

Abstract
Introduction
Genome‐wide association studies have led to numerous genetic loci associated with Alzheimer's disease (AD). Whole‐genome sequencing (WGS) now permits genome‐wide analyses to identify rare variants contributing to AD risk.
Methods
We performed single‐variant and spatial clustering–based testing on rare variants (minor allele frequency [MAF] ≤1%) in a family‐based WGS‐based association study of 2247 subjects from 605 multiplex AD families, followed by replication in 1669 unrelated individuals.
Results
We identified 13 new AD candidate loci that yielded consistent rare‐variant signals in discovery and replication cohorts (4 from single‐variant, 9 from spatial‐clustering), implicating these genes: FNBP1L, SEL1L, LINC00298, PRKCH, C15ORF41, C2CD3, KIF2A, APC, LHX9, NALCN, CTNNA2, SYTL3, and CLSTN2.
Discussion
Downstream analyses of these novel loci highlight synaptic function, in contrast to common AD‐associated variants, which implicate innate immunity and amyloid processing. These loci have not been associated previously with AD, emphasizing the ability of WGS to identify AD‐associated rare variants, particularly outside of the exome.
Keywords: Alzheimer's disease, family‐based association study, LOAD, neuronal development, rare variants, RVAS, synaptic function, whole‐genome sequencing
1. INTRODUCTION
Alzheimer's disease (AD) is the most common neurodegenerative disorder and one of the most challenging societal problems in the industrialized world. Susceptibility to AD is determined by both monogenic and polygenic risk factors as well as by environmental exposure. Monogenic AD most often presents as early onset (<60 years) familial AD (EOFAD), constituting less than 5% of all cases, and caused by any of hundreds of very rare mutations in at least three genes: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). Most AD cases are sporadic or familial late onset (>60 years) AD (LOAD) and are characterized by both a complex polygenic background and nongenetic factors. The identification of genetic determinants underlying polygenic AD has been the aim of more than 1000 genetic association studies, 1 including more than 75 genome‐wide association studies (GWAS) on AD and related traits as outcomes (according to European Bioinformatics Institute's (EBI) GWAS catalog). The largest AD GWAS 3 to date was conducted on over 600,000 individuals with an AD‐by‐proxy phenotype and highlighted a total of 29 independent genome‐wide significant (P < 5×10–8) AD risk loci. 4 Another recent GWAS by Kunkle et al. 5 found 25 loci in their analyses of clinically diagnosed LOAD in over 90,000 individuals. Essentially, these and other AD GWAS focused on common (typically with a minor allele frequency [MAF] ≥1%) variants either directly assayed or imputed using high‐density reference panels. The few exceptions to these common‐variant studies utilized either microarray‐based or next‐generation sequencing (NGS)–based genotyping limited to exonic variants and identified rare (MAF <1%) missense variants either increasing (TREM2, PLCG2, ABI3, ADAM10) or decreasing (APP, CD33) risk for AD. 6 , 7 , 8 , 9
In this study we used deep (> 40x) whole‐genome sequencing (WGS) to search for novel AD variants in 2247 individuals from 605 multiplex AD families from the National Institute on Mental Health (NIMH) 10 and National Institute on Aging (NIA) Alzheimer's Disease Sequencing Project (ADSP) 11 data sets. Analyses were focused on rare variants with MAF <1% (based on the non‐Finnish European subset of gnomAD v3.0, 12 unless stated otherwise) and entailed single‐variant and spatial clustering–derived (ie, “region‐based”) testing. Suggestive findings (P < 5×10–4) were validated in publicly available WGS (NIA ADSP case‐control population 13 ) data on more than 1650 independent AD cases and controls. In total, we highlight four single‐variant and nine region‐based findings exhibiting consistent rare‐variant association with AD across the discovery and replication phases in our study. None of the newly implicated loci were highlighted previously in any of the common‐variant AD GWAS. Functionally, our results extend existing knowledge on the underlying disease pathways highlighted by common variants and converge upon a role for neuroplasticity and synaptic function, emphasizing the power of WGS in the context of rare‐variant–based gene discovery efforts.
2. RESULTS
2.1. Description of general sequencing metrics
After sample quality control (QC) (see Methods), WGS data from 2247 individuals (NIMH n = 1393 and NIA ADSP families n = 854); hereafter referred to as the “discovery sample” (Supplementary Table 1; see Figure 1 for an overview of the study design) were available for subsequent analyses. Median read depth across the genome in NIMH was 40.4‐fold (mean 41.2). Within the discovery sample, we identified a total of 54,669,406 sequence variants, of which 40,542,616 were listed in the non‐Finnish European subset of the Genome Aggregation Database (gnomAD 12 ; v3.0, n = 32,399, Supplementary Figure 1). Of these, 907,273 (2%) were located in protein‐coding exons (Supplementary Figure 2). Of all identified variants, the vast majority, that is, 31,200,539 (77%) were “rare” (MAF ≤1%), whereas 2,855,054 (7%) were “low‐frequency” (≤5% MAF >1%), and 6,487,023 (16%) were “common” (MAF > 5%). Overall, we captured a large proportion of the “common” (95.8%) and “infrequent” (90.9%) variant space, using gnomAD as reference. As expected, the captured proportion was smaller for “rare” variants (11.7%), which can be attributed to the difference in sample sizes. After variant QC (Methods), 18,263,694 variants, 11,012,452, of which were rare, were used in subsequent analyses.
FIGURE 1.
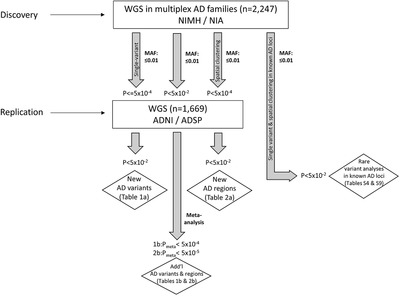
Data analysis workflow
2.2. Single‐variant AD association results
To probe for association between single markers and AD status, we used the FBAT Toolkit 14 in the family‐based discovery data set and logistic regression in the case‐control replication data (Methods). These analyses revealed a total of 24,301 rare variants showing association with AD at P < 0.01. As can be seen from the corresponding QQ plot (Figure 2), we observed a deflation of test statistics starting from P < 0.05. This deflation can be attributed to the fact that the FBAT statistics is conservative in the case of a small number of informative families and/or low allele frequencies. Of the variants showing association at P < 0.01, none have reached conventional genome‐wide significance (ie, P < 5×10–8). A total of 271 variants attained P < 5×10–4 (Supplementary Table 2) and were prioritized for validation assessments in the independent WGS case‐control data set (NIA ADSP non‐Hispanic whites (NHW), ntotal = 1669 (ncases = 983), hereafter referred to as “replication data set”; Figure 1). These assessments converged on two variants in two regions (rs74065194 ≈200 kb downstream from SEL1L [MAF = 0.0066; P meta = 0.011] and rs192471919 intronic of FNBP1L [MAF = 0.0054; P meta = 0.017]) to show at least nominal replication with the same direction of effect as in the discovery datasets (Figure 3a, Supplementary Figure 3‐4 , Table 1a). Notably, rs192471919 had a nominally significant association (P = 0.008), with same effect direction in Jansen et al. 3 where it was present only in the PGC‐ALZ cohort (n = 16,350). In addition, we highlight four variants that yielded P = 0.000538, that is, just above our screening threshold, located ≈100 kb downstream of STK31 (MAF = 0.0067; P meta = 0.0035) (Supplementary Figure 5).
FIGURE 2.
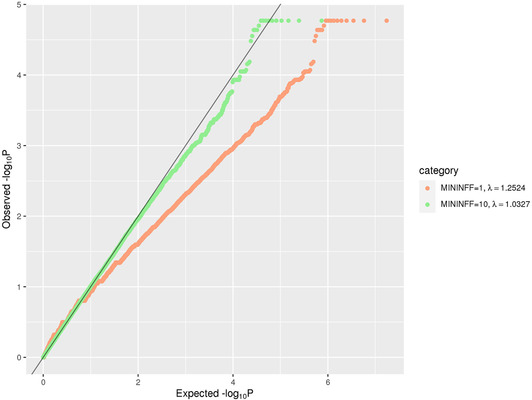
QQ plot of rare (MAF < = 1%) single‐variant association results in the family‐based discovery data set (NIMH and NIA cohorts). The red line corresponds to all statistics, where at least one informative family is observed. The green line corresponds to statistics with at least ten informative families
FIGURE 3.
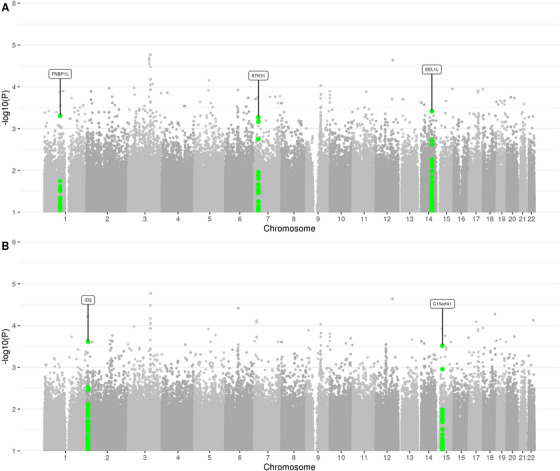
Manhattan plot of rare (MAF < = 1%) single‐variant association results in the family‐based discovery data set (NIMH and NIA cohorts). Genes that correspond to replicated variants as described in the workflow (Figure 1) are highlighted
TABLE 1.
