y Identification of Yeast Rho1p GTPase as a Regulatory Subunit of 1,3-Beta-Glucan Synthase. Science 1996, 272, 279–81. [DOI] [PubMed] [Google Scholar]
- (136).Walker LA; Gow NA; Munro CA Elevated Chitin Content Reduces the Susceptibility of Candida Species to Caspofungin. Antimicrob. Agents Chemother 2013, 57, 146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Walker LA; Munro CA; de Bruijn I; Lenardon MD; McKinnon A; Gow NA Stimulation of Chitin Synthesis Rescues Candida albicans from Echinocandins. PLoS Pathog 2008, 4, No. e1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Lee KK; Maccallum DM; Jacobsen MD; Walker LA; Odds FC; Gow NA; Munro CA Elevated Cell Wall Chitin in Candida albicans Confers Echinocandin Resistance In Vivo. Antimicrob. Agents Chemother 2012, 56, 208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Stevens DA; Ichinomiya M; Koshi Y; Horiuchi H Escape of Candida from Caspofungin Inhibition at Concentrations Above the MIC (Paradoxical Effect) Accomplished by Increased Cell Wall Chitin; Evidence for Beta-1,6-Glucan Synthesis Inhibition by Caspofungin. Antimicrob. Agents Chemother 2006, 50, 3160–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Munro CA; Selvaggini S; de Bruijn I; Walker L; Lenardon MD; Gerssen B; Milne S; Brown AJ; Gow NA The PKC, HOG and Ca2+ Signalling Pathways Co-Ordinately Regulate Chitin Synthesis in Candida albicans. Mol. Microbiol 2007, 63, 1399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Cota JM; Grabinski JL; Talbert RL; Burgess DS; Rogers PD; Edlind TD; Wiederhold NP Increases in SLT2 expression and Chitin Content are Associated with Incomplete Killing of Candida glabrata by Caspofungin. Antimicrob. Agents Chemother 2008, 52, 1144–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Singh-Babak SD; Babak T; Diezmann S; Hill JA; Xie JL; Chen YL; Poutanen SM; Rennie RP; Heitman J; Cowen LE Global Analysis of the Evolution and Mechanism of Echinocandin Resistance in Candida glabrata. PLoS Pathog 2012, 8, No. e1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Steinbach WJ; Reedy JL; Cramer RA Jr.; Perfect JR; Heitman J Harnessing Calcineurin as a Novel Anti-Infective Agent Against Invasive Fungal Infections. Nat. Rev. Microbiol 2007, 5, 418–30. [DOI] [PubMed] [Google Scholar]
- (144).Caplan T; Polvi EJ; Xie JL; Buckhalter S; Leach MD; Robbins N; Cowen LE Functional Genomic Screening Reveals Core Modulators of Echinocandin Stress Responses in Candida albicans. Cell Rep 2018, 23, 2292–2298. [DOI] [PubMed] [Google Scholar]
- (145).Lamoth F; Juvvadi PR; Fortwendel JR; Steinbach WJ Heat Shock Protein 90 is Required for Conidiation and Cell Wall Integrity in Aspergillus fumigatus. Eukaryotic Cell 2012, 11, 1324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Bruno VM; Kalachikov S; Subaran R; Nobile CJ; Kyratsous C; Mitchell AP Control of the C. albicans Cell Wall Damage Response by Transcriptional Regulator Cas5. PLoS Pathog 2006, 2, No. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Xie JL; Qin L; Miao Z; Grys BT; Diaz JC; Ting K; Krieger JR; Tong J; Tan K; Leach MD; et al. The Candida albicans Transcription Factor Cas5 Couples Stress Responses, Drug Resistance and Cell Cycle Regulation. Nat. Commun 2017, 8, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Mesa-Arango AC; Trevijano-Contador N; Roman E; Sanchez-Fresneda R; Casas C; Herrero E; Argüelles JC; Pla J; Cuenca-Estrella M; Zaragoza O The Production of Reactive Oxygen Species is a Universal Action Mechanism of Amphotericin B Against Pathogenic Yeasts and Contributes to the Fungicidal Effect of This Drug. Antimicrob. Agents Chemother 2014, 58, 6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Gonzalez-Parraga P; Sanchez-Fresneda R; Zaragoza O; Argüelles JC Amphotericin B Induces Trehalose Synthesis and Simultaneously Activates an Antioxidant Enzymatic Response in Candida albicans. Biochim. Biophys. Acta, Gen. Subj 2011, 1810, 777–783. [DOI] [PubMed] [Google Scholar]
