

Abstract
CYP121 of Mycobacterium tuberculosis (Mtb) is an essential target for the development of novel potent drugs against tuberculosis (TB). Besides known antifungal azoles, further compounds of the azole class were recently identified as CYP121 inhibitors with antimycobacterial activity. Herein, we report the screening of a similarity‐oriented library based on the former hit compound, the evaluation of affinity toward CYP121, and activity against M. bovis BCG. The results enabled a comprehensive SAR study, which was extended through the synthesis of promising compounds and led to the identification of favorable features for affinity and/or activity and hit compounds with 2.7‐fold improved potency. Mode of action studies show that the hit compounds inhibit substrate conversion and highlighted CYP121 as the main antimycobacterial target of our compounds. Exemplified complex crystal structures of CYP121 with three inhibitors reveal a common binding site. Engaging in both hydrophobic interactions as well as hydrogen bonding to the sixth iron ligand, our compounds block a solvent channel leading to the active site heme. Additionally, we report the first CYP inhibitors that are able to reduce the intracellular replication of M. bovis BCG in macrophages, emphasizing their potential as future drug candidates against TB.
Keywords: CYP121, structure-activity relationships, Mycobacterium tuberculosis, complex structures, biological activity
Similarity‐oriented library screen: Infections caused by Mycobacterium tuberculosis (Mtb) still remain among the most difficult to treat, owing to the pathogen's special characteristics within the realm of pathogenic bacteria. This report describes the development of potent inhibitors targeting an essential cytochrome P450 enzyme of Mtb, which are also able to reduce the intracellular replication of mycobacteria within macrophages.
Introduction
Tuberculosis is still one of the leading infectious diseases worldwide with around 10 million new infections and 1.6 million cases of deaths every year. [1] The disease gained more and more attention in the last years due to arising resistances of its pathogen Mycobacterium tuberculosis (Mtb) against currently applied first‐line drugs and its high prevalence in immune deprived patients such as those infected with HIV.[ 1 , 2 , 3 ] With the sequence of the Mtb genome deciphered in 1998, there were a lot of new possible targets identified. [4] However, the cell wall of Mtb is highly complex and forms a wax‐like barrier with partly unknown permeability behavior. This exceptional property impedes the discovery of new inhibitors as they could possibly fail to reach their target and thus appear biologically inactive. [5]
One of these identified targets is the cytochrome‐P450 enzyme 121 (CYP121) which was shown to be essential for the viability of Mtb. [6] The gene encoding CYP121 is, besides the Mycobacterium tuberculosis complex, only found in some Streptomyces strains with maximum 64 % identity. [7] The elucidation of its function was encouraged by the identification of the role of its neighbouring gene in Mtb which encodes a cyclodityrosine (cYY) synthetase. [8] CYP121 was subsequently identified to utilize cYY as substrate and to catalyze its conversion to mycocyclosin via a C−C coupling reaction. [9] However, the biological role of substrate and product remain unknown up to now.
McLean et al. identified known antifungal azoles such as econazole or ketoconazole as the first inhibitors of CYP121. [10] Interestingly, the compounds also showed biological activity against Mtb correlating with their respective affinities to CYP121. [6] Furthermore, econazole exhibited activity against multi‐drug resistant Mtb, murine TB in vivo as well as latent TB in vitro and in vivo.[ 11 , 12 , 13 ] In several subsequent studies, fragment‐based approaches were employed and small molecules binding to CYP121 were identified.[ 14 , 15 , 16 , 17 ] Additionally, some substrate analogues were discovered to inhibit CYP121.[ 18 , 19 ] However, none of these compounds was shown to exhibit any in vitro activity toward Mtb (or M. bovis BCG). Recently, we described a screening approach based on a CYP‐inhibitor library which resulted in the identification of the first potent inhibitors of CYP121 with activity against M. bovis BCG and Mtb. [20]
Based on this breakthrough in the search for potent CYP121 inhibitors, we herein employed the screening of a similarity‐oriented library in order to evaluate the possible structural diversity of the hit scaffold and to identify favourable features for affinity and/or activity. Furthermore, we investigated the ability of our compounds to inhibit the enzymatic conversion in vitro and their inhibitory effect on M. bovis BCG. Mode of action studies after addition of the substrate cYY demonstrated the importance of CYP121 inhibition for the antimycobacterial effect of our compounds. Complex crystal structures of CYP121 were elucidated to gain further insights into enzyme inhibition and reveal crucial enzyme interactions that could be investigated for further compound development. Additionally, the influence of several CYP121 and CYP125 inhibitors on the intracellular replication of M. bovis BCG in macrophages was evaluated.
Results and Discussion
Library generation and screening results
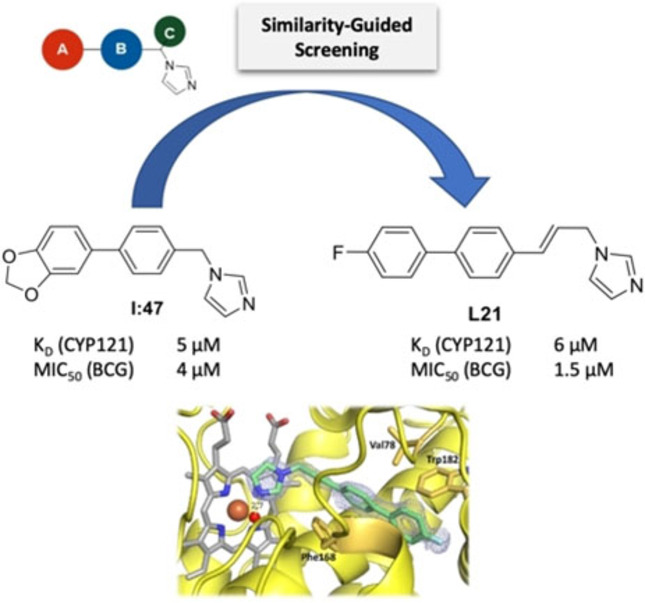
As a starting point for our screening approach, we chose the hit compound I:47, which was recently identified in a screening of a diversity oriented CYP‐inhibitor library. [20] With its high affinity to CYP121, biological activity against M. bovis BCG and Mtb as well as its structure, which offers various optimization possibilities, it serves as an ideal basis for further exploration of the binding and activity properties of this structural class. The created screening library was composed of 94 compounds which all can be assigned to a general structure scheme derived from I:47 (Figure 1). The imidazole moiety appeared to be essential for the binding to the heme iron of the enzyme and thus was conserved. [20] Additionally, the template consists of a substituted methylene bridge (Figure 1, motif C) or a propylene linker, which connects the imidazole ring with the middle aromatic moiety (Figure 1, motif B) and a Western part aromatic moiety (Figure 1, motif A), which is directly linked to the aforementioned one. The library covered various structural motifs for each of the different parts, ensuring a broad analysis of structure‐activity‐relationships and offering the possibility to discover highly potent hits. The structures of all library compounds (L1 to L94) are displayed in the respective Supporting Information (SI, Table S1).
Figure 1.
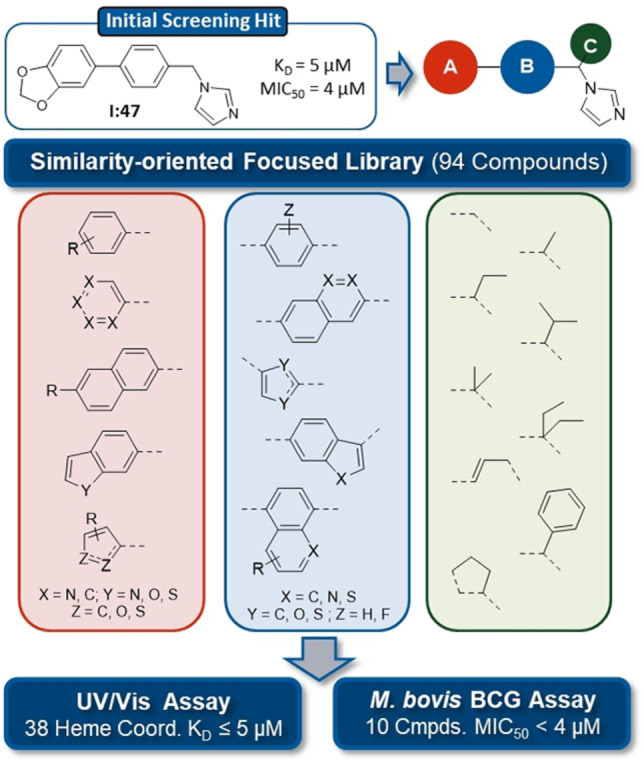
Similarity‐guided screening of a focused library. Compounds were selected based on the former screening hit I:47 with variations in the three displayed structure motifs. KD values were determined for 79 positive hits showing a visible shift of absorbance maximum at concentrations ≤20 μM. MIC50 values were determined for 29 compounds, which showed inhibition ≥60 % at 10 μM.
The screening library was examined in a UV/Vis heme binding assay regarding affinity toward CYP121 as previously described. [20] The compounds were screened at concentrations of 100 μM and 20 μM. For 79 compounds, which showed a visible shift of the absorption maximum at concentrations of 20 μM, the binding constant KD was determined by concentration dependent titration of the enzyme (respective KD values are listed in SI, table S1). All of these binders showed a type‐II shift identical to the previously determined I:47, [20] as expected because of the structural similarity (discussion on actual binding mode in “complex crystallization” paragraph). 38 of the approved binders (hit rate 40 %) exhibited a binding constant below or equal to I:47 (KD=5 μM). The highest affinity was shown by compounds L89, L16, L78 with KD values of 0.3, 0.5, and 0.6 μM, respectively. Interestingly, these compounds have the methoxynaphthalene moiety in position A in common, which seems to be particularly favorable for binding.
In parallel to affinity determination, the library was analyzed regarding biological activity against M. bovis BCG in vitro. The compounds were initially tested at a concentration of 100 μM and inhibitors showing inhibition higher than or equal to 80 % were further examined at 10 μM. For 29 compounds with inhibition higher than or equal to 60 % at 10 μM, MIC50 values were determined as described previously. [20] I:47 (MIC50=4.0 μM) was used as a reference. Ten compounds (hit rate 11 %) showed better activity than I:47. Interestingly, the two compounds with a propylene linker (L15 and L21) exhibited the highest activity (MIC50=1.8 μM and 1.5 μM).
Chemical synthesis
Based on the two most potent inhibitors identified in the screening, L15 and L21, compounds with a novel substituent at the methylene linker were designed. As the ethyl substitution was a promising motif regarding affinity, it was combined with the double bond feature of the propylene linker resulting in an ethenyl substituted methylene bridge.
Compounds S1–S10 were prepared according to Scheme 1, starting with a Suzuki coupling of the corresponding boronic acid and 4‐bromobenzaldehyde or 4‐bromo‐2‐fluorobenzaldehyde, respectively. For compounds S9 and S10, the protected boronic acids were synthesized beginning with a tert‐butyldimethylsilyl (TBDMS) protection of the hydroxyl function of 4‐bromophenol or the amino group of 5‐bromo‐1H‐indole. The boronic acid function was subsequently introduced by a substitution reaction with n‐butyllithium and triisopropylborate (Scheme 2). The coupled aldehydes were reduced in a Grignard reaction with vinyl magnesium bromide to the corresponding alcohols with simultaneous introduction of the ethenyl substituent. Consequently, the resulting alcohols were converted to the respective imidazoles through a SNt reaction with carbonyldiimidazole (CDI). The compounds were obtained as racemates and generally, no separation of the enantiomers was performed. For the protected compounds, the final step was the cleavage of the TBDMS group with tetrabutylammonium fluoride. In contrast, compound S11, a fluorinated derivative of hit I:47, was synthesized as described in Scheme 3. The first step, a SNt reaction of 4‐bromo‐2‐fluorobenzylbromide with imidazole, was followed by a Suzuki coupling of the intermediate with 3,4‐methylenedioxyphenylboronic acid resulting in the desired compound. The respective KD values of the synthesized derivatives S1–S11 are listed in table S3 in the supporting information.
Scheme 1.

Reagents and conditions: (i) method A: Pd(PPh3)4, Na2CO3, toluene/EtO/H2O=5 : 3 : 2, 100 °C, 1–4 h; (ii) method B: VinylMgBr, THF abs., −15 °C→rt, 1–2 h; (iii) method C: CDI, MeCN, 110 °C, 1–4 h.