Top single‐variant AD association results
| a | b | |||||
|---|---|---|---|---|---|---|
| Chromosome | 14 | 1 | 7 | 2 | 15 | |
| rsID (additional variants in LD showing the same results) | rs74065194 | rs192471919 | rs112941445 (rs112910644, rs111839960, rs113210961) | rs147918541 | rs141228575 (rs147002962) | |
| Nearest protein‐coding gene | SEL1L | FNBP1L | STK31 | LINC00298 | C15orf41 | |
| Allele frequency, non‐Finnish Europeans, gnomAD v3 | 0.0065 | 0.0054 | 0.0067 | 0.0072 | 0.0069 | |
| Effect allele | T | C | T | A | T | |
| Discovery dataset (NIMH + NIA families) | Z‐score | 3.551 | ‐3.485 | ‐3.461 | ‐1.98 | 2.048 |
| P‐value | 3.84E‐04 | 4.93E‐04 | 5.38E‐04 | 4.78E‐02 | 4.06E‐02 | |
| NINFF | 9 | 6 | 7 | 4 | 7 | |
| Replication dataset NHW ADSP | Z‐score | 2.275 | ‐2.182 | ‐2.698 | ‐3.576 | 3.488 |
| P‐value | 2.29E‐02 | 2.91E‐02 | 6.97E‐03 | 3.49E‐04 | 4.86E‐04 | |
| Sample size | 1669 | 1669 | 1668 | 1669 | 1669 | |
| Meta‐analysis | Effect direction | ++ | ‐‐ | ‐‐ | ‐‐ | ++ |
| Z‐score | 2.529 | ‐2.387 | ‐2.916 | ‐3.668 | 3.613 | |
| P‐value | 1.14E‐02 | 1.70E‐02 | 3.54E‐03 | 2.44E‐04 | 3.03E‐04 | |
| Jansen et al. | Z‐score | 1.134 | ‐2.656 | ‐0.319 | NA | ‐0.919 |
| P‐value | 2.57E‐01 | 7.92E‐03 | 7.50E‐01 | NA | 3.58E‐01 | |
| Kunkle et al. | Z‐score | 1.181 | NA | 1.589 | NA | ‐0.571 |
| P‐value | 2.37E‐01 | NA | 1.12E‐01 | NA | 5.68E‐01 | |
| UCSC | All mRNA | 0 | 13 | 3 | 7 | 6 |
| UCSC/ENCODE | TFBS clusters | 1 | 0 | 0 | 0 | 0 |
| GWAVA | DNase cluster | 0 | 0 | 9 | 0 | 1 |
| Ensembl | TFBS | 0 | 0 | 118 | 0 | 0 |
| GWAVA | GC content | 0.45 | 0.4 | 0.57 | 0.33 | 0.43 |
| Location | Upstream | Intronic | Downstream | Intronic | Intronic | |
| Mayo cohort | Expression in AD temporal cortex | No change | No change | Sig. up | Not tested | No change |
| Illumina bodyMap2 transcriptome | Tissue expression | Ubiquitous expression | Ubiquitous expression | Mostly expressed in testes | Mostly expressed in brain | Mostly expressed in heart |
| 3DSNP | Open chromatin | Fetal heart | Brain cingulate gyrus, liver cells and monocytes | 0 | Digestive tissue | 0 |
| Inferno | Closest enhancers cell type | CL:0000097_mast_cell | . | CL:0000047_neuronal_stem_cell | CL:0000576_monocyte,CL:0000775_neutrophil | CL:0000540_neuron,CL:0002620_skin_fibroblast |
Overlapping/GREAT‐assigned gene did not differ from the nearest gene assignment. For comparison, results from Jansen et al. (2019) and Kunkle et al. (2019) are included. NINFF ‐ number of informative families, TFBS ‐ transcription factor binding site.
a: Single‐variant AD association results with P < 0.0005 (P < 0.0006) in the discovery dataset and consistent (ie, P < 0.05 and same direction of effect) association in the ADSP NHW WGS replication dataset.
b: Single‐variant AD association results with consistent (ie, Pdiscovery < 0.05 and same direction of effect in discovery and replication dataset) association at P < 0.0005 after meta‐analysis.
RESEARCH IN CONTEXT
Systematic review: We performed an extensive literature review from PubMed and preprint servers, such as biorxiv and medrxiv. Previous work has established AD‐associated loci using either genotyping and imputation or whole exome sequencing. Few studies with whole genome sequencing data with limited sample sizes have been reported. This article is the first and, to the best of our knowledge, the currently largest systematic WGS‐based genetics study in the AD field.
Interpretation: We highlight 13 rare‐variant signals (4 from single‐variant, 9 from spatial‐clustering analyses) exhibiting association with AD across the discovery (families) and replication (case‐control) cohorts and related to synaptic function and neuronal development.
Future directions: These results should be confirmed in additional data sets and using biological validation. Additional research is needed in clarifying the role of rare variants in non‐coding regions. Currently, our results suggest different functional pathways (neuronal/synaptic) for rare variants, as compared to common‐variant functional assignments (microglial/innate immunity).
In a second filtering paradigm, we selected variants showing consistent (ie, P discovery < 0.05 and the same direction of effect in discovery and replication data sets) association at P < 0.0005 following meta‐analysis. This revealed three additional single variant associations in two loci (ie, rs147918541 intronic of LINC00298 and ≈700 kb upstream of ID2 (MAF = 0.0072; P meta = 2.44×10–4), and rs147002962 and rs141228575, both intronic of C15orf41 (MAF = 0.0069; P meta = 3.03×10–4); Figure 3B; Table 1b). Furthermore, we assessed the recently described 9 “exome‐chip”‐based rare‐variant genome‐wide and suggestive association signals (Supplementary Table 3). This revealed significant association with one of the two TREM2 variants (rs75932628 (MAF = 0.0021; P meta = 0.0329) as well as suggestive support for rs72824905 in PLCG2 in the discovery sample only (MAF = 0.0087; P discovery = 0.0546, P meta = 0.259). In contrast, we did not observe evidence for an association with the second TREM2 variant (rs143332484) or rs616338 in ABI3 in either the discovery or the replication samples. In addition, one HGFAC variant (rs114303452 (MAF = 0.012; Pdiscovery = 0.026, Pmeta = 0.7) from the extended set of suggestive variants, reported in Sims et al., was significant in our discovery sample only. Finally, we identified at least 786 nominally (P < 0.05) significant rare‐variant signals in genes corresponding to loci previously associated with AD in common‐variant GWAS 3 , 5 (Supplementary Table 4), suggesting that at least some of the common‐variant signals in these loci can be attributed to rare sequence variation (in line with earlier findings 15 , 16 ). For comparison, we also plotted single‐variant association results in the discovery cohorts without MAF restriction, that is, for both rare and common variants (Supplementary Figure 6 and Supplementary Fig. 7) and compared these with the 29 GWAS SNPs from Jansen et al. 3 (Supplementary Table 5) and 25 GWAS SNPs from Kunkle et al. 5 (Supplementary Table 6). As expected, these analyses revealed a pronounced, genome‐wide significant (P < 5×10–8) signal with markers in the apolipoprotein E (APOE) region on chromosome 19q13 as well as suggestive signals with several of the other common‐variant GWAS signals, such as BIN1, TREM2, CD2AP, PICALM, and ALPK2. In addition, we observed a borderline genome‐wide significant signal driven by low frequency variants in PTPRN2 (rs17837786: MAF = 0.043; P discovery = 1.36×10–7, P meta = 0.187); however, this signal was not replicated in the case‐control data set.
2.3. Spatial‐clustering AD‐association results
Our second analysis arm computed aggregated results on consecutive runs of rare variants in the discovery data set. In principle, this is similar to “gene‐based” testing (such as performed by VEGAS 17 or MAGMA 18 ) except the approach applied here 19 utilizes all available variants, including those located between genes that are otherwise typically omitted from this type of analysis, for example, VEGAS. These analyses revealed a total of 1756 regions showing an association with AD at P < 0.01; however, no region reached a Bonferroni threshold level of P < 2.15×10–7 (for a Manhattan and QQ plot of all spatial‐clustering‐based rare‐variant results see Figure 4 A and B and Figure 5). Using P < 5×10–4 as a suggestive threshold yielded signals in 47 regions in the discovery data sets (Supplementary Table 7), 4 of which also showed at least nominal evidence for independent replication in the NHW ADSP data set (PRKCH [P meta = 8.17×10–6], C2CD3 [P meta = 5.12×10–5], KIF2A [P meta = 1.00×10–4], APC [P meta = 1.79×10–4]; Table 2a, Supplementary Figure 8‐11). A further six (five of which were novel) candidate gene regions (PRKCH, LHX9, NALCN, CTNNA2, SYTL3, CLSTN) were highlighted in the secondary analyses focusing on top meta‐analysis results (P meta < 5×10–5 and P discovery < 0.05) only, yielding association signals with P‐values ranging from 3.27×10–5 to 8.17×10–6 (Table 2b).
FIGURE 4.
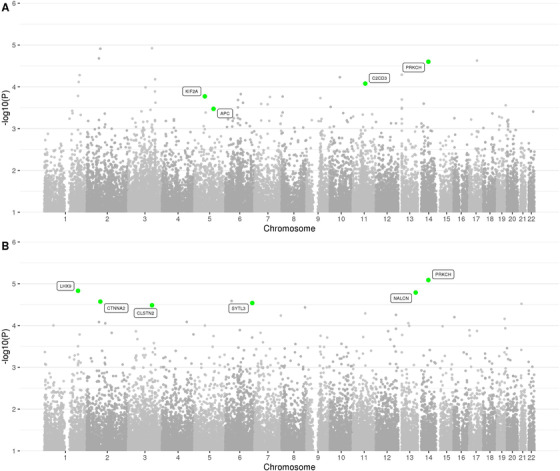
Manhattan plot of spatial clustering association results based on rare (MAF < = 1%) variants in the family‐based discovery data set (NIMH and NIA cohorts). Highlighted are genes, which correspond to replicated regions, described in the workflow (Figure 1)
FIGURE 5.
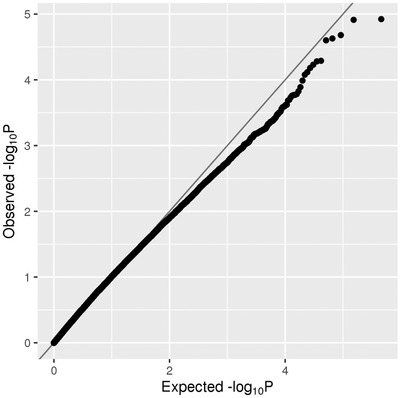
QQ plot of spatial clustering association results based on rare (MAF < = 1%) variants in the family‐based discovery data set (NIMH and NIA cohorts)
TABLE 2.
