

SUMMARY
The characterization of the genetic basis of maize (Zea mays) leaf development may support breeding efforts to obtain plants with higher vigor and productivity. In this study, a mapping panel of 197 biparental and multiparental maize recombinant inbred lines (RILs) was analyzed for multiple leaf traits at the seedling stage. RNA sequencing was used to estimate the transcription levels of 29 573 gene models in RILs and to derive 373 769 single nucleotide polymorphisms (SNPs), and a forward genetics approach combining these data was used to pinpoint candidate genes involved in leaf development. First, leaf traits were correlated with gene expression levels to identify transcript–trait correlations. Then, leaf traits were associated with SNPs in a genome‐wide association (GWA) study. An expression quantitative trait locus mapping approach was followed to associate SNPs with gene expression levels, prioritizing candidate genes identified based on transcript–trait correlations and GWAs. Finally, a network analysis was conducted to cluster all transcripts in 38 co‐expression modules. By integrating forward genetics approaches, we identified 25 candidate genes highly enriched for specific functional categories, providing evidence supporting the role of vacuolar proton pumps, cell wall effectors, and vesicular traffic controllers in leaf growth. These results tackle the complexity of leaf trait determination and may support precision breeding in maize.
Keywords: expression Quantitative Trait Loci mapping, Genome Wide Association Studies, forward genetics, maize, leaf development, Weighted Gene Co‐expression Network Analysis, multiparental populations, Zea mays
Significance Statement
Innovative genomics‐ and transcriptomics‐based forward genetic approaches can improve our understanding of the molecular basis of leaf development in maize (Zea mays). By combining transcript–trait correlations, genome‐wide associations, and expression quantitative trait locus mapping, we identified 25 candidate genes that may have a key role in early leaf growth.
INTRODUCTION
In the post‐genomic era, the ability to identify gene functions remains one of the main challenges in molecular biology. In the past decades, gene models have been mined in several plant genomes (Seaver et al., 2018; Van Bel et al., 2012), contributing to our understanding of the molecular basis of the determination of complex traits (Civelek and Lusis, 2014). This knowledge is pivotal for breeding efforts aiming at the development of more productive crops (Varshney et al., 2018), and may contribute to the virtuous cycle linking the characterization of plant genomes with the production of novel varieties (Poland, 2015).
Among cereal crops, the global production of maize (Zea mays) is the highest, approaching 1.15 billion tons in 2019 (http://www.fao.org/faostat/). Besides serving as food and animal feed and for energy production, maize is also firmly established as a model species for monocots since the early characterization of its genome sequence (Schnable et al., 2009). Maize studies benefit from an ever expanding genetic toolbox with increasingly precise genomic information (Jiao et al., 2017), pan‐genome sequences (Hirsch et al., 2014; Sun et al., 2018), advanced segregant populations (Dell’Acqua et al., 2015; McMullen et al., 2009), and genome editing protocols (Feng et al., 2018; Liu et al., 2020). The integration of these resources, which are unmatched in crop species, supports maize as the cornerstone of precision breeding efforts (Hilscher et al., 2017), contributing to global food security (Ma et al., 2018).
Any approach aimed at the manipulation and exploitation of functional variation from a breeding perspective relies on the capacity to identify the causative variants controlling complex traits. In maize, reverse genetics was extensively used to connect traits to genetic variants by the inactivation and manipulation of gene sequences using targeted or untargeted mutagenesis, by way of transposons (May et al., 2003), targeting‐induced local lesions in genomes (Till et al., 2004), RNA suppression (Mark Cigan et al., 2005), and genome editing (Svitashev et al., 2016). A complementary approach to explore gene functions is that of forward genetics, employing statistical methods combining trait values and allelic variation in segregant populations to characterize quantitative trait loci (QTLs), by either QTL mapping or genome‐wide association (GWA) studies (Mackay et al., 2009). In maize, these approaches contributed to the characterization of the genetic basis of several traits of agronomic relevance (Buckler et al., 2009; Li et al., 2016; Wallace et al., 2014a; Xue et al., 2016).
The determination of the genetic basis of quantitative traits, however, is hampered by the complex interrelation of genetic variants and regulators of expression (Albert and Kruglyak, 2015; Wallace et al., 2014b), including structural variation (Marroni et al., 2014; Wang et al., 2015). The ultimate characterization of causative variants is a challenging task (Eichler et al., 2010), and validation of individual gene functions at a system‐wide scale calls for the characterization of gene expression (Krouk et al., 2013), protein networks (Walley et al., 2016), and metabolomic profiles (Krumsiek et al., 2016). In maize, expression QTL (eQTL) mapping was successfully applied to reduce the gap between allele variants, expression variance, and maize phenotypes, improving the confidence in candidate gene identification (Christie et al., 2017; Kremling et al., 2019; Liu et al., 2017; Pang et al., 2019; Wang et al., 2018).
In this framework, one strategy to improve the ability to identify genes relevant for breeding is to rely on a combination of precision phenotyping (Baute et al., 2015; Zhang et al., 2017), advanced genetic materials (Dell’Acqua et al., 2015), and genomic tools (Kremling et al., 2019). Numerous loci and interactions contribute to the genetic architecture of maize leaf phenotypes (Monir and Zhu, 2018; Tian et al., 2011). It was shown that early leaf organ growth is controlled by a complex transcriptional network (Baute et al., 2015, 2016) involving genes that have an effect on seed yield (Sun et al., 2017). A synthesis of forward genetics methods in a system genetics framework (Civelek and Lusis, 2014) bears the promise to accelerate the identification of candidate genes involved in leaf growth dynamics and support genome editing from a breeding perspective.
In the present study, we employed a forward genetics approach integrating transcriptomics, phenotyping, and GWAs to prioritize genomic loci and candidate genes involved in the determination of maize leaf traits (Figure 1). The first step was to characterize the associations between expression levels and leaf phenotypes with an approach in concordance with Baute et al. (2016). The second step was to identify marker–trait associations (MTAs) with GWAs. The final step was to derive eQTLs and relate them with trait–phenotype associations and with MTAs. We then conducted a network analysis to capture the broader picture of gene interactions controlling the expression of leaf traits. We describe a complex interaction network of biological pathways contributing to leaf development, identifying 25 candidate genes involved in leaf trait determination. We discuss these results considering previous literature, shedding new light on the molecular mechanisms underlying maize leaf length, division zone (DZ) size, and leaf elongation dynamics.
Figure 1.
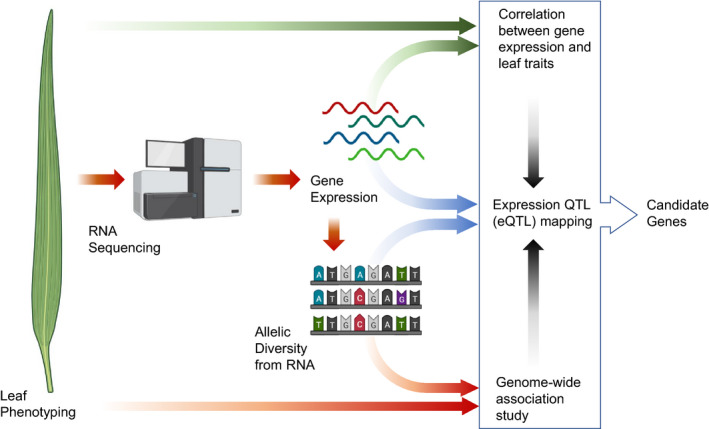
Outline of the research strategy. Developing leaves of maize were phenotyped and harvested for RNA sequencing in previous studies (Baute et al., 2015, 2016). RNA sequencing reads were used to derive gene expression levels and alleles at SNPs. Candidate genes were identified integrating three approaches: (i) correlation of phenotypes and gene expression levels, (ii) association between SNP markers and gene expression levels (eQTL), and (iii) association between SNP markers and phenotypes (GWA). eQTL, expression quantitative trait locus; GWA, genome‐wide association; SNP, single nucleotide polymorphism.
RESULTS
Genotypic features of the mapping panel
RNA sequencing (RNA‐seq) data of recombinant inbred lines (RILs) derived from biparental and MAGIC multi‐parental maize crosses originated from previous experiments (Baute et al., 2015, 2016). Raw reads were trimmed for quality, yielding more than 20 million reads per sample (Table S1). After read mapping to the maize genome, 29 573 transcripts were present in at least 60% of the genotypes and showed an expression variance above 5%. Mapping of RNA‐seq reads yielded 373 769 biallelic single nucleotide polymorphisms (SNPs) mostly in telomeric regions (Figure 2a). A genetic diversity analysis showed that each RIL set clustered between their corresponding founder lines, with the MAGIC population showing a higher diversity (Figure 2b). The partially overlapping genetic background of the two populations determined a weak structure, with the two main principal components (PCs) accounting for 14.5% of the genotypic variance. To account for this structure, downstream correlation analyses considered the RIL sets individually, while GWA and eQTL mapping used a covariate and kinship correction. The linkage disequilibrium (LD) decay, estimated on the basis of pairwise r 2 measures, dropped below r 2 = 0.2 (considered a threshold for null LD) in 0.94 million base pairs (Mb) on average, with the slowest LD decay observed on chromosome (Chr) 2 (1.34 Mb) and the fastest LD decay on Chr 10 (0.66 Mb) (Figure 2c; Table S2). Long‐range LD was generally low, but it was consistently higher in pericentromeric regions (Figure S1).
Figure 2.
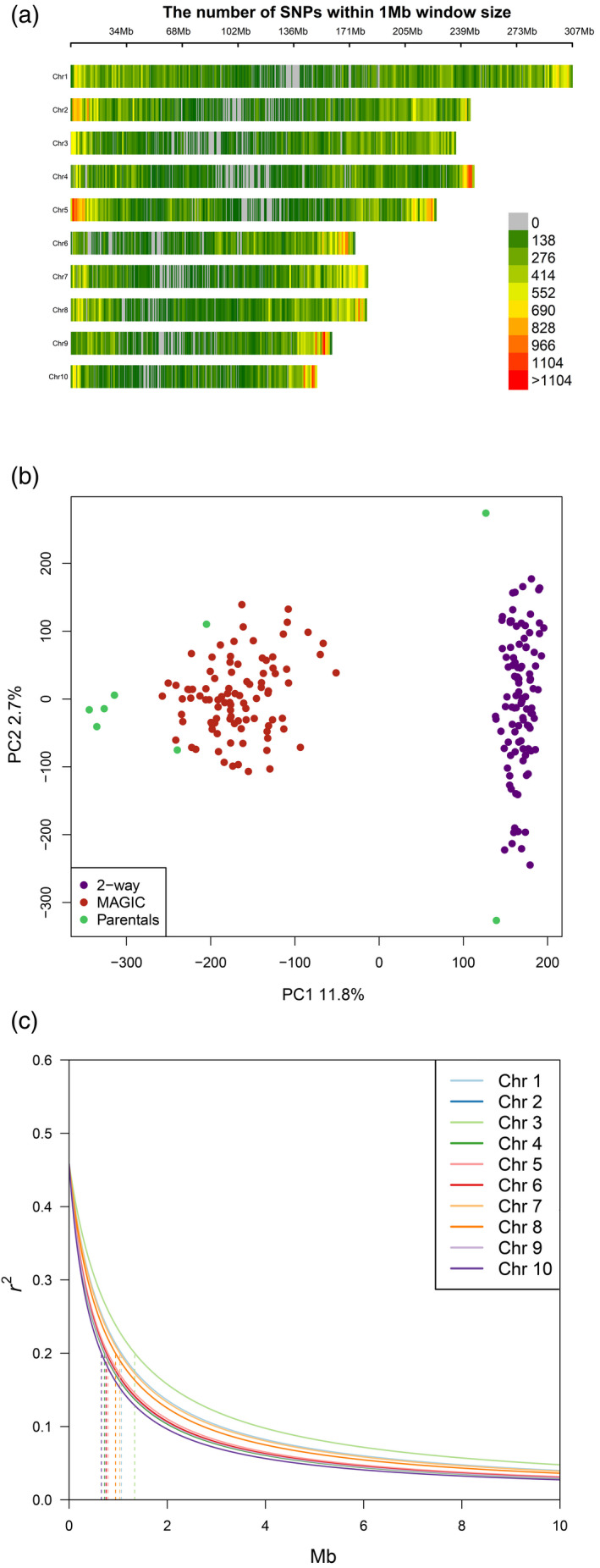
Genotypic diversity and LD decay in the mapping panel. (a) Chromosome‐specific SNPs density in 1‐Mb genomic intervals. The number of SNPs is represented in a green to red scale. (b) Structure in the mapping panel. Parental lines and RILs are represented on the first two PCs. Genotypic diversity between parental lines is roughly even, but their differentiation on the PCA space is distorted by that of RILs. (c) Chromosome‐specific LD decay. Vertical dashed bars project to the x‐axis (Mb distance) the 0.2 r 2 threshold representing null LD. LD, linkage disequilibrium; PC, Principal component; RIL, recombinant inbred line; SNP, single nucleotide polymorphism.