- (150).Selmecki A; Forche A; Berman J Genomic Plasticity of the Human Fungal Pathogen Candida albicans. Eukaryotic Cell 2010, 9, 991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Chen G; Rubinstein B; Li R Whole Chromosome Aneuploidy: Big Mutations Drive Adaptation by Phenotypic Leap. BioEssays 2012, 34, 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Harrison BD; Hashemi J; Bibi M; Pulver R; Bavli D; Nahmias Y; Wellington M; Sapiro G; Berman J A Tetraploid Intermediate Precedes Aneuploid Formation In Yeasts Exposed to Fluconazole. PLoS Biol 2014, 12, No. e1001815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Coste A; Selmecki A; Forche A; Diogo D; Bougnoux ME; d’Enfert C; Berman J; Sanglard D Genotypic Evolution of Azole Resistance Mechanisms in Sequential Candida albicans Isolates. Eukaryotic Cell 2007, 6, 1889–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Selmecki A; Forche A; Berman J Aneuploidy and Isochromosome Formation in Drug-Resistant. Science 2006, 313, 367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Selmecki A; Gerami-Nejad M; Paulson C; Forche A; Berman J An Isochromosome Confers Drug Resistance In Vivo by Amplification of Two Genes, ERG11 and TAC1. Mol. Microbiol 2008, 68, 624–41. [DOI] [PubMed] [Google Scholar]
- (156).Selmecki AM; Dulmage K; Cowen LE; Anderson JB; Berman J Acquisition of Aneuploidy Provides Increased Fitness During the Evolution of Antifungal Drug Resistance. PLoS Genet 2009, 5, No. e1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Anderson MZ; Saha A; Haseeb A; Bennett RJ A Chromosome 4 Trisomy Contributes to Increased Fluconazole Resistance in a Clinical Isolate of Candida albicans. Microbiology 2017, 163, 856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Hill JA; Ammar R; Torti D; Nislow C; Cowen LE Genetic and Genomic Architecture of the Evolution Of Resistance to Antifungal Drug Combinations. PLoS Genet 2013, 9, No. e1003390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Hirakawa MP; Chyou DE; Huang D; Slan AR; Bennett RJ Parasex Generates Phenotypic Diversity de Novo and Impacts Drug Resistance and Virulence in Candida albicans. Genetics 2017, 207, 1195–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Mount HO; Revie NM; Todd RT; Anstett K; Collins C; Costanzo M; Boone C; Robbins N; Selmecki A; Cowen LE Global Analysis of Genetic Circuitry and Adaptive Mechanisms Enabling Resistance to the Azole Antifungal Drugs. PLoS Genet 2018, 14, No. e1007319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Marichal P; Vanden Bossche H; Odds FC; Nobels G; Warnock DW; Timmerman V; Van Broeckhoven C; Fay S; Mose-Larsen P Molecular Biological Characterization of an Azole-Resistant Candida glabrata Isolate. Antimicrob. Agents Chemother 1997, 41, 2229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Polakova S; Blume C; Zarate JA; Mentel M; Jorck-Ramberg D; Stenderup J; Piskur J Formation of New Chromosomes as a Virulence Mechanism in Yeast Candida glabrata. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 2688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Forche A; Abbey D; Pisithkul T; Weinzierl MA; Ringstrom T; Bruck D; Petersen K; Berman J Stress Alters Rates and Types of Loss of Heterozygosity in Candida albicans. mBio 2011, 2, No. e00129–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Niimi K; Monk BC; Hirai A; Hatakenaka K; Umeyama T; Lamping E; Maki K; Tanabe K; Kamimura T; Ikeda F; et al. Clinically Significant Micafungin Resistance in Candida albicans Involves Modification of a Glucan Synthase Catalytic Subunit GSC1 (FKS1) Allele Followed by Loss of Heterozygosity. J. Antimicrob. Chemother 2010, 65, 842–52. [DOI] [PubMed] [Google Scholar]