Top spatial clustering–based AD association results
| a | b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | 14 | 11 | 5 | 5 | 14 | 1 | 13 | 2 | 6 | 3 | |
| Position of the first SNV | 61176726 | 74025114 | 61972635 | 112819545 | 61176726 | 197917553 | 101143359 | 79853746 | 158677635 | 140398927 | |
| Position of the last SNV | 61188023 | 74031365 | 61975793 | 112827357 | 61188023 | 197929226 | 101164882 | 79856892 | 158683441 | 140449630 | |
| Nearest protein‐coding gene | PRKCH | C2CD3 | KIF2A | APC | PRKCH | LHX9 | NALCN | CTNNA2 | SYTL3 | CLSTN2 | |
| Discovery dataset (NIMH + NIA families) | Number of SNVs in the region | 53 | 47 | 13 | 25 | 53 | 44 | 106 | 15 | 29 | 233 |
| P‐value | 2.51E‐05 | 8.36E‐05 | 1.69E‐04 | 3.37E‐04 | 2.51E‐05 | 7.62E‐03 | 2.58E‐03 | 1.22E‐05 | 1.77E‐02 | 1.19E‐05 | |
| Replication dataset NHW ADSP | Number of SNVs in the region | 53 | 42 | 16 | 29 | 53 | 39 | 134 | 18 | 28 | 273 |
| P‐value | 2.11E‐02 | 4.54E‐02 | 4.65E‐02 | 4.37E‐02 | 2.11E‐02 | 1.31E‐04 | 4.27E‐04 | 1.54E‐01 | 1.16E‐04 | 1.96E‐01 | |
| Meta‐analysis | P‐value | 8.17E‐06 | 5.12E‐05 | 1.00E‐04 | 1.79E‐04 | 8.17E‐06 | 1.48E‐05 | 1.63E‐05 | 2.67E‐05 | 2.90E‐05 | 3.27E‐05 |
| Location | Upstream | Intronic and exonic | Upstream | Intronic and exonic | Upstream | Intronic and exonic | Intronic and exonic | Intronic | Intronic and exonic | Intronic and exonic | |
| Mayo | Expression in AD temporal cortex | Sig. lower | No change | No change | No change | Sig. lower | Not tested | No change | No change | Not tested | No change |
| Illumina bodyMap2 transcriptome | Tissue expression | Mostly expressed in WBC | Ubiquitous expression | Mostly expressed in WBC and brain | Mostly expressed in brain | Mostly expressed in WBC | Mostly expressed in testes | Mostly expressed in brain | Mostly expressed in brain | Ubiquitous expression | Mostly expressed in ovaries |
| FANTOM | TFBS | 86 | 0 | 0 | 0 | 86 | 429* | 0 | 0 | 0 | 0 |
| FANTOM | Transcription start sites | 6 | 3 | 0 | 5 | 6 | 24 | 2 | 0 | 0 | 9 |
| Ensembl | BindingMotifs | 14 | 0 | 0 | 0 | 14 | 1 | 2 | 1 | 0 | 7 |
| Ensembl | Active enhancer | 10 | 0 | 0 | 10 | 10 | 0 | 26 | 6 | 0 | 32* |
| Ensembl | Open chromatin region active | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 2 |
| Ensembl | Active promoter | 61* | 15 | 0 | 0 | 61* | 104 | 0 | 0 | 0 | 57 |
| Ensembl | Active TFBS | 0 | 0 | 0 | 0 | 0 | 15* | 19* | 0 | 0 | 0 |
| Ensembl | CTCF binding site active | 9 | 2 | 0 | 0 | 9 | 31 | 73* | 19 | 0 | 16 |
| UCSC | Cell‐specific TFBS | 81 | 12 | 4 | 15 | 81 | 117 | 31 | 7 | 6 | 40 |
| UCSC | CpG islands | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 |
| UCSC | DNase cluster | 17 | 8 | 1 | 5 | 17 | 15 | 18 | 3 | 3 | 44 |
The PRKCH region was identified in both arms of the regional study, hence appears in both: a and b. Overlapping/GREAT‐assigned gene did not differ from the nearest gene assignment. SNV ‐ single nucleotide variant, TFBS ‐ transcription factor binding site, WBC ‐ white blood cells. *Significantly more annotations than expected by chance after correcting for multiple testing.
a: Replicated spatial clustering–based AD association results for regions showing P < 0.0005 in the discovery data set.
b: Top spatial clustering–based AD association results based on meta‐analysis (P meta < 5×10‐5 and P discovery < 0.05).
Next, using our discovery WGS data set, we checked recently described burden rare variant associations with AD from a large WES study. 20 By implementing a similar filtering scheme in exons and splitting the case‐control phenotype into three categories (Methods), we identified nominally significant associations in ABCA7 (P = 0.016), TREM2 (P = 0.025) and SORL1 (P = 0.039) (Supplementary Table 8). We also note that many damaging rare variants were absent in our data set due to the lower sample size.
Finally, we also performed gene‐based burden testing on rare variants in known AD genes (ie, APP, PSEN1, PSEN2 as well as those recently highlighted as genome‐wide significant loci in GWAS (Jansen et al. 3 and Kunkle et al. 5 ). This revealed two nominally significant association signals in ZCWPW1 (P = 0.028) and PICALM (P = 0.03), and two suggestive association signals in ALPK2 (P = 0.053) and MS4A6A (P = 0.084), upon meta‐analysis (Supplementary Table 9).
For comparison, we also plotted spatial clustering–based association results without MAF restriction in the discovery cohorts, that is, for both rare and common variants and, as expected, the top‐associated region in these analyses maps to the APOE locus on chromosome 19q13.32 (Supplementary Figure 12 and 13, and Supplementary Table 10).
Taken together, our WGS‐based association results revealed 13 novel potential AD loci (4 from single‐variant, 9 from spatial‐clustering analyses) with consistent rare‐variant signals in both discovery and replication cohorts. It is important to note that none of the identified loci have been highlighted previously in any common variant or WES/exome‐chip association study in the field, emphasizing the added resolution and power afforded by genome‐wide sequencing performed outside coding regions. Notwithstanding, some of the loci highlighted here may reflect spurious associations due to type I error; thus any future consideration of our results should await further validation in independent samples.
2.4. In silico functional implications of the single‐variant association findings
The leading single nucleotide variant (SNV) associations from the discovery (Table 1a) and the meta‐analysis (Table 1b) include rs74065194, which is upstream of SEL1L, located within a transcription factor–binding site cluster. The SNV rs192471919 is situated within the intron of FNBP1L and open chromatin specific to the brain cingulate gyrus, liver cells, and monocytes. Three SNVs are intronic to LINC00298 (rs147918541), a long non‐coding RNA gene expressed mostly in brain, and C15orf41 (rs147002962; rs141228575) which is expressed mostly in heart. The four variants assigned to STK31, which almost reach our P‐value threshold, show significantly higher expression in the temporal cortex of AD patient samples when compared to controls (P adj. = 1.1×10–5). Of these SNVs, rs112941445 is the most likely to be the causal variant given that it has the most epigenetic support (Table 1a).
The most highly significant SNV‐associated meta‐analysis gene was LINC00298. This long intergenic non‐coding RNA (lincRNA) has no known function. Of the 73 rare variant–associated genes co‐expressed with LINC00298 in our study, 17 are included in protein‐protein interactions with our newly AD‐associated genes, including APC (corr 0.449) and CTNNA2 (corr 0.381) (Figure 6), and also the known AD and frontal lobe dementia–associated gene encoding tau protein, MAPT (corr 0.379). Functional enrichment for LINC00298‐correlated expression of genes found in our study results in one significant enrichment for the HIPPO signaling pathway (P adj. = 2.2×10–7; Supplementary Table 11) and weaker correction‐adjusted significance for GO processes synapse organization, spindle formation, cell‐cell adhesion, and neuron projection morphogenesis.
FIGURE 6.
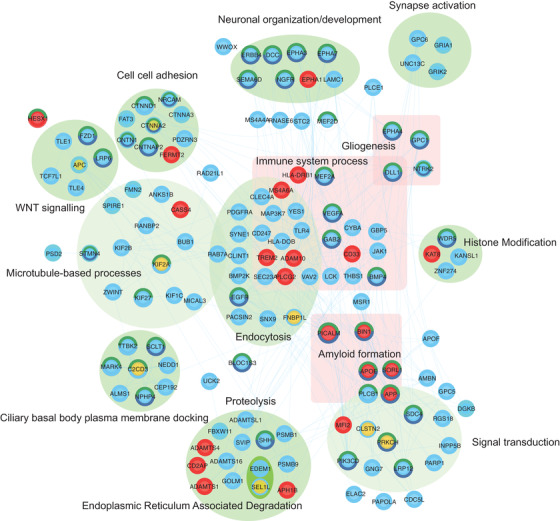
Network of direct interactions between highly ranked SNV and regional genes and known AD‐associated genes. Direct protein‐protein relationships (blue links) between reference AD genes (red), Table 1 and Table 2 (yellow), Supplementary Table 12 and 15 (blue) protein‐coding genes. LINC00298 co‐regulated expression of directly interacting genes is highlighted (turquoise border). Proteins that are in direct interaction with genes from Tables 1 and 2 have been grouped where possible according to shared GO biological processes (green ellipse). Proteins that may not be directly interacting but are found commonly enriched in immune‐related processes are grouped (pink square). Proteins with dark green–colored borders are enriched in GO:BP nervous system development, whereas a navy blue border is enriched for generation of neurons. Gene‐gene relationships are listed in Supplementary Table 24. The network can be interactively explored via the NDEX project website 106
Functional enrichment of the genes associated with the highest‐ranked 1000 SNVs from the meta‐analysis (Supplementary Table 12) identified 151 processes and pathways after correcting for multiple testing. The most highly enriched terms included flavonoid glucuronidation (P adj. = 1.09×10–7) (involved in removal of xenobiotics), and many neuroplastic/developmental‐associated processes including synapse organization (P adj. = 1.32×10–7), axon guidance (P adj. = 6.51×10–6), development and elongation, and also cell adhesion (P adj. = 0.001; Supplementary Table 13). Only two pathways were significantly co‐enriched with the GO/pathway gene set enrichment for genes associated with common variants reported in the GWAS by Jansen et al.3: cell adhesion molecules and herpes simplex infection (Supplementary Table 14). In contrast to the broad diversity of functions, such as immune‐related and amyloid processing, found to be enriched by genes annotated in the GWAS by Jansen et al., 3 10 of the 21 top‐level functions showing enrichment in our rare‐variant analysis had roles related to the maintenance and development of neurons, cardiac tissue and synapses, and neuroplasticity‐related terms including synaptogenesis, activity and synaptic integrity, neurogenesis, sensory organ development, cardiac development, tissue morphogenesis, and limb development. None of the enriched pathways, here, exhibited amyloid or immune‐related roles.