Trait associations with transcript levels
In both biparental and MAGIC RILs, the leaf elongation rate (LER), size of the cell division zone (DZ size), and leaf length were positively correlated with each other. Leaf elongation duration (LED) was negatively correlated with LER (Figure 3a,b). Trait levels for each RIL set are reported in Table S3. Pearson correlation coefficients (PCCs) were calculated between transcript levels of all retained gene models with trait values (Table S4) using permutation thresholds (Table S5) to derive genes whose expression was significantly correlated with phenotypes, hereafter referred to as PCC genes (Figure S2; Table S6). By this procedure a total of 1327 unique PCC genes were identified, which in seldom cases were correlated to multiple traits at once (Figure 3c). A total of 178 PCC genes were jointly identified by LED and leaf length measures, with 75 additional PCC genes also associated with DZ size. LER and leaf length, the phenotypes showing the highest correlation, shared only seven PCC genes (44% of the LER PCC genes) and just two PCC genes were identified in common by the three phenotypes having the highest correlation (LER, leaf length, and DZ size). Gene ontology (GO) analysis revealed that PCC genes for DZ size were highly enriched in plastid translation, peptidyl‐histidine phosphorylation (>45 fold), and the cytokine‐activated signaling pathway (>20 fold) in the biological process category and in nucleic acid binding in the molecular function category, with most proteins acting in the chloroplast according to the cellular component category (File S1). Other traits did not show specific enrichment, except for leaf length, where genes were enriched in DNA‐binding transcription factor activity and transcription regulator activity (File S1).
Figure 3.
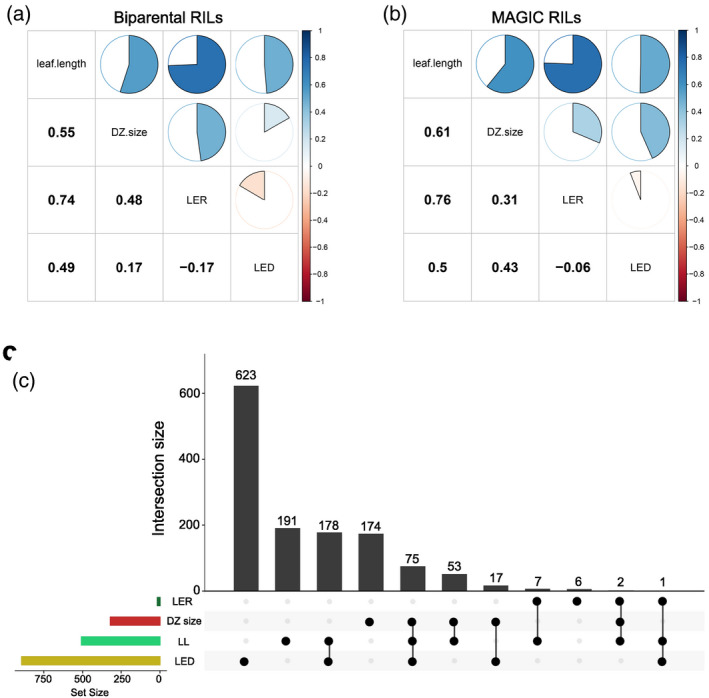
PCCs between traits and transcripts. PCCs between traits in biparental (a) and multi‐parental (b) populations are represented by pie charts. The size of pie charts is proportional to the number of PCCs, whose numerical value is reported in the lower portion of the figure. (c) Overlap of PCC genes among traits. In the lower left panel, traits are sorted by the number of PCC genes, and sharing patterns of PCC genes are represented by connected dark points. In the top panel, the number of PCC genes is reported for each pattern of traits, as per the lower panel, with decreasing PCC gene set sizes. PCC, Pearson correlation coefficient.
Trait associations with SNP markers
A GWA study was conducted between SNPs and trait values to identify MTAs (Figures S3 and S4). The 90th percentile of the permuted P‐value distributions was used to define GWA significance thresholds for each trait, that is 6.77 × 10−7 for leaf length, 5.15 × 10−7 for DZ size, 1.07 × 10−6 for LER, and 4.63 × 10−7 for LED. In total, 33 significant, non‐overlapping MTAs were identified (Table S7). Leaf length resulted in nine MTAs at multiple locations on Chr 1, Chr 3, Chr 4, Chr 8, and Chr 10, while DZ size was associated with loci on Chr 1 at around 175 Mb. LER was associated with nine MTAs, one of which was located on Chr 2, seven clustered around 190 Mb on Chr 4, and one was located on Chr 7. For LED, six MTAs were reported at multiple locations on Chr 1, Chr 2, Chr 3, Chr 4, Chr 5, and Chr 8 (Figure S3). LD decay information was used to reduce redundancy between MTAs in linkage with each other, so that unique QTLs were derived, selecting only the most significant MTAs among those mapping within a chromosome‐specific LD decay distance. By this criterion, the four traits mapped to 24 unique QTLs (Table S8).
eQTL analysis
eQTL mapping was performed considering all SNPs and all gene expression values, resulting in 8 714 461 significant associations. The genetic structure between the two RIL sets was corrected by using as covariates the first 10 PCs from transcript levels (accounting for 50% expression variance) and the first 10 PCs from SNP data (Figure S5). All SNPs derived from RNA‐seq hence belonged to coding regions of the genome. However, depending on local LD patterns, SNPs are expected to be in linkage with intergenic loci, including non‐coding DNA. eQTLs clustered in correspondence to pericentromeric regions (Figure S6), possibly contributed by LD among SNP markers. The LD decay information was used on the set of eQTLs to reduce redundancy between SNPs in linkage to each other. For each gene, only the most significant association within the LD decay distance was considered, resulting in a filtered number of 158 202 LD‐reduced eQTLs, including 17 466 unique genes and 76 852 unique SNPs. Conventionally, associations between SNPs and transcripts closer than 1 Mbp are classified as local, while the remaining eQTLs are considered distant. By this definition, most of the associations found were distant eQTLs (52%). While local eQTL genes were typically controlled by a small number of SNPs, eQTL genes in the distant class could be controlled by more than 150 loci (Figure S7).
To prioritize eQTLs with functional relevance in relation to leaf phenotypes, the list of significant eQTLs was narrowed using information with respect to PCC genes and SNPs with associations with leaf traits (Figure 1). The subset of eQTLs including genes whose expression was correlated with trait levels (PCC‐eQTLs) is reported in Table S9. The subset of eQTLs including SNPs associated to phenotypes (GWA‐eQTLs) is reported in Table S10. Altogether, PCC‐eQTLs and GWA‐eQTLs targeted 1025 genes controlled by 3968 SNPs (Table S11). Distant eQTLs were also prevalent in this subset (Figure S8a), and the β estimates of the eQTL model, that is, the change in transcription levels resulting from allelic variants at each eQTL, were similar for all traits except for LED, where distant eQTLs had a stronger negative effect (Figure S8b).
The highest numbers of PCC‐eQTLs and GWA‐eQTLs, involving 2617 unique SNPs and 601 unique genes, were obtained for LED. For this trait, an individual SNP could influence the expression level of 51 transcripts. Conversely, individual transcripts could be associated with up to 44 SNPs (Table S12). LER was the phenotype associated with the lowest number of PCC‐eQTLs and GWA‐eQTLs, with 154 associations between transcripts and MTAs involving 77 unique SNPs and 72 unique genes (Figure 4).
Figure 4.
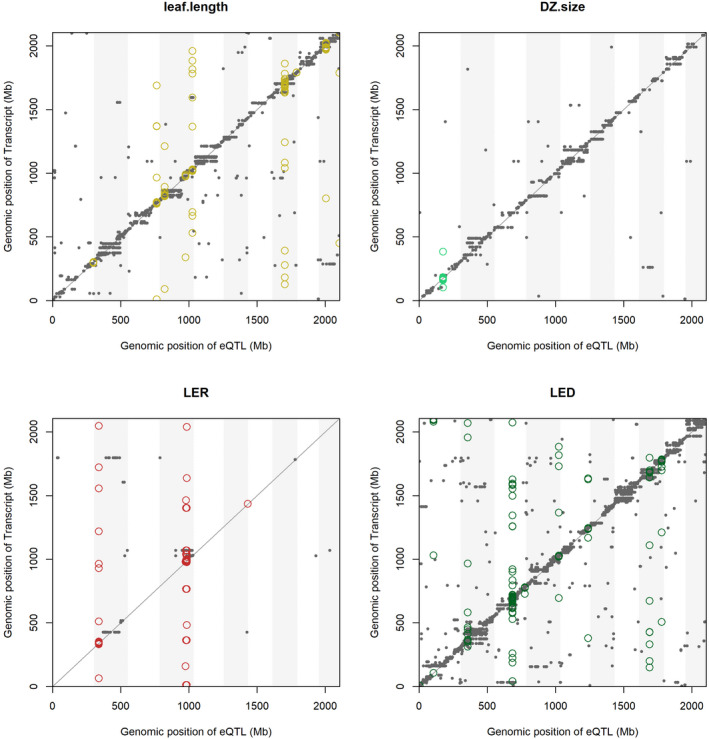
Genomic position of SNPs and transcripts in GWA‐expression quantitative trait loci (GWA‐eQTLs). Each panel reports the outcome of the GWA‐eQTLs for one of the traits. On the x‐axis, the genomic position of significant GWA‐eQTLs is shown in genomic Mb. On the y‐axis, the genomic position of transcripts targeted by GWA‐eQTLs are shown. Chromosomes are represented by alternating white and gray vertical shades. Significant GWA‐eQTLs are represented by gray dots, and their position is highlighted by vertical dashed lines. Transcripts that are identified in both GWA‐eQTLs and PCC‐eQTLs are highlighted by colored open circles. eQTL, expression quantitative trait locus; GWA, genome‐wide association; SNP, single nucleotide polymorphism.