Scheme 2.

Reagents and conditions: (ia) method D a): TBDMSCl, imidazole, CH2Cl2, rt, 16 h; (ib) method D b): TBDMSCl, NaH, CH2Cl2, 0 °C→ rt, 16 h; (ii) method E: nBuLi, B(OiPr)3, THF, −78 °C→rt, 3 h; (iii) method F: TBAF, K2HPO4, THF, rt, 16 h.
Scheme 3.
Reagents and conditions: (i) imidazole, NaH, 60 °C, 1.5 h; (ii) method A: 3,4‐methylene‐dioxyphenylboronic acid, Pd(PPh3)4, Na2CO3, toluene/EtOH/H2O=5 : 3 : 2, 110 °C, 2.5 h.
Structure‐affinity relationship
Based on the determined binding constants, the compounds were analyzed regarding their structural features in order to identify favorable motifs that contribute positively to affinity. The different parts of the binding compounds were, as far as possible, considered separately to avoid interference of the influence of several features.
First of all, the methylene bridge between imidazole ring and aromatic moiety B tolerated various substituents such as methyl, ethyl, i‐propyl, n‐butyl, ethenyl or phenyl (L75, L80, L92, L93, S7, L1). Additionally, a decoration with two methyl (L27) or ethyl (L35) substituents or the exchange with a propylene linker (L15) was accepted as well. Moreover, the rigidification we recently addressed in our docking study of I:47, [20] which was performed by connecting the methylene bridge to the adjacent phenyl moiety via an ethylene linker, was also possible without loss of affinity (L90). The affinity increased slightly (around twofold compared to ethyl) for space filling substituents such i‐propyl, n‐butyl, phenyl or two ethyl substituents indicating the involvement of hydrophobic interactions. In contrast, a hydroxyethyl substituent (L13) led to weaker binding compared to its ethyl analogue (L80), which supports the aforementioned hypothesis.
Compounds with a substituted methylene bridge were evaluated as racemic mixtures. It is known that two enantiomers commonly show strong differences in binding affinity or activity. However, as seen in the docking study of I:47, the binding pocket of CYP121 principally offers space to harbor both orientations of substituents. [20] To confirm this hypothesis, the enantiomers were separated for three exemplarily chosen ethenyl derivatives (SI, table S4). As expected, only minor differences could be observed between the two enantiomers of a racemate.
The used library contained compounds with various aromatic ring systems in the middle part (Figure 1, motif B). According to previous findings, compounds with a pyridine ring in this position show only weak binding. [20] A furan ring (L70, KD=34 μM) was rather unfavorable compared to an unsubstituted benzene whereas a thiophene (2,4‐ or 2,5 connected) led in some cases to similar affinities (L42, L60), in other cases, however, to weaker affinities (L62, L64). Furthermore, other motives such as fluorinated benzene, indole, benzothiophene, naphthalene or quinolone (L47, L48, L86, L79, L82) were tolerated as well with increased affinities (>twofold) for the latter two scaffolds.
Additionally, a range of motifs for the Western part aromatic ring (Figure 1, motif A) was evaluated including differently substituted phenyls, thiophene and furan rings and bicyclic ring systems such as benzofuran, benzothiophene, indole and naphthalene. Nearly all of those were tolerated with the exception of nitrogen‐containing 6‐membered rings (L43, L59). However, some appeared to be particularly favorable such as the bicyclic moieties with 6‐methoxynaphthalene leading to the highest affinities (L16: KD=0.5 μM, L78: KD=0.6 μM). Further motifs with increased affinities (KD≤6 μM) were electron deprived phenyls such as a meta‐ and para‐difluorinated phenyl (L34) and para‐trifluoromethyl (L44) or para‐trifluoromethoxy phenyls (L22). Additionally, motifs with substituents capable of hydrogen bond formation such as methylthio‐decorated phenyl or thiophene (L23, L37) as well as phenyls with a hydroxyl (L87) or a N‐acetamide group (L7) in para position also led to increased affinities.
Structure–activity relationship
A good correlation between affinity and activity against M. bovis BCG cannot necessarily be expected as the antimycobacterial activity is influenced by additional cellular factors such as permeability, efflux and metabolism. The features of compounds exhibiting high or weak activity were therefore regarded in detail to identify those which impair biological activity.
It is striking that, apart from the propylene linked structures (L15 and L21), the highly active compounds are either not substituted at the methylene bridge (L10, L44) or possess a methyl substituent (L73) or a rigid 5‐membered ring structure (L94). Other substituents such as ethyl (L9), ethenyl (S10) or i‐propyl (L92) led only to weak activity. Interestingly, the attempt to combine the properties of the propylene linker with the ethyl substituent of the methylene bridge via the introduction of an ethenyl substituent did not lead to the desired highly biologically active compound. In comparison with their non‐substituted analogues, the ethenyl derivatives show a loss in activity while their affinity is similar (compare I:47 and S11 with S5 and S8).
Concerning the middle aromatic moieties (Figure 1, motif B), benzene, fluorinated benzene, thiophene and indole were able to maintain biological activity (L44, L14, L66, L48). Rather unfavorable were bulky center parts such as naphthalene (L79) and quinolone (L81). Concerning the Western part aromatic moiety (Figure 1, motif A), trifluoromethylphenyl (L44) as well as fluorinated phenyls (L14, L45) were accepted as well as various bicyclic moieties, para‐methoxyphenyl (L94) and 2‐methylthio(thiophene) (L76). In contrast, hydroxyl groups significantly impaired activity (L2, S9). Furthermore, it should be noted that some motifs of the Western part aromatic moiety (naphthalene, benzothiophene, trifluoromethoxyphenyl, thiomethylphenyl, acetanilide) could not be definitely classified regarding biological activity as the respective evaluated compounds bear substituents at the methylene bridge which already impair activity.
Figure 2 highlights the structural features identified as favorable for affinity and/or activity. In total, L15 and L21 were the most active compounds identified in this study with MIC50 values of 0.7 and 0.5 μg/mL, respectively (Table 1). The other eight compounds with superior activity to I:47 (Table 1) were similarly active with MIC50 values between 0.8 or 1.2 μg/mL. Noticeably, all these compounds are not substituted at the methylene bridge except L94 which contains a 5‐membered ring structure. The respective ethyl analogues of L46, L14 and L76 (compounds L18, L20 and L37) showed similar affinities as their parent compounds, but failed to significantly inhibit mycobacterial growth.
Figure 2.
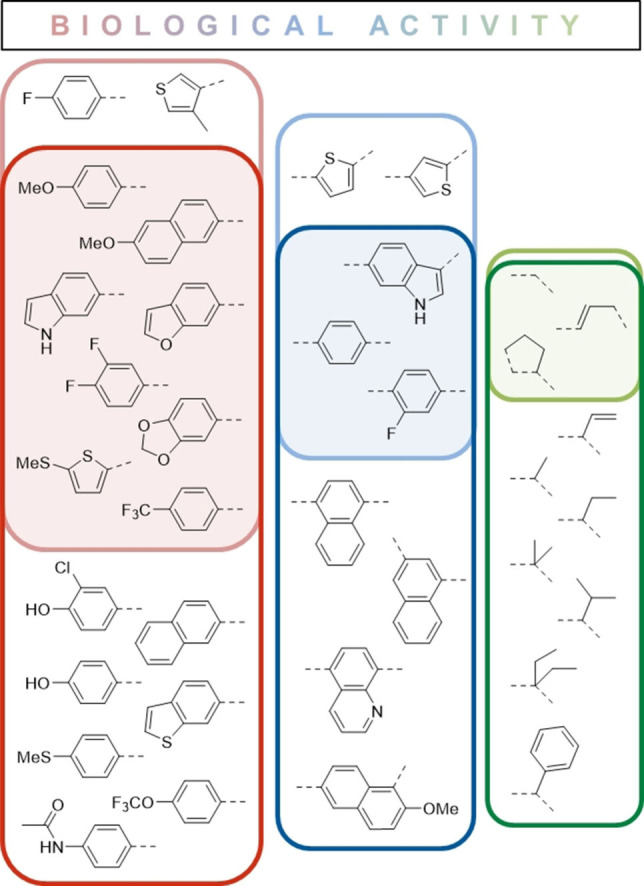
Structure‐affinity/activity‐relationships. Favorable features for affinity (KD) and activity (MIC50) were identified through comparison of similar compounds. Features leading to good affinity are framed in dark green, blue or red respective to the structure template and oriented to the bottom. In contrast, features related to good biological activity are framed in light green, blue or red and are oriented to the top. The overlap of these favorable features is shaded in the respective color.
Table 1.
KD and MIC50 values of compounds showing better activity than I:47.
|
| |||
|---|---|---|---|
|
Compound |
KD±STD [μM] |
MIC50 [mg/L] |
MIC50 [μM] |
|
Econazole |
2.8±0.2[a] |
2.6 |
12.7 |
|
I:47 |
5.4±1.0[a] |
1.4 |
4.0 |
|
L10 |
2.9±0.4 |
0.9 |
2.6 |
|
L14 |
6.4±0.9 |
0.9 |
2.7 |
|
L15 |
4.3±0.8 |
0.7 |
1.8 |
|
L21 |
5.9±0.6 |
0.5 |
1.5 |
|
L44 |
5.6±0.9 |
0.8 |
2.4 |
|
L45 |
12.6±2.0 |
1.2 |
3.4 |
|
L46 |
9.2±2.0 |
1.0 |
2.9 |
|
L76 |
6.6 ±1.0 |
1.1 |
3.2 |
|
L78 |
0.6±0.2 |
0.8 |
2.4 |
|
L94 |
4.7±0.8 |
1.1 |
3.3 |
[a] Determined previously. [20] KD±STD was calculated from four biological replicates.
In contrast, the ethyl analogue of L78, namely L16, was relatively active with a MIC50 value of 7.4 μM (KD=0.5 μM) similar to the ethenyl analogue S1 (KD=0.8 μM, MIC50=9.7 μM). For these compounds, the discrepancy between KD and MIC50 was high and can be associated to the methoxynaphthalene motif, which led to high affinities that were not translated into the expected biological activity. For the ethyl analogue L16, the activity was probably further reduced by the substituent as observed for similar compound pairs.
One of the most active compounds was the benzofuran analogue (L10) of compound I:47 (Table 1). L10 showed a slightly higher affinity and activity. Compound L78 resembles the latter two compounds with thiophene as a bioisosteric substitute for phenyl and its methylthio group replacing a part of the attached furan motif. The same applies to the methoxyphenyl motif of compound L94. Their affinities and activities are likewise similar to those of their analogues.
In contrast, the potent compounds L14, L44, L45 and L46 share an electron deprived phenyl ring at the Western part aromatic motif (Table 1). Furthermore, L14, L45 and L46 contain a fluorinated benzene ring at their middle aromatic position. Although the affinities of L45 and L46 are slightly lower compared to L14 and L44, the activities within this group are similar amongst each other and comparable to the aforementioned group of compounds.
Overall, a good a correlation between affinity and activity was observed for twelve compounds (S7, S11, L10, L47, L48, L49, L60, L66, L75, L90, L91, L94). For 13 compounds, a bad correlation was explained by unfavorable structural features (L8, L9, L16, L19, L41, L77, L78, L79, L84, L89, S1, S2, S9). For further seven compounds (S3, S4, S5, S6, S8, S10, L12), a correlation was not observed (MIC50 >2×KD) probably due to structural drawbacks that were not identified so far. Further compounds have to be evaluated to confirm this hypothesis.
Interestingly, eight compounds (L14, L15, L21, L44, L45, L46, L73, L76) showed higher activity than expected based on their affinities (KD≈2–3×MIC50). One explanation for this discrepancy is that the KD values might be actually lower than determined with our method. For econazole, we determined a KD value of 2.8 μM, although the KD value was previously reported to be 0.02 μM. [6] Apart from that, the inhibition of other targets cannot be excluded. In this context, it has been demonstrated that antifungal azoles show binding to other CYP enzymes of Mtb. [21]
Complex crystallization of CYP121 with bound inhibitors
As a CYP121 crystal structure with bound inhibitor would enable rational structure‐based drug design and possibly link physicochemical properties to structural features, complex crystallization of CYP121 was performed. Given the observed differences various western part aromatic motifs as well as linker substitutions had on substrate affinity, a structurally diverse set of inhibitors was chosen for complex crystallization. It should be noted, however, that compounds containing naphthalene, indole or benzodioxole moieties at either the western or middle aromatic part showed low occupancy in the crystal structure, which is probably related to low water solubility.
