

Summary
We aimed at molecularly dissecting the anatomical heterogeneity of small lymphocytic lymphoma (SLL), by analysing a cohort of 12 patients for whom paired DNA from a lymph node biopsy and circulating cells, as well as plasma‐circulating tumour DNA (ctDNA) was available. Notably, the analyses of the lymph node biopsy and of circulating cells complement each other since a fraction of mutations (20·4% and 36·4%, respectively) are unique to each compartment. Plasma ctDNA identified two additional unique mutations. Consistently, the different synchronous sources of tumour DNA complement each other in informing on driver gene mutations in SLL harbouring potential prognostic and/or predictive value.
Keywords: small lymphocytic lymphoma, liquid biopsy, multiregional sequencing
Introduction
Spatial heterogeneity is a hallmark of metastatic solid cancer, while it is less obvious in leukaemic B‐cell tumours that systemically involve secondary lymphoid organs and circulate in blood. 1 , 2 Small lymphocytic lymphoma (SLL) is a model that could inform on whether spatial heterogeneity exists in leukaemic B‐cell tumours, as it is the sole entity that markedly involves both blood and lymph nodes in all cases. 3 , 4 Multiregional sequencing resolves the spatial intraclonal heterogeneity in several cancer types, but it is unknown if it may also inform on intratumoural heterogeneity of leukaemic B‐cell tumours, which have so far been investigated extensively in one single disease compartment, usually peripheral blood.
The genomic profiling of lymphoma currently requires invasive tissue biopsies. The tissue biopsy provides information limited to a single point in space and time, therefore failing to capture the complex tumour heterogeneity and it is inherently limited by sampling error. In patients with cancer, the plasma collects tumour‐derived cell‐free DNA (cfDNA) fragments, termed circulating tumour DNA (ctDNA), from all cancer sites. Therefore, the analysis of ctDNA isolated from plasma has advantages over multiregional sequencing of multiple synchronous biopsies and is able to detect differences from all tumour deposits.
We show that different synchronous sources of tumour DNA complement each other in informing on driver gene mutations in SLL, including prognostic and predictive genetic biomarkers, and that the broadest mutational landscape is achieved when genomic DNA (gDNA) from lymph nodes and circulating tumour cells and plasma ctDNA are concomitantly genotyped.
Materials and methods
Patients and samples
Consecutive patients with a diagnosis of SLL according to the International Workshop on Chronic Lymphocytic Leukemia (iwCLL) guidelines and WHO criteria were recruited from 2014 to 2020. 3 , 4 All patients were provided with synchronous samples representative of different anatomical compartments, including: (a) tumour gDNA extracted from fresh frozen lymph node cells or formalin‐fixed paraffin‐embedded (FFPE) lymph node biopsies; (b) tumour gDNA extracted from sorted peripheral blood (PB) CD19+/CD5+ cells; (c) ctDNA from plasma; and (d) germline gDNA extracted from CD3+ T‐cells for comparative purposes. The extraction of cfDNA from plasma aimed at obtaining at least 30 ng, corresponding to ˜5000 genome equivalents (GE), to allow a stoichiometric sensitivity for the detection of 0·1% mutated alleles. Further details are provided in Data S1. The study was approved by the Ethical Committee of the Ospedale Maggiore della Carità di Novara associated with the Università del Piemonte Orientale (study number CE 120/19).
Multiregional sequencing and ctDNA genotyping
The LyV4.0 CAncer Personalized Profiling by deep Sequencing Assay was used for the study and comprised a panel of 124 genes relevant in B‐cell malignancies 5 (Table SI). The number of libraries loaded in the sequencer was tailored to obtain at least a coverage >2000× in >80% of the region of interest. A background error‐suppressed approach was used for variant calling. The limit of quantification of the LyV4.0 CAPP‐seq assay was 0·09%, which represented the analytical background noise threshold over which the assay produced a signal distinguishable from ‘blank’. The analytical sensitivity of the LyV4.0 CAPP‐seq was 0·1%, representing the smallest detectable allele frequency. 6 , 7 , 8
Results and discussion
The detailed characteristics of SLL patients are reported in Table I. The median clonal lymphocyte count in PB was 928·5/μl. Seven (58·3%) patients harboured unmutated and five (41·7%) mutated immunoglobulin heavy chain genes (IGHV). 17p deletion occurred in one patient (8·3%), 11q deletion and trisomy 12 in two (16·7%) each, and 13q deletion in three (25·0%). Two patients did not require therapy, two patients were treated with ibrutinib first‐line, and eight patients received chemoimmunotherapy. Biological samples were collected at the time of diagnosis in 11/12 (91·7%) patients, and at the time of relapse in one patient (8·3%).