Co‐expression network analysis
In order to capture the broader picture of the molecular mechanisms driving leaf development, a weighted gene co‐expression network analysis (WGCNA) was conducted in each RIL set and then combined with a consensus approach. A soft threshold power revealed a network with a scale‐free topology with R 2 > 0.85 for each RIL set (Figure S9). In the consensus network, genes were divided in 38 co‐expression modules (Figure S10) with a size range from 31 (plum1 module) to 5460 (turquoise module) genes (Table S13). Leaf length was correlated with six modules, LER with one module, LED with eight modules, and DZ size with six modules (P < 0.05). Only the magenta, tan, and cyan modules were associated with all traits except with LER (Figure 5a). The GO annotation of candidate modules showed significant enrichment (false discovery rate [FDR] < 0.05) for secondary cell wall biogenesis (magenta and dark‐orange modules), cell cycle arrest and auxin signal transduction (tan module), lignin catabolic process (dark‐orange module), peptide biosynthetic process (light‐cyan module), and transcription co‐activator activity (cyan module) (Files S2 and S3).
Figure 5.
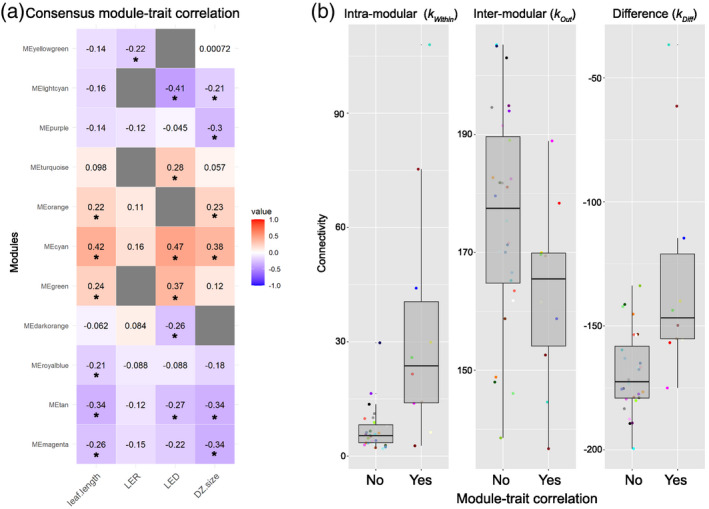
Co‐expression analysis. (a) Consensus module–trait associations. Each row corresponds to a consensus module eigengene, each column corresponds to a trait; numbers in each cell contain the correlation value. Asterisks denote significant correlations. Only modules with at least one significant correlation are reported. Gray cells indicate discordance between correlations in the two RIL sets. (b) Connectivity measures of consensus modules. The first panel corresponds to intra‐modular connectivity (k Within), the second to the inter‐modular connectivity (k Out), and the third to the difference between intra‐ and inter‐modular connectivity (k Diff). In each panel, the boxplot to the left reports connectivity measures for modules not correlated with traits, while the boxplot to the right refers to modules correlated with traits. The color of each dot corresponds to the module name. RIL, recombinant inbred line.
Candidate modules correlating with traits had higher intra‐modular mean connectivity (k Within, i.e., the degree of connection of a given gene with genes of the same module) and lower inter‐modular mean connectivity (k Out, i.e., the degree of connection of a given gene with genes outside of the module) than modules not associated with traits. All modules exhibited a negative mean connectivity difference (k Diff), that is, the difference between intra‐ and inter‐modular connectivity, with only 5% of the total genes having a positive k Diff value (Figure 5b). eQTLs were examined in relation to network connectivity considering their genomic location (Figure S11). Distant eQTL genes had a higher gene module membership (MM), that is, correlation between the gene expression level and the first PC of the assigned module (module eigengene), and a higher total connectivity within the whole network. Genes that were controlled by local eQTLs showed a higher correlation between expression levels and the traits, meaning that local eQTLs could influence the phenotype more directly than distant eQTL genes, which instead might have a higher impact on the whole network.
Candidate gene discovery
A total of 25 genes produced transcripts whose expression level was simultaneously correlated with one of the traits (PCC genes) and controlled by one or more MTAs for the same trait (GWAs) (Table 1). A complex pattern of expression control emerged for several such candidate genes. For example, a SNP located at 50.5 Mb on Chr 2 increased the expression of two PCC genes located at 5.19 Mb on Chr 2 (Zm00001d002064) and at 177.54 Mb on Chr 4 (Zm00001d052047), while it decreased that of a PCC gene located at 141.2 Mb on Chr 2 (Zm00001d004822). RILs with the homozygous alternative allele at this locus showed lower values of DZ size, LED, and leaf length, reinforcing a possible role of the targeted transcripts in the determination of the phenotypes (Figure 6). According to PLAZA 4.0, Zm00001d002064 encodes a glycosyltransferase, Zm00001d004822 encodes a putative bZIP transcription factor, and Zm00001d052047 does not have a functional description. The eQTL candidate genes belonged to 11 co‐expression modules (Table 1). The turquoise and blue modules featured the highest number of candidate genes, six and four, respectively. Thirteen eQTL candidate genes appeared in modules associated with traits and could be linked to traits beyond those correlating with the co‐expression module.
Table 1.
Candidate genes identified by the combined approach
| Chr SNP | Mb SNP | eQTL gene ID | Chr gene | Mb gene | LER | LED | Leaf length | DZ size | WGCNA_module |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 175 266 | Zm00001d030812 | 1 | 161 357 | – |

|
– |
|
Light yellow |
| 2 | 50 479 | Zm00001d002064 | 2 | 5192 | – |
|
|
|
Magenta |
| 2 | 50 479 | Zm00001d004822 | 2 | 141 289 | – |
|
– | – | Green |
| 2 | 50 479 | Zm00001d052047 | 4 | 177 540 | – |
|
|
– | Blue |
| 3 | 133 576 | Zm00001d041358 | 3 | 114 163 | – |
|
|
– | Blue |
| 3 | 133 576 | Zm00001d042191 | 3 | 155 498 | – |
|
– | – | Turquoise |
| 3 | 213 973 | Zm00001d043881 | 3 | 212 800 | – |
|
|
– | Blue |
| 3 | 224 595 | Zm00001d044255 | 3 | 223 099 | – |
|
– | – | Brown |
| 4 | 189 323 | Zm00001d052416 | 4 | 189 489 | – | – |
|
– | Turquoise |
| 4 | 192 085 | Zm00001d014166 | 5 | 34 841 |
|
– | – | – | NA |
| 4 | 198 624 | Zm00001d014166 | 5 | 34 841 |
|
– | – | – | NA |
| 4 | 198 624 | Zm00001d052992 | 4 | 207 906 |
|
|
– | – | Turquoise |
| 4 | 238 223 | Zm00001d053442 | 4 | 231 583 | – |
|
|
– | Tan |
| 4 | 238 223 | Zm00001d053633 | 4 | 237 652 | – |
|
– | – | Dark red |
| 4 | 239 602 | Zm00001d021760 | 7 | 162 497 | – | – |
|
– | Blue |
| 4 | 239 602 | Zm00001d053716 | 4 | 239 768 |
|
– |
|
– | Green |
| 5 | 205 947 | Zm00001d017728 | 5 | 205 372 | – |
|
– | – | Grey |
| 5 | 205 947 | Zm00001d017756 | 5 | 206 004 | – |
|
– | – | Yellow |
| 5 | 205 947 | Zm00001d017770 | 5 | 206 299 | – |
|
– | – | Red |
| 5 | 205 947 | Zm00001d017866 | 5 | 209 133 | – |
|
– | – | Turquoise |
| 8 | 77 011 | Zm00001d008966 | 8 | 27 722 | – |
|
– | – | Red |
| 8 | 77 011 | Zm00001d009439 | 8 | 64 588 | – |
|
– | – | Turquoise |
| 8 | 89 591 | Zm00001d008742 | 8 | 18 823 | – | – |
|
– | Red |
| 8 | 164 886 | Zm00001d010366 | 8 | 111 285 | – |
|
– |
|
Magenta |
| 8 | 164 886 | Zm00001d011745 | 8 | 160 627 | – |
|
– | – | Turquoise |
| 8 | 164 886 | Zm00001d011924 | 8 | 164 685 | – |
|
|
– | Green |
For each eQTL gene (eQTL gene ID), the associated SNP is reported with chromosome (Chr SNP) and position in Mb (Mb SNP). Gene chromosome (Chr gene) and start position in Mb (Mb gene) are also given. The trait(s) associated to each eQTL are reported with a tick mark under the trait name (LER, LED, leaf length, and DZ size). The consensus module to which each eQTL gene belongs is given when available (WGCNA module). DZ, division zone; eQTL, expression quantitative trait locus; LED, leaf elongation duration; LER, leaf elongation rate; SNP, single nucleotide polymorphism; WGCNA, weighted gene co‐expression network analysis.
Figure 6.
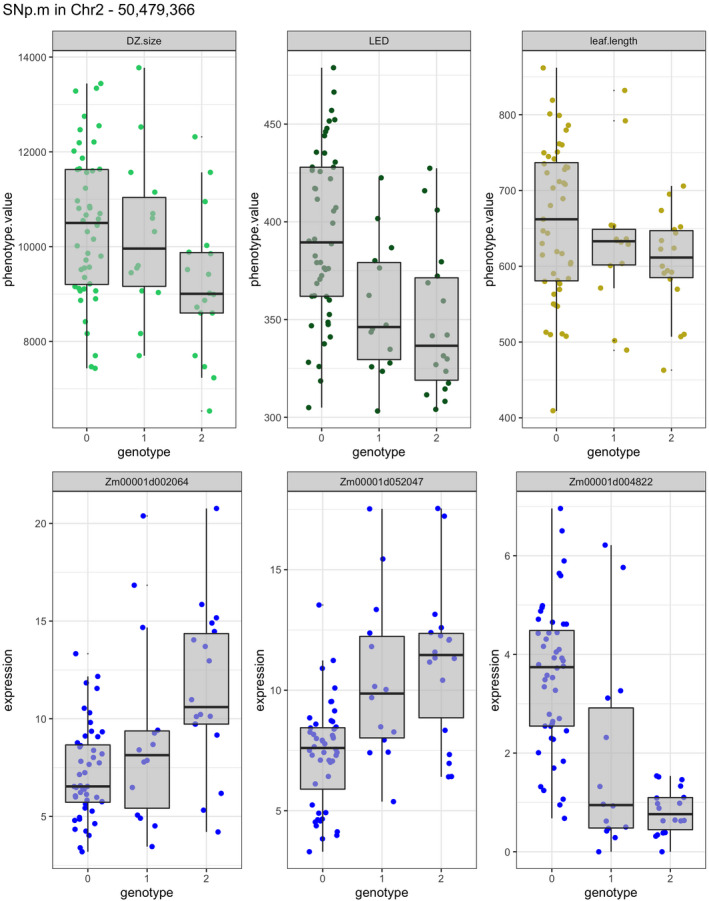
Example of the effects of an eQTL on gene expression and phenotypic value. The SNP located on Chr 2 at position 50 479 366 influences the transcript levels of three genes (Zm00001d002064 encoding a glycosyltransferase, Zm00001d052047 encoding putative bZIP transcription factor, and Zm00001d004822 encoding a protein with unknown function) and the phenotypic values of DZ size, LED, and leaf length. Genotype 0 is the homozygous reference, genotype 1 is heterozygous, and genotype 2 is the homozygous alternative. Heterozygous genotypes may be derived from residual heterozygosity still present in RIL populations. DZ, division zone; eQTL, expression quantitative trait locus; LED, leaf elongation duration; RIL, recombinant inbred line.
