

Abstract
Calcific aortic valve disease (CAVD) is dramatically increasing in global burden, yet no therapy exists outside of prosthetic replacement. The increasing proportion of younger and more active patients mandates alternative therapies. Studies suggest a window of opportunity for biologically based diagnostics and therapeutics to alleviate or delay CAVD progression. Advancement, however, has been hampered by limited understanding of the complex mechanisms driving CAVD initiation and progression towards clinically relevant interventions.
CAVD is a complex, multifaceted disease involving widespread inflammation and transdifferentiation of resident valvular cells in a mechanically active environment. Many early in-vitro studies on CAVD focused on 2D monocultures of valvular interstitial cells (VIC) and valvular endothelial cells (VEC) cells. However, these cells do not act in isolation but rather in concert with each other and inflammatory cells in the valve, notably in the case of NFκB signaling.
New experimental technologies that interrogate multiple valvular cells in 3D have yielded important insights into how they communicate with each other and their environment. Studies have demonstrated that VEC and VIC communicate using nitric oxide and cytokine signaling, and there is a large opportunity for discovery of additional communication mechanisms. Though it is known that inflammatory cells are present in diseased valves, they embody a protective and pathogenic role in valve disease through mechanisms that have yet to be elucidated. Mechanically-active experimental systems have demonstrated that VIC and VEC respond to altered mechanical stimuli with disease-like properties. This review synergizes understanding of these critical areas of research underpinning promise for the development of valve specific molecular diagnostics and biologically based therapeutics.
Subject Terms: Cardiovascular Disease, Inflammation, Treatment, Vascular Disease
Keywords: Cardiovascular Disease, Tissue Engineering, Inflammation, Mechanobiology, Drug Discovery, Global Health
I. The aortic valve: Built to Last
Aortic valve structure and mechanical function in homeostasis
The human heart is responsible for delivering blood containing oxygen, nutrients, chemical messengers, cells, and much more to the organs of the body. It achieves this by pumping 3–5 liters of blood every ~60 seconds1. Thus, it must be strong, coordinated, and durable enough to last 80+ years over a human lifetime. Blood flows through the heart unidirectionally, such that all oxygenated blood is efficiently pumped to the body and does not backflow into the heart. In order to accomplish this feat, the body has developed four one-way valves that separate the chambers of the heart from each other, the pulmonary artery, and aorta. The aortic valve is the valve that separates the aorta from the left ventricle of the heart. This cardiac chamber is responsible for pumping oxygenated blood out of the heart to the rest of the body. Thus, its function is especially crucial to sustain life in humans (Figure 1a).
Figure 1. Anatomy and Physiology of the Aortic Valve in homeostasis and disease.
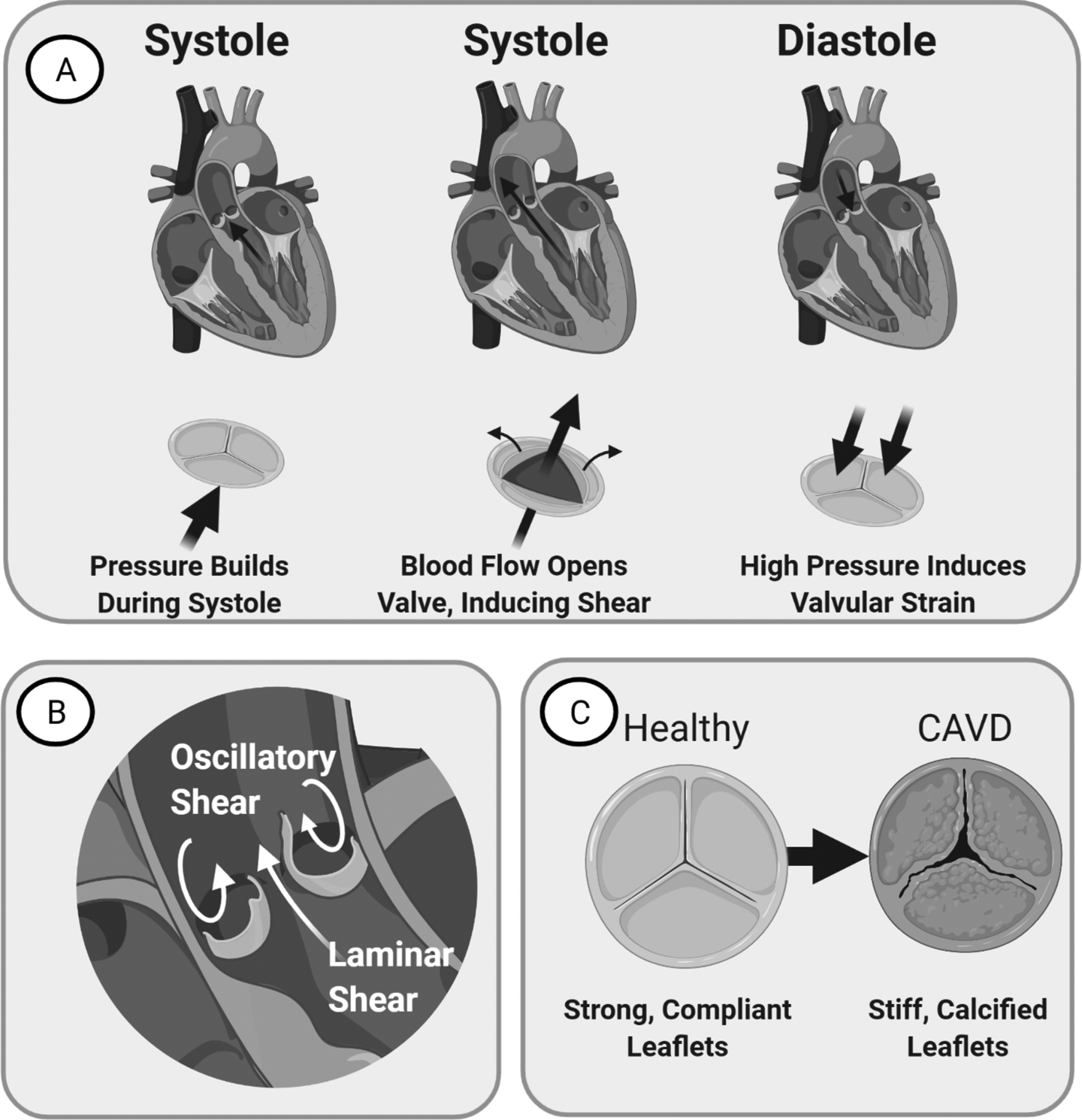