2.5. In silico functional implications of the spatial‐clustering association findings
Four of the nine leading regions associated with AD are significantly enriched for regulatory annotation (Table 2). The CLSTN2 and PRKCH regions are respectively enriched for enhancers and promoters across a number of cell types, whereas the LHX9 and NALCN loci significantly overlap with transcription factor binding sites. NALCN additionally is enriched for active CTCF binding sites. Unlike the SNVs, these nine regions mostly cover intronic and exonic locations. The four genes APC, CTNNA2, KIF2A, and NALCN are all primarily expressed in brain tissue, whereas PRKCH expression is significantly reduced in the temporal cortex of patients with AD (P adj. = 0.0001).
Functional enrichment of genes associated with the highest‐ranked 1000 spatial clustering–based results (Supplementary Table 15) revealed 127 significantly enriched pathways after correcting for multiple testing. The most highly enriched terms included neuron projection guidance (P adj. = 1.6×10–5), kidney development (P adj. = 2.32×10–5), cell‐cell adhesion (P adj. = 6.53×10–5), negative chemotaxis (P adj. = 2.17×10–4), brain development (P adj. = 4.23×10–4), and synapse organization (P adj. = 7.02×10–4). Seven of the 20 most enriched terms were related to development, and 81 of the total significantly 127 enriched terms were related to development or neuroplasticity (Supplementary Table 16). Meanwhile, no process of 422 was significantly enriched in common with the study of Jansen et al. 3 Protein localization to membrane (P adj. = 0.0126, Jansen, P adj. = 0.0631, regional gene set; Supplementary Table 17) was the closest to reaching significance.
2.6. Common functional themes between single‐variant and regional analysis
A total of 90 genes were found in common between the most highly ranked 1000 single‐variant and regional findings. These include the three highlighted genes LINC00298, SEL1L, and STK31 and genes that rank highly in both gene lists: ROBO1, PRDM9, LINC02439, and TMEM132C. One hundred fifty‐two processes and pathways reached significance for the co‐enrichment of regional‐ and SNV‐associated genes. The top five terms enriched were positive regulation of nervous system development (P adj. = 0.0025, SNV; P adj. = 0.000079, regional), heart development (P adj. = 0.0015, SNV; P adj. = 0.0039, regional), sensory organ development (P adj. = 0.0005, SNV; P adj. = 0.01, regional), trans‐synaptic signaling (P adj. = 0.0015, SNV; P adj. = 0.0031 regional), and tissue morphogenesis (P adj. = 0.002, SNV; P adj. = 7×10–5 regional). Of the 19 significantly co‐enriched terms, 10 were related to development or neuroplasticity; the remainder addressed maintenance and cellular activity–related functions such as cell‐cell adhesion, negative chemotaxis, signaling by receptor tyrosine kinases, and organelle localization (Supplementary Table 18; Supplementary Figure 14).
To investigate the impact of selecting only variants within transcribed gene boundaries on the functional enrichment results, we restricted our analysis to only those SNVs and regions occurring within a gene transcript, that is, intronic or exonic variants. The most highly enriched categories (P adj. < = 0.035) included organelle localization, cell‐cell adhesion, cell morphogenesis in neuron differentiation, synapse organization, modulation of chemical transmission, and protein localization to the centrosome (Supplementary Figure 15).
2.7. Identification of cell‐specific signatures
To assess whether our prioritized variants show an association with single cell–restricted states, we applied an Expression Weighted Cell Type Enrichment (EWCE) test 21 to genes from our prioritized SNV and regional analysis results. EWCE is used to predict the primary cell origins of a disease. Using single‐cell mouse data, primarily from the hippocampus and hypothalamus, we discovered an enriched signal of our SNVs in pyramidal CA1 neurons (Supplementary Table 19). In contrast, common loci associated with AD 22 have been enriched significantly in microglia (Figure 7).
FIGURE 7.
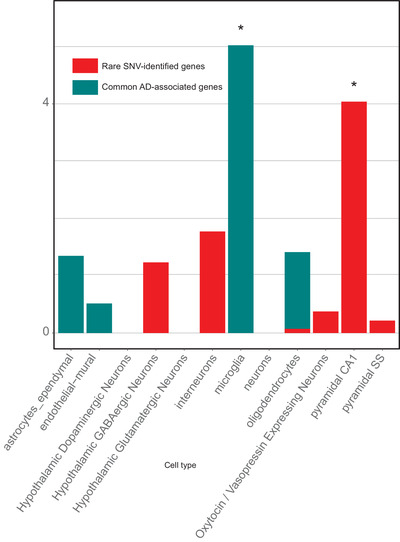
Cell‐specific enrichment results from the EWCE tool. We compared genes identified in our rare‐variant analysis to common variants published in AD 22 and which cell type each is significantly enriched in. Zero represents the mean expression in each cell based on 10,000 permutations of gene lists of the same size. The data for this figure can be found in Supplementary Table 19
2.8. Network generation of shared functions and relationships with known AD‐associated genes and processes
Using known protein‐protein interactions as a guide, a network of interactions was constructed between a total of 1274 interacting proteins, which include known AD‐associated genes, 22 our single‐variant, and regional‐associated genes. Of the 14 leading genes we pinpointed in this study, 8 (protein‐coding) were linked directly by protein‐protein interaction to additional AD‐associated genes discovered within this study or to 21 known AD‐associated genes in a subnetwork (Figure 6). Highlighted genes that interact with known AD genes include FNBP1L, which interacts directly with the validated GWAS AD genes, PICALM and BIN1, as well as KIF2A, which interacts directly with the AD gene HLA‐DRB1. Seventeen genes in the subnetwork also co‐express with the highlighted gene, LINC00298.
Functional enrichment of the subnetwork of directly interacting proteins revealed 196 enriched GO process/KEGG pathway terms (Supplementary Table 20). The three highest ranked GO processes (nervous system development, 236 genes, FDR 1.32×10–9; neurogenesis 168 genes, FDR 4.74×10–7; developmental process, 460 genes, FDR 3.74×10–7) reflected neuroplasticity/developmental processes of 90 processes enriched for development, differentiation, or biogenesis. Neurogenesis, a GO process term that annotates 1519 genes, was co‐enriched with PRKCH, LHX9, and CTNNA2 from our pinpointed genes, and SORL1, PICALM, CNTNAP2 and APOE, and BIN1 from our reference list of known AD genes (PICALM is a known AD gene also discovered in our top 1000 regional analysis‐associated genes). Co‐expression analysis using pathway co‐activation mapping (PCXN 23 ) revealed that nervous system development and several of the associated enriched GO terms show significant correlated gene expression activity, even when there was low gene overlap between enriched term gene sets (Supplementary Table 21).
3. DISCUSSION
Based on WGS of 2247 subjects from 605 multiplex AD families and a case‐control cohort of >1650 individuals, we have identified 13 rare‐variant signals (four from single‐variant, nine from spatial‐clustering analyses) exhibiting association with AD across the discovery (families) and replication (case‐control) cohorts. Our work represents one of the first and, to the best of our knowledge, the currently largest, systematic WGS‐based genetics study in the AD field. In AD, we are only aware of two published WGS‐based studies, 24 , 25 both utilizing different analyses paradigms and much smaller sample sizes. Of note, data from the latter of these WGS projects were utilized in the current study for purposes of independent replication.
The top signals emerging from our single variant–associated analyses were associated with the genes FNBP1L and SEL1L (and STK31), whereas the secondary analysis pointed to LINC00298 and C15orf41. All genes directly overlapped with the single variant associations except for SEL1L, which encodes the suppressor/enhancer of lin‐12‐like (Sel1L) adaptor protein for an E3 ligase involved in endoplasmic reticulum‐associated degradation (ERAD) for protein quality control. Of interest, ERAD has been reported to regulate the generation of amyloid beta (Aβ) by gamma secretase. 26 Deficiency of SEL1L has also been shown to activate ER stress and promote cell death. 27 In addition, an SNV in intron 3 of SEL1L has previously been reported to confer susceptibility to AD. 28
The FNBP1L gene, which encodes the formin‐binding protein 1‐like protein, has been associated with adult 29 and childhood intelligence. 30 FNBP1L has also been reported to be essential for autophagy of intracellular pathogens, such as Salmonella typhimurium, which serves to curb intracellular growth. 31 This is particularly interesting given the emerging evidence for the role of microbes in driving AD neuropathology. 32 FNBP1L, also known as TOCA‐1, is implicated in neurite elongation and axonal branching. 33 Thus FNBPL1 may play a role in neuroplasticity‐related AD pathology.
The STK31 gene encodes the cell cycle kinase, serine/threonine kinase 31, which is known to promote PDCD5‐mediated apoptosis in p53‐dependent human colon cancer cells. 34 It is tempting to speculate as to whether this kinase might also affect phosphorylation of tau and neurofibrillary tangle formation in AD. However, we note that variants in this gene technically did not fulfill the significant thresholds and are highlighted here as additional results.
LINC00298 is a long intergenic non‐coding RNA and does not code for a protein. Its functional role is not known, 35 but it exhibits CNS‐specific expression, with a 50‐fold and 24‐fold enrichment in the nervous system and brain samples in FANTOM 5 CAT (P = 2.9×10‐23 and 4.6×10‐21, respectively). 36 It contains an experimentally supported target for the brain‐expressed non‐coding miRNA mir‐7, 37 which has been associated with AD and other brain diseases. 38 , 39 LINC00298 can be more broadly functionally characterized by where and when it is expressed and the genes with which its expression is correlated. LINC00298 is co‐expressed with 33 SNV‐associated, and 40 regional‐associated genes (lnchub 40 ). LINC00298's co‐expressed genes appear to be enriched for developmentally associated processes: Its bias for expression in the brain; its association with HIPPO pathway, which has a role in development; co‐expression with genes involved in neuronal differentiation; and expression in neuronal induced pluripotent stem cells (iPSC) suggest that one of its roles may be in regulation involved in neuronal plasticity. C15orf41 encodes the codanin 1‐Interacting nuclease gene (CDIN1), which is highly expressed in the heart, with much lower expression in the brain. CDIN1 is associated with erythrocyte differentiation and has genetic associations with congenital dyserythropoietic anemia type I. 41
Spatial clustering–based analyses highlighted a total of four independent genomic regions (Table 2a). One of these regions was in the gene encoding the protein kinase C receptor eta subunit (PRKCH). Of interest, we have previously reported three highly penetrant rare mutations in another protein kinase C subunit alpha (PRKCA) that segregates with AD in five families. All three AD‐linked PRKCA mutations displayed increased catalytic activity (by live imaging) versus wild‐type PRKCA, and potentiated the ability of Aβ to suppress synaptic activity in hippocampal slices. 42 It will be interesting to determine whether mutations in PRKCH have similar aberrant effects on receptor activity.