A GO enrichment analysis was conducted by traits for the 25 candidate genes identified. For the biological process annotation terms, 42% of candidate genes were enriched (P < 0.05) in transport activities (two of which specifically in calcium ion transport), 40% in cell recognition, 8% in organelle assembly, and 8% in recognition of pollen and in pollen–pistil interaction (Table S14). Expression levels of candidate genes as reported in the maize RNA‐seq gene atlas and the Electronic Fluorescent Pictograph browser are reported in File S3.
DISCUSSION
Associations between leaf traits and molecular diversity in the collection
The genetic control of plant organ growth is complex. Previous studies revealed that different organs in maize partially share their genetic architecture and control (Dignat et al., 2013). In maize, early leaf growth is relevant from a breeding perspective as it can be put in relation with adult plant yield (Sun et al., 2017) and heterotic potential (Feys et al., 2018). With this study, we aimed at establishing the links between DNA variants, RNA transcript levels, and trait measures in early leaf phenotypes.
RNA‐seq‐derived SNP markers are representative of the functional space of the genome, and are a reliable source of information about molecular diversity and uniqueness of typed accessions (Zhao et al., 2019). When deriving SNPs from mRNA, all variants fall in coding regions. However, depending on the recombination landscape, they may be in linkage with intergenic regions, including regulatory elements and non‐coding DNA. Hence, the association signals derived from SNPs included in this study may capture causative variants outside gene bodies. When used in GWA studies or eQTL mapping, RNA‐seq‐derived SNPs may target causative variants in LD with them, most probably in upstream promoter regions (Li et al., 2012). It has been shown that variants in open chromatin regions explain a large proportion of phenotypic variation in maize (Rodgers‐Melnick et al., 2006). In this frame, the extensive genotyping based on RNA‐seq analysis provides a uniform representation of RIL haplotypes (Figure 2a). The genetic structure of the mapping panel is reminiscent of the two populations of which it is comprised (Figure 2b). It is expected that the two‐way RILs have slower LD decay than the MAGIC RILs (Dell’Acqua et al., 2015); this may reduce the mapping definition of the panel, contributing to areas of higher LD (Figure S1). This is also reflected in our eQTL mapping results, which show extensive portions of local signals in close vicinity and localizing in pericentromeric regions (Figure 4). It is likely that increased pericentromeric LD induces some redundancy in the PCC‐eQTL identification procedure (Figure S6) since transcripts are tested with all SNPs regardless of their prior association with traits.
Using the PCC approach, we identified thousands of genes whose expression was correlated with trait values (Table S6). The number of PCC genes shared across different phenotypes was not proportional with the number of genes correlated with any given phenotype, revealing a complex effect of the RIL genetic background on the gene expression patterns. These figures are in agreement with those reported in Baute et al. (2015, 2016), which rely on the same genetic materials and trait values. In the present study, we identified 183 PCC genes in common with these earlier studies. Discrepancies among gene lists can result from the different set of phenotypes considered, from the fact that two RIL sets are used as a combined mapping panel in this study, and from the fact that the studies employ different maize genome annotations.
In GWA studies, sample size is crucial to achieve statistical power (Visscher et al., 2017). This is especially true as it becomes increasingly clear that the genetic control of maize traits, among other species, is dependent on a plethora of loci with small effects (Wallace et al., 2014b). In the present study, we detected 26 significant QTLs, some of which were pleiotropic among the tested traits (Figure S3; Table S8). It is likely that these are not the sole loci controlling leaf traits (Tian et al., 2011), but rather are the MTAs with the highest effect that could be detected segregating in the mapping panel given the sample size. The QTLs identified by MTAs are in agreement with previous reports, as multiple QTLs for maize leaf growth were repeatedly mapped in genetic positions compatible with our findings on Chr 1, Chr 2, Chr 4, Chr 7, and Chr 10 (Ku et al., 2010; Reymond et al., 2003). One QTL for LED at 205 Mb on Chr 5 co‐maps with an MTA reported for early maize growth (Muraya et al., 2017), and the hotspots for GWAs (Figure S3) and eQTLs (Figure 4) identified on Chr 4 and Chr 10 may correspond to QTLs reported in maize for leaf length and for leaf angle at similar positions (Wang et al., 2017).
Transcriptional mechanisms underlying leaf trait determination
PCC‐eQTLs were more abundant than GWA‐eQTLs, because by the PCC approach we identified a large number of transcripts, targeting several genomic loci controlling their expression. PCC genes may represent genes whose expression is not controlled by variants in LD with RNA‐seq‐derived SNPs, or whose expression is controlled by SNPs whose allele frequency cannot be associated with measured phenotypes through GWA. Likewise, MTAs may target genes whose expression variance is not captured by the tissue‐specific and/or timepoint‐specific RNA expression samples, or may affect phenotypes without causing changes in gene transcript levels, for example, by way of non‐coding RNAs (Wang et al., 2015). eQTLs were overwhelmingly acting as distant for all traits (Figures S7 and S8), that is, derived from markers in distant gene bodies and intergenic regions in LD with them. Previous studies reported that distant eQTLs may explain a higher proportion of expression variance than local eQTL in maize (Liu et al., 2017; Swanson‐Wagner et al., 2009), as hotspots of distant eQTL may act as key regulators of phenotypes (Wang et al., 2018). Local eQTL display a mode of gene expression regulation in cis, with co‐regulated gene clusters. Conversely, distant eQTLs tend to form hotspots in the genome, controlling genes significantly enriched in specific functional categories and being trans‐regulators for a variety of metabolic pathways (Wang et al., 2018). Still, trans‐regulation may occur via downstream effects of cis‐regulatory elements, like in the case of transcription factors (Rockman and Kruglyak, 2006). Further studies may focus on the trans eQTLs reported here as putative hotspots containing master regulators controlling leaf development.
The WGCNA approach aimed at capturing the broader picture of the complex gene interactions contributing to trait values (Ardlie et al., 2015), with a consensus clustering procedure that is aimed at reducing confounding effects when dealing with sub‐groups of samples (Shahan et al., 2018; Wu et al., 2002). Negatively correlated genes were considered unconnected, leading to the construction of a signed network. The overall network was scale‐free (Figure S9) and was comprised of distinct modules, as expected for biological networks (Barabási and Oltvai, 2004). Co‐expression clusters had a similar behavior for leaf length, LED, and DZ size, sharing modules with high correlation values and suggesting the activation of the same pathways (Figure 5a). LER was the only trait significantly correlated with only one module (yellowgreen) with a small number of genes (Table S13), in accordance with the fact that this trait showed the smallest number of eQTLs (Table S11). The co‐expression analysis revealed that genes controlled by local eQTLs were less connected to the whole network, but they had a higher intra‐modular connectivity. This suggests that local eQTLs are more likely to influence specific biological pathways and might be less crucial for the network stability. Conversely, genes that were influenced by distant eQTLs showed a higher inter‐modular connectivity and a lower gene–trait association, indicating that they might have a broad effect, acting in different pathways across candidate modules (Figure S11).
Candidate genes
The list of gene models resulting from the intersection of the two approaches provides several compelling candidates for leaf traits, showing that the combination of eQTLs with trait measures may support the identification of breeding targets (Table 1). The genomic positions of SNPs associated to expression levels of candidate genes overlapped with the genomic positions of several associations previously reported for leaf traits, including: (i) MTAs reported for leaf length (Chr 3 and Chr 4), leaf area (Chr 8), and leaf width (Chr 4, Chr 5, and Chr 8) on the maize Nested Association Mapping population (Tian et al., 2011), (ii) multi‐environment meta‐QTLs reported for leaf architecture traits (Chr 3, Chr 4, and Chr 8) (Zhao et al., 2018), (iii) QTLs associated with leaf rolling for leaf length (Chr 4 and Chr 5) and leaf width (Chr 1) (Gao et al., 2019), (iv) QTLs for ear leaf length specific to sowing density (Chr 4) (Wang et al., 2017), and (v) QTLs for leaf orientation (Chr 8) (Zhang et al., 2021).
The list of candidate genes includes several putative DNA‐binding domain proteins, like in the case of the three eQTLs associated to DZ size, LED, and leaf length (Figure 6). In this group of genes, Zm00001d004822 encodes a C2H2‐like zinc finger protein whose lower expression is associated with lower values of LED. The same eQTL SNP is responsible for the upregulation of the gene Zm00001d002064, whose expression level is associated to three phenotypes at the same time (Table 1); the gene encodes a glycosyltransferase, likely involved in cell wall biogenesis and organization (Liepman et al., 2010; Suliman et al., 2013; Yin et al., 2010) and in plant growth (Burn et al., 2002; Yao et al., 2019). Glycosyltransferases are thought to be involved in pericarp development in maize (Chateigner‐Boutin et al., 2016) and possibly in drought tolerance (Shikha et al., 2017). The function of the third gene in the eQTL group is unknown (Table 1). The information sourced from maize expression atlases can support a biological interpretation of this eQTL (File S3). According to the MaizeGDB RNA‐seq expression data atlas, the glycosyltransferase‐encoding gene Zm00001d002064 is highly expressed in immature leaves, while Zm00001d052047 (unknown function) is highly expressed in pollen in the background of B73 (which bears the low‐expression allele in our study, as shown in Figure 6) and, to a lesser extent, in leaves (File S3). The closest Arabidopsis ortholog for this gene (AT4G30780) encodes a helicase and suggests an involvement in DNA accessibility. The zinc‐finger gene Zm00001d004822 has an expression pattern opposite to that of the other genes at this eQTL and may have a trans‐regulatory effect on their expression, though further studies are necessary to fully uncover the relation between these candidate genes.
The gene Zm00001d053716 encodes a Tic21 translocon component that is localized in the chloroplast inner membrane. Tic21 (AT2G15290) null mutants in Arabidopsis thaliana suggest that it might be important in later stages of leaf development due to its crucial function as part of the inner membrane protein‐conducting channel (Teng et al., 2006). Seven unlinked genes encoding proteins involved in proton transport and signal transduction were involved in multiple traits (Table 1), supported by the corresponding GO enrichment (Table S14). According to PLAZA 4.0, the candidate gene Zm00001d017770 encodes a soluble inorganic pyrophosphatase 2 involved in acidification of vacuoles. Lower amounts of the pyrophosphate‐energized vacuolar membrane proton pump AVP1 in A. thaliana AVP1 (AT1G15690) mutants result in a reduction in leaf size caused by a decrease in cell number without a concomitant change in cell size (Li et al., 2005), and overexpression of AVP1 results in enlarged leaves (Gonzalez et al., 2010). Lack of expression of this gene has been related to developmental damage in A. thaliana (Fukuda et al., 2016). In maize, vacuolar pyrophosphatases were also related with leaf development in different evapotranspiration regimes (Devi and Reddy, 2018) and under drought tolerance (Wang et al., 2016), and may enhance organ size by a mechanism similar to that of AVP1 (Schilling et al., 2017). The gene Zm00001d014166, located on Chr 5 and controlled by two distant eQTLs on Chr 4, encodes a putative calcium‐transporting ATPase (Table S14), while the gene Zm00001d010366 on Chr 8 encodes a putative IQ‐25 domain‐binding protein overexpressed in samples with the alternative SNP and involved in DZ size and LED. The A. thaliana ortholog of the latter (AT3G16490) is a calmodulin‐binding protein (CaM) that requires calcium ions as intermediate for signal transduction. Ca2+‐ATPases play an important role in Ca2+ homeostasis by restoring the ion concentration to resting levels (Snedden and Fromm, 2001; Zielinski, 1988).