(A) Left: Valve just before systole. Blood flow assists opens the valve. Middle: Valve during Systole. Leaflets ends bend to create an opening for blood flow. Right: Valve during Diastole. Valve provides seal that resists back-flow pressure, inducing strain. (B) Image of the valve demonstrating laminar flow on the ventricularis side and oscillatory flow on the fibrosa side. (C) CAVD manifests as valvular thickening with stiffening and calcification that reduces leaflet mobility over time.
The unique structure of the aortic valve allows it to provide minimal resistance to blood flow in systole, when the ventricle pumps blood, and a leak-tight seal during diastole, when the ventricle relaxes. It has a tri-leaflet structure, and each leaflet is concave to the aorta side. When aortic pressure is equal to ventricular pressure, the valve begins to open2. As the ventricle pumps, blood flow forces the leaflets to fold back towards the walls of the aorta2. This results in a large opening to the aorta for blood flow. Vortexes that form behind each leaflet assist in valvular closure towards the end of systole1.Once closed, during diastole, blood cannot flow back through the aorta to the left ventricular side, preventing mixing of the blood (Figure 1a, 1b).
In calcific aortic valve disease (CAVD), the aortic valve undergoes pathological matrix remodeling that thickens the valve and eventually exhibits calcified lesions that reduce its ability to open and close over the cardiac cycle (Figure 1c). As CAVD progresses, stiffening of the valve due to remodeling in addition to increased pressure/strain due to reduced valve mobility result in altered valvular mechanical stimuli. These mechanical stimuli include shear stress, cyclic strain, and stiffness. This hindered ability of the valve to open and close over the cardiac cycle also increases left ventricular pressure3. To compensate, the left ventricle increases muscle mass over time, called left ventricular hypertrophy (LVH), which can ultimately lead to heart failure3,4. Through CAVD was initially thought to be a degenerative process, seminal work in the field has discovered that the progression of CAVD is not merely a pathological occurrence of degredation but a mechanobiological manifesatation controlled by key cellular regulators of osteogenesis and inflammatory factors5–8
Failure of the Aortic Valve: CAVD Prevalence, Risk Factors, and Burden
12.6 million cases of CAVD have been reported globally in 2017, and an estimated 102,700 of these cases resulted in death9. There are no current therapeutic treatments for CAVD, and all patients seeking treatment must get surgical valve replacement. It is predicted that CAVD patients will undergo 80,000 surgical procedures related to valve disease per year by 205010.
Current surgical treatments include transcatheter aortic valve replacement (TAVR) or surgical aortic valve replacement (SAVR)11
In industrialized countries, the mean age of patients with heart valve related diseases is 55, compared to 20–25 for patients in low-and-middle-income countries12. In these countries, diseases such as Rheumatic Fever (RF) lead to aortic valve complications13. However, neither of the two available surgical methods are accessible in low-and-middle-income countries, especially those that lack a cardiac surgeon. In industrialized countries with more accessible healthcare, other limitations to surgical treatment include immunogenic responses and tissue rejection. Thus, cost effective and accessible alternatives to current treatments for aortic valve disease must be developed to meet the ever-increasing demands of the growing population and demographic discrepancies.