The three other genes implicated in the spatial clustering–based analyses included C2CD3, which encodes the C2 domain containing 3 centriole elongation regulator that is expressed at relatively high levels in the brain. Mutations in human C2CD3 cause skeletal dysplasia, caused by defective assembly of the primary cilium, a microtubule‐based cellular organelle involved in developmental signaling. 43 KIF2A encodes the kinesin family member 2A, which is required for normal mitotic spindle activity and normal brain development, most likely via its ATP dependent MT‐depolymerase activity. 44 Like C2CD3, KIF2A has also been implicated to affect ciliogenesis, relating to its role in the cell cycle. KIF2A‐related cortical development defects have been attributed to decoupling between ciliogenesis and cell cycle. 44 A KIF2A His321Asp missense mutation was identified in a subject with defective cortical development owing to impairment of KIF2A microtubule depolymerase activity. 45 Several members of the kinesin family are overexpressed in the brains of AD patients, 46 and KIF2A expression is specifically upregulated in axons, spinal neurons, and oligodendrocytes adjacent to spinal cord injuries 47 Finally, APC encodes the Adenomatosis Polyposis Coli Regulator of WNT Signaling Pathway (as a negative regulator) and serves as a major tumor suppressor. The WNT signaling pathway plays an important role in the development of the central nervous system, including axonal pathfinding and synaptic plasticity, and has been linked to AD pathogenesis. 48 Aβ neurotoxicity in AD has been reported to downregulate WNT signaling, 49 and WNT signaling, in turn, has been shown to regulate β‐secretase cleavage of APP. 50 Collectively, these findings indicate that inhibition of WNT signaling may play a role in the generation and neurotoxicity of Aβ. Thus APC may influence AD neuropathogenesis via regulation of the WNT signaling pathway.
In addition to these four loci, an additional five candidate regions were identified in the secondary analyses, based on the top meta‐analysis results (P < 5×10–5; Table 2b). These included LHX9, NALCN, CTNNA2, SYTL3, and CLSTN2. LHX9 is a LIM homeobox gene family member and is involved in the development of the forebrain. 51 This gene has also exhibited genetic association with “self‐reported educational attainment.” 52 NALCN encodes a voltage‐gated sodium and calcium channel that is expressed in neurons. Of interest, the calcium‐sensing receptor, CaSR, which has been reported to regulate NALCN, has been previously implicated as an important signaling molecule in AD. 53 CTNNA2 encodes the neural version of α‐catenin (αN‐catenin), a mechano‐sensing protein that links cadherins with the cytoskeleton; as such, they are required for proper neuronal migration and neuritic outgrowth. 54 SYTL3 encodes the Rab effector protein, synaptotagmin‐like 3, which plays a role in vesicle trafficking, 55 and has been genetically associated with lipoprotein (a) levels. 56 CLSTN2 encodes Calsyntenin 2, which modulates calcium‐mediated postsynaptic signaling in the brain. Absence of CLSTN2 impairs synaptic complexes in mice, 57 and has been associated with episodic memory function in human subjects. 58
Pathway analyses based on our highlighted rare variant–associated genes, emphasize functional roles in neuroplasticity, synaptic function and integrity, axonal maintenance, neuronal development, and heart tissue development. In contrast, genes identified through common‐variant associations by GWAS have been more involved with pathways linked to immune‐system response, lipid metabolism, and Aβ deposition. This stark difference in enrichment profiles may represent an essential contribution of rare variants to the development of AD based more on neuronal and synaptic function. This finding is further substantiated by examining our SNV‐associated genes and published common AD‐associated genes for cell‐specific biases in expression. We found that hippocampal CA1 neurons were significantly enriched for our rare signature, whereas common genes from AD GWAS have primarily highlighted microglia as the likely primary cell type of effect (Figure 7). Synaptic loss and disruption of neuronal plasticity is considered as an early event in AD pathology. 59 A recent study proposing tau pathology as one of the initial factors in LOAD, suggests that selected pyramidal cells are particularly vulnerable to calcium signaling disruption. 60 Aβ can directly affect synaptic integrity and also alter calcium homeostasis. Our findings suggest that the rare‐variant genetic profile could trigger an early stage calcium dysregulation event in line with the Calcium Hypothesis. 61
Using whole‐genome sequencing, we have performed a whole‐genome global screen to search for association of rare variants with AD. It is noteworthy that our most significantly SNV‐associated gene, LINC00298, is non‐coding and of unknown function. Furthermore, all nine regions of the genome we have identified to be associated with AD risk, overlap with regulatory annotations, of which four are significantly enriched. Thus our study emphasizes the importance of focusing on the non‐coding part of the genome for a better understanding of the genetic and functional basis of Alzheimer's disease.
The methodologies applied and the results obtained are not without limitations. First and foremost, we note that the size (n∼2300) of the discovery sample is relatively small compared to common‐variant GWAS in the field. This is due to the limited availability of samples (ie., multiplex AD families) and funds (ie, costs for generating WGS data are still 1‐2 orders of magnitude higher than for common‐variant GWAS, which rely on microarray‐based genotype calls). This comes at the price of reduced statistical power (Supplementary Table 22), which we addressed by adjusting the discovery and meta‐analysis significance thresholds. As a result, our top findings show P‐values ranging between ≈0.01 and ≈8×10–6, which is still almost two orders of magnitude above a recommended threshold (P < 1×10–8) for rare variant–based studies in European‐based samples. 62 We carefully selected suggestive P‐value thresholds to balance our ability to detect novel rare variants while keeping the false‐positive rate under control. We tried to alleviate the limitation of low discovery power by utilizing validation data from an independent case‐control WGS data set (NIA ADSP), but all of the main findings highlighted here should be considered preliminary until validated in additional data sets. Eventually, only the generation and analysis of additional data sets, specifically WGS, investigating these and other rare variants in relation to AD susceptibility will allow us to distinguish true from false‐positive findings.
Second, most variants highlighted to be associated with AD risk in our analyses are located in non‐coding regions of the genome. Although this is to be expected given the proportions of coding (∼2%) versus non‐coding (≈98%) sequence variation in humans, it aggravates efforts to validate and functionally annotate our top findings. However, efforts like ENCODE, 63 the NIH Epigenomics Roadmap Consortium, 64 or the International Human Epigenome Consortium 65 continue to provide compelling evidence that an increasing fraction of disease‐associated variation maps to the regions between genes, providing a strong argument for using whole‐genome in addition to whole‐exome approaches to capture the full rare‐variant architecture underlying AD.
Finally, unlike genetic association analyses in case‐control settings, our family‐based approach is robust against common genetic confounders due to population substructure. However, given the fact that more than 80% of our discovery family–based sample were individuals of European ancestry, we limited our replication sample to individuals of the same ancestry. Family‐based and case‐control data sets consisting of subjects with non‐European ancestry are smaller and likely more diverse. In addition, effect sizes might vary among different populations. Thus future efforts are necessary to expand AD WGS sequencing and analysis in samples of non‐European ancestries.
In summary, here, we describe the first WGS‐based rare‐variant association study in AD, and highlight several novel variants and regions found to be associated with disease risk. Subsequent functional annotation assessments imply several molecular pathways to be relevant in AD based on rare variant analysis, for example, neuronal development and synaptic integrity. This contrasts with innate immune, amyloid, and lipid pathways previously implicated by network analyses of AD GWAS based on common variants. Together with the results of common‐variant AD risk GWAS, our study highlights several novel promising routes of AD research and provides new potential targets for therapeutic interventions aimed at the early treatment or prevention of AD.
4. METHODS
4.1. Sample descriptions
The discovery cohort was composed of two WGS familial cohorts with 1393 (NIMH; AD: n = 966) and 854 (NIA ADSP families; AD: n = 543) individuals. A subject was considered to be affected if he/she was included in these categories: “definite AD,” “probable AD,” or “possible AD.” Unaffected subjects were taken from one of the following categories: no dementia (667 subjects), suspected dementia (46 subjects) or non‐AD dementia (10 subjects). It is important to note that NIA ADSP families by design did not include individuals with two APOE ε4 alleles. Because our discovery cohort consisted mostly of individuals of European ancestry, we used a matching subset (non‐Hispanic whites [NHW]) from the replication cohort (NIA ADSP unrelated, n = 1669). A total of 564 individuals (AD: n = 307) were obtained with RNA‐Seq data in the temporal cortex from Mayo Clinic Alzheimer's Disease Genetics Studies (MCADGS 66 ). All data sets are described in Supplementary Table 1.
4.2. Whole‐genome sequencing methods
Plated DNA was obtained from the Rutgers Cell Repository and sent to Illumina Inc (San Diego, CA, USA) and used to create short‐insert paired‐end libraries. Paired‐end libraries are manually generated from 500 ng to 1 ug of gDNA using the Illumina TruSeq DNA Sample Preparation Kit. Samples are fragmented and libraries were size‐selected targeting 300 bp inserts and sequenced using the HiSeq 2000 System. Illumina‐provided BAM files were re‐aligned to the human reference genome (GRh38) with bwa‐mem 67 (v0.7.7, default parameters). Reads were marked for duplication using samtools 68 (v0.1.19). Germline variants were jointly called for each family using FreeBayes 69 (v0.9.9.2‐18) and GATK 70 (v3.0) best practices method 71 as part of the bcbio‐nextgen workflow 72 before being squared‐off with bcbio.recall 73 across the whole cohort to distinguish reference calls from no variant calls. Library and read quality was assessed using FastQC (v0.10.1 74 and Qualimap 75 (v0.7.1). Variant calls in vcf format for the NIA ADSP cohort were obtained from the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS) under accession number NG00067.