The glycosydase superfamily protein encoded by Zm00001d42191 may be related to DNA repair, specifically in the presence of oxidative stress. This in turn may put this candidate gene in relation to cell division efficiency as well as the abscisic acid response (Jiang and Zhang, 2001), similarly to the serine/threonine protein kinases encoded by Zm00001d052416 and Zm00001d008966, which are involved in leaf length and LED, respectively, and are both involved in signal transduction and in the abscisic acid pathway.
Network metrics show a largely negative difference between intra‐ and inter‐modular connectivity (k Diff), suggesting that the impact of genes correlated to modules may be higher on the whole network than inside each module (Figure 5b). The large inter‐modular connectivity (k Out) of candidate genes suggests that there is overlap in biological functions across modules, as expected from a complex phenotype like leaf trait determination. Network studies in humans (Battle et al., 2014) and plants (Mähler et al., 2017) showed that genes with higher connectivity (e.g., hub genes) are less likely to be related to detectable eQTLs, likely because hub genes are more constrained against changes in gene expression as compared to genes in the periphery of the network. In our study, genes belonging to modules correlated with phenotypes share a higher degree of internal connectivity (k Within) (Figure 5b). These results imply that when targeting specific traits from a breeding perspective, it may be more critical to focus on genes that have a lower whole‐network connectivity, but play a key role in controlling the phenotypic variation of the trait of interest (Langfelder et al., 2013) as reported for eQTLs.
CONCLUSIONS
Our results show that the forward genetics approach in maize may leverage transcriptomic and phenotypic data produced on a relatively small set of maize RILs to provide compelling evidence to identify candidate genes involved in leaf growth. Other studies focusing on different species, including poplar (Populus deltoides) (Balmant et al., 2020), strawberry (Fragaria × ananassa) (Barbey et al., 2020), maize (Kremling et al., 2019), and sweet potato (Ipomoea batatas L.) (Zhang et al., 2020), and traits devised alternative ways to integrate phenotyping, transcriptomic, and GWA data to prioritize candidate genes. These studies demonstrated that combined approaches, when applied on large datasets in some cases already available in the literature, may effectively advance our understanding of the molecular mechanisms underlying complex traits and provide candidate genes to prioritize for validation and breeding. Indeed, further studies in reverse genetics are needed to fully validate candidate genes and their interactions.
The small number of candidate genes that we report for leaf traits, strongly supported by predicted gene function and previous reports, are manageable targets for multi‐locus genome editing (Liu et al., 2020) to confirm their role and possibly manipulate their expression levels. The value of our forward genetics approach lies in the valorization of transcriptomic data in segregant populations to derive information about gene networks and their interactions in the determination of their roles in traits of interest. This approach can be applied on virtually any organism featuring a matched transcriptomic and phenotypic dataset. When put in relation to plant traits, the rapidly expanding body of knowledge on eQTL control and determination will accelerate our understanding of complex trait determination in crops, contributing to breaking new ground in precision breeding.
EXPERIMENTAL PROCEDURES
Plant material and phenotyping
This study relies on previously published phenotyping and transcriptomic data (Baute et al., 2015, 2016). Data were produced on two segregant populations with a partially overlapping pedigree: (i) a biparental population composed of 103 biparental RILs derived from the intercross between the inbred lines B73 and H99 (Marino et al., 2009) and (ii) 94 RILs randomly sampled from the MAGIC maize population and derived from the intercross of the inbred lines A632, B73, B96, F7, H99, HP301, Mo17, and W153R (Dell’Acqua et al., 2015). The random sampling of MAGIC RILs ensures a balanced representation of the eight founder haplotypes thanks to the lack of genetic structure in these mapping populations (Dell’Acqua et al., 2015; Scott et al., 2020). All plant materials were phenotyped with the same method, with full details given in Baute et al. (2015, 2016). In short, RILs were grown in completely randomized order, one plant per pot, 20 pots per RIL, in a controlled environment (24°C, 55% relative humidity, light intensity of 170 mmol m−2 sec−1 photosynthetically active radiation, in a 16 h/8 h day/night cycle). Phenotyping was focused on leaf four of the seedlings and included the following traits: final leaf length (mm), DZ size (µm), LER (mm h−1), and LED (h). DZ size is defined as the distance between the base of the leaf and the most distal mitotic cell in the epidermis according to 4′,6‐diamidino‐2‐phenylindole staining. LER was determined measuring the length of leaf four from soil to tip on a daily basis from emergence to full maturity and deriving the average growth rate during the steady growth stage as in Rymen et al. (2007). LED was measured in days until full maturity. Traits were measured on 18–20 plants per RIL, except for DZ size, which was determined on three plants per RIL due to the complexity of the phenotyping procedure. Traits were consistent and repeatable: heritability (h 2) was 0.65 for leaf length, 0.81 for DZ size, 0.52 for LER, and 0.61 for LED (Baute et al., 2015, 2016).
SNP detection from transcriptomic data and quantification of transcript abundances
In previous studies, paired‐end Illumina RNA‐seq reads with a length of 100 bp were produced from proliferative tissues of the fourth leaf of RIL seedlings, with the same procedure on the two populations. Three biological and three technical replicates were performed for parental lines, and one biological replicate was performed for RILs. For details, see Baute et al. (2015, 2016).
Raw RNA‐seq data were retrieved from ENA‐ERP009123, ENA‐ERP011069, and ENA‐ERP012784. The data were filtered for quality using the erne‐filter tool from the ERNE2 package (version 2.1.1, http://erne.sourceforge.net/) (Vezzi et al., 2012). Bases with a Phred score of >30 were retained and reads shorter than 50 nt were discarded. To identify SNPs and quantify gene expression levels, post‐processed reads from each sample were mapped independently to the reference B73 assembly version 4.0 (Jiao et al., 2017) using the aligner STAR v. 2.5.3a (Dobin et al., 2013) in 2‐pass protocol, with default parameters. The annotation was provided during the read mapping step and the order of the multi‐mapping alignments for each read was chosen to be random. A bam file was produced for each sample and used to extract and to normalize gene transcript abundances into transcript per million values using StringTie v.1.3.2 with default parameters (Pertea et al., 2016). Only genes expressed in at least 60% of the samples and with expression variance above 5% across RILs were considered for downstream analysis.
SNPs were obtained following the GATK (McKenna et al., 2010) best practices for variant discovery in RNA‐seq datasets, starting from the raw data clean‐up (http://www.broadinstitute.org/gatk/guide/best‐practices). Briefly, the workflow includes (i) removal of duplicates with PicardTools‐MarkDuplicates (https://broadinstitute.github.io/picard/), (ii) splitting into component reads and trimming of reads with N operators in the CIGAR string (Split’N’Trim – GATK v. 3.8), and (iii) local realignment around insertions or deletions in the sample’s genome compared to the reference (RealignerTargetCreator + IndelRealigner – GATK v 3.8). SNPs were called with the tool UnifiedGenotyper from GATK v 3.8 in the multi‐sample mode. Since the RILs were derived from at most eight parents, RIL SNP calls were restricted to those present in the parental genomes. The tool was run with the default parameters and heterozygosity set to 0.01. Only biallelic SNPs were allowed. Markers heterozygous in the parental lines, markers with more than 20% of missing data, and markers with a minor genotype frequency of <0.1 were filtered out to avoid noise and spurious association in the downstream analysis.
LD analysis
LD was calculated on a subset of high‐quality SNPs filtered for minor allele frequency above 30%, using LDcorsv (Mangin et al., 2012). The r 2 metric was obtained for each pairwise marker comparison and LD heatmap plots were produced with R/BigLD (Kim et al., 2018) (available at https://github.com/sunnyeesl/BigLD). The LD decay was estimated using the Hill and Weir equation (Hill and Weir, 1988) interpolating estimated LD values with physical distance between markers (Brunazzi et al., 2018; Marroni et al., 2011) with custom R scripts. An estimated r 2 of 0.2 was considered null LD and used to derive a chromosome‐specific LD decay distance.
Trait associations with transcript levels and SNP markers
Transcript levels were correlated with trait values using PCCs in R. Transcript–trait correlation analysis was conducted separately for the biparental RILs and MAGIC maize RILs to account for population structure. Maintaining the populations separated allowed to fully exploit their features, in particular (i) the balanced allele frequency within each RIL set, (ii) faster LD decay in the MAGIC RILs, and (iii) different segregating haplotypes, and hence stronger breeding significance of the candidate genes jointly identified. The significance threshold for each trait correlation was determined permuting the phenotype 7000 times and extracting the top and bottom 5/1000th quantiles of the permuted PCC distribution. The intersections of transcripts with correlation coefficients exceeding the significance threshold in both biparental RILs and MAGIC maize RILs were termed PCC genes and used for downstream analyses. A GO enrichment analysis was conducted with the Protein ANalysis THrough Evolutionary Relationships (PANTHER) classification system (Mi et al., 2010; Thomas et al., 2003), reporting terms with an FDR of <0.05 (Fisher exact test).
RNA‐seq‐derived SNP markers were used in a GWA analysis to test their associations with leaf traits with R/MVP (available at https://github.com/xiaolei‐lab/rMVP). Estimation of variance components was performed with the EMMA method (Kang et al., 2008), and mapping was conducted with the FarmCPU module (Liu et al., 2016). Population structure was corrected using PC covariates from the genetic diversity in the RIL set. The number of PC covariates to be included for each trait was iteratively assessed by running the GWA with a varying number of PCs from 1 to 15 and selecting the retained model via visual assessment of model fit through quantile–quantile (Q–Q) plots. The threshold for each GWA scan was determined by 199 permutations of the trait values and selecting the 90th percentile of the permuted P‐values with the procedure implemented in R/MVP. As multiple MTAs in LD were likely to target the same locus, a custom procedure was used to collapse significant signals based on LD decay. All MTAs falling within twice the LD decay distance from the signal with the highest significance were merged in a single QTL. For each QTL, the most significant MTA was the sole reported and used in further analyses.
eQTL mapping and derivation of candidate genes
eQTL mapping was conducted to establish associations between gene expression levels and SNP markers. The R package Matrix eQTL v2.2 (Shabalin, 2012) was used with default parameters in the linear model mode. To identify the optimal number of covariates to control for structure in the dataset, a random subset of 1000 SNPs and 1000 transcripts was used to map eQTLs using 1–20 PCs derived from gene expression values and from population structure. The model fit was checked on Q–Q plots. The full model was run with the optimal number of covariates considering all SNPs and all transcripts used for PCC and GWA analyses. For each transcript tested, a stringent Bonferroni correction was employed to reduce Type II errors, dividing a nominal P‐value of 0.01 by the total number of SNPs tested. Accordingly, eQTLs with a P‐value of <2.7 × 10−8 were considered significant for downstream analyses. Similarly to MTAs, LD decay information was used to collapse significant eQTL hits on the basis of chromosome‐specific LD extension: for each tested transcript, all markers significantly associated with it were clustered on the basis of chromosome‐specific LD decay distance, so that the only eQTL reported is the marker with the highest significance. eQTLs were classified as local when the SNP was below 1 Mb upstream or downstream from the associated gene and as distant otherwise.