There are many risk factors for calcific aortic valve disease, including genetic mutation, smoking, hypertension/vascular stiffening, hyperlipidemia, diabetes, infection, sex, presence of kidney disease, and age, recently reviewed by Chen et al14–16. How these risk factors contribute to the disease is in many cases inferred by atherosclerotic literature and due to aberrant paracrine and autocrine nitric oxide signaling, inflammatory adhesion molecules, and production of pro-inflammatory destructive reactive oxygen species. These are summarized in Table 1.
Table 1. Risk factors linked to valve deterioration and disease.
Per risk factor, in-vivo, in-vitro, and human observations are presented from various studies.
| Risk Factor | How it could cause CAVD | In-Vivo | In-Vitro | Human |
|---|---|---|---|---|
| Genetic MutationsNotch1, LPA gene, PALMD, TEX41 |
|
|
|
|
| Smoking |
|
Potential area for further study. | Potential area for further study. | Strong association between Degenerative Aortic Valve Disease in a study of 756 healthy male workers26. |
| Hypertension (HTN) /Increased Valve Resistance |
|
|
Potential area for further study. | . |
| Hyperlipidemia |
|
|
||
| Diabetes/Hyperglycemia | Potential area for further study. |
|
|
|
| Infection 16,37,17,38,17,38 |
|
Potential area for further study. |
|
|
| Sex |
|
Potential area for further study. | Potential area for further study. |
|
| Kidney Disease | Abnormal phosphate, calcium, and vitamin D levels, which ↑ formation of calcific deposits39. | Potential area for further study. | Potential area for further study. |
|
| Age | The valve thickens and stiffens with increasing age40. | Potential area for further study. | Potential area for further study. |
|
CAVD Diagnosis and Prognosis
It has been known for many years that survival is poor after valve disease symptoms appear, yet logically patients often do not seek medical evaluation for AS until they experience symptoms17. Ross and Brunwald showed in their seminal graph of aortic disease timecourse that after onset of symptoms, survival declines rapidly due to angina, syncope and heart failure18,19. The absence of sensitive and specific diagnostic tools for early-stage AS prior to irreversible cardiac damage poses a challenge for the development of effective therapeutics, as many therapeutics may only be effective if administered at specific stages of disease. Thus, there is a need and great opportunity for the development of targeted-CAVD molecular diagnostics based on pathways specific to CAVD initiation and progression. Studies have previously been conducted to evaluate biomarkers for CAVD, reviewed by Beckmann et al. and more recently Small et al., and this remains an area of ongoing research20,21. Looking forward, novel bioinformatics technologies such as the use of cell-free DNA and proteomics to identify circulating factors associated with CAVD could provide a disease-specific method to identify CAVD in early stages, as evidenced in promising work published by Kossar et al.22.
In addition to understanding pathways specific to CAVD initiation and progression, it is important to consider how these pathways diverge from the homeostatic valve for the development of diagnostics and therapeutics. Features of the aortic valve in homeostasis are discussed in the following sections.
Aortic Valve Mechanics in Homeostasis
To facilitate folding and unfolding of the leaflet structure, the aortic valve leaflet has specialized extracellular matrix (ECM) structures on the fibrosa, or aortic side, of the valve and the ventricularis, or ventricle side of the valve (Figure 2). The fibrosa side has collagen is oriented parallel to the bending axis (circumferential). It has a corrugated appearance when the leaflet folds, and a smooth appearance in diastole to support backpressure from the blood1,41., On the flip side, the ventricularis ECM consists mostly of disorganized collagen and elastin that is oriented radially along the leaflet41. Separated between these two layers is the spongiosa, which consists mostly of glycosaminoglycans (GAGs) to absorb shock in compression and provide lubrication from shear between the fibrosa and ventricularis42. Resident cells in the valve, called valvular interstitial cells, repair and maintain these ECM structures over the lifetime of the leaflet. Lining the valve are endothelial cells, which provide protection to the valve and can sense mechanics/hemodynamics on the leaflet. The mechanical properties of the valvular ECM structure is crucial to its function, as the valve must bend and extend approximately 3 billion times over its lifetime43. The rest of this section will focus on the mechanical forces experienced by the valve.
Figure 2. The aortic valve during homeostasis (cross-section).
