

Abstract
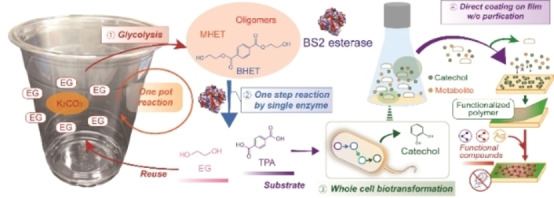
Chemo‐biological upcycling of poly(ethylene terephthalate) (PET) developed in this study includes the following key steps: chemo‐enzymatic PET depolymerization, biotransformation of terephthalic acid (TPA) into catechol, and its application as a coating agent. Monomeric units were first produced through PET glycolysis into bis(2‐hydroxyethyl) terephthalate (BHET), mono(2‐hydroxyethyl) terephthalate (MHET), and PET oligomers, and enzymatic hydrolysis of these glycolyzed products using Bacillus subtilis esterase (Bs2Est). Bs2Est efficiently hydrolyzed glycolyzed products into TPA as a key enzyme for chemo‐enzymatic depolymerization. Furthermore, catechol solution produced from TPA via a whole‐cell biotransformation (Escherichia coli) could be directly used for functional coating on various substrates after simple cell removal from the culture medium without further purification and water‐evaporation. This work demonstrates a proof‐of‐concept of a PET upcycling strategy via a combination of chemo‐biological conversion of PET waste into multifunctional coating materials.
Keywords: biocatalysis, catechol, esterase, poly(ethylene terephthalate), upcycling
PETit four: A proof‐of‐concept upcycling strategy to convert poly(ethylene terephthalate) (PET) into multifunctional catechol coating materials is presented: (1) PET glycolysis without purification; (2) one‐step enzymatic hydrolysis of glycolyzed products into terephthalic acid (TPA); (3) whole‐cell biotransformation of TPA into catechol using an engineered Escherichia coli; (4) application of crude catechol solution as a multifunctional coating material without purification.
Introduction
Over 360 million tons of plastics are produced annually worldwide; [1a] however, the recycling rate accounts for only less than 9 % of the produced plastics because the value of recycled plastics is relatively low due to poor properties. [1b] Plastic wastes are hardly recycled, and most of them ends up in landfills or oceans, leading to deleterious effects on the environment. A massive amount of accumulated plastics intensifies the destruction of the ecosystem because plastic wastes are hardly decomposed in the environment.[ 1b , 1c ] To address the plastic crisis, considerable research achievements were made including biological degradation and upcycling of polyesters and the discovery of plastic‐eating organisms.[ 1d , 1e ] In particular, biodegradable plastics have been vigorously developed, [2] but their commercial production is very low compared with nondegradable plastics. [1e] The environmental impact of biodegradable plastics is still marginal. Thus, plastic upcycling to valuable materials is of considerable interest for a sustainable society. Poly(ethylene terephthalate) (PET), a polymer synthesized from terephthalic acid (TPA) and ethylene glycol (EG), accounts for 5 % of the total plastic production and is used as a common thermoplastic for fibers and packaging materials. In contrast to hydrocarbon plastics, such as polyethylene (PE) and polypropylene (PP), the ester bond linkage of PET is relatively easy to decompose through hydrolysis. [3] PET has the advantage of being upcycled because the PET hydrolysates, TPA and EG, can be converted to high‐value‐added chemicals. Recently, PET upcycling strategies have been developed to produce value‐added chemicals, such as medium‐chain‐length polyhydroxyalkanoate (PHA), hydroxyalkanoyloxy alkanoates, catechol, gallic acid, vanillic acid, muconic acid, 2‐pyrone‐4,6‐dicarboxylic acid,[ 4a , 4b , 4c , 4d , 4e , 4f , 4g ] from the depolymerized monomers (i. e., TPA and EG). Reclaimed PET bottles are upcycled to higher‐value, long‐lifetime materials, fiber‐reinforced plastics via combination with bio‐based monomers. [4h] Antimicrobial compounds can be produced from PET using a catalyst‐free polyaddition polymerization process.[ 4i , 4j ] In addition, it has been reported that polyurethanes or poly(ester‐amide)s are synthesized using terephthalamide diol monomers derived from the aminolysis of PET waste.[ 4k , 4l , 4m , 4n , 4o , 4p ]
Nonetheless, in order to promote the industrialization of PET recycling and upcycling processes, it is desirable that the production cost of recycled or upcycled chemicals approaches that of virgin analogues. The high production cost of upcycled chemicals from PET is associated with the energy‐ and time‐consuming processes, such as (1) depolymerization of PET, (2) valorization of TPA, and (3) purification of upcycled products and water evaporation. In particular, the economic feasibility of PET upcycling is highly dependent on the efficiency of depolymerization, but various thermochemical and biological depolymerization methods reported until now have respective drawbacks. [5] The thermochemical methods readily depolymerize PET into a mixture of EG, TPA, bis(2‐hydroxyethyl) terephthalate (BHET), mono(2‐hydroxyethyl) terephthalate (MHET), and PET oligomers at a high temperature (≈450 °C) and high pressure (≈15 MPa). However, these processes often result in uncontrollable reactions, such as carbonization and over‐decomposition (e. g., ring‐opening), leading to the formation of byproducts. [6] In contrast, biological methods employing enzymes and microorganisms not only depolymerize PET under mild conditions but also produce value‐added chemicals from depolymerized monomers. However, the enzymatic hydrolysis of PET by a combination of PETase and MHETase results in low substrate loading and slow degradation of PET compared to thermochemical methods. [7] Recently, an engineered leaf‐branch compost cutinase (LCC) achieved high efficiency of PET depolymerization (more than 90 % in 10 h), useful to make new PET. [5c] Therefore, a chemo‐biological method with advantages of respective chemical and biological depolymerization processes has been suggested, in which a chemical pretreatment, such as glycolysis, degrades PET into hydrolysates with low molecular weight, including BHET, MHET, and PET oligomers, without over‐decomposition. [8] Furthermore, owing to improved enzyme accessibility, the enzymatic production of TPA from those hydrolysates is more efficient than the direct production from PET polymers. [9] Another critical step in PET upcycling is the purification of value‐added products. The biological valorization of TPA produces not only target chemicals but also byproducts and metabolites as impurities in aqueous solutions; thus, the purification of target chemicals, including the removal of impurities and water, is regarded as the cost‐determining method. The removal of impurities can be conducted via a combination of purification technologies such as membrane filtration, crystallization, extraction, or distillation. In addition, the target chemicals are generally produced in diluted concentrations, and excessive water content decreases the efficiency of transportation and drying. Costly and slow drying methods, such as freeze‐drying and spray‐drying, are required; however, these can also damage the target chemicals.