Table I.
Clinical data of small lymphocytic lymphoma (SLL) patients (n = 12).
| Characteristics | Values |
|---|---|
| Sex | |
| Male | 7 (58·3%) |
| Female | 5 (41·7%) |
| Median age (years) | 69·0 |
| Median lymphocyte count/μl | 4020 |
| Median clonal lymphocyte count/μl | 928·5 |
| Median haemoglobin levels (g/l) | 142 |
| Median platelet count/μl | 225 000 |
| Matutes score ≥3 | 12 (100%) |
| IGHV unmutated | 7 (58·3%) |
| IGHV mutated | 5 (41·7%) |
| 17p deletion | 1 (8·3%) |
| No 17p deletion | 11 (91·7%) |
| 11q deletion | 2 (16·7%) |
| No 11q deletion | 10 (83·3%) |
| 13q deletion | 3 (25·0%) |
| No 13q deletion | 9 (75·0%) |
| Trisomy 12 | 2 (16·7%) |
| No Trisomy 12 | 10 (83·3%) |
| Treatment status | |
| Previously treated | 1 (8·3%) |
| Treatment‐naïve | 11 (91·7%) |
| First‐line treatment | |
| Watch and wait | 2 (16·7%) |
| Ibrutinib | 2 (16·7%) |
| Chemoimmunotherapy | 8 (66·7%) |
IGHV, immunoglobulin heavy chain gene.
Overall, the analysis of the three SLL compartments analysed (i.e. lymph node biopsy, circulating PB CD19+ cell compartment, and plasma ctDNA) identified a total of 46 mutations in genes recurrently mutated in B‐cell malignancies. At least one mutation was found in 11 out of 12 (91·7%) patients with a median number of three mutations per patient (range 0–6). The median variant allele frequency (VAF) was 6·48% (range 1·43–99·67%) for mutations identified in the circulating PB CD19+ cell compartment; 14·10% (range 1·83–58·38%) for mutations identified in the lymph node biopsy; and 2·40% (range 0·85–38·16%) for mutations identified in the plasma ctDNA (Table SII). The median haploid genome equivalents per ml (hGE/ml) of plasma ctDNA was 33·9. The most frequently mutated genes were TRAF3 and ASXL1 in 3/12 (25·0%) patients each, followed by NOTCH1, EGR2, and SF3B1 in 2/12 (16·7%) patients each (Fig 1A). TP53 was mutated in one single patient in all three compartments. ASXL1 mutations are frequently involved in clonal haematopoiesis 9 but the analysis of paired granulocytes did not detect ASXL1 mutations that could therefore be ascribed to the SLL clone. Interestingly, a BIRC3 mutation that may harbour potential predictive value, 8 , 10 has been identified in one patient only in the circulating PB CD19+ cell compartment. Overall, the mutational analysis of SLL patients is consistent with the mutational landscape expected in the disease, including an enrichment of mutations affecting the NF‐κB pathway. 11 , 12 , 13
Fig 1.
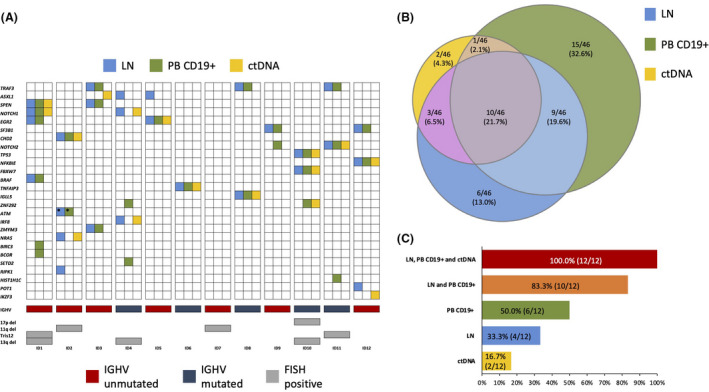
Comparison of mutation types (A) and prevalence (B, C) among anatomical compartments of small lymphocytic lymphoma (SLL). (A) Case level mutational profiles of 12 SLL patients clustered according to the different anatomical compartments. Each column represents one tumour sample, each row represents one gene. Mutations identified in lymph node biopsies (LN) are represented by light blue boxes, mutations identified in CD19+ peripheral blood (PB) cells by green boxes and mutations identified in circulating tumour DNA (ctDNA) by yellow boxes. IGHV mutational status, and fluorescent in‐situ hybridization (FISH) karyotype are plotted below the heatmap. *denotes that different ataxia‐telangiectasia mutated mutations have been identified in LN and in CD19+ cells. (B) Venn diagram representing the comparison of different mutations among the three anatomical compartments. Dimensions of the diagrams are proportional to the number of mutations identified in each compartment. (C) Histogram representing the percentage of patients in which the analysis of single or multiple compartments is able to identify all SLL gene mutations that are present among the ones included in our panel.