- (165).Yang F; Kravets A; Bethlendy G; Welle S; Rustchenko E Chromosome 5 Monosomy of Candida albicans Controls Susceptibility to Various Toxic Agents, Including Major Antifungals. Antimicrob. Agents Chemother 2013, 57, 5026–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Yang F; Teoh F; Tan ASM; Cao Y; Pavelka N; Berman J Aneuploidy Enables Cross-Adaptation to Unrelated Drugs. Mol. Biol. Evol 2019, 36, 1768–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Bravo Ruiz G; Ross ZK; Holmes E; Schelenz S; Gow NAR; Lorenz A Rapid and Extensive Karyotype Diversification in Haploid Clinical Candida auris Isolates. Curr. Genet 2019, 65, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Bhattacharya S; Holowka T; Orner EP; Fries BC Gene Duplication Associated with Increased Fluconazole Tolerance in Candida auris Cells of Advanced Generational Age. Sci. Rep 2019, 9, 5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Maligie MA; Selitrennikoff CP Cryptococcus neoformans Resistance to Echinocandins: (1,3)Beta-Glucan Synthase Activity is Sensitive to Echinocandins. Antimicrob. Agents Chemother 2005, 49, 2851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Ferrari S; Sanguinetti M; Torelli R; Posteraro B; Sanglard D Contribution of CgPDR1-Regulated Genes in Enhanced Virulence of Azole-Resistant Candida glabrata. PLoS One 2011, 6, No. e17589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Borst A; Raimer MT; Warnock DW; Morrison CJ; Arthington-Skaggs BA Rapid Acquisition of Stable Azole Resistance by Candida glabrata Isolates Obtained Before the Clinical Introduction of Fluconazole. Antimicrob. Agents Chemother 2005, 49, 783–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Ferrari S; Sanguinetti M; De Bernardis F; Torelli R; Posteraro B; Vandeputte P; Sanglard D Loss of Mitochondrial Functions Associated with Azole Resistance in Candida glabrata Results in Enhanced Virulence in Mice. Antimicrob. Agents Chemother 2011, 55, 1852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Zavrel M; Hoot SJ; White TC Comparison of Sterol Import Under Aerobic and Anaerobic Conditions in Three Fungal Species, Candida albicans, Candida glabrata, and Saccharomyces cerevisiae. Eukaryotic Cell 2013, 12, 725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Lamping E; Ranchod A; Nakamura K; Tyndall JD; Niimi K; Holmes AR; Niimi M; Cannon RD Abc1p is a Multidrug Efflux Transporter that Tips the Balance in Favor of Innate Azole Resistance in Candida krusei. Antimicrob. Agents Chemother 2009, 53, 354–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Orozco AS; Higginbotham LM; Hitchcock CA; Parkinson T; Falconer D; Ibrahim AS; Ghannoum MA; Filler SG Mechanism of Fluconazole Resistance in Candida krusei. Antimicrob. Agents Chemother 1998, 42, 2645–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Pfaller MA; Diekema DJ; Gibbs DL; Newell VA; Ng KP; Colombo A; Finquelievich J; Barnes R; Wadula J; Global Antifungal Surveillance Group.. Geographic and Temporal Trends in Isolation and Antifungal Susceptibility of Candida parapsilosis: A Global Assessment from the ARTEMIS DISK Antifungal Surveillance Program, 2001 to 2005. J. Clin. Microbiol 2008, 46, 842–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Cheung C; Guo Y; Gialanella P; Feldmesser M Development of Candidemia on Caspofungin Therapy: A Case Report. Infection 2006, 34, 345–8. [DOI] [PubMed] [Google Scholar]