Catechol is one of the value‐added chemicals that can be produced through a biosynthetic pathway from PET waste‐derived TPA. The catechol‐mediated surface functionalization exhibits material‐independent surface‐coating ability. [10] Catechol groups are simultaneously oxidized in an aqueous solution and then covalently coupled with other catechols, as well as amines and thiols via Michael addition. [11] This reaction deposits multifunctional thin layers on plastic surfaces that are hardly coated. [12] TPA can be biologically converted to a catechol aqueous solution. It was reasoned that the catechol solution may form a functional coating on substrate surfaces even in the presence of rich impurities and excessive water.
In this study, we developed a proof‐of‐concept PET upcycling approach through chemo‐ and bio‐cascades including PET glycolysis into BHET by a biocompatible K2CO3 catalyst, BHET hydrolysis into TPA by Bs2Est esterase, whole‐cell biotransformation of TPA into catechol, and utilization of catechol as a coating agent.
Results and Discussion
PET glycolysis into BHET
PET glycolysis was conducted to produce BHET for enzymatic hydrolysis (Figure 1a,b). Zn acetate is a well‐known catalyst for PET glycolysis, and typical PET conversion in glycolysis is 50–66 %. [13] However, a biocompatible catalyst for PET glycolysis is necessary to effectively perform enzymatic hydrolysis of BHET and whole‐cell biotransformation of TPA into catechol. In this study, K2CO3 was selected as a catalyst for PET glycolysis because K2CO3 can catalyze PET glycolysis[ 14a , 14b ] and is commonly used as a buffering agent in biotransformation reactions.[ 14c , 14d , 14e , 14f , 14g , 14h ] A PET glycolysis efficiency of K2CO3 was as high as that of Zn acetate. [14] Reaction time (1–5 h), temperature (180–210 °C), and K2CO3 loading (0.05–0.4 g) were optimized for glycolysis of 5 g PET (Figures 1b, S1, and S2). The PET glycolysis conversion [Eq. (2)], that is, the amount of PET reacted, was enhanced with the increase in all three factors. However, the BHET yield was the highest (73.5 %, [Eq. (3)]) at the optimal conditions where temperature, reaction time, and catalyst content were 200 °C, 3 h, and 0.1 g, respectively for glycolysis of 5 g PET. The compositions of BHET, MHET, and PET oligomers at optimal conditions were 84.8, 7.7, and 8.7 %. Further PET glycolysis reactions were performed under these conditions. The glycolyzed PET products were purified via filtration and recrystallization to remove PET oligomers and EG/MHET, respectively. Any potential impurities in purified BHET may interfere with enzymatic hydrolysis of BHET or whole‐cell biotransformation of TPA. The Fourier‐transform infrared (FTIR) spectroscopy, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, and differential scanning calorimetry (DSC) analysis of the purified BHET product were identical to those of the standard BHET. These results confirmed the high purity of BHET (Figures 1c,e,f and S3a).
Figure 1.

Preparation of TPA from PET by a combination with glycolysis and enzymatic hydrolysis. (a) Three different PET‐to‐TPA hydrolysis pathways: (1) path 1 (enzymatic hydrolysis of pure BHET after crystallization), (2) path 2 (enzymatic hydrolysis of BHET mixture after filtration, (3) path 3 (enzymatic hydrolysis of BHET mixture without purification). (b) Temperature dependency of glycolysis. (c) FTIR spectra of the glycolyzed BHET in the PET‐to‐BHET conversion, reagent‐grade EG, BHET, and PET pieces. (d) Mass spectroscopy data of purified BHET after filtration. (e) 1H and (f) 13C NMR data of purified BHET after recrystallization. (g–i) Enzymatic hydrolysis profiles in paths 1–3, respectively. Among three different PET‐to‐TPA hydrolysis pathways, the one‐pot enzymatic hydrolysis of glycolyzed mixtures from path 3 without filtration and recrystallization achieves a high TPA yield (116.3 %). Paths 1–3 were performed at 30 °C and 1000 rpm in 100 mm sodium phosphate (pH 7.5) buffer for 10 h. The definition of enzyme unit used in this study is described in the Supporting Information.
Enzymatic hydrolysis of the glycolyzed products into TPA by Bacillus subtilis esterase
BHET substrates were prepared from the glycolyzed products by three pathways, that is, path 1 (pure BHET after crystallization), path 2 (BHET mixture after filtration), and path 3 (BHET mixture without purification). If the TPA yield of path 3 is equivalent to that of path 1, path 3 would be preferable to reduce the purification steps and maximize substrate utilization. Thus, four commercial esterases from Bacillus subtilis, Paenibacillus barcinonensis, Rhizopus oryzae, and Methylobacterium populi were examined to investigate their BHET‐hydrolytic activity on the glycolyzed products obtained from these three pathways (Figure S4). Among the four commercial esterases tested, only Bacillus subtilis esterase (Bs2Est) could completely hydrolyze BHET into TPA without the formation of MHET. [15] Biochemical characterization experiments for Bs2Est, including heterologous expression (Figure S5), reaction optimization (Figures S6–S8), the effect of potential inhibitors (Figures S9–S18), and kinetic parameters (Table S1), were applied to establish the enzymatic hydrolysis of BHET.