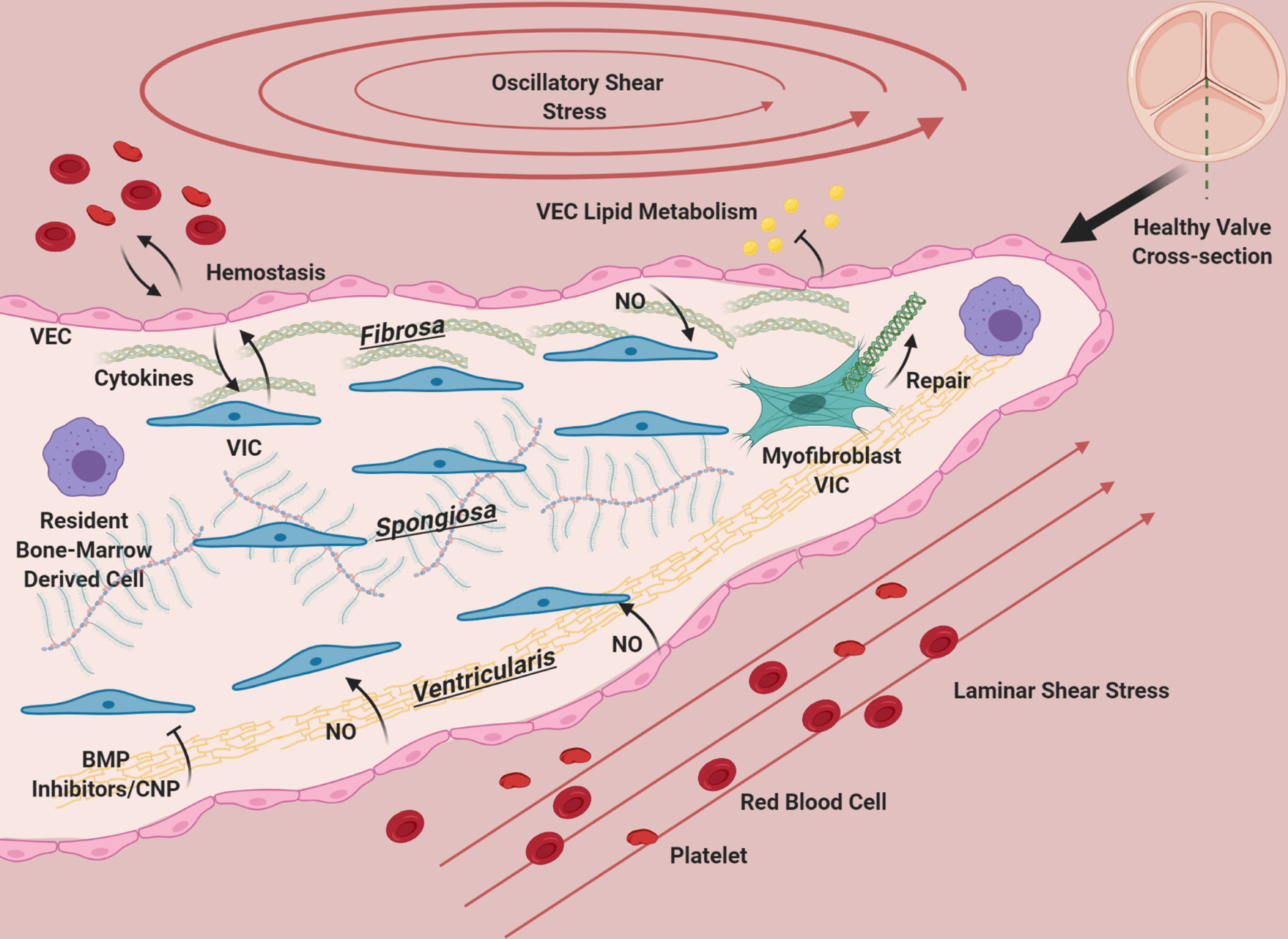
The aortic valve has three layers: the fibrosa, consisting primarily of circumferentially oriented collagen, the spongiosa, consisting primarily of GAGs, and the ventricularis containing radially-aligned elastin. A diverse population of interstitial cells broadly defined as VIC inhabit these layers in the interstitium of the valve (blue) and become activated to remodel the ECM of the valve as needed (green). VEC line the valve and provide a barrier from the blood, preventing clotting, mediating infiltration of lipids, nutrients, and modulating extravasation of inflammatory cells. VEC (pink) promote quiescence in VIC through nitric oxide signaling, and VIC and VEC also communicate with cytokines. Osteogenic inhibitors such as BMP inhibitors and CNP from the ventricularis endothelium likely maintain homeostasis by preventing VIC activation. The homeostatic valve also contains resident immune cells (purple), whose functions are just beginning to be uncovered. The ventricularis of the valve experiences laminar shear, while the fibrosa has oscillatory flow. The valve is constantly under tension/compression as the valve opens and closes, and resists pressure. The ECM composition of the valve and cellular maintenance ensures that it remains flexible, strong and compliant so that it is durable over a human lifespan.