CYP121 crystallized in space group P 65 2 2 and crystals diffracted to a maximum of 1.3 Å resolution. High resolution datasets were obtained for complex structures with inhibitors L21, L44 and S2, and were solved by molecular replacement with the deposited apo structure of CYP121 as a search model (PDB ID 1n40). The full data collection and refinement statistics including the aforementioned occupancies of the inhibitors can be found in Supplementary Table S5. The complex structure of L21 revealed the compound being bound in a solvent channel leading from the protein surface to the active site heme b. The inhibitor adopts a planar conformation with only a slight rotation between the two phenyl rings, except for the imidazole that is extending toward the heme macrocycle. The imidazole nitrogen is forming a hydrogen bond with the water ligand of the heme b iron while the western and middle part aromatic moieties are engaging in hydrophobic interactions with hydrophobic side chains, especially of Phe168 and Trp182, flanking the solvent channel (Figure 3A). Previous studies on the coordination of cYY in the CYP121 crystal structure have demonstrated that cYY binding relies on an extensive water‐mediated H‐bonded network involving the sixth iron ligand. [9] Thus, hydrogen bonding of L21 to the heme water ligand prevents the formation of this network in addition to occluding the solvent channel, making the heme non‐accessible for cYY.
Figure 3.

Complex crystal structures of CYP121. A Coordination of inhibitor L21 in the CYP121 crystal structure. The protein is shown as a yellow cartoon, inhibitor, heme b and interacting residues are shown as sticks. The heme iron is shown as a brown, the water ligand as a red sphere. The difference electron density map of L21 (FO–FC) was contoured at 3σ with phases calculated from a model that was refined in the absence of L21 and is shown as a blue isomesh. The hydrogen bond distance between the water ligand and the imidazole nitrogen is 2.7 Å and shown as a yellow dashed line. B Clipped surface representation of the CYP121‐L21 complex, highlighting the position of the active site heme b and inhibitor L21 in the crystal structure. C Overlay of the complex structures of L21 (green), L44 (magenta) and S2 (cyan). A black arrow is highlighting the location of the entrance to the CYP121 active site.
Our inhibitor shares a common binding mode with the well‐known antifungal drug fluconazole, for which hydrogen bonding of its triazole nitrogen to the water ligand has been reported as a novel drug azole‐P450 binding mode. Interestingly, fluconazole does also exhibit a type‐II shift in UV‐Vis studies (as our compounds) which has formerly been linked to a direct azole‐iron interaction. [22] The clipped surface representation of CYP121 (Figure 3B) reveals the active site heme b being buried deep inside the enzyme, with L21 occupying a bottleneck‐like cleft leading to the cofactor. The overlay of all co‐crystallized inhibitors L21, L44 and S2 (Figure 3C) shows a common binding site located at the entrance to the CYP121 active site in between α‐helices F and G (helix numbering according to previously published structure). [23]
A summarized overview of the inhibitor structures together with their respective KD and MIC50 values is shown in Table 2. The complex structures of L44 and S2 with the respective difference electron density maps are displayed in figure S5. L44 shows a similar binding mode to L21, with an imidazole‐water hydrogen bonding distance of 2.9 Å (Figure S5A). However, the position of the two phenyls is shifted due to a shorter linker bridge. The compound shows lower occupancy than L21 in the crystal structure, which is possibly the result of less than optimal hydrophobic interactions with Phe168 and Trp182, leading to a lower rigidity of the molecule. Within the ethenyl substituted compound class, S2 was chosen for complex crystallization due to sufficient water solubility. Even though the enantiomers of S2 were not separated prior to crystallization, the observed electron density hints at a predominantly (R)‐configured enantiomer being bound. In accordance with the complex structure (figure S5B), the observed impaired in vivo activity can be related to the binding mode of the compound. A shorter linker bridge in combination with the position of the western and middle part aromatic structure mimicking the orientation of L21 leads to an inability of forming a hydrogen bond (distance 4.7 Å) with the water ligand of the heme b iron. The orientation of this inhibitor is dictated by a strong hydrogen bond between the para‐hydroxy group and an aspartate side chain (Asp185) of CYP121 (Figure S6), possibly trapping it close to α‐helix G and therefore preventing it from reaching the heme b water ligand. As such, this compound can possibly be displaced by an incoming substrate in an in vivo situation as it does not inhibit the formation of the water mediated H‐bonded network of cYY.
Table 2.
Structures of co‐crystallized compounds with KD and MIC50 values.
|
Compound |
KD ± STD [μM] |
MIC50 [μM] |
|
|---|---|---|---|
|
L21 |
|
5.9±0.6 |
1.5 |
|
L44 |
|
5.6±0.9 |
2.4 |
|
S2 |
|
8.3±1.7 |
50 |
Inhibition of cYY conversion in vitro
In order to evaluate the influence of the identified heme binders on the inhibition of CYP121, we used our in vitro assay consisting of CYP121, two electron transfer proteins (etp1fd and Arh1_A18G), NADPH as electron donor and an electron regenerating system. [20] Under the defined conditions, the substrate cYY was converted to mycocyclosin within approximately 10 minutes (Figure 4). The presence of a heme binder is expected to inhibit this enzymatic conversion in a concentration dependent manner. Indeed, we could observe the anticipated behavior for four exemplarily chosen compounds (I:47, L10, L15, L21) and the reference econazole. The IC50 values ranged between 19 and 36 μM in the presence of 20 μM substrate (SI, Table S7). The IC50 values were determined for both, substrate depletion and product formation and resulted in similar values (SI, Figure S7). Compared to the determined binding constants, the IC50 values are significantly higher. This is probably caused by the high substrate concentration which was necessary for LC‐MS/MS analysis.[ 24 , 25 ] As expected, there is a correlation between binding affinities of the compounds and their IC50 values. In contrast, the reference compound econazole has a twofold lower IC50 value despite of having a similar binding constant. An explanation for this finding could be the described inhibitory effect of econazole on NADPH‐dependent reductases and G6P dehydrogenase, which are present in the assay.[ 26 , 27 ]
Figure 4.
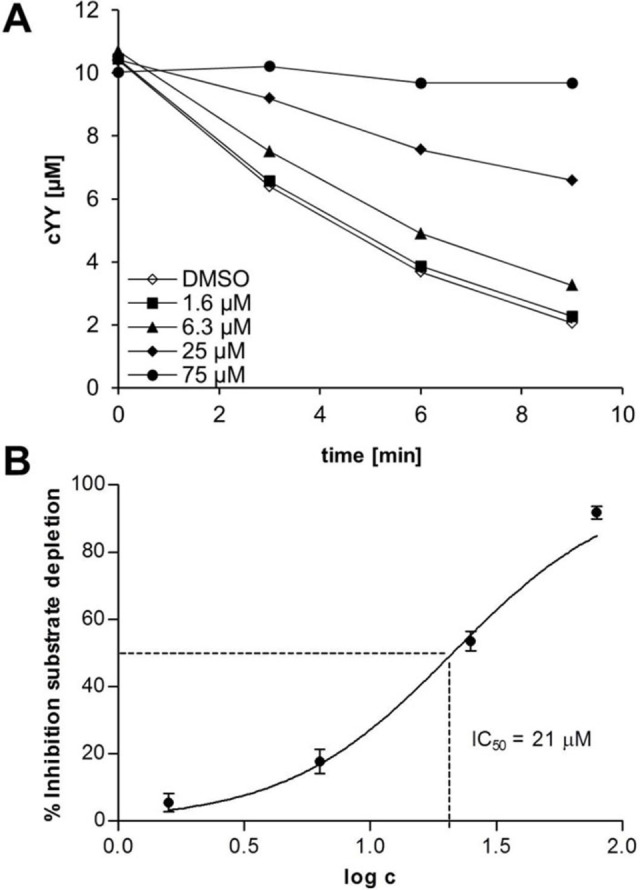
Influence of L21 on the conversion of cYY to mycocyclosin by CYP121. CYP121 was incubated with L21, electron transfer system (Arh1, etp1fd), regenerative system (G6P‐DH, G6P, MgCl2) and substrate cYY. The conversion was stopped by the addition of MeOH with ISTD and analyzed via LC‐MS/MS. DMSO was used as control. A: time‐dependent concentration of cYY for different inhibitor concentrations. The relative inhibition was calculated via determination of the respective reaction velocity. IC50 values were determined through non‐linear regression of inhibition vs. log c (B). The error bars represent the standard deviation of at least 2 replicate measurements.
Effect of cYY on M. bovis BCG growth and CYP121 inhibition
To investigate whether the reduced growth of M. bovis BCG in presence of our compounds was indeed caused by inhibition of CYP121, we evaluated the growth inhibition effect of I:47 in presence of cYY. In the control without inhibitor, the growth was significantly accelerated (Figure 5A). CYP121 was previously shown to be essential for viability, however, the reason for this was not yet elucidated. [6] Our observation suggests that a potential toxic effect of accumulated cYY is rather unlikely and the reaction product mycocyclosin may be essential for growth. In presence of I:47, the growth enhancing effect of cYY was completely blocked at inhibitor concentrations higher than 0.6 μM, whereas at lower concentration the stimulating effect of cYY was partly reduced (Figure 5B). These findings support the hypothesis that the binding of our compounds to the heme iron of CYP121, which is blocking the formation of mycocyclosin, is the main reason for the antimycobacterial activity.
Figure 5.
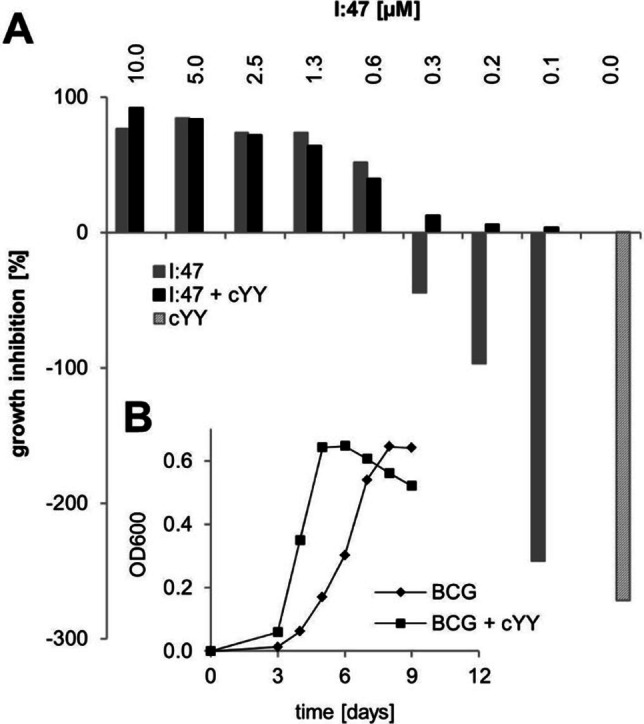
Effect of CYP121 substrate cYY on the growth of M. bovis BCG and its inhibition by I:47. The addition of 50 μM cYY accelerated the growth of M. bovis BCG (B). The resulting growth enhancement of cYY (A, grey striped bar) can be reversed through the CYP121 inhibitor I:47 in a concentration‐dependent manner (grey bars) in accordance to its growth‐inhibiting effect (black bars).
Inhibition of intracellular replication of macrophages
Mtb has the ability to survive and replicate within the macrophages of the host.[ 28 , 29 ] Therefore, we determined whether our compounds can influence the intracellular replication of M. bovis BCG in macrophages. The effects of three recently identified CYP121 inhibitors (I:16, I:48, I:47), [20] two previously described inhibitors of CYP125 (C36, C23) [30] and one dual inhibitor of CYP121 and CYP125 (C43) [30] were evaluated (SI, table S9). CYP125 is part of the igr operon which was identified as a key player for intracellular survival of mycobacteria in macrophages. [31] It catalyses the oxidation of cholest‐4‐en‐3‐one as part of the cholesterol detoxification and is a further promising target for TB treatment. [32]
First of all, the compounds were evaluated regarding their cytotoxicity toward macrophages. None of them showed any significant reduction of viability (SI, Figure S8). Regarding antimycobacterial activity, among the two evaluated CYP125 inhibitors, only C23 showed a strong reduction of intracellular replication (Figure 6). In contrast, C36 showed only little efficacy. This marked difference in antimycobacterial activity could be due to impaired permeability of C36. The compounds have to cross the macrophage cell membrane as well as the mycobacterial cell envelope to reach their target and the structure differences of the two compounds could cause different permeabilities. Furthermore, C23 also binds to CYP121, the selectivity toward CYP125, however, is more than 6‐fold higher. [30]
Figure 6.