4.3. Quality control of WGS‐derived variant calls
We first performed individual‐based quality control. Based on genotyping rate and inbreeding coefficient, we removed three outliers in the NIMH data set. Further 12 duplicates and 24 individuals with wrong family assignments, as per estimated identity by descent (IBD) sharing, were removed as well (Supplementary Table 23). One thousand three hundred ninety‐three clean individuals from NIMH were combined with 854 individuals from NIA and analysis was performed only on variants present in both data sets. This was done to ensure a consistent discovery data set for region‐based rare‐variant analysis. Next, family‐based discovery data sets were filtered for monomorphic variants, singletons, variants with a missingness rate higher than 5%, Mendelian errors, and variants that had a Hardy‐Weinberg equilibrium P < 1×10–8. Only variants that had a filter “PASS” in the vcf file were included in the analysis.
In the case‐control replication data sets, variant‐based filtering was performed as in family‐based data sets, that is, monomorphic variants, singletons, variants with a missingness rate higher than 5%, and variants that had a Hardy‐Weinberg equilibrium P < 1×10–8 were excluded. Only variants that had a filter “PASS” in the vcf file were included in the analysis. We kept only unrelated individuals of European ancestry in order to closely match our discovery dataset population. Principal components were calculated based on rare variants using the Jaccard index. 76 Outliers based on principal components were excluded.
4.4. External minor allele frequency reference dataset (gnomAD)
We have downloaded v3.0 of the Genome Aggregation database (gnomAD), 12 which included 71,702 whole genomes (32,399 non‐Finnish European). For minor allele frequency (or MAF) we used the AF NFE field, which corresponds to allele frequency in the non‐Finnish European population. Variants were considered rare if AF NFE was less than 1% or more than 99%.
4.5. Single‐variant association analyses
In the family‐based discovery data sets we used the FBAT Toolkit 14 to perform association analysis on variants seen in at least one informative family in combined NIMH/NIA data set. We used an offset of 0.15, which corresponds approximately to the population prevalence of disease. Although sample preparation and sequencing for NIMH and NIA families were performed at multiple centers, members of the same family were always sequenced at the same center, which minimizes the impact of batch effects on the FBAT approach. QQ plots (Figures 2 and 5) show no evidence of batch effects leading to inconsistent Mendelian transmission patterns.
In the case‐control replication data sets we performed a logistic regression (with option “firth‐fallback”) for case/control status as implemented in PLINK 2. 77 We included sex, age, sequencing center, and 5 Jaccard principal components 76 with standardized variance as covariates. We next performed a fixed‐effects meta‐analysis of two data sets. The meta‐analysis was performed with the METAL toolkit 78 with a sample‐size–based weighting scheme. Quantile‐quantile plots were drawn in R for all results and for variants with at least 10 informative families.
4.6. Spatial‐clustering/region‐based association analyses
In the family‐based discovery data set, we systematically grouped the whole‐genome sequencing data into non‐overlapping regions using a spatial‐clustering approach. Briefly, variants are grouped together into regions assuming an inhomogeneous Poisson process based on the physical positions of single variants and include only those that are in proximity to one another. We included only variants seen in at least two families. After we partitioned the chromosomes into non‐overlapping windows, there were 232,188 regions with a mean number of 45 variants in each region (median = 22). We ran FBAT‐RV, 79 a multimarker test with MAF weighting, which was used to test identified non‐overlapping regions in the combined family‐based data set. First, only rare variants were included in the analysis. Next, we performed a second run including all variants.
In the case‐control replication data sets, joint variant testing was performed on rare variants using the burden test as implemented in the SKAT package. 80 We next used SKAT‐RC 81 to incorporate all variants with no MAF threshold. We used the same set of covariates as in the single‐variant analysis. For consistency, we tested the same non‐overlapping regions, which were identified in the combined NIMH/NIA data set. This allowed us to perform a meta‐analysis of the identified regions, using Fisher's combined probability test.
4.7. Power calculations
We have calculated power to detect a significant association of a variant with a MAF of 0.01 for a range of effect sizes (Supplementary Table 22). Alpha level was set to 0.0005, which corresponds to our discovery P‐value threshold, and prevalence was set to 0.15. We used PBAT 82 to estimate power in a family‐based study design and assumed 605 families with two affected and one unaffected offspring and missing parents, which approximates the structure in the real data set. We used the GAS Power Calculator 83 to estimate power in a case‐control design. We assumed a sample size of 4000 with a case to control ratio of 3:2, which approximates a sample size of our meta‐analysis.
As expected, this revealed that we only had modest power in the WGS discovery sample for variants with an odds ratio (OR) ≈4.5 (50%) and high power for variants with an OR ≈6 (82%). We had reasonable power to detect lower‐effect alleles in the combined “meta‐analysis” arm of our study (87% for variants exerting an OR = 2.5).
4.8. Burden association testing in exons
To assess the associations described in Holstege et al., 20 we closely followed their filtering scheme. We used LOFTEE and REVEL to annotate exonic variants and create deleteriousness categories. We tested only genes in their top categories from their table 1. Burden of rare variants was tested using FBAT‐RV with EOAD cases (age at onset < = 65) coded as 1, LOAD cases coded as 2, and controls coded as −0.2.
4.9. Variant and regional association with genes
Disease‐associated variants are often assigned to genes by their proximity, where only genes overlapping or closely flanking the reported SNVs are considered. The overlap‐only strategy excludes other potentially causal genes within the associated haplotype. However, expanding gene association to include non‐overlapping SNVs or regions is complicated by the current diversity and inconsistency of annotation for non‐coding regions of the genome. As regulatory regions proximal and distal to a gene are becoming extensively annotated, 84 we have leveraged the functional significance of sets of cis‐regulatory regions of the vertebrate genome. We applied The Genomic Regions Enrichment of Annotations Tool (GREAT) to leverage functional cis‐regulatory regions identified by localized measurements of DNA‐binding events across the genome. 85 GREAT assigned additional genes to both SNVs and regions when applied to loci with no direct gene overlap.
4.10. Differential gene expression
A mixed‐effect linear regression was performed on the RNA‐Seq output with Bioconductor (v3.7) using CQN 86 and limma 87 adjusting for clinical and technical variations. A multiple testing correction was applied.
4.11. Annotation and geneset enrichment
Prioritized variants and regions were annotated for relationships to eQTLs (GTEX 88 ), CpG islands, DNase hypersensitivity, RNA gene locations, and RNA‐binding sites (UCSC 89 ), enhancers, promoters, transcription start sites, transcription factor binding sites, and other regulatory features (Ensembl 90 ; FANTOM5 91 ), histone marks and GC‐content (GWAVA 92 ), 3D genomic interactions and open chromatin (3DSNP 93 ), cell‐specific enhancers (INFERNO 94 ), and the Illumina bodyMap2 transcriptome (GSE30611).
4.12. Regulatory enrichment within spatial‐clustering/region‐based association
To test whether the top regions of interest were overpopulated with regulatory annotations, we computed 103 random permutations per region across the genome of the same length to count the number of overlapping annotations. These regions were restricted to regions with similar numbers of genes. A Fisher exact test was used to compare annotations within the top leading regions against these permuted regions. Multiple testing correction was applied for every region x annotation that was tested.
4.13. Cell‐specific enrichments
We performed Expression Weighted Cell Type Enrichment with EWCE 21 using mouse single‐cell transcriptomic data from the cortex and hippocampus. 95 EWCE aims to identify the cellular origins of a disorder by examining where a disease‐associated gene list is primarily expressed and testing this against a distribution obtained from 10,000 permutations of random lists. We selected four gene lists to be tested: the leading SNV/region‐associated genes from Tables 1 and 2 (n = 5), SNV‐associated genes (P meta < 0.01; n = 185), region‐associated genes (P meta < 0.0005; n = 55), and published common‐variant AD‐associated genes 22 (n = 32). Seventy‐eight percent of these genes had a mouse homolog, which was then used in the analysis.
4.14. Functional enrichment analysis for associated genes
Functional enrichment for the SNV‐ and regional‐associated genes or for genes found to be co‐expressed with LINC00298, was performed via the Metascape server, 96 which applies the hypergeometric test 97 and Benjamini‐Hochberg P‐value correction algorithm 98 to identify terms (all GO ontologies, Reactome, and KEGG pathways) that contain a statistically greater number of genes in common with an input list than expected by chance. Enriched terms were filtered at an FDR < = 0.1.
4.15. Network relationships with known AD genes
First, we set out to understand novel but direct relationships between genes associated with our identified variants and regions and already published Alzheimer's‐associated genes. These known Alzheimer's genes were selected from a recently published review 22 and include genes that cause familial forms of the disease (eg, APP, PSEN1, and PSEN2) as well as genes that have the highest association in GWAS. 3 , 5 , 9 , 99 , 100 We used the StringDB 101 protein‐protein interaction resource using only identified protein‐protein interactions. Using this background that agglomerates protein‐protein interaction data sets, combining evidence from several experimentally derived, curated interaction databases, we identified direct (curated AD genes directly interacting with our associated genes) associations in a global network, which contained 22 known AD genes, 73 regional‐associated genes, and 59 SNV‐associated genes (Supplementary Table 24). This network was reviewed for direct interactions between known AD genes and SNV/regional‐associated genes. Genes related to each other in this manner were then visualized using Cytoscape. 102 Genes in this network co‐expressed with LINC00298 were highlighted when correlated in expression as defined according to pre‐calculated correlations available at the lncHUB server 103 (Supplementary Table 25). The server provides gene‐lncRNA Pearson correlation computed from 11,284 TCGA normalized samples processed by recount241.
Functional enrichment within this network was performed using the remote StringDB server linked to Cystoscape “String App Enrichment function,” 104 producing enrichments using the hypergeometric test, with P‐values corrected for multiple testing using the method of Benjamini and Hochberg in known molecular pathways and GO terms as described in Frenceschini et al. 105 Enriched GO/pathway terms were considered at an FDR < = 0.05. Genes from our study and known Alzheimer's genes coding for proteins directly interacting with proteins identified by genes from Table 1 and Table 2 were examined for common enrichment and grouped around the genes we highlighted in these tables into functional clusters where possible. Genes from our study or known AD genes that show protein‐protein interaction links with Table 1‐ and Table 2‐identified genes were grouped most closely in the common annotation clusters. The top GO enrichment classes (nervous system development and generation of neurons) were annotated to nodes using the String enrichment color palette function to produce highlighted node borders. Immune‐related functions that showed enrichment for currently known AD‐related genes were used to group both known AD genes and regional‐associated and SNV‐associated into annotation clusters.