Shear stress:
As the aortic valve opens and closes over a cardiac cycle, friction from the blood induces a shear stress on the fibrosa and ventricularis44. Yap et al. have conducted a series of in-vitro experiments using explanted porcine valves in a heart simulator to directly measure the shear stress experienced by the homeostatic valve. They found that the aortic leaflet approaches approximately 20dyn/cm2 maximum shear stress and that the ventricularis shear stress peaks at 64–91dyn/cm2. On the aortic side of the leaflet, they found that reducing stroke volume tended to reduce peak shear stress at the same heart rate, and that increasing heart rate also decreased peak shear stress45,46. Lower stroke volume could be attributed to a stenotic pathogenic state, but few studies have experimentally or computationally investigated how the shear profile on the valve is altered in CAVD. Additionally, the ventricularis side of the leaflet is exposed to a laminar shear stress, while the fibrosa side experiences an oscillatory one47.Oscillatory shear stress as well as decreased magnitude of shear stress has been shown to promote atherosclerotic plaque formation in vessels, and this paradigm is also assumed to be the case in CAVD, as calcification typically presents on the fibrosa side of the valve48,49.
One of the most well-known ways that valvular endothelial cells communicate to interstitial cells during homeostasis in response to shear is through nitric oxide (NO). Laminar shear stress in vessels is known to induce the production of NO, which promotes homeostasis of the vessels50. Studies have investigated endothelial NO production, in addition to the general valvular response to altered shear magnitudes and profiles, and are discussed below.
Cyclic Strain:
As the valve opens and closes in response to blood pressure, it experiences a cyclic strain in both the radial and circumferential direction, and its associated stiffness accounts for this, as discussed below. The average strain has been shown to be 23%−40% in the radial direction and 10% in the circumferential direction1,44,51. Unpublished work from the Yoganathan group indicated that cyclic stretch increases about 5% for every increase of 40mmHg in pressure52. Thus, many studies discussed below investigate 10% strain as physiologic, and 15–20% strain as pathologic strain induced by higher pressure in CAVD.
Stiffness:
In the homeostatic porcine valve, both the ventricularis and fibrosa portions were found to be stiffer in the circumferential than the radial direction53. The ventricularis was found to have a stiffness of 7.41kPa in the circumferential direction and 3.68kPa in the radial direction53. Additionally, the fibrosa was found to have a stiffness of 13.02kPa in the radial and 4.65kPa in the circumferential direction53. In aortic valve sclerosis and aortic stenosis (AS), ECM remodeling causes valve fibrosis and changes in stiffness over time, affecting the valve’s ability to open and close over the cardiac cycle. Santoro et al. found using atomic force microscopy (AFM) on an aortic valve leaflet that the local stiffness of the valve significantly increases near calcific lesions, indicating a spatial heterogeneity of matrix changes54. Recent studies have used computational models to aid in determining how this stiffness changes over disease progression. Maleki et al. were able to develop a model that determines a relative stiffness parameter in hopes of diagnosing disease severity in-vivo55. They found a relative increase in stiffness from 0.001 to 7.38MPa, and a 0.94 correlation between the valves tested in the study and electron beam computed tomography (EBCT) scores, which is a way to measure calcification in patients56.
II. Homeostatic mechanisms of resident valvular cells
Two primary cells inhabit the valve: valvular interstitial cells (VIC) which are embedded in the fibosa, spongiosa, and ventricularis, and valvular endothelial cells (VEC) that line the outside of the valve leaflets (Figure 2). The valve interstitial cells aid in maintaining structural ECM components of the valve, through the secretion of structural proteins as well as proteinases. Endothelial cells provide a barrier between the valvular tissue and blood. These cells work together to sense their environment, respond to damage, and restore homeostasis in the valve. Studies have also discovered resident valvular immune cells, and their contributions to valvular homeostasis is beginning to be uncovered57–59.
VEC Functions in Homeostasis
Barrier function/transport:
Like the vascular endothelium, the valvular endothelium provides a barrier between the blood and interstitial tissue. It also directs transport of oxygen and nutrients to the inside of the valve, which is crucial because the valve is avascular, unlike large vessels that have the vasovasorum to supply nutrients. Despite these important functions, there have been limited studies investigating the homeostatic barrier function in valvular endothelial cells. One study investigating this feature has found that diffusion of LDL is much higher through the valvular endothelium in comparison to the arteries, and that the valve endothelium had higher permeability to LDL than the aorta60. In a follow up study, Zeng et al. modeled the valve leaflet as a non-continuous monolayer with leaky cells dispersed and a basement intimal lining. They found evidence of dispersed leaks in-vitro, and concluded that their model accounting for leakiness and parallel transport in the sub-endothelial space was able to explain the earlier results of Tompkins et al.61,62. These unique transport and lipid diffusion properties demonstrate a distinct feature of valvular barrier function and sub-endothelial space.