Effect on intracellular replication in macrophages. Macrophages were infected with M. bovis BCG and maintained in the presence of selected CYP125 inhibitors C36 and C23, CYP121 inhibitors I:16, I:47 and I:48 or dual inhibitor C43. At 72 h post infection, macrophages were plated on agar to determine intracellular CFU. Penicillin‐Streptomycin and DMSO were used as positive and negative controls, respectively. Significance of results for treated versus untreated cells (DMSO). **p<0.001; ***p<0.0001.
The treatment with CYP121 inhibitors I:47 and I:48 significantly reduced the replication of M. bovis BCG inside macrophages. However, the effect of I:16 was more pronounced. This finding can be explained by additional inhibition of another CYP enzyme present in the mycobacteria. As the imidazole moiety of the compounds is known to be a typical interaction partner for CYP enzymes, inhibition of another CYP cannot be excluded.[ 4 , 33 ]
Interestingly, compound C43 could block the intracellular replication comparable to the penicillin‐streptomycin control. C43 binds to CYP121 and CYP125 with similar affinities and its strong effect is probably caused by these two activities. Thus, the dual inhibition of the two CYP enzymes was shown to play an important role in viability and persistence is a successful strategy to prevent the proliferation of the mycobacteria in macrophages.[ 6 , 31 , 34 ]
Conclusion
The recently published screening of a CYP‐inhibitor library led to the identification of hit compound I:47 with micromolar affinity toward CYP121. Observed activity against Mtb highlighted CYP121 as a possible target for treatment of Mtb infections. [20] We herein presented a continuation of the former study by the screening of a similarity‐oriented library based on the frontrunner compound I:47. The screening approach was comprised of the evaluation of the respective compound class regarding affinity toward CYP121 and biological activity against M. bovis BCG. 38 compounds (hit rate 40 %) showed binding constants lower than or equal to I:47 (KD=5 μM) and a type‐II shift determined in a UV‐Vis heme coordination assay. Despite the visible type‐II shift that has been linked to a direct azole‐iron interaction, co‐crystallisation of our inhibitors revealed a water‐mediated coordination of the heme iron comparable to the results for fluconazole by Seward et al.. [22] Hence, we classify the interaction as fluconazole‐like binding mode.
In contrast, the hit rate regarding activity, which was measured by means of growth inhibition of M. bovis BCG in vitro, was comparatively low (11 %) emphasizing that further criteria like permeability or stability toward mycobacterial metabolism and efflux must be met. Based on the analysis of the structural features for biological activity, we enlarged our library by synthesis of further promising compounds to ensure a comprehensive SAR study. Ten compounds with an ethenyl substituted methylene linker that differ in their Western part aromatic motifs were synthesized accompanied by a fluorine analogue of the former hit I:47.
The quantity of determined binding affinities enabled a broad analysis of structure‐affinity‐relationships. This analysis revealed the tolerance of various substituents at the methylene bridge with increased affinities for hydrophobic space‐filling groups. As the library compounds were principally screened as racemates, the enantiomers were separated for three exemplarily chosen ethenyl derivates. The binding constants revealed only minor differences between the enantiomers and thus a separation was of less importance. Concerning the middle aromatic motif, a negative influence on affinity could be observed for pyridine, furan and partly for thiophene. In contrast, benzene, fluorinated phenyl, indole, benzothiophene, naphthalene or quinolone were accepted with the tendency of increasing affinity for large moieties, indicating that hydrophobic interactions might be involved. For the Western part aromatic motif, a wide range of different aromatic ring systems was tolerated with highest affinities for compounds with a 6‐methoxynaphthalene motif.
Besides the elucidation of the binding mode, we were also interested in the correlation between affinity and antimycobacterial activity. A direct correlation between these two properties would be a straightforward way to validate our target. However, there are several limitations that impair this approach such as the mycobacterial cell wall, which has special permeability properties. There are some models for permeability described in literature, though principal rules for the design of compounds with good penetration properties do not exist.[ 35 , 36 ] Additionally, other factors such as the mycobacterial metabolism of the compound or the involvement of efflux pumps could also impede the correlation. Notably, it was reported that Mtb and M. bovis BCG possess efflux systems, which are able to reduce the intracellular level of econazole in resistant mutants and could possibly also transport other azoles. [37]
Based on the results of our screening, we were able to identify structural features that impair the correlation between affinity and activity. First of all, the substituent at the methylene linker has strong influence on antimycobacterial activity. Only compounds with either no substituent or a methyl or ethenyl substituent as well as compounds with an interconnecting five‐membered ring between linker and adjacent phenyl ring showed an inhibitory effect on growth. Other substituted compounds did not show appropriate growth inhibition despite their high affinities. Similar negative effects had bulky middle parts such as naphthalene or quinolone, which is in accordance to previously described permeability simulation results. [35]
The Western part aromatic motif tolerated diverse functions with the exception of hydroxyl groups, which also impair a good correlation. In accordance with the complex crystal structure of S2, a hydrogen bond donor in para‐position of this motif leads to hydrogen bonding to an aspartate side chain (Asp185) of CYP121, trapping the compound too far away from the heme b water ligand. Compared to the complex crystal structures of L21 and L44, imidazole hydrogen bonding of S2 to the sixth water ligand is thus not possible, leaving it available for the unique binding mode of cYY. A follow‐up study should consider a combination of a western part aromatic motif substituted with a para‐hydroxy group in combination with a longer linker bridge, therefore possibly enabling double hydrogen bonding, which could result in sub‐μM affinity.
In general, the three inhibitors show a similar binding mode, adopting a planar conformation in the solvent channel leading from the protein surface to the active site heme. This solvent channel formed by the F/G loop and their respective helices has been proposed to be the substrate access channel for several P450 enzymes. [38] Indeed, it has been shown to undergo conformational changes upon substrate binding, typically leading to an increase in active site volume. [38] Given the position of cYY and cYY derivatives in substrate‐bound complex structures of CYP121 pointing toward the same entrance channel, it is reasonable to assume an inability of substrate binding due to our inhibitors. [18]
The hit compounds of this screening, which exhibited superior (maximum 2.7‐fold) activity to the former hit I:47, can be structurally divided into three groups: Firstly, the two most active compounds (L15 and L21) possess a propylene linker and a rather simple structure with two phenyl rings connected to each other (the Western part phenyl being fluorinated in L21). The complex crystal structure of L21 provides a probable explanation for this result. The combination of two phenyl rings with a propylene linker leads to an ideal distance between both aromatic motifs for optimal stacking of hydrophobic interactions with Phe168 and Trp182, while allowing for imidazole hydrogen bonding to the heme b water ligand. The second group (L10, L76, L78, L94) has a methylene linker, a benzene ring in the middle part and a Western part aromatic ring with an electron donor function similar to I:47. Finally, the third group of compounds (L45, L46, L14, L44) has a non‐substituted methylene bridge and an electron deprived Western part aromatic ring caused by fluorine or fluorine‐containing substituents. Overall, the compounds offer the possibility for further optimization based on the gained insights into structure‐activity‐relationships.
In order to validate CYP121 as the main target of the described compound class, further mode of action studies were performed. First of all, four exemplarily chosen hit compounds were shown to inhibit the enzymatic conversion of cYY to mycocyclosin in vitro. Furthermore, we evaluated the effect of a potent CYP121 inhibitor (I:47) on the growth of M. bovis BCG in the presence of cYY. In absence of inhibitor, cYY caused a significant growth enhancement of the mycobacteria. However, in presence of I:47, this effect was only observed at very low inhibitor concentrations, indicating that the conversion to mycocyclosin is essential for the intensified growth. These results strengthen the hypothesis that inhibition of CYP121 is the main reason for the antimycobacterial effect of our compounds.
Finally, we were also able to show that compounds targeting CYP121 and/or CYP125 can reduce the intracellular replication of M. bovis BCG in macrophages. The most promising compounds exhibiting this effect appear to be the dual inhibitor C43 and the CYP121 inhibitor I:16. Both could block the intracellular proliferation of mycobacteria, although the results indicate a possible involvement of an additional target for the latter compound. The inhibition of other CYP enzymes cannot be excluded, especially since the genome of Mtb encodes 20 of them, which are in general prone to inhibition by azoles.[ 4 , 39 ] Noticeably, the dual inhibition of two of these CYP enzymes seems to be a successful strategy to address Mtb infections.
In summary, we described a screening approach for the discovery of CYP121 inhibitors which led to a deeper understanding of the binding properties of the respective compound class and revealed possibilities for optimization. Furthermore, we gained insights into structure‐activity‐relationships, highlighted CYP121 inhibition as the main reason for the antimycobacterial activity of our compounds and introduced the concept of dual CYP inhibition as a promising strategy for the treatment of TB. The results of this study lay the foundation for the development and optimization of CYP121 inhibitors as potent drugs against Mtb infections.
Experimental Section
Bacterial strains and growth conditions: Bacterial strains used in this study were Mycobacterium bovis DSM‐43990 (BCGT) and E. coli K12 BL21. Mycobacteria were cultured in Middlebrook 7H9 broth complemented with ADC Enrichment (Middlebrook).
Protein expression and purification: E. coli K12 BL21 (DE3) cells were transformed with plasmid harboring cyp121 gene (pHAT2/cyp121). [16] The enzymes were expressed and purified as described previously.[ 10 , 20 ] For crystallization, eluted CYP121 protein from the 5 mL HisTrap was passed over a HiPrep 26/10 desalting column (GE Healthcare) pre‐equilibrated in 100 mM NaCl, 10 mM HEPES pH 8.0. The total protein amount was estimated from the primary amino acid sequence by UV‐Vis spectroscopy at 280 nm using a NanoDrop 2000 (Thermo Fisher Scientific). The C‐terminal His6‐Tag was removed by addition of a 1 : 500 ratio of Thermolysin and incubation on ice for 1 h. Subsequently, the enzyme was passed over a 5 mL HisTrap and loaded onto a 5 mL HisTrap Q HP column (GE Healthcare) pre‐equilibrated in 100 mM NaCl, 10 mM HEPES pH 8.0. CYP121 was eluted from the Q HP column around a concentration of 200 mM NaCl using a 90 mL gradient elution with a final concentration of 500 mM NaCl. Lastly, CYP121 was passed over a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) pre‐equilibrated in 10 mM Tris pH 7.5. Protein purity was assessed by SDS‐PAGE as well as intact mass spectrometry. Pure protein was concentrated using a 30 kDa cutoff filter and diluted to 700 μM for crystallization, determined by UV‐VIS spectroscopy at 280 nm. The enzyme was flash‐frozen in liquid N2 and stored at −80 °C until further use.
UV/Vis heme P450 binding assay: Optical titration experiments were performed and analyzed as described previously.[ 20 , 40 ]
Determination of BCG MIC50 by OD600 assay: A pre‐culture of M. bovis BCG was grown in 7H9 medium supplemented with ADC Enrichment until OD600 reached a value of 0.7 and was then used for susceptibility testing in the following 72 h. The assay was performed in 48 well plates (Greiner, Kremsmünster, AT). Prior to culture addition, compounds were serially diluted in DMSO to fit a final DMSO concentration of 1 %. For compound susceptibility testing, the pre‐culture was diluted with fresh medium (7H9+ADC enrichment) to a concentration of 1–5×105 cfu/mL. After 4 to 6 days of incubation (until OD600 of positive control approached its maximum) at 37 °C and 80 % air moisture, bacterial growth was measured by determination of OD600. Absorption data was recorded on a Fluostar Omega Multidetection Plate Reader (BMG LABTECH, Ortenberg, DE). The relative inhibition compared to the positive control was plotted against concentration with GraphPad Prism and MIC50 values were fitted using OneSite Log IC50 model provided by the software with the following constraints: bottom=0 and top=100. MIC50 was defined as the concentration at which 50 percent of growth was detected in accordance with previous methods used. [41]
Crystallization: CYP121 was crystallized by mixing 2 μL of 700 μM enzyme with 2 μL of a 500 μL well solution containing 200 mM calcium acetate, 100 mM sodium acetate pH 4.5 and 30 % PEG400 using the Hanging Drop vapour diffusion method. Single crystals appeared after incubating the plates at 4 °C for 3 d, but could be grown to around 400 μm after a 10 d incubation. For complex crystallization, stock solutions of 200 mM compound were prepared in 100 % DMSO. Single crystals of CYP121 were soaked in a 20 mM compound solution containing 10 % DMSO for 24 h at 4 °C, before being mounted in a cryo loop for data collection.