By comparing the representation of gene mutations in the different anatomical compartments investigated in SLL, 10/46 (21·7%) mutations were identified in all three compartments (lymph node biopsy, circulating PB CD19+ cell compartment and plasma ctDNA), whereas the remaining mutations were differently distributed among the three examined compartments (Fig 1B). More precisely, 6/46 (13·0%) mutations were exclusive for the lymph node biopsy, 15/46 (32·6%) were exclusive for the circulating PB CD19+ cell compartment, and only a small fraction of mutations (2/46; 4·3%) was detectable uniquely to the plasma ctDNA (Fig 1B). Notably, SLL genotyping on the liquid biopsy does not identify all SLL genetic lesions and does not identify a large fraction of mutations (30/46, 65·2%) that are present in the lymph node biopsy and/or in the circulating PB CD19+ cell compartment. These findings are in contrast with the role of ctDNA genotyping in diffuse large B‐cell lymphoma (DLBCL) and in Hodgkin lymphoma (HL), in which liquid biopsy recapitulates the disease genetics to a large extent. 6 , 7 The different performance of liquid biopsy in disease genotyping among these lymphomas may reflect the notion that, due to its indolent behaviour, slow growth and limited apoptosis, SLL cells release ctDNA at lower levels in the blood stream compared to DLBCL and HL.
Considering the 44 mutations identified in the lymph node biopsy and in the circulating PB CD19+ cell compartment, their distribution did not overlap between these two anatomical sites. More precisely, 20·4% of mutations were unique to the lymph node biopsy, 36·4% were unique to the PB CD19+ cell compartment, and only 43·2% were shared between the lymph node biopsy and the circulating PB CD19+ cell compartment (Fig 1B). These findings suggest that the lymph node biopsy and the PB CD19+ cell compartment of SLL may harbour unique genetic lesions and that both compartments complement each other in comprehensive genotyping analysis.
In addition to gene mutations, copy number variations (CNVs) play an important role in the pathogenesis of CLL/SLL. 14 , 15 Therefore, a bioinformatic algorithm has been applied to eight patients in order to identify potential additional differences between the lymph node biopsy and the PB CD19+ (Data S1). Due to intrinsic limitations imposed by the algorithm, it was not possible to evaluate the CNVs on plasma ctDNA. Fluorescent in‐situ hybridization (FISH) analysis of 11q deletion and trisomy 12 cross‐validated the bioinformatic algorithm in all three patients harbouring the above‐mentioned genetic lesions. Moreover, in three out of eight patients (37·5%), the bioinformatic algorithm allowed the detection of ≥1 SV (structural variant) that was detectable in only one of the two anatomical compartments analysed (Figure S1).
In SLL, the identification of all disease mutations with potential predictive value is essential for treatment choice in the individual patient. 3 This study documents that multiregional sequencing of different anatomical compartments increases the possibility of detecting gene mutations in individual SLL patients. The genetics of SLL is recapitulated in 2/12 (16·7%) patients by the sole analysis of ctDNA, in 4/12 (33·3%) patients by the sole analysis of the lymph node biopsy, and in 6/12 (50·0%) patients by the sole analysis of the circulating PB CD19+ cell compartment (Fig 1C). Conversely, analysis of both the lymph node biopsy and of the PB CD19+ cells increases the possibility of detecting disease mutations in individual patients and identifying all the disease genetic lesions in 10/12 (83·3%) patients (Fig 1C).
These results suggest that multiregional sequencing of the different anatomical compartments of SLL is essential to gain a comprehensive view of the disease’s mutational landscape. Consistently, mutational analysis of the SLL lymph node biopsy should be coupled to the analysis of the circulating PB CD19+ cell compartment, and eventually to ctDNA analysis, to identify all SLL genetic lesions. This observation may have clinical relevance when treatment tailoring, like in SLL, is based on specific gene mutations used as molecular predictors that might be present only in one specific anatomical compartment of the disease.