- (178).Blankenship JR; Mitchell AP How to Build a Biofilm: A Fungal Perspective. Curr. Opin. Microbiol 2006, 9, 588–94. [DOI] [PubMed] [Google Scholar]
- (179).Douglas LJ Candida Biofilms and their Role in Infection. Trends Microbiol 2003, 11, 30–6. [DOI] [PubMed] [Google Scholar]
- (180).Mayer FL; Wilson D; Hube B Candida albicans Pathogenicity Mechanisms. Virulence 2013, 4, 119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Mukherjee PK; Chandra J; Kuhn DM; Ghannoum MA Mechanism of Fluconazole Resistance in Candida albicans Biofilms: Phase-Specific Role of Efflux Pumps and Membrane Sterols. Infect. Immun 2003, 71, 4333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Ramage G; Bachmann S; Patterson TF; Wickes BL; Lopez-Ribot JL Investigation of Multidrug Efflux Pumps in Relation to Fluconazole Resistance in Candida albicans biofilms. J. Antimicrob. Chemother 2002, 49, 973–80. [DOI] [PubMed] [Google Scholar]
- (183).Nett J; Lincoln L; Marchillo K; Massey R; Holoyda K; Hoff B; VanHandel M; Andes D Putative Role of Beta-1,3 Glucans in Candida albicans Biofilm Tesistance. Antimicrob. Agents Chemother 2007, 51, 510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Nett JE; Crawford K; Marchillo K; Andes DR Role of Fks1p and Matrix Glucan in Candida albicans Biofilm Resistance to an Echinocandin, Pyrimidine, and Polyene. Antimicrob. Agents Chemother 2010, 54, 3505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Nett JE; Sanchez H; Cain MT; Andes DR Genetic Basis of Candida Biofilm Resistance Due to Drug-Sequestering Matrix Glucan. J. Infect. Dis 2010, 202, 171–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Robbins N; Uppuluri P; Nett J; Rajendran R; Ramage G; Lopez-Ribot JL; Andes D; Cowen LE Hsp90 Governs Dispersion and Drug Resistance of Fungal Biofilms. PLoS Pathog 2011, 7, No. e1002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Spitzer M; Robbins N; Wright GD Combinatorial Strategies for Combating Invasive Fungal Infections. Virulence 2017, 8, 169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Onyewu C; Blankenship JR; Del Poeta M; Heitman J Ergosterol Biosynthesis Inhibitors Become Fungicidal when Combined with Calcineurin Inhibitors Against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother 2003, 47, 956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Baym M; Stone LK; Kishony R Multidrug Evolutionary Strategies to Reverse Antibiotic Resistance. Science 2016, 351, No. aad3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Costanzo M; VanderSluis B; Koch EN; Baryshnikova A; Pons C; Tan G; Wang W; Usaj M; Hanchard J; Lee SD; et al. A Global Genetic Interaction Network Maps a Wiring Diagram of Cellular Function. Science 2016, 353, No. aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Costanzo M; Baryshnikova A; Myers CL; Andrews B; Boone C Charting the Genetic Interaction Map of a Cell. Curr. Opin. Biotechnol 2011, 22, 66–74. [DOI] [PubMed] [Google Scholar]
- (192).Costanzo M; Baryshnikova A; Bellay J; Kim Y; Spear ED; Sevier CS; Ding H; Koh JL; Toufighi K; Mostafavi S; et al. The Genetic Landscape of a Cell. Science 2010, 327, 425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Nishikawa JL; Boeszoermenyi A; Vale-Silva LA; Torelli R; Posteraro B; Sohn YJ; Ji F; Gelev V; Sanglard D; Sanguinetti M; et al. Inhibiting Fungal Multidrug Resistance by Disrupting an Activator-Mediator Interaction. Nature 2016, 530, 485–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Whitesell L; Robbins N; Huang DS; McLellan CA; Shekhar-Guturja T; LeBlanc EV; Nation CS; Hui R; Hutchinson A; Collins C; et al. Structural Basis for Species-Selective Targeting of Hsp90 in a Pathogenic Fungus. Nat. Commun 2019, 10, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Huang DS; LeBlanc EV; Shekhar-Guturja