Data collection and refinement: Full, high resolution datasets of the L21, L44 and S2 complexes were collected at ESRF Beamline ID 23‐2 (L21) in France, SLS Beamline Xo6DA (L44) and SLS Beamline X10SA (S2) in Switzerland, respectively. Data reduction using an appropriate resolution cutoff was performed using XDS, [42] Pointless, [43] Aimless [44] and Ctruncate [45] within the CCP4 suite. [46] The structures were solved by molecular replacement with a previously published high resolution structure (PDB ID 1n40) as the search model using Phaser [47] within the Phenix suite. Multiple rounds of manual model building in COOT [48] followed by refinement using phenix.refine [49] were carried out. Ligand structures were modeled with phenix.eLBOW [50] using simple geometry optimization. The structures were validated using MolProbity. [51] All images presented were created using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Determination of influence of cYY on growth and inhibition of I:47: The assay was performed as described above with the following adjustments: The final DMSO concentration was set to 2 % as I:47 was serially diluted in DMSO and eventually supplied with cYY. The readout time point for cultures supplemented with cYY occurred after 3 to 5 days as the addition of cYY induced an earlier onset of exponential growth phase.
CYP121 in vitro enzyme inhibition assay: The enzyme inhibition assay was performed in 200 μL PBS buffer pH 7.2. Compounds were used in concentrations of 1.6, 6.3, 25 and 50, 75 or 100 μM dependent on the respective solubility. The final DMSO concentration was set to 7.5 %. The compounds were incubated with 0.5 μM CYP121 for 10 minutes at 30 °C. After incubation the electron transfer system consisting of Arh1_A18G (2.5 μM), Etp1fd (7.5 μM) and NADPH+H+ (200 μM) as well as a regeneration system consisting of glucose‐6‐phosphate (2 mM), glucose‐6‐phosphate dehydrogenase (1 unit/200 μL) and MgCl2 (1 mM) were added. The reaction was started with the addition of cYY (20 μM) and stopped after 0, 3, 6 and 9 min by addition of 200 μL methanol with internal standard dichloro‐cYY (4 μM final concentration, addition included).
The characterization of CYP121 activity was conducted by a UHPLC‐MS/MS analysis carried out on a TSQ Quantum Access Max mass spectrometer equipped with an ESI‐II source and a triple quadrupole mass detector (Thermo Scientific, Dreieich, Germany). Compounds were separated on an Accucore RP‐MS 150×2.1 mm 2.6 μm column (Thermo Fisher, Waltham, US) by a methanol/water gradient (from 1.4 min–3.5 min 50 % methanol to 3.5 min–5.0 min 90 % methanol) with a flow of 400 μL/min. Compounds were ionized in negative mode by electrospray ionization. Ionization was assisted by a post‐column addition of 2 mM ammonia in methanol with an automated syringe at 1.25 μL/min. Monitored ions were (mother ion [m/z], product ion [m/z], scan time [s], scan width [m/z], collision energy [V], tube lens offset [V], polarity): mycocyclosin: 323.101, 111.100, 0.30, 0.01, 28, 95, negative; CYY: 325.129, 113.043, 0.3, 0.01. 29, 85, negative; internal standard (diiodo‐cYY): 576.878, 126.930, 0.3, 0.01, 43, 77, negative. Samples were injected in a volume of 20 μL. Xcalibur software was used for data acquisition.
For quantification, the ratios of the area under the curve of the educt and the product were converted to respective concentrations on the basis of determined calibration curves of mycocyclosin and cYY which were synthesized as previously described.[ 20 , 52 ] The initial velocity v0 of the cYY conversion was determined by linear fit of the respective concentrations of mycocyclosin or cYY over time (0–9 min) with Microsoft Excel software. The IC50 values were calculated using GraphPad Prism by fitting the relative inhibition of v0 versus concentration with the OneSite Log IC50 model provided by the software with the following constraints: bottom=0 and top=100.
Macrophage cytotoxicity and intracellular macrophage replication: Strain and growth conditions: M. bovis BCG Pasteur strain was grown at 37 °C in Middlebrook 7H9 broth (BD Biosciences) supplemented with 10 % Middlebrook OADC enrichment medium (BD Biosciences), 0.002 % glycerol (Roth), and 0.05 % Tween 80 (Roth). Midlog phase cultures were harvested, aliquoted, and frozen at −80 °C. Bacteria were prepared from frozen stocks by thawing at 37 °C, resuspension in PBS and passage through a 27G needle. Determination of optical density at 580 nm was done to adjust the amount of the bacteria required in cell culture medium without antibiotics.
Cells and media: Bone marrow derived macrophages were prepared from femurs and tibiae remove from C57BL/6 mice using a standard protocol. Briefly, bone marrow cells were cultured for 7 days in complete RPMI (RPMI 1640 plus Glutamax (Gibco), fetal calf serum 10 % (HyClone), 100 U/mL penicillin and 100 μg/mL streptomycin (Biochrom) and 50 μM β‐mercaptoethanol (Gibco)) supplemented with 10 % L929 cell‐conditioned medium as a source of murine M–CSF. On day 7, fully differentiated macrophages were transferred to 96‐well plates. Antibiotics were omitted from cell culture medium 24 hours prior to and during the experiments.
Mycobacteria infection: Macrophages at 200.000 cells/well were seeded on 96 flat well plates (100 μL) 24 hours previous infection in DMEM 10 % L929 CM. After 2.5 hours of infection with M. bovis BCG Pasteur at multiplicity of infection (MOI) 10 : 1, cells were extensively washed with RPMI to remove extracellular mycobacteria. Infected macrophages were maintained in cell culture medium at 37 °C and 5 % CO2 in the presence or absence of the compounds at the indicated concentrations. Penicillin‐Streptomycin and DMSO were used as control at the indicated concentrations.
To monitor the bacterial intracellular survival, cells were lysed with a sterile solution of 0.1 % (vol/vol) Triton X‐100 in H2O, and serial dilutions of lysates were rapidly plated on 7H9 agar plates to enumerate CFU. This procedure was done at 72 hours after compound incubation. Experiments were carried out in triplicate. Each assay was repeated in at least three independent experiments.
Statistical analysis: Statistical analysis was performed with one‐way analysis of variance, followed by a post hoc Tukey test, using Graph‐ Pad Prism 5.0 software. The data are represented as means ± standard errors of the means (SEM).
In vitro cytotoxicity assay: In vitro cytotoxicity assays were performed with murine macrophages generated as mentioned before. Microtiter plates (96 wells) were loaded with at 200.000 cells/well suspended in 100 μL of RPMI 1640 plus Glutamax (GIBCO) supplemented with 10 % heat inactivated fetal calf serum 10 % (HyClone) and 50 μM β‐mercaptoethanol (Gibco) in presence or absence of the compounds at the indicated concentrations. Isoniazid, rifampicin and DMSO were used as control at the indicated concentrations. The plates were incubated for 48 h at 37 °C in 5 % CO2. Cell viability was assessed by measuring using LIVE/DEAD Fixable Aqua Dead Cell Stain Kit by FACS. The data were analyzed using FlowJo (Treestar). Experiments were carried out in triplicate. Each assay was repeated in at least three independent experiments.
Chemical synthesis: Chemical synthesis and analytical characterization: Chemicals were purchased from commercial suppliers and used without further purification. Column flash chromatography was performed on silica gel (40–63 μm), and reaction progress was monitored by TLC on TLC Silica Gel 60 F254 plates (Merck, Darmstadt, Germany). All moisture‐sensitive reactions were performed under nitrogen atmosphere using anhydrous solvents. 1H and 13C NMR spectra were recorded on Bruker Fourier spectrometers (300 MHz) at ambient temperature with the chemical shifts recorded as δ values in ppm units by reference to the hydrogenated residues of deuterated solvent as internal standard. Coupling constants (J) are given in Hertz (Hz), and signal patterns are indicated as follows: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; m, multiplet, br, broad signal. The purity of the final compounds was >95 % measured by HPLC with UV detection at 254 nm. The SpectraSystem LC system consisted of a pump, an autosampler, and a UV/Vis detector (ThermoFisher, Dreieich, Germany). Mass spectrometry was performed on an LC‐coupled Surveyor MSQ electrospray mass spectrometer (ThermoFisher, Dreieich, Germany). The system was operated by the Xcalibur software. A RP C18 NUCLEODUR ec 100‐5 125×3 mm 5 μm column (Macherey‐Nagel GmbH, Düren, Germany) was used as the stationary phase. All solvents were HPLC grade. In a gradient run, the percentage of acetonitrile (containing 0.1 % trifluoroacetic acid) was initially kept constant for 2 min at 30 % and was consequently increased from 30 % at 2 min to 100 % at 8 min, kept at 100 % for 2 min and finally at 30 % for 2 min. The injection volume was 10 μL, and flow rate was set to 700 μL/min. MS analysis was carried out in ESI ionization mode at a spray voltage of 3800 V, a cone voltage of 55 V and a probe temperature of 350 °C. Spectra were acquired in positive mode from 100 to 600 m/z.
Method A: Suzuki coupling
The aldehyde (1 eq.) and the boronic acid (1.3 eq.) were dissolved in toluene (7.5 mL/mmol) and ethanol (4.5 mL/mmol). After addition of 2 M Na2CO3 solution (3 mL/mmol) the mixture was flushed with N2 for 10 min. Subsequently, Pd(PPh3)4 (0.05 eq.) was added and the mixture flushed for further 5 min with N2. The mixture was heated to 100 °C for 1 to 4 h until TLC control showed complete conversion. The reaction was quenched with water (20 mL) and extracted with ethyl acetate (3×20 mL). The combined organic phases were washed with brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by chromatography on silica gel.
1.1: 4‐(6‐methoxynaphthalen‐2‐yl)benzaldehyde (S1b)
Synthesized according to method A using 4‐bromobenzaldehyde (1.35 mmol; 250 mg) and 6‐Methoxy‐2‐naphthaleneboronic acid (1.73 mmol; 350 mg). S1b was obtained as a white solid (330 mg, 93 %): Rf=0.58 (hexane/EtOAc 8 : 2); 1H NMR (300 MHz, CDCl3): δ=3.97 (s, 3H), 7.17–7.25 (m, 2H), 7.75 (dd, J=8.1, 2.2 Hz, 1H), 7.80–7.92 (m, 4H), 7.99 (d, J=8.4 Hz, 2H), 8.05 (d, J=1.5 Hz, 1H), 10.08 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=55.4, 105.6, 119.5, 125.6, 126.4, 127.6, 127.6, 129.0, 129.9, 130.3, 147.2, 158.3, 191.9 ppm; LC–MS (ESI): Rt=9.54 min, m/z: 263.01 [M+H]+.
3’‐chloro‐4’‐hydroxy‐[1,1’‐biphenyl]‐4‐carbaldehyde (S2b)
Synthesized according to method A using 4‐bromobenzaldehyde (1.35 mmol; 250 mg) and 3‐Chloro‐4‐hydroxyphenylboronic acid (1.45 mmol; 250 mg). S2b was obtained as a white solid (112 mg, 36 %): Rf=0.27 (hexane/EtOAc 8 : 2); 1H NMR (300 MHz, CDCl3): δ=5.71 (br. s., 1H), 7.14 (d, J=8.5 Hz, 1H), 7.49 (dd, J=8.5, 2.1 Hz, 1H), 7.64 (d, J=2.1 Hz, 1H), 7.70 (d, J=8.3 Hz, 2H), 7.95 (d, J=8.2 Hz, 2H), 10.06 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=117.2, 121.0, 127.6, 127.9, 128.2, 130.8, 133.7, 135.5, 145.8, 152.2, 192.2 ppm; LC–MS (ESI): Rt=7.22 min, m/z: 232.99 [M+H]+.