Co‐expression network and identification of trait‐associated modules
WGCNA was performed using the R package WGCNA v1.63 (Langfelder and Horvath, 2008). A consensus clustering approach was used to identify robust modules across RIL sets, so that modules represent groups of co‐expressed genes that are preserved in the two populations. This approach, which is an extension of standard co‐expression network analysis, is commonly used to account for sub‐groups of samples in animal and plant studies (Monti et al., 2003; Shahan et al., 2018; Wu et al., 2002). Genes that were strongly negatively correlated were considered unconnected (since they could belong to different biological categories), leading to a signed network.
The signed consensus network was constructed through the function blockwiseConsensusModules(). The adjacency matrix was calculated with a soft threshold power of 8 for both RIL sets and consensus modules were forced to contain at least 30 genes. Connectivity values were obtained through the function intramodularConnectivity() starting from the adjacency matrix. The function moduleEigengenes() was used to calculate the most important eigengenes of the expression matrix to quantify co‐expression similarity of each module. Modules whose eigengenes were correlated with a value above 0.8 were clustered together. MM was used as a fuzzy measure correlating the gene expression profile with the module eigengene. After the trait measurements were imported into the co‐expression network, gene trait significance and the gene trait significance P‐value were calculated for each gene in the two RIL populations separately. For each trait, only modules with the same correlation coefficient sign in both populations were considered for the downstream analysis, choosing the lowest absolute value for each module–trait pair.
The function intramodularConnectivity() was applied to the adjacency matrix separately for each population to calculate the network connectivity values k Tot, k Within, k Out, and k Diff, where k Tot is the connectivity of each gene to all other genes in the whole network, corresponding to the sum of all edge weights; k Within is the connectivity of each gene within a single module (i.e., intra‐modular connectivity); k Out (calculated as k Tot − k Within) is the inter‐modular connectivity; and k Diff is the difference between k Within and k Out.
Identification and characterization of candidate genes
The eQTL results were intersected with the list of PCC genes and with the list of QTLs derived with the GWA study. eQTL genes were defined as candidate genes when they were (i) significantly correlated with one of the traits (PCC genes) and (ii) controlled by a SNP marker that was also associated with one of the traits (MTAs) (Figure 1). Candidate genes were characterized with PLAZA Monocot 4.0 (Van Bel et al., 2018), using the integrative orthology tool and the GO enrichment tool. Statistical overrepresentation analysis of the WGCNA modules was conducted through the PANTHER classification system (Mi et al., 2010; Thomas et al., 2003). Only terms with FDR < 0.05 (Fisher exact test) were considered significant. The expression levels of candidate genes were manually inspected on the RNA‐seq gene atlas (Walley et al., 2016) and on the Electronic Fluorescent Pictograph browser (eFP Atlas Browser), both part of MaizeGDB (maizegdb.org; Portwood et al., 2019), to describe peculiar features of their expression patterns.
AUTHOR CONTRIBUTIONS
DI, MEP, and MD conceptualized the research and supervised the work. MM and MD analyzed the data and interpreted results. MBH conducted LD analyses. FM conducted bioinformatics data analyses. HN produced the data and supervised the work. MM and MD drafted the manuscript and produced figures. MD agrees to serve as the corresponding author and ensures communication. All authors read and approved the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
Figure S1. Pairwise LD measures, by chromosome.
Figure S2. Pearson correlation coefficients (PCCs) between traits and transcripts.
{kind=link}
Figure S3. GWA scan of leaf traits.
Figure S4. Quantile–quantile plots for GWA scans.
Figure S5. Representative quantile–quantile plots for eQTL scans, with 1, 10, and 20 PCs as covariates.
Figure S6. Localization of PCC‐eQTLs and GWA‐eQTLs.
Figure S7. Characterization of eQTL associations by localization. (a) Number of SNPs that control each gene. (b) Distribution of genes regulated by local and/or distant SNPs.
Figure S8. (a) eQTL distribution and (b) eQTL effects, by phenotype.
Figure S9. Analysis of network topology for various soft‐thresholding powers.
Figure S10. Consensus clustering dendrograms of genes.
Figure S11. Relationship between network connectivity and eQTL genomic location.
Table S1. RNA sequencing statistics.
Table S2. Chromosome‐specific LD decay information.
Table S3. Phenotypic values of each individual.
Table S4. Correlation coefficients between gene expression levels and trait values.
Table S5. Permutation thresholds for Pearson correlation coefficients (PCCs).
Table S6. Output of the PCC scans for each trait.
Table S7. Complete GWA study results, with P‐value and effect for each tested marker.
Table S8. QTLs derived from marker–trait associations (MTAs).
Table S9. Expression QTLs derives from Pearson correlation coefficient analysis (PCC‐eQTLs).
Table S10. Expression QTLs derived from marker–trait associations (GWA‐eQTLs).
Table S11. Expression QTLs derived from Pearson correlation coefficient analysis (PCC‐eQTLs) and from marker–trait associations (GWA‐eQTLs).
Table S12. Summary statistics for PCC‐eQTL and GWA‐eQTL analysis.
Table S13. List of genes and consensus module.
Table S14. Outcome of the gene ontology (GO) enrichment analysis on candidate genes.
File S1. Outcome of the gene ontology (GO) enrichment analysis on PCC genes.
File S2. Outcome of the gene ontology (GO) enrichment analysis on genes for each putative module.
File S3. Functional information for candidate genes according to MaizeGDB databases.
ACKNOWLEDGMENTS
This research was supported by the European Research Council under the European Community’s Seventh Framework Program (FP7/2007‐2013) under ERC Grant agreement 339341‐AMAIZE. We thank the three anonymous reviewers for insightful comments.
DATA AVAILABILITY STATEMENT
All relevant data can be found in online repositories. Raw RNA‐seq reads are stored in the European Nucleotide Archive (www.ebi.ac.uk/ena) database under the following accession numbers: ENA‐ERP009123 (parental lines), ENA‐ERP011069 (biparental RILs), and ENA‐ERP012784 (MAGIC maize RILs). Scripts are available at https://github.com/mdellh2o/maize_eQTL. Other data used for the experiments may be retrieved at Figshare (10.6084/m9.figshare.13702846).
REFERENCES
- Albert, F.W. & Kruglyak, L. (2015) The role of regulatory variation in complex traits and disease. Nature Reviews Genetics, 16(4), 197–212. [DOI] [PubMed] [Google Scholar]
- Ardlie, K.G. , Deluca, D.S. , Segre, A.V. , Sullivan, T.J. , Young, T.R. , Gelfand, E.T. et al. (2015) The Genotype‐Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 348(6235), 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmant, K.M. , Noble, J.D. , Alves, F.C. , Dervinis, C. , Conde, D. , Schmidt, H.W. et al. (2020) Xylem systems genetics analysis reveals a key regulator of lignin biosynthesis in Populus deltoides . Genome Research, 30, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási, A.L. & Oltvai, Z.N. (2004) Network biology: understanding the cell’s functional organization. Nature Reviews Genetics, 5(2), 101–113. [DOI] [PubMed] [Google Scholar]
- Barbey, C. , Hogshead, M. , Schwartz, A.E. , Mourad, N. , Verma, S. , Lee, S. et al. (2020) The genetics of differential gene expression related to fruit traits in strawberry (Fragaria × ananassa). Frontiers in Genetics, 10, 1317. 10.3389/fgene.2019.01317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle, A. , Mostafavi, S. , Zhu, X. , Potash, J.B. , Weissman, M.M. , McCormick, C. et al. (2014) Characterizing the genetic basis of transcriptome diversity through RNA‐sequencing of 922 individuals. Genome Research, 24(1), 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baute, J. , Herman, D. , Coppens, F. , De Block, J. , Slabbinck, B. , Dell’Acqua, M. et al. (2015) Correlation analysis of the transcriptome of growing leaves with mature leaf parameters in a maize RIL population. Genome Biology, 16(1), 10.1186/s13059-015-0735-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baute, J. , Herman, D. , Coppens, F. , De Block, J. , Slabbinck, B. , Dell’Acqua, M. et al. (2016) Combined large‐scale phenotyping and transcriptomics in maize reveals a robust growth regulatory network. Plant Physiology, 170(3), 1848–1867. 10.1104/pp.15.01883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunazzi, A. , Scaglione, D. , Talini, R.F. , Miculan, M. , Magni, F. , Poland, J. et al. (2018) Molecular diversity and landscape genomics of the crop wild relative Triticum urartu across the Fertile Crescent. The Plant Journal. 10.1111/tpj.13888 [DOI] [PubMed] [Google Scholar]
- Buckler, E.S. , Holland, J.B. , Bradbury, P.J. , Acharya, C.B. , Brown, P.J. , Browne, C. et al. (2009) The genetic architecture of maize flowering time. Science, 325(5941), 714–718. [DOI] [PubMed] [Google Scholar]
- Burn, J.E. , Hocart, C.H. , Birch, R.J. , Cork, A.C. & Williamson, R.E. (2002) Functional analysis of the cellulose synthase genes CesA1, CesA2, and CesA3 in Arabidopsis. Plant Physiology, 129, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateigner‐Boutin, A.L. , Ordaz‐Ortiz, J.J. , Alvarado, C. , Bouchet, B. , Durand, S. , Verhertbruggen, Y. et al. (2016) Developing pericarp of maize: a model to study arabinoxylan synthesis and feruloylation. Frontiers in Plant Science, 7, 1476. 10.3389/fpls.2016.01476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, N. , Myburg, A.A. , Joubert, F. , Murray, S.L. , Carstens, M. , Lin, Y.‐C. et al. (2017) Systems genetics reveals a transcriptional network associated with susceptibility in the maize‐grey leaf spot pathosystem. The Plant Journal, 89(4), 746–763. [DOI] [PubMed] [Google Scholar]
- Civelek, M. & Lusis, A.J. (2014) Systems genetics approaches to understand complex traits. Nature Reviews Genetics, 15(1), 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Acqua, M. , Gatti, D.M. , Pea, G. , Cattonaro, F. , Coppens, F. , Magris, G. et al. (2015) Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in Zea mays . Genome Biology, 16(1). 10.1186/s13059-015-0716-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi, M.J. & Reddy, V.R. (2018) Effect of temperature under different evaporative demand conditions on maize leaf expansion. Environmental and Experimental Botany, 155, 509–517. [Google Scholar]