T; Robbins N; Krysan DJ; Pizarro J; Whitesell L; Cowen LE; Brown LE Design and Synthesis of Fungal-Selective Resorcylate Aminopyrazole Hsp90 Inhibitors. J. Med. Chem 2020, 63, 2139–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Juvvadi PR; Fox D 3rd; Bobay BG; Hoy MJ; Gobeil SMC; Venters RA; Chang Z; Lin JJ; Averette AF; Cole DC; et al. Harnessing Calcineurin-FK506-FKBP12 Crystal Structures from Invasive Fungal Pathogens to Develop Antifungal Agents. Nat. Commun 2019, 10, 4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Borisy AA; Elliott PJ; Hurst NW; Lee MS; Lehar J; Price ER; Serbedzija G; Zimmermann GR; Foley MA; Stockwell BR; et al. Systematic Discovery of Multicomponent Therapeutics. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 7977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Zhang L; Yan K; Zhang Y; Huang R; Bian J; Zheng C; Sun H; Chen Z; Sun N; An R; et al. High-Throughput Synergy Screening Identifies Microbial Metabolites as Combination Agents for the Treatment of Fungal Infections. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 4606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Lehar J; Zimmermann GR; Krueger AS; Molnar RA; Ledell JT; Heilbut AM; Short GF 3rd; Giusti LC; Nolan GP; Magid OA; et al. Chemical Combination Effects Predict Connectivity in Biological Systems. Mol. Syst. Biol 2007, 3, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Lo YC; Senese S; Li CM; Hu Q; Huang Y; Damoiseaux R; Torres JZ Large-Scale Chemical Similarity Networks for Target Profiling of Compounds Identified in Cell-Based Chemical Screens. PLoS Comput. Biol 2015, 11, No. e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Spitzer M; Griffiths E; Blakely KM; Wildenhain J; Ejim L; Rossi L; De Pascale G; Curak J; Brown E; Tyers M; et al. Cross-Species Discovery of Syncretic Drug Combinations that Potentiate the Antifungal Fluconazole. Mol. Syst. Biol 2011, 7, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Caplan T; Lorente-Macias A; Stogios PJ; Evdokimova E; Hyde S; Wellington MA; Liston S; Iyer KR; Puumala E; Shekhar-Guturja T; et al. Overcoming Fungal Echinocandin Resistance through Inhibition of the Non-essential Stress Kinase Yck2. Cell Chem. Biol 2020, 27, 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Baltz RH Daptomycin: Mechanisms of Action and Resistance, and Biosynthetic Engineering. Curr. Opin. Chem. Biol 2009, 13, 144–51. [DOI] [PubMed] [Google Scholar]
- (204).Clatworthy AE; Pierson E; Hung DT Targeting Virulence: A New Paradigm for Antimicrobial Therapy. Nat. Chem. Biol 2007, 3, 541–8. [DOI] [PubMed] [Google Scholar]
- (205).Gauwerky K; Borelli C; Korting HC Targeting Virulence: A New Paradigm For Antifungals. Drug Discovery Today 2009, 14, 214–22. [DOI] [PubMed] [Google Scholar]
- (206).Noble SM; Gianetti BA; Witchley JN Candida albicans Cell-Type Switching and Functional Plasticity in the Mammalian Host. Nat. Rev. Microbiol 2017, 15, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Noble SM; French S; Kohn LA; Chen V; Johnson AD Systematic Screens of a Candida albicans Homozygous Deletion Library Decouple Morphogenetic Switching and Pathogenicity. Nat. Genet 2010, 42, 590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Fazly A; Jain C; Dehner AC; Issi L; Lilly EA; Ali A; Cao H; Fidel PL Jr.; Rao RP; Kaufman PD Chemical Screening Identifies Filastatin, A Small Molecule Inhibitor of Candida albicans Adhesion, Morphogenesis, and Pathogenesis. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 13594–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Vargas-Blanco D; Lynn A; Rosch J; Noreldin R; Salerni A; Lambert C; Rao RP A Pre-Therapeutic Coating for Medical Devices that Prevents the Attachment of Candida albicans. Ann. Clin. Microbiol. Antimicrob 2017, 16, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).O’Meara TR; Veri A; Ketela T; Jiang B; Roemer T; Cowen LE Global Analysis of Fungal Morphology Exposes Mechanisms of Host Cell Escape. Nat. Commun 2015, 6, 6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Shapiro RS; Uppuluri P; Zaas AK; Collins C; Senn H; Perfect JR; Heitman J; Cowen LE Hsp90 Orchestrates Temperature-Dependent Candida albicans Morphogenesis via Ras1-PKA Signaling. Curr. Biol 2009, 19, 621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Cordeiro RA; Evangelista AJJ; Serpa R; Marques FJF; Melo CVS; Oliveira JS; Franco JDS; Alencar LP; Bandeira T; Brilhante RSN; et al. Inhibition of Heat-Shock Protein 90 Enhances the Susceptibility to Antifungals and Reduces the Virulence of Cryptococcus neoformans/Cryptococcus gattii Species Complex. Microbiology 2016, 162, 309–317. [DOI] [PubMed] [Google Scholar]
- (213).Xie JL; Grahl N; Sless T; Leach MD; Kim SH; Hogan DA; Robbins N; Cowen LE Signaling through Lrg1, Rho1 and Pkc1 Governs Candida albicans Morphogenesis in Response to Diverse Cues. PLoS Genet 2016, 12, No. e1006405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Tafesse FG; Rashidfarrokhi A; Schmidt FI; Freinkman E; Dougan S; Dougan M; Esteban A; Maruyama T; Strijbis K; Ploegh HL Disruption of Sphingolipid Biosynthesis Blocks Phagocytosis of Candida albicans. PLoS Pathog 2015, 11, No. e1005188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Rittershaus PC; Kechichian TB; Allegood JC; Merrill AH Jr.; Hennig M; Luberto C; Del Poeta M Glucosylceramide Synthase is an Essential Regulator of Pathogenicity of Cryptococcus neoformans. J. Clin. Invest 2006, 116, 1651–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Levery SB; Momany M; Lindsey R; Toledo MS; Shayman JA; Fuller M; Brooks K; Doong RL; Straus AH; Takahashi HK Disruption of the Glucosylceramide Biosynthetic Pathway in Aspergillus nidulans and Aspergillus fumigatus by Inhibitors of UDP-Glc:Ceramide Glucosyltransferase Strongly Affects Spore Germination, Cell Cycle, and Hyphal Growth. FEBS Lett 2002, 525, 59–64. [DOI] [PubMed] [Google Scholar]
- (217).Zhong W; Jeffries MW; Georgopapadakou NH Inhibition of Inositol Phosphorylceramide Synthase by Aureobasidin A in Candida and Aspergillus Species. Antimicrob. Agents Chemother 2000, 44, 651–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Fortwendel JR; Juvvadi PR; Rogg LE; Asfaw YG; Burns KA; Randell SH; Steinbach WJ Plasma Membrane Localization is Required For RasA-Mediated Polarized Morphogenesis and Virulence of Aspergillus fumigatus. Eukaryotic Cell 2012, 11, 966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Hast MA; Nichols CB; Armstrong SM; Kelly SM; Hellinga HW; Alspaugh JA; Beese LS Structures of Cryptococcus neoformans Protein Farnesyltransferase Reveal Strategies for Developing Inhibitors that Target Fungal Pathogens. J. Biol. Chem 2011, 286, 35149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Song JL; White TC RAM2: An Essential Gene in the Prenylation Pathway of Candida albicans. Microbiology 2003, 149, 249–59. [DOI] [PubMed] [Google Scholar]
- (221).Vallim MA; Fernandes L; Alspaugh JA The RAM1 Gene Encoding a Protein-Farnesyltransferase Beta-Subunit Homologue is Essential in Cryptococcus neoformans. Microbiology 2004, 150, 1925–1935. [DOI] [PubMed] [Google Scholar]
- (222).Qiao J; Gao P; Jiang X; Fang H In Vitro Antifungal Activity of Farnesyltransferase Inhibitors Against Clinical Isolates of Aspergillus and Candida. Ann. Clin. Microbiol. Antimicrob 2013, 12, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Armstrong-James D; Brown GD; Netea MG; Zelante T; Gresnigt MS; van de Veerdonk FL; Levitz SM Immunotherapeutic Approaches to Treatment of Fungal Diseases. Lancet Infect. Dis 2017, 17, E393–E402. [DOI] [PubMed] [Google Scholar]