Interestingly, the solubility of TPA in aqueous buffer solution has an important effect on BHET hydrolysis of Bs2Est. EG (up to 40 mm, Figure S9) and TPA disodium salt (up to 16 mm, Figure S9) did not inhibit BHET hydrolytic activity of Bs2Est. However, 8 mm BHET and 8 mm MEHT significantly inhibited the hydrolytic activity of Bs2Est (Figure S11). The solubility of TPA was dependent on the type of buffer and TPA exhibited higher solubility in sodium phosphate buffer than in Tris‐HCl (Figure S12). High solubility of TPA in sodium phosphate buffer resulted in the improved hydrolytic activity of Bs2Est at high concentrations BHET (Figures S13, S15, S16). Enzyme inhibition experiments reveal that the acid form of TPA over maximal solubility (e. g., more than 24 mm of TPA in 50 mm sodium phosphate buffer) causes a deleterious effect on the hydrolytic activity of Bs2Est (Figure S13e,f).
After sequential optimization of Bs2Est‐catalyzed reactions, 2 u ml −1 (41.8 μg ml −1 ) of Bs2Est could hydrolyze the glycolyzed products obtained from three pathways with high yields at 30 °C within 10 h (Figure 1g). The yield (124.8 % [mol mol−1]) of TPA obtained from path 3 was highest in comparison with those from path 1 (97.7 % [mol mol−1]) and path 2 (102.4 % [mol mol−1]). This result indicates that the excessive yields of TPA from path 2 and 3 compared to that of path 1 can be attributed to the formation of TPA from the glycolyzed mixtures other than BHET. EG and PET oligomers seem to have no deleterious effect on the hydrolytic activity of Bs2Est. The TPA hydrolysis yield (97.7 % [mol mol−1]) for BHET from path 1 was similar to that for commercial reagent‐grade BHET (96.2 % [mol mol−1]) (Figure S7). The resulting TPA exhibited reagent‐grade purity, as confirmed using 1H and 13C NMR spectroscopy (Figures 1j,k, and S3c). Paths 2 and 3 contained more glycolyzed products other than BHET because of their simple purification steps compared with path 1 (Figure 1a). The enzymatic hydrolysis developed for path 1 was applied first to the glycolyzed mixture obtained from path 2 containing EG, BHET, and MHET. When the glycolyzed mixture containing 19.69 mm BHET and 0.47 mm MHET was allowed to react with 2 u ml −1 of Bs2Est in 100 mm sodium phosphate buffer (Figure 1h), 20.17 mm of TPA was produced without residual BHET within 10 h. The TPA yield based on the initial BHET loading (100 %) of path 2 was higher than that of path 1, containing purified BHET, due to additional hydrolysis of MHET in the path 2 mixture. This result indicates that the hydrolytic activity of Bs2Est was not inhibited by the impurities in the path 2 mixture, including EG or unknown products derived from PET (Figure S9).
Finally, the path 3 glycolyzed mixture of EG, BHET (18.82 mm), MHET (1.37 mm), and PET oligomers without any purification were hydrolyzed using 2 u ml −1 of Bs2Est (Figure 1i). Bs2Est successfully produced 23.49 mm TPA (124.8 % [mol mol−1]) without residual MHET and BHET. The production yield of TPA was higher than the expected TPA yield (20.19 mm) from soluble fractions including BHET and MHET in the path 3 mixture, indicating that Bs2Est hydrolyzed PET oligomers to release TPA. The applicability of chemo‐enzymatic PET depolymerization approach developed in this study was further examined with other commercially available PET bottles (Figure S19) resulting in the similar PET depolymerization yields for all samples tested. PET has different physical properties depending on its application. In general, PET for bottle has a higher molecular weight, tensile strength, and elastic modulus than PET for other applications such as fiber and film. [16] Although PET bottles were successfully depolymerized by chemo‐enzymatic depolymerization approach developed in this study, further experimental analysis will be necessary to investigate the effect of physical properties of PET on chemo‐biological upcycling.
Biocatalytic properties of Bs2Est activity toward PET oligomers, BHET, and MHET
Utilization of incompletely converted products (e. g., insoluble fraction in the glycolyzed mixtures) during chemical or biological conversion processes is an important issue to maximize substrate utilization and production yield, as well as reduce the purification burden. In the one‐pot hydrolysis reaction of the Bs2Est glycolysis mixtures (path 3), a higher amount of excess TPA was produced than that of BHET and MHET. This indicated that PET oligomers in the glycolyzed mixtures were hydrolyzed to release TPA, contributing to the enhanced TPA yield in path 3. To investigate the biocatalytic properties of Bs2Est toward PET oligomers, the composition of PET oligomers was analyzed using 1H NMR spectroscopy, gel‐permeation chromatography (GPC), and FTIR spectroscopy (Figure S20), and changes in the composition of PET oligomers during the enzymatic hydrolysis were monitored using high‐pressure liquid chromatography (HPLC) and matrix‐assisted laser desorption ionization‐time of flight (MALDI‐TOF) analysis (Figures 2a,b, and S21). When 1 g L−1 of PET oligomers (4.4 mm calculated based on dimer) ranged from DP2 to DP4 (mainly DP2, Figures 2b, S20, and S21) reacted with 2 U mL−1 of Bs2Est, 3.46 mm TPA was produced without BHET accumulation and putative reaction intermediates, such as TPA‐EG‐TPA or EG‐TPA‐EG‐TPA (Figure 2a,b). From MALDI‐TOF analysis, it was observed that the dimer disappeared, but DP3 and DP4 were detected at the end of hydrolysis reactions. This result indicates that DP3 and DP4 are poor substrates for Bs2Est. In particular, DP3 and DP4 may have poor solubility in the sodium phosphate buffer and thus, these oligomeric substrates may be better enzymatically depolymerized in the presence of organic solvents such as DMSO. [17] To verify the exo‐ and endo‐cleavage activities of Bs2Est, the hydrolysis reaction was performed in the presence of lower amounts of enzyme (0.5 and 1 U mL−1, Figure S21) and the reaction mixture profile was analyzed using MALDI‐TOF. Based on these results, we assumed that the dimer in PET oligomers was first hydrolyzed into BHET and MHET