- Dignat, G. , Welcker, C. , Sawkins, M. , Ribaut, J.M. & Tardieu, F. (2013) The growths of leaves, shoots, roots and reproductive organs partly share their genetic control in maize plants. Plant, Cell and Environment, 36(6), 1105–1119. [DOI] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C.A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. et al. (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler, E.E. , Flint, J. , Gibson, G. , Kong, A. , Leal, S.M. , Moore, J.H. et al. (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nature Reviews Genetics, 11(6), 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, C. , Su, H. , Bai, H. , Wang, R. , Liu, Y. , Guo, X. et al. (2018) High‐efficiency genome editing using a dmc1 promoter‐controlled CRISPR/Cas9 system in maize. Plant Biotechnology Journal. 10.1111/pbi.12920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feys, K. , Demuynck, K. , De Block, J. , Bisht, A. , De Vliegher, A. , Inzé, D. et al. (2018) Growth rate rather than growth duration drives growth heterosis in maize B104 hybrids. Plant, Cell and Environment, 41(2), 374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, M. , Segami, S. , Tomoyama, T. , Asaoka, M. , Nakanishi, Y. , Gunji, S. et al. (2016) Lack of H+‐pyrophosphatase prompts developmental damage in Arabidopsis leaves on ammonia‐free culture medium. Frontiers in Plant Science, 7, 819. 10.3389/fpls.2016.00819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L. , Yang, G. , Li, Y. , Fan, N. , Li, H. , Zhang, M. et al. (2019) Fine mapping and candidate gene analysis of a QTL associated with leaf rolling index on chromosome 4 of maize (Zea mays L.). Theoretical and Applied Genetics, 132(11), 3047–3062. [DOI] [PubMed] [Google Scholar]
- Gonzalez, N. , De Bodt, S. , Sulpice, R. , Jikumaru, Y. , Chae, E. , Dhondt, S. et al. (2010) Increased leaf size: different means to an end. Plant Physiology, 153(3), 1261–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W.G. & Weir, B.S. (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theoretical Population Biology, 33(1), 54–78. [DOI] [PubMed] [Google Scholar]
- Hilscher, J. , Bürstmayr, H. & Stoger, E. (2017) Targeted modification of plant genomes for precision crop breeding. Biotechnology Journal, 12(1), 1600173. [DOI] [PubMed] [Google Scholar]
- Hirsch, C.N. , Foerster, J.M. , Johnson, J.M. , Sekhon, R.S. , Muttoni, G. , Vaillancourt, B. et al. (2014) Insights into the maize pan‐genome and pan‐transcriptome. The Plant Cell, 266(1), 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, M. & Zhang, J. (2001) Effect of abscisic acid on active oxygen species, antioxidative defence system and oxidative damage in leaves of maize seedlings. Plant and Cell Physiology, 42(11), 1265–1273. [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Peluso, P. , Shi, J. , Rank, D.R. & Ware, D. (2017) Improved maize reference genome with single molecule technologies. bioRxiv. 10.1101/079004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H.M. , Zaitlen, N.A. , Wade, C.M. , Kirby, A. , Heckerman, D. , Daly, M.J. et al. (2008) Efficient control of population structure in model organism association mapping. Genetics, 178(3), 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.A. , Cho, C.‐S. , Kim, S.‐R. , Bull, S.B. & Yoo, Y.J. (2018) A new haplotype block detection method for dense genome sequencing data based on interval graph modeling of clusters of highly correlated SNPs. Bioinformatics, 34(3), 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremling, K.A.G. , Diepenbrock, C.H. , Gore, M.A. , Buckler, E.S. & Bandillo, N.B. (2019) Transcriptome‐wide association supplements genome‐wide association in Zea mays . G3: Genes, Genomes, Genetics, 9, 3023–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krouk, G. , Lingeman, J. , Colon, A.M. , Coruzzi, G. & Shasha, D. (2013) Gene regulatory networks in plants: learning causality from time and perturbation. Genome Biology, 14(6), 123. 10.1186/gb-2013-14-6-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumsiek, J. , Bartel, J. & Theis, F.J. (2016) Computational approaches for systems metabolomics. Current Opinion in Biotechnology, 39, 198–206. [DOI] [PubMed] [Google Scholar]
- Ku, L.X. , Zhao, W.M. , Zhang, J. , Wu, L.C. , Wang, C.L. , Wang, P.A. et al. (2010) Quantitative trait loci mapping of leaf angle and leaf orientation value in maize (Zea mays L.). Theoretical and Applied Genetics, 121(5), 951–959. [DOI] [PubMed] [Google Scholar]
- Langfelder, P. & Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9(1), 559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P. , Mischel, P.S. & Horvath, S. (2013) When is hub gene selection better than standard meta‐analysis? PLoS One, 8(4), e61505. 10.1371/journal.pone.0061505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Yang, H. , Peer, W.A. , Richter, G. , Blakeslee, J. , Bandyopadhyay, A. et al. (2005) Plant science: Arabidopsis H+‐PPase AVP1 regulates auxin‐mediated organ development. Science, 310(5745), 121–125. [DOI] [PubMed] [Google Scholar]
- Li, X. , Zhu, C. , Yeh, C.‐T. , Wu, W. , Takacs, E.M. , Petsch, K.A. et al. (2012) Genic and nongenic contributions to natural variation of quantitative traits in maize. Genome Research, 22(12), 2436–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y.‐X. , Li, C. , Bradbury, P.J. , Liu, X. , Lu, F. , Romay, C.M. et al. (2016) Identification of genetic variants associated with maize flowering time using an extremely large multi‐genetic background population. The Plant Journal, 86(5), 391–402. 10.1111/tpj.13174 [DOI] [PubMed] [Google Scholar]
- Liepman, A.H. , Wightman, R. , Geshi, N. , Turner, S.R. & Scheller, H.V. (2010) Arabidopsis ‐ a powerful model system for plant cell wall research. The Plant Journal, 61(6), 1107–1121. [DOI] [PubMed] [Google Scholar]
- Liu, H.‐J. , Jian, L. , Xu, J. , Zhang, Q. , Zhang, M. , Jin, M. et al. (2020) High‐throughput CRISPR/Cas9 mutagenesis streamlines trait gene identification in maize. The Plant Cell, 32(5), 1397–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Luo, X. , Niu, L. , Xiao, Y. , Chen, L.u. , Liu, J. et al. (2017) Distant eQTLs and non‐coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize. Molecular Plant, 10(3), 414–426. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Huang, M. , Fan, B. , Buckler, E.S. & Zhang, Z. (2016) Iterative usage of fixed and random effect models for powerful and efficient genome‐wide association studies. PLoS Genetics, 12(2), e1005767. 10.1371/journal.pgen.1005767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X. , Mau, M. & Sharbel, T.F. (2018) Genome editing for global food security. Trends in Biotechnology, 36(2), 123–127. [DOI] [PubMed] [Google Scholar]
- Mackay, T.F.C. , Stone, E.A. & Ayroles, J.F. (2009) The genetics of quantitative traits: challenges and prospects. Nature Reviews Genetics, 10(8), 565–577. [DOI] [PubMed] [Google Scholar]
- Mähler, N. , Wang, J. , Terebieniec, B.K. , Ingvarsson, P.K. , Street, N.R. & Hvidsten, T.R. (2017) Gene co‐expression network connectivity is an important determinant of selective constraint. PLoS Genetics, 13(4), e1006402. 10.1371/journal.pgen.1006402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangin, B. , Siberchicot, A. , Nicolas, S. , Doligez, A. , This, P. & Cierco‐Ayrolles, C. (2012) Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity, 108(3), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino, R. , Ponnaiah, M. , Krajewski, P. , Frova, C. , Gianfranceschi, L. , Pè, M.E. et al. (2009) Addressing drought tolerance in maize by transcriptional profiling and mapping. Molecular Genetics and Genomics, 281(2), 163–179. [DOI] [PubMed] [Google Scholar]
- Mark Cigan, A. , Unger‐Wallace, E. & Haug‐Collet, K. (2005) Transcriptional gene silencing as a tool for uncovering gene function in maize. The Plant Journal, 43(6), 929–940. [DOI] [PubMed] [Google Scholar]
- Marroni, F. , Pinosio, S. & Morgante, M. (2014) Structural variation and genome complexity: is dispensable really dispensable? Current Opinion in Plant Biology, 18, 31–36. [DOI] [PubMed] [Google Scholar]
- Marroni, F. , Pinosio, S. , Zaina, G. , Fogolari, F. , Felice, N. , Cattonaro, F. et al. (2011) Nucleotide diversity and linkage disequilibrium in Populus nigra cinnamyl alcohol dehydrogenase (CAD4) gene. Tree Genetics and Genomes, 7(5), 1011–1023. [Google Scholar]
- May, B.P. , Liu, H. , Vollbrecht, E. , Senior, L. , Rabinowicz, P.d. , Roh, D. et al. (2003) Maize‐targeted mutagenesis: a knockout resource for maize. Proceedings of the National Academy of Sciences United States of America, 100(20), 11541–11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen, M.d. , Kresovich, S. , Villeda, H.s. , Bradbury, P. , Li, H. , Sun, Q. et al. (2009) Genetic properties of the maize nested association mapping population. Science 325(5941), 737–740. [DOI] [PubMed] [Google Scholar]
- Mi, H. , Dong, Q. , Muruganujan, A. , Gaudet, P. , Lewis, S. & Thomas, P.D. (2010) PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Research, 38(Suppl_1), D204–D210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monir, M.M. & Zhu, J. (2018) Dominance and epistasis interactions revealed as important variants for leaf traits of maize NAM population. Frontiers in Plant Science, 9, 627. 10.3389/fpls.2018.00627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti, S. , Tamayo, P. , Mesirov, J. & Golub, T. (2003) Consensus clustering: a resampling‐based method for class discovery and visualization of gene expression microarray data. Machine Learning, 52, 91–118. [Google Scholar]
- Muraya, M.M. , Chu, J. , Zhao, Y. , Junker, A. , Klukas, C. , Reif, J.C. et al. (2017) Genetic variation of growth dynamics in maize (Zea mays L.) revealed through automated non‐invasive phenotyping. The Plant Journal, 89, 366–380. [DOI] [PubMed] [Google Scholar]
- Pang, J. , Fu, J. , Zong, N. , Wang, J. , Song, D. , Zhang, X. et al. (2019) Kernel size‐related genes revealed by an integrated eQTL analysis during early maize kernel development. The Plant Journal, 98, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Kim, D. , Pertea, G.M. , Leek, J.T. & Salzberg, S.L. (2016) Transcript‐level expression analysis of RNA‐ seq experiments with HISAT, StringTie and Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nature Protocols, 11(9), 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland, J. (2015) Breeding‐assisted genomics. Current Opinion in Plant Biology, 24, 119–124. [DOI] [PubMed] [Google Scholar]
- Portwood, J.L. , Woodhouse, M.R. , Cannon, E.K. , Gardiner, J.M. , Harper, L.C. , Schaeffer, M.L. et al. (2019) Maizegdb 2018: the maize multi‐genome genetics and genomics database. Nucleic Acids Research, 47(D1), D1146–D1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond, M. , Muller, B. , Leonardi, A. , Charcosset, A. & Tardieu, F. (2003) Combining quantitative trait loci analysis and an ecophysiological model to analyze the genetic variability of the responses of maize leaf growth to temperature and water deficit. Plant Physiology, 131(2), 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman, M.V. & Kruglyak, L. (2006) Genetics of global gene expression. Nature Reviews Genetics, 7(11), 862–872. [DOI] [PubMed] [Google Scholar]
- Rodgers‐Melnick, E. , Vera, D.L. , Bass, H.W. & Buckler, E.S. (2016) Open chromatin reveals the functional maize genome. Proceedings of the National Academy of Sciences United States of America, 113(22), 3177–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymen, B. , Fiorani, F. , Kartal, F. , Vandepoele, K. , Inze, D. & Beemster, G.T.S. (2007) Cold nights impair leaf growth and cell cycle progression in maize through transcriptional changes of cell cycle genes. Plant Physiology, 143(3), 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling, R.K. , Tester, M. , Marschner, P. , Plett, D.C. & Roy, S.J. (2017) AVP1: one protein, many roles. Trends in Plant Science, 22(2), 154–162. [DOI] [PubMed] [Google Scholar]