3’,4’‐dimethoxy‐[1,1’‐biphenyl]‐4‐carbaldehyde (S3b)
Synthesized according to method A using 4‐bromobenzaldehyde (1.35 mmol; 250 mg) and 3,4‐Dimethoxyphenylboronic acid (1.76 mmol; 320 mg). S3b was obtained as a white solid (342 mg, 100 %): Rf=0.48 (hexane/EtOAc 7 : 3); 1H NMR (300 MHz, CDCld3): δ=3.86–4.02 (m, 6H), 6.98 (d, J=8.3 Hz, 1H), 7.15 (d, J=1.9 Hz, 1H), 7.18–7.29 (m, 1H), 7.73 (d, J=8.1 Hz, 2H), 7.90–7.97 (m, 2H), 10.04 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=56.4, 110.8, 111.9, 120.4, 127.6, 130.7, 132.9, 135.2, 147.4, 149.8, 150.1, 192.2 ppm; LC–MS (ESI): Rt=7.08 min; m/z: 242.99 [M+H]+.
4’‐methoxy‐3’‐methyl‐[1,1’‐biphenyl]‐4‐carbaldehyde (S4b)
Synthesized according to method A using 4‐bromobenzaldehyde (1.35 mmol; 250 mg) and 4‐Methoxy‐3‐methylphenylboronic acid (1.75 mmol; 291 mg). S4b was obtained as a white solid (317 mg, 100 %): Rf=0.54 (hexane/EtOAc 9 : 1); 1H NMR (300 MHz, CDCl3): δ=2.31 (s, 3H), 3.90 (d, J=1.2 Hz, 3H), 6.94 (d, J=8.1 Hz, 1H), 7.41–7.52 (m, 2H), 7.65–7.77 (m, 2H), 7.87–7.97 (m, 2H), 10.04 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=16.4, 55.4, 110.3, 125.8, 127.0, 127.3, 129.6, 130.3, 131.6, 134.6, 147.0, 158.4, 191.9 ppm; LC–MS (ESI): Rt=9.04 min, m/z: 227.04 [M+H]+.
4‐(benzo[d][1,3]dioxol‐5‐yl)benzaldehyde (S5b)
Synthesized according to method A using 4‐bromobenzaldehyde (1.35 mmol; 250 mg) and 3,4‐methylenedioxyphenylboronic acid (1.75 mmol; 290 mg). S5b was obtained as a white solid (322 mg, 100 %): Rf=0.66 (hexane/EtOAc 8 : 2); 1H NMR (300 MHz, CDCl3): δ=6.04 (s, 2H), 6.92 (dd, J=7.7, 0.7 Hz, 1H), 7.10–7.17 (m, 2H), 7.64–7.71 (m, 2H), 7.88–7.97 (m, 2H), 10.04 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=101.4, 107.6, 108.8, 121.3, 127.3, 130.3, 133.9, 134.9, 146.8, 148.1, 148.4, 191.8 ppm; LC–MS (ESI): Rt=8.20 min, m/z: 227.01 [M+H]+.
3’‐fluoro‐4’‐methoxy‐[1,1’‐biphenyl]‐4‐carbaldehyde (S6b)
Synthesized according to method A using 4‐bromobenzaldehyde (1.35 mmol; 250 mg) and 3‐Fluoro‐4‐methoxybenzeneboronic acid (1.75 mmol; 297 mg). S6b was obtained as a white solid (289 mg, 93 %): Rf=0.46 (hexane/EtOAc 8 : 2); 1H NMR (300 MHz, CDCl3): δ=3.96 (s, 3H), 7.01–7.13 (m, 1H), 7.34–7.45 (m, 2H), 7.70 (d, J=8.5 Hz, 2H), 7.91–7.99 (m, 2H), 10.05 (s, 1H); 13C NMR (75 MHz, CDCl3): δ=56.4 (s), 113.7 (d, J=2.2 Hz), 115.0 (d, J=18.6 Hz), 123.1 (d, J=3.7 Hz), 127.1 (s), 130.3 (s), 132.7 (d, J=6.7 Hz), 135.1 (s), 145.6 (d, J=1.5 Hz), 148.1 (d, J=10.4 Hz), 152.6 (d, J=246.6 Hz), 191.8 ppm (s); LC–MS (ESI): Rt=8.50 min, m/z: 231.05 [M+H]+.
Method B: Grignard reaction
The aldehyde (1.0 eq.) was dissolved in a heat dried flask in dry THF (2 mL/mmol) and cooled to −15 °C in an ice/acetone bath. Vinylmagnesium bromide (1.2 eq., 0.7 M in THF) was added dropwise and the reaction was stirred for 5 min before letting it warm to room temperature for 1–2 h and TLC showed complete conversion. The reaction mixture was quenched with saturated NH4Cl solution (15 mL) and extracted with ethyl acetate (3×20 mL). The combined organic phases were washed with brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by chromatography on silica gel.
1‐(4‐(6‐methoxynaphthalen‐2‐yl)phenyl)prop‐2‐en‐1‐ol (S1a)
Synthesized according to method B using 1b (1.14 mmol; 300 mg) and vinylmagnesium bromide (0.7 M in THF; 1.37 mmol; 2.0 mL). S1a was obtained as a yellow solid (201 mg, 61 %): Rf=0.33 (hexane/EtOAc 8 : 2); 1H NMR (300 MHz, CDCl3): δ=3.96 (s, 3H), 5.21–5.32 (m, 2H), 5.43 (dd, J=17.1, 1.3 Hz, 1H), 6.13 (ddd, J=16.9, 10.6, 6.1 Hz, 1H), 7.14–7.22 (m, 2H), 7.50 (d, J=8.1 Hz, 2H), 7.71 (d, J=8.1 Hz, 3H), 7.77‐7.85 (m, 2H), 7.98 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=55.8, 75.6, 106.0, 115.7, 119.6, 126.0, 126.4, 127.3, 127.7, 127.8, 130.1, 134.2, 136.4, 140.6, 141.2, 141.8, 158.2 ppm; LC–MS (ESI): Rt=8.94 min, m/z: 274.08 [M‐OH]+.
3‐chloro‐4’‐(1‐hydroxyallyl)‐[1,1’‐biphenyl]‐4‐ol (S2a)
Synthesized according to method B using 2b (0.25 mmol; 60 mg) and vinylmagnesium bromide (0.7 M in THF; 0.49 mmol; 0.7 mL). S2a was obtained as a yellow solid (44 mg, 67 %): Rf=0.44 (hexane/EtOAc 7 : 3); 1H NMR (300 MHz, CDCl3): δ=5.20–5.29 (m, 2H), 5.35–5.44 (m, 1H), 5.60 (br. s., 1H), 6.09 (ddd, J=16.9, 10.5, 6.1 Hz, 1H), 7.09 (d, J=8.5 Hz, 1H), 7.38‐7.47 (m, 3H), 7.49–7.54 (m, 2H), 7.56 ppm (d, J=2.2 Hz, 1H).
1‐(3’,4’‐dimethoxy‐[1,1’‐biphenyl]‐4‐yl)prop‐2‐en‐1‐ol (S3a)
Synthesized according to method B using 3b (1.24 mmol; 300 mg) and vinylmagnesium bromide (0.7 M in THF; 1.48 mmol; 2.1 mL). S3a was obtained as a yellow solid (230 mg, 69 %): Rf=0.37 (hexane/EtOAc 7 : 3); 1H NMR (300 MHz, CDCl3): δ=3.93 (s, 3H), 3.95 (s, 3H), 5.21–5.29 (m, 2H), 5.40 (d, J=17.1 Hz, 1H), 6.10 (ddd, J=16.9, 10.5, 6.0 Hz, 1H), 6.95 (d, J=8.3 Hz, 1H), 7.09‐7.18 (m, 2 H), 7.42–7.46 (m, 2 H), 7.53–7.59 ppm (m, 2 H); 13C NMR (75 MHz, CDCl3): δ=55.9, 56.0, 75.1, 110.4, 111.5, 115.2, 119.4, 126.7, 127.0, 133.8, 140.2, 140.6, 141.2, 148.7, 149.2 ppm; LC–MS (ESI): Rt=6.48 min, m/z: 254.04 [M‐OH]+.
1‐(4’‐methoxy‐3’‐methyl‐[1,1’‐biphenyl]‐4‐yl)prop‐2‐en‐1‐ol (S4a)
Synthesized according to method B using 4b (1.10 mmol; 250 mg) and vinylmagnesium bromide (0.7 M in THF; 1.40 mmol; 2.0 mL). S4a was obtained as a yellow solid (96 mg, 34 %): Rf=0.26 (hexane/EtOAc 9 : 1); 1H NMR (300 MHz, CDCl3): δ=2.29 (s, 3H), 3.88 (s, 3H), 5.20–5.29 (m, 2H), 5.40 (d, J=17.0 Hz, 1H), 6.11 (ddd, J=16.8, 10.4, 6.0 Hz, 1H), 6.90 (d, J=8.9 Hz, 1H), 7.37–7.45 (m, 4H), 7.53‐7.58 ppm (m, 2H); 13C NMR (75 MHz, CDCl3): δ=15.9, 55.0, 74.8, 109.8, 114.7, 124.9, 126.3, 126.5, 126.5, 129.0, 132.5, 139.8, 140.2, 140.4, 157.0 ppm; LC–MS (ESI): Rt=8.66 min, m/z: 238.09 [M‐OH]+.
1‐(4‐(benzo[d][1,3]dioxol‐5‐yl)phenyl)prop‐2‐en‐1‐ol (S5a)
Synthesized according to method B using 5b (1.24 mmol; 280 mg) and vinylmagnesium bromide (0.7 M in THF; 1.49 mmol; 2.1 mL). S5a was obtained as a yellow solid (209 mg, 66 %): Rf=0.48 (hexane/EtOAc 8 : 2;. 1H NMR (300 MHz, CDCl3): δ=5.18–5.29 (m, 2H), 5.40 (dd, J=17.1, 1.1 Hz, 1H), 6.10 (ddd, J=16.9, 10.5, 6.1 Hz, 1H), 6.85–6.92 (m, 1H), 7.03‐7.10 (m, 2H), 7.43 (d, J=8.1 Hz, 2H), 7.52 ppm (d, J=8.5 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ=75.1, 101.1, 107.6, 108.6, 115.2, 120.6, 126.7, 127.0, 135.2, 140.1, 140.4, 141.3, 147.1, 148.1 ppm; LC–MS (ESI): Rt=7.65 min, m/z: 238.04 [M‐OH]+.
Method C: CDI reaction
The alcohol (1.0 eq.) and 1,1’‐carbonyldiimidazole (3.0 eq.) were dissolved in acetonitrile (15 mL/mmol) and heated to reflux for 1–4 h until TLC control showed complete conversion. The reaction was quenched with water (15 mL) and extracted with ethyl acetate (3×20 mL). The combined organic phases were washed with brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by chromatography on silica gel.
1‐(1‐(4‐(6‐methoxynaphthalen‐2‐yl)phenyl)allyl)‐1H‐imidazole (S1)
Synthesized according to method C using 1a (100 mg, 0.34 mmol) and CDI (162 mg, 1.0 mmol). S1 was obtained as a light yellow solid (39 mg, 34 %): Rf=0.19 (EtOAc); 1H NMR (300 MHz, CDCl3): δ=3.95 (s, 3H), 5.20 (d, J=17.0 Hz, 1H), 5.47 (d, J=10.3 Hz, 1H), 5.85 (d, J=6.3 Hz, 1H), 6.33 (ddd, J=17.0 Hz, 10.2 Hz, 6.3 Hz, 1H), 6.94 (s, 1H), 7.18 (m, 3H), 7.29 (m, 2H), 7.58 (s, 1H), 7.70 (m, 3H), 7.81 (m, 2H), 7.97 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=55.8, 63.7, 106.0, 119.0, 119.7, 119.8, 126.1, 126.2, 127.8, 128.1, 128.4, 129.5, 129.9, 130.1, 134.4, 135.8, 136.2, 137.1, 137.3, 141.9, 158.4 ppm; LC‐MS (ESI): Rt=6.77 min, m/z: 341.18 [M+H]+, 237.04 [M‐Imidazole]+.