via the endo‐cleavage activity of Bs2Est. Thereafter, BHET was rapidly hydrolyzed into MHET, and then MHET was slowly hydrolyzed into TPA and EG (Table S1). To support the assumption of the specific hydrolysis of the endo‐ester bond in the dimeric substrate and the higher hydrolytic activity toward BHET than MHET, molecular modeling of Bs2Est was performed. In in‐silico docking of the dimeric substrate (EG‐TPA‐EG‐TPA‐EG), the endo‐ester docking pose located the MHET moiety in the small pocket via the formation of π–π and π–cation interactions as well as hydrogen bonds (Figure 2d), but no suitable productive binding modes were found for cleavage of the exo‐ester bond (Figure S22). In in‐silico docking of BHET and MHET, the ester group of BHET was directly oriented towards the catalytic S189, but the carboxylic group of MHET formed a hydrogen bond with the catalytic S189, leading to unfavorable substrate binding (Figure 2e,f). Further docking simulation results on the docking pose and specific interactions are discussed in the Supporting Information. Calculating based on the theoretical maximum equivalent of TPA wherein 4.54 g L−1 of PET was 23.62 mm [Eq. (S1)], PET was depolymerized into TPA with the yield of 99.5 % (mol mol−1) by chemo‐enzymatic depolymerization examined in this study (Figures 1i and S16). However, further optimization of Bs2Est‐catalyzed hydrolysis such as lowering the enzyme amount or solubilization of PET oligomers (>dimers) with buffer screening, pH‐stat reaction, or esterase engineering will be necessary for faster hydrolysis of MHET.
Figure 2.

(a) Enzymatic hydrolysis profiling on 1 g l −1 of PET oligomers. (b) MALDI‐TOF pattern analysis of the hydrolysis products for 10 min, 30 min, 1 h, and 10 h. (c) Suggested mode of action toward the dimer. Molecular docking simulations of (d) dimeric substrate (EG‐TPA‐EG‐TPA‐EG), (e) BHET, and (f) MHET. 2 U mL−1 of Bs2Est was used for the enzymatic hydrolysis.
Multifunctional coatings using water‐rich, impure catechol solutions
In biorefineries, complicated purification steps often increase the production cost of the biochemical‐derived products, hindering their commercialization. In this study, a direct application of catechol produced from PET waste to coating agents was attempted for the reduction of purification steps. To produce catechol from PET hydrolysates, a catechol biosynthetic strain was established using the combination of the TPA degradation module and catechol biosynthesis module (pKE112TphBaroY and pKM212TphAabc; Tables S2–S4) in E. coli as reported in our previous study. [4a] When 6 mm of TPA obtained from PET depolymerization was incubated with the catechol biosynthetic strain after optimization of substrate loading (Figure S23), 5.97 mm of catechol was produced within 12 h with 99.5 % yield, which was consistent with the result obtained using the reagent‐grade TPA (Figure 3). To test the direct application of catechol produced from TPA as a coating agent, various substrates, like PET, Teflon, and aluminum foil, were immersed in the crude catechol aqueous solution (6 mm) at pH 8, obtained by simple centrifugation to remove the biocatalyst. As a result, we found that the catechol was successfully coated on the substrates by spontaneous deposition of a poly‐catechol multifunctional coating (Figure 3c). To clear the surface and increase the surface polarity, PET, Teflon, and aluminum foil were exposed to UV light for 15 min. The substrates were then immersed in the catechol solution for 24 h, washed with deionized water, and dried under ambient conditions. Catechol coating was demonstrated by changes in the contact angle and FTIR spectra before and after coating (Figures S24 and S25). The poly‐catechol layer has an oxidative potential, thereby reacting with thiols and amines via Michael addition or Schiff base reactions and reducing metal ions to metal nanoparticles. [12] Thus, the immersion of poly‐catechol‐coated surfaces into a solution containing amine‐containing polymers and Ag+ provided a convenient route to secondary organic or inorganic deposition (Figures 3d–f and S26). An important well‐known fact is that functional polymers and metals are hardly deposited on PET and Teflon without an adherent layer, like the catechol coating. [18] Polylysine and chitosan secondary layers were generated by immersing the poly‐catechol‐coated PET surfaces in aqueous solutions of the two functional biopolymers. Silver nanoparticles were formed on the poly‐catechol‐coated PET through immersion in AgNO3 aqueous solution (Figure 3c). Antibacterial activities of the functional coated PET substrates were compared using a disk diffusion test against the gram‐negative Escherichia coli. A neat PET film was used as a negative control and showed no inhibition zone (7.0 mm diameter). The catechol only‐, chitosan‐, and silver nanoparticle‐coated substrates showed significant inhibition zones with diameters of 8.6±0.3, 8.6±0.6, and 10.4±0.7 mm, respectively, against E. coli, whereas no inhibition of bacterial growth was caused by polylysine. The antibacterial properties of polyphenols, chitosan, and silver nanoparticles have been well established. [19] Cell adhesion tests using the L‐929 cell line were carried out for the functional coated PET substrates. [20] Polylysine‐coated substrate improved the cell adhesion of PET by a factor of 1.6. Polylysine coating technique can be applied to improve the performance of tissue engineering scaffolds. In contrast, the silver nanoparticle‐coated substrate showed a significantly lower cell adhesion because silver nanoparticles killed the cells. To confirm the role of catechol as an adherent layer, the introduction of catechol was omitted and PET films were coated with chitosan and AgNO3 in the same way. No color change occurred when silver nanoparticles were formed (Figure S27). In contrast to the results of Figure S24, the coating without catechol did not give a significant contact angle change (Figure S28). The results suggest that catechol can act as an adherent layer for further functional coatings.