- Schnable, P.S. , Ware, D. , Fulton, R.S. , Stein, J.C. , Wei, F. , Pasternak, S. et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science, 326(5956), 1112–1115. [DOI] [PubMed] [Google Scholar]
- Scott, M.F. , Ladejobi, O. , Amer, S. , Bentley, A.R. , Biernaskie, J. , Boden, S.A. et al. (2020) Multi‐parent populations in crops: a toolbox integrating genomics and genetic mapping with breeding. Heredity, 125(6), 396–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaver, S.M.D. , Lerma‐Ortiz, C. , Conrad, N. , Mikaili, A. , Sreedasyam, A. , Hanson, A.D. et al. (2018) PlantSEED enables automated annotation and reconstruction of plant primary metabolism with improved compartmentalization and comparative consistency. The Plant Journal, 95(6), 1102–1113. 10.1111/tpj.14003 [DOI] [PubMed] [Google Scholar]
- Shabalin, A.A. (2012) Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics, 28(10), 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahan, R. , Zawora, C. , Wight, H. , Sittmann, J. , Wang, W. , Mount, S.M. et al. (2018) Consensus coexpression network analysis identifies key regulators of flower and fruit development in wild strawberry. Plant Physiology, 178(1), 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikha, M. , Kanika, A. , Rao, A.R. , Mallikarjuna, M.G. , Gupta, H.S. & Nepolean, T. (2017) Genomic selection for drought tolerance using genome‐wide SNPs in Maize. Frontiers in Plant Science, 8, 550. 10.3389/fpls.2017.00550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snedden, W.A. & Fromm, H. (2001) Calmodulin as a versatile calcium signal transducer in plants. New Phytologist, 151(1), 35–66. [DOI] [PubMed] [Google Scholar]
- Suliman, M. , Chateigner‐Boutin, A.‐l. , Francin‐Allami, M. , Partier, A. , Bouchet, B. , Salse, J. et al. (2013) Identification of glycosyltransferases involved in cell wall synthesis of wheat endosperm. Journal of Proteomics, 78, 508–521. [DOI] [PubMed] [Google Scholar]
- Sun, S. , Zhou, Y. , Chen, J. , Shi, J. , Zhao, H. , Zhao, H. et al. (2018) Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nature Genetics, 50(9), 1289–1295. 10.1038/s41588-018-0182-0. [DOI] [PubMed] [Google Scholar]
- Sun, X. , Cahill, J. , Van Hautegem, T. , Feys, K. , Whipple, C. , Novák, O. et al. (2017) Altered expression of maize PLASTOCHRON1 enhances biomass and seed yield by extending cell division duration. Nature Communications, 8(1), 10.1038/ncomms14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitashev, S. , Schwartz, C. , Lenderts, B. , Young, J.K. & Mark Cigan, A. (2016) Genome editing in maize directed by CRISPR–Cas9 ribonucleoprotein complexes. Nature Communications, 7(1), 13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson‐Wagner, R.A. , Decook, R. , Jia, Y. , Bancroft, T. , Ji, T. , Zhao, X. et al. (2009) Paternal dominance of trans‐eQTL influences gene expression patterns in maize hybrids. Science, 326(5956), 1118–1120. [DOI] [PubMed] [Google Scholar]
- Teng Y.‐S., Su Y.‐S., Chen L.‐J., Lee Y.J., Hwang I., & Li H.‐M. (2006) Tic21 is an essential translocon component for protein translocation across the chloroplast inner envelope membrane. The Plant Cell, 18(9), 2247–2257. 10.1105/tpc.106.044305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, P.D. , Campbell, M.J. , Kejariwal, A. , Mi, H. , Karlak, B. , Daverman, R. et al. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Research, 13(9), 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, F. , Bradbury, P.J. , Brown, P.J. , Hung, H. , Sun, Q. , Flint‐Garcia, S. et al. (2011) Genome‐wide association study of leaf architecture in the maize nested association mapping population. Nature Genetics, 43(2), 159–162. [DOI] [PubMed] [Google Scholar]
- Till, B.J. , Reynolds, S.H. , Weil, C. , Springer, N. , Burtner, C. , Young, K. et al. (2004) Discovery of induced point mutations in maize genes by TILLING. BMC Plant Biology, 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bel, M. , Diels, T. , Vancaester, E. , Kreft, L. , Botzki, A. , Van De Peer, Y. et al. (2018) PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Research, 46, D1190–D1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bel, M. , Proost, S. , Wischnitzki, E. , Movahedi, S. , Scheerlinck, C. , Van de Peer, Y. et al. (2012) Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiology, 158(2), 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney, R.K. , Singh, V.K. , Kumar, A. , Powell, W. & Sorrells, M.E. (2018) Can genomics deliver climate‐change ready crops? Current Opinion in Plant Biology, 45, 205–211. 10.1016/j.pbi.2018.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzi, F. , Del Fabbro, C. , Tomescu, A.I. & Policriti, A. (2012) rNA: a fast and accurate short reads numerical aligner. Bioinformatics, 28(1), 123–124. [DOI] [PubMed] [Google Scholar]
- Visscher, P.M. , Wray, N.R. , Zhang, Q. , Sklar, P. , McCarthy, M.I. , Brown, M.A. et al. (2017) 10 Years of GWAS Discovery: Biology, Function, and Translation. The American Journal of Human Genetics, 101(1), 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, J.G. , Bradbury, P.J. , Zhang, N. , Gibon, Y. , Stitt, M. & Buckler, E.S. (2014a) Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genetics, 10(12), e1004845. 10.1371/journal.pgen.1004845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, J.G. , Larsson, S.J. & Buckler, E.S. (2014b) Entering the second century of maize quantitative genetics. Heredity, 112(1), 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley, J.W. , Sartor, R.C. , Shen, Z. , Schmitz, R.J. , Wu, K.J. , Urich, M.A. et al. (2016) Integration of omic networks in a developmental atlas of maize. Science, 353, 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Liang, Q. , Li, K. , Hu, X. , Wu, Y. , Wang, H. et al. (2017) QTL analysis of ear leaf traits in maize (Zea mays L.) under different planting densities. The Crop Journal, 5(5), 387–395. 10.1016/j.cj.2017.05.001 [DOI] [Google Scholar]
- Wang, H. , Niu, Q.W. , Wu, H.W. , Liu, J. , Ye, J. , Yu, N. & et al. (2015) Analysis of non‐coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. The Plant Journal, 84(2), 404–416. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Chen, Q. , Wu, Y. , Lemmon, Z.H. , Xu, G. , Huang, C. et al. (2018) Genome‐wide analysis of transcriptional variability in a large maize‐teosinte a population. Molecular Plant, 11, 443–459. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Wang, H. , Liu, S. , Ferjani, A. , Li, J. , Yan, J. et al. (2016) Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nature Genetics, 48(10), 1233–1241. [DOI] [PubMed] [Google Scholar]
- Wu, L.F. , Hughes, T.R. , Davierwala, A.P. , Robinson, M.D. , Stoughton, R. & Altschuler, S.J. (2002) Large‐scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nature Genetics, 31(3), 255–265. [DOI] [PubMed] [Google Scholar]
- Xue, S. , Bradbury, P.J. , Casstevens, T. & Holland, J.B. (2016) Genetic architecture of domestication‐related traits in maize. Genetics, 204(1), 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, P. , Deng, R. , Huang, Y. , Stael, S. , Shi, J. , Shi, G. et al. (2019) Diverse biological effects of glycosyltransferase genes from Tartary buckwheat. BMC Plant Biology, 19(1), 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y. , Chen, H. , Hahn, M.G. , Mohnen, D. & Xu, Y. (2010) Evolution and function of the plant cell wall synthesis‐related glycosyltransferase family 8. Plant Physiology, 153(4), 1729–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Yu, Y. , Shi, T. , Kou, M. , Sun, J. , Xu, T. et al. (2020) Genome‐wide analysis of expression quantitative trait loci (eQTLs) reveals the regulatory architecture of gene expression variation in the storage roots of sweet potato. Horticulture Research, 7(1), 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Jin, Y. , Ma, Y. , Zhang, Q. , Wang, Q. , Jiang, N. et al. (2021) Identification of QTLs and candidate genes associated with leaf angle and leaf orientation value in maize (Zea mays L.) based on GBS. Tropical Plant Biology, 14(1), 34–49. [Google Scholar]
- Zhang, X. , Huang, C. , Wu, D.i. , Qiao, F. , Li, W. , Duan, L. et al. (2017) High‐throughput phenotyping and QTL mapping reveals the genetic architecture of maize plant growth. Plant Physiology, 173(3), 1554–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Fang, P. , Zhang, J. & Peng, Y. (2018) QTL mapping for six ear leaf architecture traits under water‐stressed and well‐watered conditions in maize (Zea mays L.). Plant Breeding, 137, 60–72. [Google Scholar]
- Zhao, Y. , Wang, K. , Wang, W.L. , Yin, T.T. , Dong, W.Q. & Xu, C.J. (2019) A high‐throughput SNP discovery strategy for RNA‐seq data. BMC Genomics, 20(1), 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski, R.E. (1998) Calmodulin and calmodulin‐binding proteins in plants. Annual Review of Plant Biology, 49(1), 697–725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pairwise LD measures, by chromosome.
Figure S2. Pearson correlation coefficients (PCCs) between traits and transcripts.
Figure S3. GWA scan of leaf traits.
Figure S4. Quantile–quantile plots for GWA scans.
Figure S5. Representative quantile–quantile plots for eQTL scans, with 1, 10, and 20 PCs as covariates.
Figure S6. Localization of PCC‐eQTLs and GWA‐eQTLs.
Figure S7. Characterization of eQTL associations by localization. (a) Number of SNPs that control each gene. (b) Distribution of genes regulated by local and/or distant SNPs.
Figure S8. (a) eQTL distribution and (b) eQTL effects, by phenotype.
Figure S9. Analysis of network topology for various soft‐thresholding powers.
Figure S10. Consensus clustering dendrograms of genes.
Figure S11. Relationship between network connectivity and eQTL genomic location.
Table S1. RNA sequencing statistics.
Table S2. Chromosome‐specific LD decay information.
Table S3. Phenotypic values of each individual.
Table S4. Correlation coefficients between gene expression levels and trait values.
Table S5. Permutation thresholds for Pearson correlation coefficients (PCCs).
Table S6. Output of the PCC scans for each trait.
Table S7. Complete GWA study results, with P‐value and effect for each tested marker.
Table S8. QTLs derived from marker–trait associations (MTAs).
Table S9. Expression QTLs derives from Pearson correlation coefficient analysis (PCC‐eQTLs).
Table S10. Expression QTLs derived from marker–trait associations (GWA‐eQTLs).
Table S11. Expression QTLs derived from Pearson correlation coefficient analysis (PCC‐eQTLs) and from marker–trait associations (GWA‐eQTLs).
Table S12. Summary statistics for PCC‐eQTL and GWA‐eQTL analysis.
Table S13. List of genes and consensus module.
Table S14. Outcome of the gene ontology (GO) enrichment analysis on candidate genes.
File S1. Outcome of the gene ontology (GO) enrichment analysis on PCC genes.
File S2. Outcome of the gene ontology (GO) enrichment analysis on genes for each putative module.
File S3. Functional information for candidate genes according to MaizeGDB databases.
Data Availability Statement
All relevant data can be found in online repositories. Raw RNA‐seq reads are stored in the European Nucleotide Archive (www.ebi.ac.uk/ena) database under the following accession numbers: ENA‐ERP009123 (parental lines), ENA‐ERP011069 (biparental RILs), and ENA‐ERP012784 (MAGIC maize RILs). Scripts are available at https://github.com/mdellh2o/maize_eQTL. Other data used for the experiments may be retrieved at Figshare (10.6084/m9.figshare.13702846).