Endothelial cells have also been shown to have vast immunomodulatory functions as barriers for tissues, with expression of MHCII on their surface, the ability to secrete pro- and anti-inflammatory cytokines, and express adhesion molecules that allow inflammatory cells to infiltrate the tissues they line63. Upon acute injury or infection, these mechanisms aid in restoring tissue to a homeostatic state.
Nitric Oxide Signaling:
Nitric oxide production through the eNOS pathway in vascular endothelial cells provides protection to the vasculature by preventing platelet aggregation and adhesion, inhibiting monocyte and leukocyte adhesion, promoting quiescence of smooth muscle cells, and preventing apoptosis of the endothelial cells64. There have been many studies demonstrating that valvular endothelial cells can regulate the activity of valvular interstitial cells through nitric oxide signaling through the prevention of activation and calcification. Interestingly, there is more eNOS expression found in the ventricularis than the fibrosa endothelium, and this could in part explain why the ventricularis is protected from calcification, unlike the fibrosa65. Dysregulation of NO signaling through eNOS uncoupling has been implicated in CAVD and the role of NO in VIC-VEC interactions and eNOS uncoupling is discussed in detail in later sections.
Production of nitric oxide by endothelial cells occurs through nitric oxide synthase 3, also known as eNOS. In homeostasis, co-factors BH4 and NADPH are bound to eNOS66. NADPH is attached to the reductase domain of eNOS and BH4 is attached to the oxygenase domain66. In order to produce nitric oxide, NADPH donates an electron through BH4 to the oxygenase domain, which then is used to convert O2 and L-arginine into nitric oxide and L-citrulline66,67. This nitric oxide then is able diffuse out of the endothelial cell and activate guanylate cyclase in a target cell, such as a valve interstitial cell or vascular smooth muscle cell66. Nitric oxide-activated guanylate cyclase then aids in transforming GTP to cyclic GMP in the target cell, which can interact with other signaling pathways66,67 (Figure 3).
Figure 3. Role of NO and eNOS uncoupling in CAVD.
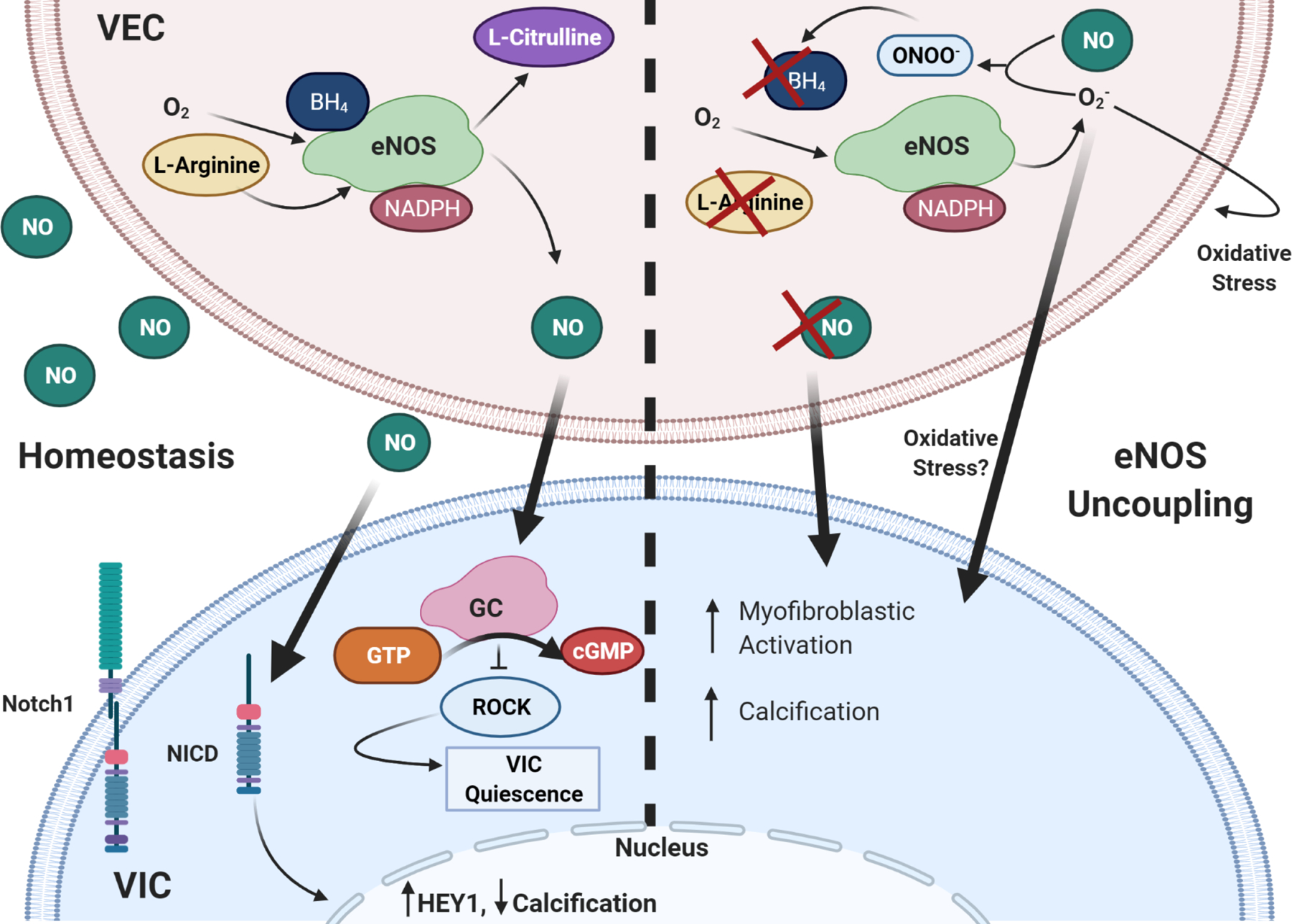
NO signaling from VEC stimulates VIC quiescence through activation of GC/cGMP pathway and subsequent downregulation of RhoA pathway. It also prevents calcification through assisting with Notch1 translocation to the nucleus. eNOS uncoupling causes endothelial dysfunction, such that there is little to no NO signaling to VIC, increasing myofibroblastic activation and calcification. ROS is known to induce cell damage, Superoxide can further cause oxidative stress in VEC and VIC. Thick arrows are VIC-VEC signaling, thin are intracellular or autocrine VEC effects.