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
R.E.T, L.B., C.L. and D.P. designed the study. K.M., R.K., O.H., and B.C performed sequencing and quality control. D.P., S.L.M., and K.M analyzed the data. S.L.M., S.A., and W.A.H performed the functional analysis. O.H., W.A.H., L.B., K.M., I.W., D.P., S.L.M., C.L., and R.E.T. contributed ideas and insights. L.B., C.L., W.A.H., and R.E.T. supervised this work. R.E.T. and W.A.H. obtained funding. L.B., D.M., and R.E.T. wrote the original draft of the paper, and all authors edited and reviewed the manuscript.
Supporting information
Supplementary information
Supplementary information
ACKNOWLEDGEMENTS
The authors would like to thank the staff from the National Institute of Mental Health (NIMH) Divisions of Clinical and Treatment Research (DCTR) and Epidemiology and Services Research (DESR), including David Shore, MD, Mary Farmer, MD, MPH, Debra Wynne, MSW, Steven 0. Moldin, PhD, Darrell G. Kirch, MD (1989‐1994), Nancy E. Maestri, PhD (1992‐1994), William Huber (1989‐1995), Pamela Wexler (1995‐), and Darrel A. Regier, MD, MPH. They would also like to thank the study staff at all three sites and the data management staff at SRA Technologies, Inc., particularly Cheryl McDonnell, PhD, for the care and attention that they paid to all aspects of the study. The authors are also extremely grateful to the families whose participation made this work possible. We would like to thank Dr. Ioannis Vlachos and Leinal Sejour, Beth Israel Hospital Non‐coding RNA precision diagnostics and therapeutics core of the Harvard Medical School Initiative for RNA Medicine, Beth Israel Deaconess Medical Center, for their help in interpretation of non‐coding genome and ncRNA genes. This study was supported by the Cure Alzheimer's Fund, and the following federal grants: U24AG026395 (NIA‐LOAD Family Study); U24AG021886 (National Cell Repository for Alzheimer's Disease); P50AG08702 (Boston University and Columbia University); P30AG028377 (Duke University); P30AG010133 (Indiana University); PO1 AG05138 (Massachusetts General Hospital; Mayo Clinic, Rochester; Mayo Clinic, Jacksonville; and Mount Sinai School of Medicine); and P30AG010124 (Northwestern University Medical School; Oregon Health and Science University; Rush University Medical Center; University of Alabama at Birmingham; David Geffen School of Medicine, University of California, Los Angeles; University of Kentucky, Lexington; University of Pennsylvania; University of Pittsburgh; University of Southern California; The University of Texas Southwestern Medical Center; University of Washington; and Washington University School of Medicine). This work was also supported in part by the National Institute for Health Research (NIHR) Sheffield Biomedical Research Centre (Translational Neuroscience)/NIHR Sheffield Clinical Research Facility and the Cure Alzheimer's Fund (Alzheimer's Disease Research Foundation) (W.A.H.). Please refer to the Supplementary Note for additional acknowledgements.
Prokopenko D, Morgan SL, Mullin K, et al. Whole‐genome sequencing reveals new Alzheimer's disease‐associated rare variants in loci related to synaptic function and neuronal development. Alzheimer's Dement. 2021;17:1509–1527. 10.1002/alz.12319
Dmitry Prokopenko and Sarah L. Morgan contributed equally.
Christoph Lange, Winston Hide, Lars Bertram, and Rudolph E. Tanzi jointly supervised this work.
REFERENCES
- 1. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta‐analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17‐23. [DOI] [PubMed] [Google Scholar]
- 2. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI‐EBI GWAS Catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertram L, Tanzi RE. Alzheimer disease risk genes: 29 and counting. Nat Rev Neurol. 2019;15:191‐192. [DOI] [PubMed] [Google Scholar]
- 5. Kunkle BW, Grenier‐Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature. 2012;488:96‐99. [DOI] [PubMed] [Google Scholar]
- 8. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat Genet. 2017;49:1373‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blacker D, Haines JL, Rodes L, Terwedow H, Go RC, Harrell LE, et al. ApoE‐4 and age at onset of Alzheimer's disease: the NIMH genetics initiative. Neurology. 1997;48:139‐147. [DOI] [PubMed] [Google Scholar]
- 11. Lee JH, Cheng R, Graff‐Radford N, Foroud T, Mayeux R. National Institute on Aging Late‐Onset Alzheimer's Disease Family Study Group. Analyses of the National Institute on Aging Late‐Onset Alzheimer's Disease Family Study: implication of additional loci. Arch Neurol. 2008;65:1518‐1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beecham GW, Bis JC, Martin ER, Choi S‐H, DeStefano AL, van Duijn CM, et al. The Alzheimer's Disease Sequencing Project: study design and sample selection. Neurol Genet. 2017;3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laird NM, Horvath S, Xu X. Implementing a unified approach to family‐based tests of association. Genet Epidemiol. 2000;19 Suppl 1:S36‐42. [DOI] [PubMed] [Google Scholar]
- 15. Zhao L, He Z, Zhang D, Wang GT, Renton AE, Vardarajan BN, et al. A rare variant nonparametric linkage method for nuclear and extended pedigrees with application to late‐onset Alzheimer disease via WGS data. Am J Hum Genet. 2019;105:822‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss‐of‐function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet. 2015;47:445‐447. [DOI] [PubMed] [Google Scholar]
- 17. Mishra A, Macgregor S. VEGAS2: software for More Flexible Gene‐Based Testing. Twin Res Hum Genet. 2015;18:86‐91. [DOI] [PubMed] [Google Scholar]
- 18. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Loehlein Fier H, Prokopenko D, Hecker J, Cho MH, Silverman EK, Weiss ST, et al. On the association analysis of genome‐sequencing data: a spatial clustering approach for partitioning the entire genome into nonoverlapping windows. Genet Epidemiol. 2017;41:332‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holstege H, Hulsman M, Charbonnier C, Grenier‐Boley B, Quenez O, Grozeva D, et al. Exome sequencing identifies novel AD‐associated genes. medRxiv. 2020. 2020.07.22.20159251. [Google Scholar]
- 21. Skene NG, Grant SGN. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front Neurosci. 2016;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertram L, Tanzi RE. Genomic mechanisms in Alzheimer's disease. Brain Pathology. 2020;30(5):966‐977. 10.1111/bpa.12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pita‐Juárez Y, Altschuler G, Kariotis S, Wei W, Koler K, Green C, et al. The Pathway Coexpression Network: revealing pathway relationships. PLoS Comput Biol. 2018;14:e1006042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beecham GW, Vardarajan B, Blue E, Bush W, Jaworski J, Barral S, et al. Rare genetic variation implicated in non‐Hispanic white families with Alzheimer disease. Neurol Genet. 2018;4:e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nho K, Horgusluoglu E, Kim S, Risacher SL, Kim D, Foroud T, et al. Integration of bioinformatics and imaging informatics for identifying rare PSEN1 variants in Alzheimer's disease. BMC Med Genomics. 2016;9 Suppl 1:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu B, Jiang L, Huang T, Zhao Y, Liu T, Zhong Y, et al. ER‐associated degradation regulates Alzheimer's amyloid pathology and memory function by modulating γ‐secretase activity. Nat Commun. 2017;8:1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonça N, et al. Sel1L is indispensable for mammalian endoplasmic reticulum‐associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A. 2014;111:E582‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saltini G, Dominici R, Lovati C, Cattaneo M, Michelini S, Malferrari G, et al. A novel polymorphism in SEL1L confers susceptibility to Alzheimer's disease. Neurosci Lett. 2006;398:53‐58. [DOI] [PubMed] [Google Scholar]
- 29. Davies G, Tenesa A, Payton A, Yang J, Harris SE, Liewald D, et al. Genome‐wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psychiatry. 2011;16:996‐1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benyamin B, Pourcain B, Davis OS, Davies G, Hansell NK, Brion M‐JA, et al. Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol Psychiatry. 2014;19:253‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huett A, Ng A, Cao Z, Kuballa P, Komatsu M, Daly MJ, et al. A novel hybrid yeast‐human network analysis reveals an essential role for FNBP1L in antibacterial autophagy. J Immunol. 2009;182:4917‐4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018;14:1602‐1614. [DOI] [PubMed] [Google Scholar]
- 33. Kakimoto T, Katoh H, Negishi M. Regulation of neuronal morphology by Toca‐1, an F‐BAR/EFC protein that induces plasma membrane invagination. J Biol Chem. 2006;281:29042‐29053. [DOI] [PubMed] [Google Scholar]
- 34. Kwak S, Lee S‐H, Han E‐J, Park S‐Y, Jeong M‐H, Seo J, et al. Serine/threonine kinase 31 promotes PDCD5‐mediated apoptosis in p53‐dependent human colon cancer cells. J Cell Physiol. 2019;234:2649‐2658. [DOI] [PubMed] [Google Scholar]
- 35. Volders P‐J, Anckaert J, Verheggen K, Nuytens J, Martens L, Mestdagh P, et al. LNCipedia 5: towards a reference set of human long non‐coding RNAs. Nucleic Acids Res. 2019;47:D135‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hon C‐C, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, et al. An atlas of human long non‐coding RNAs with accurate 5’ ends. Nature. 2017;543:199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paraskevopoulou MD, Vlachos IS, Karagkouni D, Georgakilas G, Kanellos I, Vergoulis T, et al. DIANA‐LncBase v2: indexing microRNA targets on non‐coding transcripts. Nucleic Acids Res. 2016;44:D231‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fernández‐de Frutos M, Galán‐Chilet I, Goedeke L, Kim B, Pardo‐Marqués V, Pérez‐García A, et al. MicroRNA 7 impairs insulin signaling and regulates Aβ levels through posttranscriptional regulation of the insulin receptor substrate 2, insulin receptor, insulin‐degrading enzyme, and liver X receptor pathway. Mol Cell Biol. 2019;39:e00170‐19. 10.1128/MCB.00170-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao J, Zhou Y, Guo M, Yue D, Chen C, Liang G, et al. MicroRNA‐7: expression and function in brain physiological and pathological processes. Cell Biosci. 2020;10:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lachmann A, Schilder BM, Wojciechowicz ML, Torre D, Kuleshov MV, Keenan AB, et al. Geneshot: search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019;47:W571‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Russo R, Marra R, Andolfo I, De Rosa G, Rosato BE, Manna F, et al. Characterization of two cases of congenital dyserythropoietic anemia Type I shed light on the uncharacterized C15orf41 protein. Front Physiol. 2019;10:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alfonso SI, Callender JA, Hooli B, Antal CE, Mullin K, Sherman MA, et al. Gain‐of‐function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer's disease. Sci Signal. 2016;9:ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cortés CR, McInerney‐Leo AM, Vogel I, Rondón Galeano MC, Leo PJ, Harris JE, et al. Mutations in human C2CD3 cause skeletal dysplasia and provide new insights into phenotypic and cellular consequences of altered C2CD3 function. Sci Rep. 2016;6:24083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Broix L, Asselin L, Silva CG, Ivanova EL, Tilly P, Gilet JG, et al. Ciliogenesis and cell cycle alterations contribute to KIF2A‐related malformations of cortical development. Hum Mol Genet. 2018;27:224‐238. [DOI] [PubMed] [Google Scholar]