- (224).Torosantucci A; Bromuro C; Chiani P; De Bernardis F; Berti F; Galli C; Norelli F; Bellucci C; Polonelli L; Costantino P; et al. A Novel Glyco-Conjugate Vaccine Against Fungal Pathogens. J. Exp. Med 2005, 202, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Bromuro C; Romano M; Chiani P; Berti F; Tontini M; Proietti D; Mori E; Torosantucci A; Costantino P; Rappuoli R; et al. Beta-Glucan-CRM197 Conjugates as Candidates Antifungal Vaccines. Vaccine 2010, 28, 2615–23. [DOI] [PubMed] [Google Scholar]
- (226).Pietrella D; Rachini A; Torosantucci A; Chiani P; Brown AJ; Bistoni F; Costantino P; Mosci P; d’Enfert C; Rappuoli R; et al. A Beta-Glucan-Conjugate Vaccine and Anti-Beta-Glucan Antibodies are Effective Against Murine Vaginal Candidiasis as Assessed by a Novel In Vivo Imaging Technique. Vaccine 2010, 28, 1717–25. [DOI] [PubMed] [Google Scholar]
- (227).Specht CA; Lee CK; Huang H; Tipper DJ; Shen ZT; Lodge JK; Leszyk J; Ostroff GR; Levitz SM Protection Against Experimental Cryptococcosis Following Vaccination with Glucan Particles Containing Cryptococcus Alkaline Extracts. mBio 2015, 6, No. e01905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Sui X; Yan L; Jiang YY The Vaccines and Antibodies Associated with Als3p for Treatment of Candida albicans Infections. Vaccine 2017, 35, 5786–5793. [DOI] [PubMed] [Google Scholar]
- (229).Singh S; Uppuluri P; Mamouei Z; Alqarihi A; Elhassan H; French S; Lockhart SR; Chiller T; Edwards JE Jr.; Ibrahim AS The NDV-3A Vaccine Protects Mice from Multidrug Resistant Candida auris Infection. PLoS Pathog 2019, 15, No. e1007460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Ibrahim AS; Luo G; Gebremariam T; Lee H; Schmidt CS; Hennessey JP Jr.; French SW; Yeaman MR; Filler SG; Edwards JE Jr. NDV-3 Protects Mice from Vulvovaginal Candidiasis Through T- and B-Cell Immune Response. Vaccine 2013, 31, 5549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Schmidt CS; White CJ; Ibrahim AS; Filler SG; Fu Y; Yeaman MR; Edwards JE Jr.; Hennessey JP Jr. NDV-3, a Recombinant Alum-Adjuvanted Vaccine for Candida and Staphylococcus aureus, is Safe and Immunogenic in Healthy Adults. Vaccine 2012, 30, 7594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Spellberg BJ; Ibrahim AS; Avanesian V; Fu Y; Myers C; Phan QT; Filler SG; Yeaman MR; Edwards JE Jr. Efficacy of the Anti-Candida rAls3p-N or rAls1p-N Vaccines Against Disseminated and Mucosal Candidiasis. J. Infect. Dis 2006, 194, 256–260. [DOI] [PubMed] [Google Scholar]
- (233).Edwards JE Jr.; Schwartz MM; Schmidt CS; Sobel JD; Nyirjesy P; Schodel F; Marchus E; Lizakowski M; DeMontigny EA; Hoeg J; et al. A Fungal Immunotherapeutic Vaccine (NDV-3A) for Treatment of Recurrent Vulvovaginal Candidiasis-A Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Infect. Dis 2018, 66, 1928–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Roemer T; Boone C Systems-Level Antimicrobial Drug and Drug Synergy Discovery. Nat. Chem. Biol 2013, 9, 222–31. [DOI] [PubMed] [Google Scholar]
- (235).Roemer T; Davies J; Giaever G; Nislow C Bugs, Drugs and Chemical Genomics. Nat. Chem. Biol 2012, 8, 46–56. [DOI] [PubMed] [Google Scholar]
- (236).Lipinski CA Lead- and Drug-Like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technol 2004, 1, 337–41. [DOI] [PubMed] [Google Scholar]
- (237).Wassermann AM; Lounkine E; Hoepfner D; Le Goff G; King FJ; Studer C; Peltier JM; Grippo ML; Prindle V; Tao J; et al. Dark Chemical Matter as a Promising Starting Point for Drug Lead Discovery. Nat. Chem. Biol 2015, 11, 958–66. [DOI] [PubMed] [Google Scholar]