Figure 3.

(a) Bioconversion of TPA to catechol using an engineered E. coli strain. (b) Production of catechol from hydrolyzed TPA and reagent‐grade TPA. (c) General catechol coating process for various substrates (aluminum foil, PET, and Teflon), and the introduction of secondary functional layers on the catechol‐coating: chitosan, polylysine, and silver nanoparticle (AgNP). (d) Picture and (e) E. coli inhibition zone formation of neat PET and catechol only‐, chitosan‐, polylysine‐, and AgNP‐coated PET films. (f) (left) Escherichia coli inhibition zone diameter (negative control, 7.0 mm) and (right) relative L‐929 cell adhesion capacity of neat PET and catechol only‐, chitosan‐, polylysine‐, and AgNP‐coated PET films.
Conclusion
The development of an efficient poly(ethylene terephthalate) (PET) upcycling technology is currently of great scientific and industrial importance. Here, we demonstrate chemo‐biological upcycling for application as coating materials without complicated purification steps. Although chemo‐ and bio‐cascade reactions to produce catechol from PET waste is still a proof‐of‐concept technology, a newly developed chemo‐enzymatic depolymerization approach will contribute to the establishment of cost‐effective PET depolymerization. Therefore, chemo‐enzymatic depolymerization of PET developed here can be used as a platform technology for the production of value‐added chemicals from PET waste. In addition, further optimization to improve the efficiency of Bs2Est and whole‐cell biotransformation will facilitate the development of various microbial upcycling methods.
Experimental Section
Preparation of BHET by chemical glycolysis of PET from a waste plastic cup
All reagents for experiment were obtained from commercial suppliers and details are described in the Supporting Information. Functional groups and the purity of the PET waste pieces and Sigma‐Aldrich PET films were compared by NMR and FTIR spectroscopy (Supporting Information) before the glycolysis. The glycolysis reactions of PET were set in a 250 mL round flask equipped with a reflux condenser and magnetic stirrer. 5 g of PET pieces, 20 g of EG, and different amounts of K2CO3 were added to the reaction flask. The reaction mixtures were heated to various temperatures (180–210 °C) for different times (1–5 h). After the glycolysis reaction was completed, 400 ml of hot water was added to the mixture, and PET oligomers were removed by filtration. The filtrate was concentrated to 30–40 ml by rotary evaporation at 60 °C. The concentrated mixture was recrystallized in a refrigerator at 4 °C for 18 h. White crystals obtained by filtration (i. e., g‐BHET) were dried at 90 °C for 12 h. The same method was used for preparation of BHET from granular PET (Sigma‐Aldrich, cat. #429252). The PET conversion rate from coffee cup PET and granular PET, and the BHET yield are calculated according to Equations (1)–(3), respectively. The commercially available PET granules contained 30 % glass particles and thus, only 70 % of theoretically obtainable PET was considered for the calculation of PET conversion. The theoretical weight of BHET is calculated by dividing the initial weight of PET samples by the molecular weight of the repeating unit (TPA‐EG, 192.2 g mol−1) and multiplying the molecular weight of BHET (254 g mol−1). The actual weight of BHET is the BHET actually obtained through recrystallization of the reaction mixture.
| (1) |
| (2) |
| (3) |
1H NMR (500mhz, d 6‐DMSO): δ=8.13 (s, 4H), 4.96 (t, J=5, 2H), 4.32 (t, J=5, 4H), 3.72 ppm (q, J=5, 4H).
Enzymatic hydrolysis of BHET (path 1)
For the enzyme reaction, the gene encoding BsEst was cloned into pET‐28a according to the general cloning procedure. Then, enzyme was prepared, and its properties were evaluated as stated in the Supporting Information. The enzyme reaction was carried out in 50 mm Tris‐HCl buffer (pH 7.5) at 30 °C in the presence of 2 U mL −1 of Bs2Est according to the previous study. [21] The enzyme hydrolysis was stopped by adding methanol (Figure S6b) in the ratio of 1 : 19 (v/v), and the reaction mixture was filtered by 0.22 μm nylon membrane (Choice filter, Thermo Scientific) to analyze substrate and products using HPLC system. Purified TPA was simply obtained by subsequent acidification and filtration according to the previous study. [22] Purified TPA was analyzed by 1H and 13C NMR. 1H NMR (500mhz, d6‐DMSO): δ=13.28 (s, 2H), 8.07 ppm (s, 4H).
One‐step chemo‐enzymatic depolymerization of glycolyzed products (paths 2 and 3)
To develop the one‐pot chemo‐enzymatic hydrolysis of waste PET, enzymatic reactions using the glycolyzed mixtures described in Figure 1 (paths 2 and 3) were conducted. In path 2, glycolyzed products after filtration were used while in path 3, glycolyzed products without any purification were used for enzymatic hydrolysis. For enzyme reactions, glycolyzed products were diluted into 4–24 mm of BHET in the proper buffer systems such as 50 mm Tris‐HCl buffer or 50, 100, and 200 mm sodium phosphate (pH 7.5) for the initial substrate loadings. The enzyme reactions were carried out by adding 2 U mL −1 of Bs2Est at 30 °C and 1000 rpm.
Whole‐cell biotransformation
As a whole‐cell catalysis, the recombinant E. coli strain harboring pKE112TphBaroY and pKM212TphAabc was constructed (Supporting Information). Whole‐cell biotransformation using the recombinant E. coli strain was performed as follows. For seed cultures, 10 ml LB media in a 50 ml conical tube was used with proper antibiotics and cultivated overnight at 37 °C and 200 rpm. Seed cultures were inoculated in 500 ml of LB media in 2 l flasks and incubated at 37 °C and 200 rpm until optical density at 600 nm (OD600) reached 0.4–0.6. Then, 0.1 mm isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) was added to the flask. After induction, cells were further cultivated at 16 °C and 180 rpm. Cells were harvested after 24 h by centrifugation at 6520×g for 10 min at 4 °C, and then washed twice with 50 mm Tris‐HCl buffer (pH 7.0) containing 2 % (w/v) glycerol (TG buffer). The concentration of cells was adjusted to OD600=30 in 50 ml conical tube, and then cells were re‐suspended in 5 ml of TG buffer containing the appropriate concentration of substrate (3, 6, 12, and 30 mm of TPA). Whole‐cell biotransformation was performed at 30 °C and 250 rpm for 20 h. All experiments were performed in triplicate.