An early study demonstrating the influence of endothelial eNOS on aortic valve pathology was performed by T. C. Lee et al. in 200068. They investigated the effects of eNOS-knockout on cardiovascular formation in mice, and found that there was a higher prevalence of bicuspid aortic valve in the knockout mice but no difference in aortic lumen diameter or morphology, indicating specificity to the valve68. Later studies performed in-vitro and in-vivo found that endothelial eNOS production provided a protective effect against valvular sclerosis and calcification. Co-culture of VIC with VEC has been shown to reduce pathologic alpha-smooth muscle actin (aSMA) and osteogenic expression in osteogenic-media (OGM) stimulated VIC in 3D, and has been shown to reduce myofibroblastic expression and expression of pSMAD2, a downstream target of the TGF-B pathway, in 2D culture65,69.
This effect has not shown to be valve-endothelial cell specific, as Bosse et al. demonstrated that co-culture with both human umbilical vein endothelial cells (HUVEC) and lung endothelial cells had an anti-calcific effect on porcine aortic valvular interstitial cells (PAVIC)25. Many gain and loss of function experiments have indicated that this protective effect of endothelial cell co-culture is largely due to the endothelial nitric-oxide signaling pathway outlined above25,65,70,71. Bosse et al. have further shown that in VIC cells, nitric oxide stimulus affects downstream Notch1 signaling in VIC, and this downstream Notch signaling is ultimately responsible for the anti-calcific effects of nitric oxide signaling25. Gould et al. have also investigated VIC and VEC co-culture in response to different stiffnesses, and found that NO signaling from VEC is able to reduce myofibroblastic activation of VIC on stiffer substrates72. They determined that similarly to atherosclerosis, valvular endothelial NO likely signaled to VICs through cGMP, and modulated anti-fibrotic effects through a rho-associated protein kinase (ROCK)-dependent mechanism72. They also found that addition of NO from VEC after VIC have already been activated could rescue VIC back to a quiescent state, pointing to paracrine NO as a potential therapeutic target72.
Calcific inhibitors such as bone morphogenic protein (BMP) anagonists noggin, CV-2/BMPER, SMAD-6 levels, and C-type natriuretic peptide (CNP) in VEC also likely protect against disease and aid in homeostasis. This is evidenced by increased expression in the non-calcified ventricularis endothelium, and in the case of CNP the ventricularis interstitium, compared to the calcific-prone valvular fibrosa endothelium73–75.