4’‐(1‐(1H‐imidazol‐1‐yl)allyl)‐3‐chloro‐[1,1’‐biphenyl]‐4‐ol (S2)
Synthesized according to method C using 2a (64 mg, 0.25 mmol) and CDI (121 mg, 0.75 mmol). S2 was obtained as a yellow oil (24 mg, 31 %): Rf=0.27 (EtOAc); 1H NMR (300 MHz, CDCl3): δ=5.18 (d, J=17.0 Hz, 1H), 5.47 (d, J=10.2 Hz, 1H), 5.82 (d, J=6.2 Hz, 1H), 6.30 (ddd, J=16.9 Hz, 10.4 Hz, 6.3 Hz, 1H), 6.94 (s, 1H), 7.08 (d, J=8.4 Hz, 1H), 7.15 (s, 1H), 7.25 (m, 2H), 7.34 (dd, J= 8.4 Hz, 2.2 Hz, 1H), 7.54 (m, 3H), 7.60 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=63.4, 116.9, 118.7, 119.6, 121.0, 126.6, 127.2, 127.9, 128.6, 132.9, 135.4, 136.4, 136.5, 140.2, 152.5 ppm; LC‐MS (ESI): Rt=2.46 min, m/z: 311.15 [M+H]+, 243.09 [M–Imidazole]+.
1‐(1‐(3’,4’‐dimethoxy‐[1,1’‐biphenyl]‐4‐yl)allyl)‐1H‐imidazole (S3)
Synthesized according to method C using 3a (200 mg, 0.74 mmol) and CDI (360 mg, 2.22 mmol). S3 was obtained as a yellow oil (70 mg, 30 %): Rf=0.27 (EtOAc); 1H NMR (300 MHz, CDCl3): δ=3.93 (d, J=6.89 Hz, 6H), 5.17 (d, J=17.04 Hz, 1H), 5.44 (d, J=10.24 Hz, 1H), 5.81 (d, J=6.24 Hz, 1H), 6.30 (ddd, J=16.88, 10.36, 6.29 Hz, 1H), 6.90‐6.98 (m, 2H), 7.07–7.16 (m, 3H), 7.21–7.28 (m, 2H), 7.53‐7.59 ppm (m, 3H); 13C NMR (75 MHz, CDCl3): δ=55.9, 63.2, 110.3, 111.5, 118.5, 119.2, 119.4, 127.2, 127.8, 129.3, 133.1, 135.7, 136.6, 141.2, 148.8, 149.2 ppm; LC‐MS (ESI): Rt=2.04 min, m/z: 253.13 [M‐Imidazole]+.
1‐(1‐(4’‐methoxy‐3’‐methyl‐[1,1’‐biphenyl]‐4‐yl)allyl)‐1H‐imidazole (S4)
Synthesized according to method C using 4a (95 mg, 0.37 mmol) and CDI (182 mg, 1.1 mmol). S4 was obtained as a yellow oil (51 mg, 45 %): Rf=0.33 (EtOAc); 1H NMR (300 MHz, CDCl3): δ=2.29 (s, 3H), 3.87 (s, 3H), 5.17 (d, J=17.04 Hz, 1H), 5.44 (d, J=10.24 Hz, 1H), 5.81 (d, J=6.33 Hz, 1H), 6.30 (ddd, J=16.86, 10.34, 6.33 Hz, 1H), 6.86‐6.95 (m, 2H), 7.11 (s, 1H), 7.20–7.29 (m, 2H), 7.35–7.43 (m, 2H), 7.53–7.61 ppm (m, 3H); 13C NMR (75 MHz, CDCl3): δ=16.3, 55.3, 63.2, 110.2, 118.5, 119.1, 125.3, 126.9, 127.1, 127.8, 129.2, 129.3, 132.2, 135.8, 136.2, 136.6, 141.2, 157.6 ppm. LC‐MS (ESI): Rt=4.40 min, m/z: 237.18 [M‐Imidazole]+.
1‐(1‐(4‐(benzo[d][1,3]dioxol‐5‐yl)phenyl)allyl)‐1H‐imidazole (S5)
Synthesized according to method C using 5a (200 mg, 0.79 mmol) and CDI (382 mg, 2.35 mmol). S5 was obtained as a colorless oil (65 mg, 27 %): Rf=0.26 (EtOAc/MeOH 98 : 2); 1H NMR (300 MHz, CDCl3): δ=5.17 (d, J=17.23 Hz, 1H), 5.44 (d, J=10.24 Hz, 1H), 5.80 (d, J=6.15 Hz, 1H), 6.00 (s, 2H), 6.29 (ddd, J=16.86, 10.34, 6.33 Hz, 1H), 6.84–6.94 (m, 2H), 7.01–7.13 (m, 3H), 7.22 (d, J=8.10 Hz, 2H), 7.47–7.58 ppm (m, 3H); 13C NMR (75 MHz, CDCl3): δ=63.2, 101.2, 107.5, 108.6, 118.5, 119.3, 120.7, 127.3, 127.9, 129.4, 134.6, 135.7, 136.7, 136.8, 141.2, 147.4, 148.2 ppm. LC‐MS (ESI): Rt=4.32 min, m/z: 305.14 [M+H]+, 237.09 [M‐Imidazole]+.
Method D: TBDMS protection
a) 4‐Bromophenol (1.0 eq.) and imidazole (1.1 eq.) were dissolved in dry CH2Cl2 (3 mL/mmol) before TBDMS chloride (1.1 eq.) was added. The reaction was stirred at room temperature overnight and quenched with water (30 mL) and 1 M HCl (3 mL). The aqueous phase was extracted with CH2Cl2 (2×30 mL) and the combined organic phases were washed with brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by chromatography on silica gel.
(4‐bromophenoxy)(tert‐butyl)dimethylsilane (S9e)
Synthesized according to method D a) using 4‐Bromophenol (2.89 mmol; 500 mg), imidazole (3.18 mmol; 216 mg) and TBDMSCl (3.18 mmol; 479 mg). S9e was obtained as a colorless oil (810 mg, 98 %): Rf=0.89 (hexane/EtOAc 8 : 2); 1H NMR (300 MHz, CDCl3): δ=0.19 (s, 6H), 0.99 (s, 9H), 6.70–6.75 (m, 2H), 7.30‐7.35 ppm (m, 2H); 13C NMR (75 MHz, CDCl3): δ=−4.5, 18.2, 25.6, 113.6, 121.9, 132.3, 154.9 ppm; LC‐MS (ESI): Rt=11.59 min.
b) NaH (1.2 eq.) was dissolved in acetonitrile (1 mL/mmol) and cooled to 0 °C. 5‐Bromo‐1H‐indole was dissolved in acetonitrile (1 mL/mmol) and slowly added to the NaH suspension. After stirring for 15 min TBDMS chloride (1.25 eq.) was added and the suspension was let warm to room temperature and stirred overnight. The reaction was quenched with saturated NH4Cl (10 mL) solution and extracted with CH2Cl2 (3×20 mL). The combined organic phases were washed with brine and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was purified by chromatography on silica gel.
5‐bromo‐1‐(tert‐butyldimethylsilyl)‐1H‐indole (S10e)
Synthesized according to method D b) using 5‐Bromo‐1H‐indole (2.55 mmol; 500 mg), NaH (3.06 mmol; 73.4 mg) and TBDMSCl (3.19 mmol; 480 mg). S10e was obtained as a colorless oil (267 mg, 34 %): Rf=0.95 (hexane/EtOAc 9 : 1); 1H NMR (300 MHz, CDCl3): δ=0.60 (s, 6H), 0.92 (s, 9H), 6.56 (d, J=3.2 Hz, 1H), 7.18 (d, J=3.2 Hz, 1H), 7.23 (dd, J=8.8, 2.0 Hz, 1H), 7.38 (d, J=8.8 Hz, 1H), 7.75 ppm (d, J=2.0 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ=−4.0, 19.4, 26.2, 104.3, 113.1, 115.1, 123.1, 124.1, 132.2, 133.2, 139.6 ppm; LC‐MS (ESI): Rt=11.40 min, m/z: 312.11 [M+H]+.
Method E: Boronic acid preparation
The TBDMS protected alcohol or amine (1.0 eq.) was dissolved in dry THF (1 mL/mmol) and cooled to −78 °C. nBuLi (1.25 eq., 2.5 M in THF) was added dropwise and the solution was stirred for 30 min. Triisopropylborate (2.5 eq.) was added slowly and the mixture was stirred for further 45 min at −78 °C. After letting the mixture warm to room temperature for 3 h it was quenched with ethyl acetate : 1 M HCl=1 : 1 (10 mL). The organic layer was separated, washed with brine (2×10 mL) and dried over MgSO4. The solvent was evaporated under reduced pressure and the crude product was washed with cold hexane followed by warm water and dried under reduced pressure.
(4‐((tert‐butyldimethylsilyl)oxy)phenyl)boronic acid (S9d)
Synthesized according to method E using 9e (2.78 mmol; 800 mg), nBuLi (2.5 M in THF; 3.48 mmol; 1.4 mL) and B(OiPr)3 (5.56 mmol; 970 mg). S9d was obtained as an orange solid (400 mg, 57 %): 1H NMR (300 MHz, CDCl3): δ=0.27 (s, 6H), 1.03 (s, 9H), 6.96 (d, J=8.5 Hz, 2H), 8.12 ppm (d, J=8.5 Hz, 2H); LC‐MS (ESI): Rt=9.03 min.
(1‐(tert‐butyldimethylsilyl)‐1H‐indol‐5‐yl)boronic acid (S10d)
Synthesized according to method E using 10e (2.58 mmol; 800 mg), nBuLi (2.5 M in THF; 3.23 mmol; 1.3 mL) and B(OiPr)3 (5.16 mmol; 1.05 g). S10d was obtained as a white solid (487 mg, 69 %): Rf=0.93 (hexane/EtOAc 9 : 1), 1H NMR (300 MHz, CDCl3): δ=0.69 (s, 5H), 1.00 (s, 8H), 6.81 (d, J=2.9 Hz, 1H), 7.26–7.30 (m, 1H), 7.69 (d, J=8.4 Hz, 1H), 8.16 (d, J=8.4 Hz, 1H), 8.69 ppm (s, 1H); 13C NMR (75 MHz, CDCl3): δ=−3.9, 19.6, 26.3, 105.6, 113.4, 128.4, 129.6, 131.2, 131.3, 144.0 ppm; LC–MS (ESI): Rt=8.84 min, m/z: 276.18 [M+H]+.
Method F: TBDMS deprotection
The aldehyde (1.0 eq.) was dissolved in dry THF (9 mL/mmol), tetrabutylammonium fluoride solution (0.5 eq., 1 M in THF) and 0.1 M K2HPO4 solution (100 μL/mmol, pH 7) were added and the reaction mixture was stirred overnight. The solvent was evaporated under reduced pressure and the crude product was purified by chromatography on silica gel.
5‐(4‐(1‐(1H‐imidazol‐1‐yl)allyl)phenyl)‐1H‐indole (S10)
Synthesized according to method F using 10a (106 mg, 0.25 mmol) and TBAF (0.13 mL 1 M sol., 0.13 mmol). S10 was obtained as a light brown solid (57 mg, 76 %): Rf=0.20 (EtOAc/MeOH 98 : 2); 1H NMR (300 MHz, CDCl3): δ=5.20 (dt, J=17.0, 1.1 Hz, 1H), 5.46 (dt, J=10.2, 1.0 Hz, 1H), 5.83 (d, J=6.2 Hz, 15H), 6.33 (ddd, J=16.9, 10.4, 6.3 Hz, 1H), 6.61–6.64 (m, 1H), 6.94–6.97 (m, 1H), 7.13 (s, 1H), 7.24–7.30 (m, 3H), 7.41–7.49 (m, 2H), 7.58 (s, 1H), 7.64–7.69 (m, 2H), 7.86 (d, J=0.8 Hz, 1H), 8.38 ppm (br s, 1H); 13C NMR (75 MHz, CDCl3): δ=63.5, 102.9, 111.4, 118.7, 119.2, 119.2, 121.6, 125.1, 127.8, 128.4, 128.9, 132.2, 135.5, 135.8, 135.8, 136.6, 142.9 ppm. LC‐MS (ESI): Rt=3.02 min, m/z: 300.19 [M+H]+, 232.18 [M‐Imidazole]+.