Functional coating
The PET films (or aluminum foils and Teflon films) were coated with the poly‐catechol complex. The PET films were UV‐treated for 15 min. PET films were cut into a circle and placed in the 24 well cell culture plate. The catechol aqueous solution (6 mm) produced by whole‐cell biotransformation was used without purification, and the pH of this solution was subsequently raised to 8 by adding drops of Tris‐HCl buffer. PET films were immersed in the catechol aqueous solution for 6 h, which produce poly‐catechol coated PET films. The poly‐catechol coated PET films were washed with distilled water and then dried under ambient conditions. For poly‐l‐lysine coating, the poly‐catechol coated PET films were immersed in 0.1 wt% poly‐l‐lysine aqueous solution for 3 h. After the coating, they were washed with distilled water and then dried under ambient conditions. For chitosan coating, the poly‐catechol coated PET films were immersed in 0.1 wt% chitosan aqueous solution for 3 h. After the coating, they were washed with distilled water and then dried under ambient conditions. For silver nanoparticle coating, the poly‐catechol coated PET films were immersed in a 3.25 mm AgNO3 aqueous solution. The reaction chambers were sealed and left at room temperature for 24 h for the reduction of Ag+ ions via oxidation of catechol groups. After the coating, they were washed with distilled water and then dried under ambient conditions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (MSIT) (NRF‐2020R1A5A1019631, NRF‐2020R1C1C1005719, and NRF‐2021R1A2C2011669). J.C.J. acknowledges that this study was supported by the Research Fund, 2021 of The Catholic University of Korea.
H. T. Kim, M. Hee Ryu, Y. J. Jung, S. Lim, H. M. Song, J. Park, S. Y. Hwang, H.-S. Lee, Y. J. Yeon, B. H. Sung, U. T. Bornscheuer, S. J. Park, J. C. Joo, D. X. Oh, ChemSusChem 2021, 14, 4251.
Contributor Information
Prof. Si Jae Park, Email: parksj93@ewha.ac.kr.
Prof. Jeong Chan Joo, Email: jcjoo@catholic.ac.kr.
Prof. Dongyeop X. Oh, Email: dongyeop@krict.re.kr.
References
- 1.
- 1a. Geyer R., Jambeck J. R., law K. L., Sci. Adv. 2017, 3, e1700782; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Rambonnet L., Vink S. C., Land-Zandstra A. M., Bosker T., Mar. Pollut. Bull. 2019, 145, 271–277; [DOI] [PubMed] [Google Scholar]
- 1c. Lonca G., Lesage P., Majeau-Bettez G., Bernard S., Margni M., Resour. Conserv. Recycl. 2020, 162, 105013; [Google Scholar]
- 1d. Chae Y., An Y.-J., Environ. Pollut. 2018, 240, 387–395; [DOI] [PubMed] [Google Scholar]
- 1e. Wei R., Tiso T., Bertling J., O'Connor K., Blank L. M., Bornscheuer U. T., Nat. Catal. 2020, 3, 867–871. [Google Scholar]
- 2.
- 2a. Choi T.-R., Jeon J.-M., Bhatia S. K., Gurav R., Han Y. H., Park Y. L., Park J.-Y., Song H.-S., Park H. Y., Yoon J.-J., Seo S.-O., Yang Y.-H., Biotechnol. Bioprocess Eng. 2020, 25, 279–286; [Google Scholar]
- 2b. Sohn Y. J., Kim H. T., Baritugo K.-A., Song H. M., Ryu M. H., Kang K. H., Jo S. Y., Kim H., Kim Y. J., Choi J.-I., Park S. K., Joo J. C., Park S. J., Int. J. Biol. Macromol. 2020, 149, 593–599; [DOI] [PubMed] [Google Scholar]
- 2c. Choi S. Y., Park S. J., Kim W. J., Yang J. E., Lee H., Shin J., Lee S. Y., Nat. Biotechnol. 2016, 34, 435–440; [DOI] [PubMed] [Google Scholar]
- 2d. Choi S. Y., Rhie M. N., Kim H. T., Joo J. C., Cho I. J., Son J., Jo S. Y., Sohn Y. J., Baritugo K.-A., Pyo J., Lee Y., Lee S. Y., Park S. J., Metab. Eng. 2020, 58, 47–81; [DOI] [PubMed] [Google Scholar]
- 2e. Sohn Y. J., Kim H. T., Jo S. Y., Song H. M., Baritugo K.-A., Pyo J., Choi J.-I., Joo J. C., Park S. J., Biotechnol. Bioprocess Eng. 2020, 25, 848–861. [Google Scholar]
- 3.
- 3a. Baliga S., Wong W. T., J. Polym. Sci. 1989, 27, 2071–2082; [Google Scholar]
- 3b. Karayannidis G. P., Chatziavgoustis A. P., Achilias D. S., Adv. Polym. Technol. 2002, 21, 250–259. [Google Scholar]
- 4.