Supporting information
Fig S1. Comparison of mutation types and CNVs among anatomical compartments of small lymphocytic lymphoma (SLL) clustered according to the different anatomical compartments. Each column represents one tumour sample, each row represents one genetic lesion. Genetic lesions identified in lymph node biopsies (LN) are represented by light blue boxes, mutations identified in CD19+ peripheral blood (PB) cells by green boxes. FISH karyotype is plotted in orange.
{kind=link}
Data S1. Supplementary methods.
Table SI. Target region.
Table SII. Somatic non‐synonymous mutations discovered in the three compartments.
Acknowledgements
This work was supported by: Molecular bases of disease dissemination in lymphoid malignancies to optimize curative therapeutic strategies, (5 x 1000 No. 21198), Associazione Italiana per la Ricerca sul Cancro Foundation Milan, Italy; Progetti di Rilevante Interesse Nazionale (PRIN; 2015ZMRFEA), Rome, Italy; the AGING Project — Department of Excellence — DIMET, Università del Piemonte Orientale, Novara, Italy; and Ricerca Finalizzata 2018 (project RF‐2018‐12365790), MoH, Rome, Italy; Swiss Cancer League, ID 3746, 4395 4660, and 4705, Bern, Switzerland; Research Advisory Board of the Ente Ospedaliero Cantonale, ABREOC 2019‐22514, Bellinzona, Switzerland; European Research Council (ERC) Consolidator Grant CLLCLONE, ID: 772051; Swiss National Science Foundation, ID 320030_169670/1 and 310030_192439, Berne, Switzerland; Fondazione Fidinam, Lugano, Switzerland; Nelia & Amadeo Barletta Foundation, Lausanne, Switzerland; Fond’Action, Lausanne, Switzerland; the Leukemia & Lymphoma Society, Translational Research Program, ID 6594‐20, New York, USA.
References
- 1. Wang DI, Niu X, Wang Z, Song C‐L, Huang Z, Chen K‐N, et al. Multiregion sequencing reveals the genetic heterogeneity and evolutionary history of osteosarcoma and matched pulmonary metastases. Cancer Res. 2019;79(1):7–20. [DOI] [PubMed] [Google Scholar]
- 2. Wei Q, Ye Z, Zhong X, Li L, Wang C, Myers RE, et al. Multiregion whole‐exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Ann Oncol. 2017;28(9):2135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hallek M, Cheson BD, Catovsky D, Caligaris‐Cappio F, Dighiero G, Döhner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–60. [DOI] [PubMed] [Google Scholar]
- 4. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131(22):2413–25. [DOI] [PubMed] [Google Scholar]
- 7. Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, et al. Diffuse large B‐cell lymphoma genotyping on the liquid biopsy. Blood. 2017;129(14):1947–57. [DOI] [PubMed] [Google Scholar]
- 8. Diop F, Moia R, Favini C, Spaccarotella E, De Paoli L, Bruscaggin A, et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica. 2020;105(2):448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watson CJ, Papula AL, Poon GYP, Wong WH, Young AL, Druley TE, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. 2020;367(6485):1449–54. [DOI] [PubMed] [Google Scholar]
- 10. Moia R, Patriarca A, Schipani M, Ferri V, Favini C, Sagiraju S, et al. Precision medicine management of chronic lymphocytic leukemia. Cancers. 2020;12(3):642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Landau DA, Tausch E, Taylor‐Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puente XS, Beà S, Valdés‐Mas R, Villamor N, Gutiérrez‐Abril J, Martín‐Subero JI, et al. Non‐coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–24. [DOI] [PubMed] [Google Scholar]
- 13. Martínez‐Trillos A, Pinyol M, Delgado J, Aymerich M, Rozman M, Baumann T, et al. The mutational landscape of small lymphocytic lymphoma compared to non‐early stage chronic lymphocytic leukemia. Leuk Lymphoma. 2018;59(10):2318–26. [DOI] [PubMed] [Google Scholar]
- 14. Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6. [DOI] [PubMed] [Google Scholar]
- 15. Ouillette P, Collins R, Shakhan S, Li J, Peres E, Kujawski L, et al. Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood. 2011;118(11):3051–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Comparison of mutation types and CNVs among anatomical compartments of small lymphocytic lymphoma (SLL) clustered according to the different anatomical compartments. Each column represents one tumour sample, each row represents one genetic lesion. Genetic lesions identified in lymph node biopsies (LN) are represented by light blue boxes, mutations identified in CD19+ peripheral blood (PB) cells by green boxes. FISH karyotype is plotted in orange.
Data S1. Supplementary methods.
Table SI. Target region.
Table SII. Somatic non‐synonymous mutations discovered in the three compartments.