4’‐(1‐(1H‐imidazol‐1‐yl)allyl)‐[1,1’‐biphenyl]‐4‐ol (S9)
Synthesized according to method F using 9a (55 mg, 0.14 mmol) and TBAF (0.07 mL 1 M sol., 0.07 mmol). S9 was obtained as a white solid (7.8 mg, 20 %): Rf=0.19 (EtOAc); 1H NMR (300 MHz, CD3OD): δ=4.59 (s, 1H), 5.18 (dt, J=16.9, 1.3 Hz, 1H), 5.44 (dt, J=10.2, 1.1 Hz, 1H), 6.04 (d, J=6.4 Hz, 1H), 6.42 (ddd, J=16.9, 10.3, 6.4 Hz, 1H), 6.82–6.88 (m, 2H), 7.01 (t, J=1.1 Hz, 1H), 7.10 (t, J=1.3 Hz, 1H), 7.29 (d, J=8.1 Hz, 2H), 7.43–7.49 (m, 2H), 7.55–7.62 (m, 2H), 7.70 ppm (s, 1H); 13C NMR (75 MHz, CD3OD): δ=64.7, 116.9, 119.5, 120.2, 128.0, 129.2, 129.2, 133.0, 137.7, 138.0, 138.2, 142.8, 158.7 ppm; LC‐MS (ESI): Rt=1.47 min, m/z: 277.09 [M+H]+, 209.09 [M‐Imidazole]+.
1‐(4‐Bromobenzyl)‐1H‐imidazole (S11a):
NaH (19.5 mmol, 800 mg, 60 % in paraffin oil) was suspended in 3 mL dry DMF. After cooling to 4 °C a solution of imidazole (15.0 mmol, 1.02 g) in DMF (3 mL) was added dropwise. The solution was stirred at 4 °C for 1 h. 1‐Bromo‐4‐(bromomethyl)benzene (15.0 mmol, 3.75 g) was dissolved in 5 mL DMF added dropwise to the reaction mixture which was subsequently warmed to room temperature and then to 60 °C for 1.5 h. The reaction was quenched with H2O (5 mL) and extracted with ethyl acetate (3×15 mL). The combined organic phases were washed with brine, dried over MgSO4 and the solvent was evaporated under reduced pressure. Remaining DMF was removed by repeated addition of n‐heptane and evaporation under reduced pressure. The crude product was purified by chromatography on silica gel (EtOAc : petrol ether=9 : 1). The product was obtained as white solid (2.10 g, 59 %): Rf=0.21 (EtOAc); 1H NMR (500 MHz, DMSO‐d6 ): δ=5.19 (s, 2H), 6.92 (m, 1H), 7.20 (m, 2H), 7.56 (m, 3H), 7.75 ppm (m, 1H); 13C NMR (125 MHz, DMSO‐d6 ): δ=86.2, 157.0, 158.3, 166.3, 167.1, 169.0, 174.8, 174.9 ppm; LC‐MS (ESI): Rt=4.99 min, m/z: 236.9 [M+H]+.
Enantiomer separation and analysis: The enantiomers were separated using a Daicel ChiralPak® IE column (5 μm, 10×250 mm) with a preparative HPLC (Thermo Scientific Ultimate 3000, consisting of Dionex Ultimate 3000 pump, diode array detector and automated fraction collector) monitoring the absorbance at 254 nm. An isocratic flow of 5 mL/min was applied with the following solvent composition: Compound S1 and S4: methyl‐tert‐butyl ether (MtBE) : (EtOH+0.5 % ethylendiamine)=98 : 2, Compound S5: MtBE:(EtOH+0.5 % ethylendiamine)=96 : 4.
Protein Data Bank submissions
The complex structures of CYP121 with bound L21, L44 and S2 have been submitted to the RCSB Protein Data Bank (PDB IDs 6TET, 6TEV and 6TE7, respectively).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We acknowledge the use of ESRF Beamline ID 23–2 and SLS Beamlines X06DA and X10SA. Open Access funding enabled and organized by Projekt DEAL.
I. Walter, S. Adam, M. V. Gentilini, A. M. Kany, C. Brengel, A. Thomann, T. Sparwasser, J. Köhnke, R. W. Hartmann, ChemMedChem 2021, 16, 2786.
References
- 1.WHO, Global Tuberculosis Report 2017, 2017.
- 2. Zhang Y., Yew W. W., Int J Tuberc Lung Dis 2009, 13, 1320–1330. [PubMed] [Google Scholar]
- 3. Corbett E. L., Watt C. J., Walker N., Maher D., Williams B. G., Raviglione M. C., Dye C., Arch. Intern. Med. 2003, 163, 1009–1021. [DOI] [PubMed] [Google Scholar]
- 4. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., C. E. Barry 3rd , Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G., Nature 1998, 393, 537–544. [DOI] [PubMed] [Google Scholar]
- 5. Brennan P. J., Nikaido H., Annu. Rev. Biochem. 1995, 64, 29–63. [DOI] [PubMed] [Google Scholar]
- 6. McLean K. J., Carroll P., Lewis D. G., Dunford A. J., Seward H. E., Neeli R., Cheesman M. R., Marsollier L., Douglas P., Smith W. E., Rosenkrands I., Cole S. T., Leys D., Parish T., Munro A. W., J. Biol. Chem. 2008, 283, 33406–33416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.UniProt-Consortium, “UniProtKB Protein Knowledgebase”, can be found under http://www.uniprot.org/, 2018. [DOI] [PMC free article] [PubMed]
- 8. Gondry M., Sauguet L., Belin P., Thai R., Amouroux R., Tellier C., Tuphile K., Jacquet M., Braud S., Courcon M., Masson C., Dubois S., Lautru S., Lecoq A., Hashimoto S., Genet R., Pernodet J. L., Nat. Chem. Biol. 2009, 5, 414–420. [DOI] [PubMed] [Google Scholar]
- 9. Belin P., Le Du M. H., Fielding A., Lequin O., Jacquet M., Charbonnier J. B., Lecoq A., Thai R., Courcon M., Masson C., Dugave C., Genet R., Pernodet J. L., Gondry M., Proc. Natl. Acad. Sci. USA 2009, 106, 7426–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McLean K. J., Cheesman M. R., Rivers S. L., Richmond A., Leys D., Chapman S. K., Reid G. A., Price N. C., Kelly S. M., Clarkson J., Smith W. E., Munro A. W., J. Inorg. Biochem. 2002, 91, 527–541. [DOI] [PubMed] [Google Scholar]
- 11. Ahmad Z., Sharma S., Khuller G. K., Singh P., Faujdar J., Katoch V. M., Int. J. Antimicrob. Agents 2006, 28, 543–544. [DOI] [PubMed] [Google Scholar]
- 12. Ahmad Z., Sharma S., Khuller G. K., FEMS Microbiol. Lett. 2006, 261, 181–186. [DOI] [PubMed] [Google Scholar]
- 13. Ahmad Z., Sharma S., Khuller G. K., FEMS Microbiol. Lett. 2006, 258, 200–203. [DOI] [PubMed] [Google Scholar]
- 14. Kavanagh M. E., Coyne A. G., McLean K. J., James G. G., Levy C. W., Marino L. B., de Carvalho L. P., Chan D. S., Hudson S. A., Surade S., Leys D., Munro A. W., Abell C., J. Med. Chem. 2016, 59, 3272–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kavanagh M. E., Gray J. L., Gilbert S. H., Coyne A. G., McLean K. J., Davis H. J., Munro A. W., Abell C., ChemMedChem 2016, 11, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hudson S. A., McLean K. J., Surade S., Yang Y. Q., Leys D., Ciulli A., Munro A. W., Abell C., Angew. Chem. Int. Ed. Engl. 2012, 51, 9311–9316. [DOI] [PubMed] [Google Scholar]
- 17. Hudson S. A., Surade S., Coyne A. G., McLean K. J., Leys D., Munro A. W., Abell C., ChemMedChem 2013, 8, 1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fonvielle M., Le Du M. H., Lequin O., Lecoq A., Jacquet M., Thai R., Dubois S., Grach G., Gondry M., Belin P., J. Biol. Chem. 2013, 288, 17347–17359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belin P., Lecoq A., Beaurepaire Ledu M. H., Gondry M., Pernodet J. L., Patent EP2088143 A1 2009. [Google Scholar]
- 20. Brengel C., Thomann A., Schifrin A., Allegretta G., Kamal A. A. M., Haupenthal J., Schnorr I., Cho S. H., Franzblau S. G., Empting M., Eberhard J., Hartmann R. W., ChemMedChem 2017, 12, 1616–1626. [DOI] [PubMed] [Google Scholar]
- 21. Chenge J. T., Duyet L. V., Swami S., McLean K. J., Kavanagh M. E., Coyne A. G., Rigby S. E., Cheesman M. R., Girvan H. M., Levy C. W., Rupp B., von Kries J. P., Abell C., Leys D., Munro A. W., J. Biol. Chem. 2017, 292, 1310–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seward H. E., Roujeinikova A., McLean K. J., Munro A. W., Leys D., J. Biol. Chem. 2006, 281, 39437–39443. [DOI] [PubMed] [Google Scholar]
- 23. Leys D., Mowat C. G., McLean K. J., Richmond A., Chapman S. K., Walkinshaw M. D., Munro A. W., J. Biol. Chem. 2003, 278, 5141–5147. [DOI] [PubMed] [Google Scholar]
- 24. Burlingham B. T., Widlanski T. S., J. Chem. Educ. 2003, 80, 214. [Google Scholar]
- 25.R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, Wiley, John, 2013. [PubMed]
- 26. Kurokohchi K., Nishioka M., Ichikawa Y., J. Steroid Biochem. Mol. Biol. 1992, 42, 287–292. [DOI] [PubMed] [Google Scholar]
- 27. Raab W., Hogl F., Mykosen 1981, 24, 525–533. [PubMed] [Google Scholar]
- 28. Armstrong J. A., Hart P. D., J. Exp. Med. 1975, 142, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russell D. G., Nat. Rev. Mol. Cell Biol. 2001, 2, 569–577. [DOI] [PubMed] [Google Scholar]
- 30. Brengel C., Thomann A., Schifrin A., Eberhard J., Hartmann R. W., ChemMedChem 2016, 11, 2385–2391. [DOI] [PubMed] [Google Scholar]
- 31. Chang J. C., Harik N. S., Liao R. P., Sherman D. R., J. Infect. Dis. 2007, 196, 788–795. [DOI] [PubMed] [Google Scholar]
- 32. Capyk J. K., Kalscheuer R., Stewart G. R., Liu J., Kwon H., Zhao R., Okamoto S., W. R. Jacobs, Jr. , Eltis L. D., Mohn W. W., J. Biol. Chem. 2009, 284, 35534–35542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Souter A., McLean K. J., Smith W. E., Munro A. W., J. Chem. Technol. Biotechnol. 2000, 75, 933–941. [Google Scholar]
- 34. Hudson S. A., McLean K. J., Munro A. W., Abell C., Biochem. Soc. Trans. 2012, 40, 573–579. [DOI] [PubMed] [Google Scholar]
- 35. Hong X., Hopfinger A. J., Biomacromolecules 2004, 5, 1066–1077. [DOI] [PubMed] [Google Scholar]
- 36. Hong X., Hopfinger A. J., Biomacromolecules 2004, 5, 1052–1065. [DOI] [PubMed] [Google Scholar]
- 37. Milano A., Pasca M. R., Provvedi R., Lucarelli A. P., Manina G., Ribeiro A. L., Manganelli R., Riccardi G., Tuberculosis (Edinb) 2009, 89, 84–90. [DOI] [PubMed] [Google Scholar]
- 38. Urban P., Lautier T., Pompon D., Truan G., Int. J. Mol. Sci. 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McLean K. J., Munro A. W., Drug Metab. Rev. 2008, 40, 427–446. [DOI] [PubMed] [Google Scholar]
- 40. McLean K. J., Marshall K. R., Richmond A., Hunter I. S., Fowler K., Kieser T., Gurcha S. S., Besra G. S., Munro A. W., Microbiology 2002, 148, 2937–2949. [DOI] [PubMed] [Google Scholar]
- 41. Franzblau S. G., DeGroote M. A., Cho S. H., Andries K., Nuermberger E., Orme I. M., Mdluli K., Angulo-Barturen I., Dick T., Dartois V., Lenaerts A. J., Tuberculosis (Edinb) 2012, 92, 453–488. [DOI] [PubMed] [Google Scholar]
- 42. Kabsch W., Acta Crystallogr. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Evans P. R., Acta Crystallogr. Sect. D 2011, 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Evans P. R., Murshudov G. N., Acta Crystallogr. Sect. D 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. French S., Wilson K., Acta Crystallogr. Sect. A 1978, 34, 517–525. [Google Scholar]
- 46. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D., Acta Crystallogr. Sect. D 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen V. B., W. B. Arendall, 3rd , Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cochrane J. R., White J. M., Wille U., Hutton C. A., Org. Lett. 2012, 14, 2402–2405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information