- 4a. Franden M. A., Jayakody L. N., Li W. J., Wagner N. J., Cleveland N. S., Michener W. E., Hauer B., Blank L. M., Wierckx N., Klebensberger J., Beckham G. T., Metab. Eng. 2018, 48, 197–207; [DOI] [PubMed] [Google Scholar]
- 4b. Narancic T., Salvador M., Hughes G. M., Beagan N., Abdulmutalib U., Kenny S. T., Wu H., Saccomanno M., Um J., O'Connor K. E., Jiménez J. I., Microb. Biotechnol. 2021, DOI: 10.1016/j.ymben.2021.03.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Welsing G., Wolter B., Hintzen H. M. T., Tiso T., Blank L. M., Methods Enzymol. 2021, 648,391–421; [DOI] [PubMed] [Google Scholar]
- 4d. Pardo I., Jha R. K., Bermel R. E., Bratti F., Gaddis M., McIntyre E., Michener W., Neidle E. L., Dale T., Beckham G. T., Johnson C. W., Metab. Eng. 2020, 62, 260–274; [DOI] [PubMed] [Google Scholar]
- 4e. Kim H. T., Kim J. K., Cha H. G., Kang M. J., Lee H. S., Khang T. U., Yun E. J., Lee D.-H., Song B. K., Park S. J., Joo J. C., Kim K. H., ACS Sustainable Chem. Eng. 2019, 7, 19396–19406; [Google Scholar]
- 4f. Sohn Y. J., Kim H. T., Baritugo K., Jo S. Y., Song H. M., Park S. Y., Park S. K., Pyo J., Cha H. G., Kim H., Na J.-G., Park C., Choi J.-I., Joo J. C., Park S. J., Biotechnol. J. 2020, 15, 1900489; [DOI] [PubMed] [Google Scholar]
- 4g. Kang M. J., Kim H. T., Lee M.-W., Kim K.-A., Khang T. U., Song H. M., Park S. J., Joo J. C., Cha H. G., Green Chem. 2020, 22, 3461–3469; [Google Scholar]
- 4h. Rorrer N. A., Nicholson S., Carpenter A., Biddy M. J., Grundl N. J., Beckham G. T., Joule 2019, 3, 1006–1027; [Google Scholar]
- 4i. Tan J. P. K., Tan J., Park N., Xu K., Chan E. D., Yang C., Piunova V. A., Ji Z., Lim A., Shao J., Bai A., Bai X., Mantione D., Sardon H., Yang Y. Y., Hedrick J. L., Macromolecules 2019, 52, 7878–7885; [Google Scholar]
- 4j. Fukushima K., Liu S., Wu H., Engler A. C., Coady D. J., Maune H., Pitera J., Nelson A., Wiradharma N., Venkataraman S., Huang Y., Fan W., Ying J. Y., Yang Y. Y., Hedrick J. L., Nat. Commun. 2013, 4, 3861/1-3861/9; [DOI] [PubMed] [Google Scholar]
- 4k. Shamsi R., Abdouss M., Sadeghi G. M. M., Taromi F. A., Polym. Int. 2009, 58, 22–30; [Google Scholar]
- 4l. Aslzadeh M. M., Sadeghi G. M. M., Abdouss M., Materwiss. Werksttech. 2010, 41, 682–688; [Google Scholar]
- 4m. Fonseca A. C., Gil M. H., Simoes P. N., Prog. Polym. Sci. 2014, 39, 1291–1311; [Google Scholar]
- 4n. Jamdar V., Kathalewar M., Sabnis A., J. Polym. Environ. 2018, 26, 2601–2618; [Google Scholar]
- 4o. Natarajan J., Madras G., Chatterjee K., ACS Appl. Mater. Interfaces 2017, 9, 28281–28297; [DOI] [PubMed] [Google Scholar]
- 4p. Elsaeed S. M., Farag R. K., J. Appl. Polym. Sci. 2009, 112, 3327–3336. [Google Scholar]
- 5.
- 5a. Yang Y., Lu Y., Xiang H., Xu Y., Li Y., Polym. Degrad. Stab. 2002, 75, 185–191; [Google Scholar]
- 5b. Nikles D. E., Farahat M. S., Macromol. Mater. Eng. 2005, 290, 13–30; [Google Scholar]
- 5c. Tournier V., Topham C. M., Gilles A., David B., Folgoas C., Moya-Leclair E., Kamionka E., Desrousseaux M.-L., Texier H., Gavalda S., Cot M., Guémard E., Dalibey M., Nomme J., Cioci G., Barbe S., Chateau M., André I., Duquesne S., Marty A., Nature 2020, 580, 216–219; [DOI] [PubMed] [Google Scholar]
- 5d. Carniel A., Valoni É., Nicomedes Junior J., da C. Gomes A., de Castro A. M., Process Biochem. 2017, 59, 84–90; [Google Scholar]
- 5e. Barth M., Honak A., Oeser T., Wei R., Belisário-Ferrari M. R., Then J., Schmidt J., Zimmermann W., Biotechnol. J. 2016, 11, 1082–1087. [DOI] [PubMed] [Google Scholar]
- 6. Carta D., Cao G., D'Angeli C., Environ. Sci. Pollut. Res. Int. 2003, 10, 390–394. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Palm G. J., Reisky L., Böttcher D., Müller H., Michels E. A. P., Walczak M. C., Berndt L., Weiss M. S., Bornscheuer U. T., Weber G., Nat. Commun. 2019, 10, 1717; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Joo S., Cho I. J., Seo H., Son H. F., Sagong H.-Y., Shin T. J., Choi S. Y., Lee S. Y., Kim K.-J., Nat. Commun. 2018, 9, DOI: 10.1038/s41467-018-02881–1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Knott B. C., Erickson E., Allen M. D., Gado J. E., Graham R., Kearns F. L., Pardo I., Topuzlu E., Anderson J. J., Austin H. P., Dominick G., Johnson C. W., Rorrer N. A., Szostkiewicz C. J., Copié V., Payne C. M., Woodcock H. L., Donohoe B. S., Beckham G. T., McGeehan J. E., Proc. Natl. Acad. Sci. USA 2020, 117, 25476–25485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. de Castro A. M., Carniel A., Biochem. Eng. J. 2017, 124, 64–68; [Google Scholar]
- 8b. de Castro A. M., Carniel A., Nicomedes Junior J., da C. Gomes A., Valoni É., J. Ind. Microbiol. Biotechnol. 2017, 44, 835–844. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Knotta B. C., Erickson E., Allen M. D., Gado J. E., Graham R., Kearns F. L., Pardo I., Topuzlu E., Anderson J. J., Austin H. P., Dominick G., Johnson C. W., Rorrer N. A., Szostkiewicz C. J., Copié V., Payne C. M., Woodcock H. L., Donohoe B. S., Beckham G. T., McGeehan J. E., Proc. Natl. Acad. Sci. USA 2020, 117, 25476–25485; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Qiu L., Yin X., Liu T., Zhang H., Chen G., Wu S., J. Basic Microbiol. 2020, 60, 699–711. [DOI] [PubMed] [Google Scholar]
- 10. Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Science 2007, 318, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Yang B., Ayyadurai N., Yun H., Choi Y. S., Hwang B. H., Huang J., Lu Q., Zeng H., Cha H. J., Angew. Chem. Int. Ed. 2014, 53, 13360–13364; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13578–13582; [Google Scholar]
- 11b. Kim B. J., Cheong H., Hwang B. H., Cha H. J., Angew. Chem. Int. Ed. 2015, 127, 7426–7430. [Google Scholar]
- 12.