- 45. Gilet JG, Ivanova EL, Trofimova D, Rudolf G, Meziane H, Broix L, et al. Conditional switching of KIF2A mutation provides new insights into cortical malformation pathogeny. Hum Mol Genet. 2020;29:766‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hares K, Miners JS, Cook AJ, Rice C, Scolding N, Love S, et al. Overexpression of Kinesin superfamily motor proteins in Alzheimer's disease. J Alzheimers Dis. 2017;60:1511‐1524. [DOI] [PubMed] [Google Scholar]
- 47. Seira O, Liu J, Assinck P, Ramer M, Tetzlaff W. KIF2A characterization after spinal cord injury. Cell Mol Life Sci. 2019;76:4355‐4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Ferrari GV, Avila ME, Medina MA, Perez‐Palma E, Bustos BI, Alarcon MA. Wnt/β‐catenin signaling in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2014;13:745‐754. [DOI] [PubMed] [Google Scholar]
- 49. De Ferrari GV, Chacón MA, Barría MI, Garrido JL, Godoy JA, Olivares G, et al. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta‐amyloid fibrils. Mol Psychiatry. 2003;8:195‐208. [DOI] [PubMed] [Google Scholar]
- 50. Tapia‐Rojas C, Burgos PV, Inestrosa NC. Inhibition of Wnt signaling induces amyloidogenic processing of amyloid precursor protein and the production and aggregation of Amyloid‐β (Aβ) peptides. J Neurochem. 2016;139:1175‐1191. [DOI] [PubMed] [Google Scholar]
- 51. Rétaux S, Rogard M, Bach I, Failli V, Besson M‐J. Lhx9 : a novel LIM‐Homeodomain gene expressed in the developing forebrain. J Neurosci. 1999;19:783‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al. Gene discovery and polygenic prediction from a genome‐wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Armato U, Bonafini C, Chakravarthy B, Pacchiana R, Chiarini A, Whitfield JF, et al. The calcium‐sensing receptor: a novel Alzheimer's disease crucial target?. J Neurol Sci. 2012;322:137‐140. [DOI] [PubMed] [Google Scholar]
- 54. Schaffer AE, Breuss MW, Caglayan AO, Al‐Sanaa N, Al‐Abdulwahed HY, Kaymakçalan H, et al. Biallelic loss of human CTNNA2, encoding αN‐catenin, leads to ARP2/3 complex overactivity and disordered cortical neuronal migration. Nat Genet. 2018;50:1093‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fukuda M, Mikoshiba K. Synaptotagmin‐like protein 1‐3: a novel family of C‐terminal‐type tandem C2 proteins. Biochem Biophys Res Commun. 2001;281:1226‐1233. [DOI] [PubMed] [Google Scholar]
- 56. Li J, Lange LA, Sabourin J, Duan Q, Valdar W, Willis MS, et al. Genome‐ and exome‐wide association study of serum lipoprotein(a) in the Jackson Heart Study. J Hum Genet. 2015;60:755‐761. [DOI] [PubMed] [Google Scholar]
- 57. Ranneva SV, Maksimov VF, Korostyshevskaja IM, Lipina TV. Lack of synaptic protein, calsyntenin‐2, impairs morphology of synaptic complexes in mice. Synapse. 2020;74:e22132. [DOI] [PubMed] [Google Scholar]
- 58. Preuschhof C, Heekeren HR, Li S‐C, Sander T, Lindenberger U, Bäckman L. KIBRA and CLSTN2 polymorphisms exert interactive effects on human episodic memory. Neuropsychologia. 2010;48:402‐408. [DOI] [PubMed] [Google Scholar]
- 59. Jackson J, Jambrina E, Li J, Marston H, Menzies F, Phillips K, et al. Targeting the synapse in Alzheimer's disease. Front Neurosci. 2019;13:735. 10.3389/fnins.2019.00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arnsten AFT, Datta D, Tredici KD, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimers Dement. 2021;17:115‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alzheimer's Association Calcium Hypothesis Workgroup. Calcium hypothesis of Alzheimer's disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 2017;13:178‐182.e17. [DOI] [PubMed] [Google Scholar]
- 62. Fadista J, Manning AK, Florez JC, Groop L. The (in)famous GWAS P‐value threshold revisited and updated for low‐frequency variants. European Journal of Human Genetics. 2016;24:1202‐1205. 10.1038/ejhg.2015.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, et al. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Consortium RoadmapEpigenomics, A Kundaje, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bujold D, de Lima Morais DA, Gauthier C, Côté C, Caron M, Kwan T, et al. The International Human Epigenome Consortium Data Portal. Cell Syst. 2016;3:496‐499.e2. [DOI] [PubMed] [Google Scholar]
- 66. Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, et al. Human whole genome genotype and transcriptome data for Alzheimer's and other neurodegenerative diseases. Sci Data. 2016;3:160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. 2013) 1‐3. arXiv.1303.3997.
- 68. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Garrison E, Marth G. Haplotype‐based variant detection from short‐read sequencing. 2012. arXiv.1207.3907.
- 70. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Website n.d. https://software.broadinstitute.org/gatk/best‐practices/ (accessed December 21, 2020).
- 72. bcbio. bcbio/bcbio‐nextgen n.d. https://github.com/bcbio/bcbio‐nextgen (accessed December 21, 2020).
- 73. bcbio. bcbio/bcbio.variation.recall n.d. https://github.com/bcbio/bcbio.variation.recall (accessed December 21, 2020).
- 74. Babraham Bioinformatics ‐ FastQC A Quality Control tool for High Throughput Sequence Data n.d. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed December 21, 2020).
- 75. García‐Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, et al. Qualimap: evaluating next‐generation sequencing alignment data. Bioinformatics. 2012;28:2678‐2679. [DOI] [PubMed] [Google Scholar]
- 76. Prokopenko D, Hecker J, Silverman EK, Pagano M, Nöthen MM, Dina C, et al. Utilizing the Jaccard index to reveal population stratification in sequencing data: a simulation study and an application to the 1000 Genomes Project. Bioinformatics. 2016;32:1366‐1372. 10.1093/bioinformatics/btv752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. The American Journal of Human Genetics. 2007;81:559‐575. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. De G, Yip W‐K, Ionita‐Laza I, Laird N. Rare variant analysis for family‐based design. PLoS One. 2013;8:e48495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, et al. Optimal unified approach for rare‐variant association testing with application to small‐sample case‐control whole‐exome sequencing studies. Am J Hum Genet. 2012;91:224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ionita‐Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence Kernel Association Tests for the Combined Effect of Rare and Common Variants. The American Journal of Human Genetics. 2013;92:841‐853. 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lange C, Laird NM. Power calculations for a general class of family‐based association tests: dichotomous traits. Am J Hum Genet. 2002;71:575‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Home n.d. https://csg.sph.umich.edu/abecasis/gas_power_calculator/index.html (accessed December 27, 2020).
- 84. Frankish A, Diekhans M, Ferreira A‐M, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol. 2010;28:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA‐seq data using conditional quantile normalization. Biostatistics. 2012;13:204‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Consortium GTEx. The Genotype‐Tissue Expression (GTEx) project. Nat Genet. 2013;45:580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018;46:D762‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, et al. Ensembl 2017. Nucleic Acids Res. 2017;45:D635‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kawaji H, Severin J, Lizio M, Forrest ARR, van Nimwegen E, Rehli M, et al. Update of the FANTOM web resource: from mammalian transcriptional landscape to its dynamic regulation. Nucleic Acids Res. 2011;39:D856‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ritchie GRS, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nature Methods. 2014;11:294‐296. 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lu Y, Quan C, Chen H, Bo X, Zhang C. 3DSNP: a database for linking human noncoding SNPs to their three‐dimensional interacting genes. Nucleic Acids Research. 2017;45:D643‐9. 10.1093/nar/gkw1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Amlie‐Wolf A, Tang M, Mlynarski EE, Kuksa PP, Valladares O, Katanic Z, et al. INFERNO: inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res. 2018;46:8740‐8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zeisel A, Muñoz‐Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single‐cell RNA‐seq. Science. 2015;347:1138‐1142. [DOI] [PubMed] [Google Scholar]
- 96. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10:615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zar JacksonDABiostatisticalAnalysisJerroldH. The Quarterly Review of Biology. 2000;75:501‐502. 10.1086/393742. [DOI] [Google Scholar]
- 98. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811‐818. [DOI] [PubMed] [Google Scholar]
- 99. Lambert JC, Ibrahim‐Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry. 2018;8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res. 2019;47:D607‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. lncHUB n.d. https://amp.pharm.mssm.edu/lnchub/ (accessed December 23, 2020).
- 104. Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: network Analysis and Visualization of Proteomics Data. J Proteome Res. 2019;18:623‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein‐protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. NDEx WebApp v2.5.0 n.d. https://tinyurl.com/y6p9xjlw (accessed December 21, 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information