- 12a. Oh D. X., Shin S., Yoo H. Y., Lim C., Hwang D. S., Korean J. Chem. Eng. 2014, 31, 1306–1315; [Google Scholar]
- 12b. Guo J., Ping Y., Ejima H., Alt K., Meissner M., Richardson J. J., Yan Y., Peter K., von Elverfeldt D., Hagemeyer C. E., Caruso F., Angew. Chem. Int. Ed. 2014, 126, 5652–5657; [DOI] [PubMed] [Google Scholar]
- 12c. Ejima H., Richardson J. J., Liang K., Best J. P., van Koeverden M. P., Such G. K., Cui J., Caruso F., Science 2013, 341, 154–157. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Chen J.-W., Chen L.-W., Cheng W.-H., Polym. Int. 1999, 48, 885–888; [Google Scholar]
- 13b. López-Fonseca R., Duque-Ingunza I., de Rivas B., Arnaiz S., Gutiérrez-Ortiz J. I., Polym. Degrad. Stab. 2010, 95, 1022–1028. [Google Scholar]
- 14.
- 14a. López-Fonseca R., Duque-Ingunza I., de Rivas B., Arnaiz S., Gutiérrez-Ortiz J. I., Polym. Degrad. Stab. 2010, 95, 1022–1028; [Google Scholar]
- 14b. Pham D. D., Cho J., Green Chem. 2021, 23, 511–525; [Google Scholar]
- 14c. Kidwai M., Lal M., Mishra N. K., Jahan A., Green Chem. Lett. Rev. 2013, 6, 63–68; [Google Scholar]
- 14d. Ouk S., Thiébaud S., Borredon E., Le Gars P., Green Chem. 2002, 4, 431–435; [Google Scholar]
- 14e. Tundo P., Trotta F., Moragliob G., J. Chem. Soc.-Perkin Trans. 1989, 1070–1071; [Google Scholar]
- 14f. Tundo P., Pure Appl. Chem. 2000, 72, 1793–1797; [Google Scholar]
- 14g. Kidwai M., Saxena S., Rahman Khan M. K., Thukral S. S., Bioorg. Med. Chem. Lett. 2005, 15, 4295–4298; [DOI] [PubMed] [Google Scholar]
- 14h. Lin Y., Lü F., Shao L., He P., Bioresour. Technol. 2013, 137, 245–253. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Heinze B., Kourist R., Fransson L., Hult K., Bornscheuer U. T., Protein Eng. Des. Sel. 2007, 20, 125–131; [DOI] [PubMed] [Google Scholar]
- 15b. Schmidt M., Henke E., Heinze B., Kourist R., Hidalgo A., Bornscheuer U. T., Biotechnol. J. 2007, 2, 249–253. [DOI] [PubMed] [Google Scholar]
- 16. de B. Sanches N., Dias M. L., Pacheco E. B. A. V., Polym. Eng. Sci. 2008, 10, 1953–1962. [Google Scholar]
- 17.
- 17a. Batra R., Gupta M. N., Biotechnol. Lett. 1994, 16, 1059–1064; [Google Scholar]
- 17b. Doukyu N., Ogino H., Biochem. Eng. J. 2010, 48, 270–282; [Google Scholar]
- 17c. Zhang L.-Q., Zhang Y.-D., Xu L., Li X.-L., Yang X., Xu G.-L., Wu X.-X., Gao H.-Y., Du W.-B., Zhang X.-T., Zhang X.-Z., Enzyme Microb. Technol. 2001, 29, 129–135; [Google Scholar]
- 17d. Schumacher J., Eckstein M., Kragl U., Biotechnol. J. 2006, 1, 574–581. [DOI] [PubMed] [Google Scholar]
- 18. Huang J., Tian C., Wang J., Liu J., Li Y., Liu Y., Chen Z., Appl. Surf. Sci. 2018, 458, 734–742. [Google Scholar]
- 19.
- 19a. Nguyen H. L., Jo Y. K., Cha M., Cha Y. J., Yoon D. K., Sanandiya N. D., Prajatelistia E., Oh D. X., Hwang D. S., Polymer 2016, 8, 102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Payra D., Naito M., Fujii Y., Nagao Y., Chem. Commun. 2016, 52, 312–315. [DOI] [PubMed] [Google Scholar]
- 20. Park S. A., Jeon H., Kim H., Shin S. H., Choy S., Hwang D. S., Koo J. M., Jegal J., Hwang S. Y., Park J., Oh D. X., Nat. Commun. 2019, 10, 2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu L., Yin X., Liu T., Zhang H., Chen G., Wu S., J. Basic Microbiol. 2020, 60, 699–711. [DOI] [PubMed] [Google Scholar]
- 22. Wu S.-C., Cheng Z.-M., Wang S.-D., Shan X.-L., Chem. Eng. Technol. 2011, 34, 1614–1618. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information