VIC Functions in Homeostasis
Sources of VIC Cells:
Though VIC cells are generally considered a fibroblastic population, they have been shown to exhibit great plasticity and have much variation both in their phenotype and cellular origins. Li et al. report in their review on valvular injury that there are at least five types of VIC in the cardiac valve: progenitor mesenchymal/endothelial cells, VIC progenitors, activated (myofibroblast) VICs, quiescent VICs, and osteoblastic VICs76. Smooth muscle cells have also been identified in the aortic valve, and Chen et al. have demonstrated that there are VIC multipotent progenitors that have the potential to differentiate into myofibroblast, osteoblast, chondrocyte, and adipogenic phenotypes, further demonstrating the heterogeneity and plasticity of interstitial cells in the valve77,78. In development, endothelial cells in the cardiac jelly undergo a mesenchymal transition (EndMT), and invade the underlying matrix, becoming valvular interstitial cells79. EndMT in endothelial cells has been shown to depend on many pathways, including transforming growth factor-beta (TGFB), BMP, Notch1, and Wnt signaling79. In adult maintenance, Hadju et al. have also discovered that circulating bone-marrow derived cells can engraft into heart valves and differentiate into VIC, as evidenced by Hsp47 and Periostin staining80. Adult VEC EndMT has been postulated as another source of VIC turnover, however lineage tracing by our group has demonstrated that this likely only occurs in disease conditions81.
Remodeling and maintenance of ECM in the valve:
There have been few studies performed to directly investigate how VIC remodel and repair valvular tissue in homeostasis, and the studies that have investigated this have largely used the mitral valve82–84. In a mitral valve wound model, Lester et al. found that the interstitial cells seemed to be the main cell type driving repair84.
In homeostasis, myocardial fibroblasts are known to deposit ECM, and also secrete MMP’s to maintain the tissue structure of the cardiac tissue85. These fibroblasts proliferate upon injury, and become activated with aSMA expression and secretion of ECM proteins85. Aortic valvular fibroblasts are generally assumed exhibit a similar response to injury, as they also have been shown to differentiate to a myofibroblastic aSMA+ state that increases collagen production and MMP production upon activation86. However, in disease valvular fibroblasts also have a high propensity for calcification, which points to a divergent phenotype from cardiac fibroblasts. This in addition to the lack of studies in the aortic valve and more modern experimental technologies prompts further investigation into the role of VIC’s in ECM regulation and turnover during homeostasis.
The Immune System in Valvular Homeostasis
Though much is still unknown about the contribution of the immune system to valvular homeostasis, a few studies have investigated the presence and identity of hematopoietic cells in the non-diseased adult aortic valve. Anstine et al. found that as mice age from postnatal to 16 months, the percentage of CD45+ hematopoietic cells increases with age from ~1.2 to ~10.9%57. In 6-week old mice, they did not find a substantial proportion of F40/80+ macrophages, indicating that these cells were largely undifferentiated. Further, in irradiated mice transplanted with reporter hematopoietic cells and depleted of macrophages, they did not see a substantial population of new F40/80+ macrophages in the valve57. Hulin et al. similarly found an increase in leukocyte density in aortic valves, rising from 5% of the valve at birth to 12% at eight weeks58. They conversely found that 60% of CD4+ cell in the valve were macrophages and 20% were dendritic cells, and that there was a higher percent recruitment of macrophages from one week to four weeks, indicating that further clarity into the role of macrophage engraftment in adult homeostasis is warranted58. Raddatz et al. also investigated the role of macrophages in perpetuating CAVD in WT and NOTCH−/+ mice59. They found a substantial number of F40/80+ macrophages at 10–12 weeks of age in WT and NOTCH+/− valves, and found that diseased valves likely home macrophages to the valve, and not the other way around. This indicates that in homeostatic conditions, macrophages are likely not recruited to the valve, but rather become pro-inflammatory upon interaction with a diseased valve. In a hyperlipemic disease model, Calin et al. found that depletion of macrophages perpetuated, rather than ameliorated lipid deposition and collagen secretion87. This indicates that macrophages homing to the valve may be attempting to serve a protective role against disease progression87. Further studies into the effects of macrophage depletion in homeostatic conditions are warranted to elucidate their role in valvular maintenance. Circulating bone-marrow progenitors have additionally been found to engraft into the valve and differentiate to valvular cells, as described above80.
III. VIC, VEC, and Cellular Interactions during Initiation and Progression of CAVD
CAVD has two clinical stages: sclerosis, which is valve thickening and some calcification that does not affect functional properties, and stenosis which is substantial obstruction by a heavily remodeled, stiff valve resulting in hemodynamic deficiency88. An exact cause of CAVD disease is unknown, however, there are many risk factors that have been discussed in earlier sections and are presented in Table 1. In early stages of the disease, there are local areas of thickening, matrix disruption, and lipid deposition under the endothelial layer on the fibrosa side of the valve, accompanied by some micromineralization, usually found at the base of the valve7. These lesions typically have calcium and lipid deposits deeper in the leaflet with inflammatory cells on top7. Severe cases present with large calcific nodules on the fibrosa side of the valve that associate with inflammatory cells and can form mature bone structures and new blood vessels in the valve89. (Figure 4)
Figure 4. The Aortic Valve in CAVD.
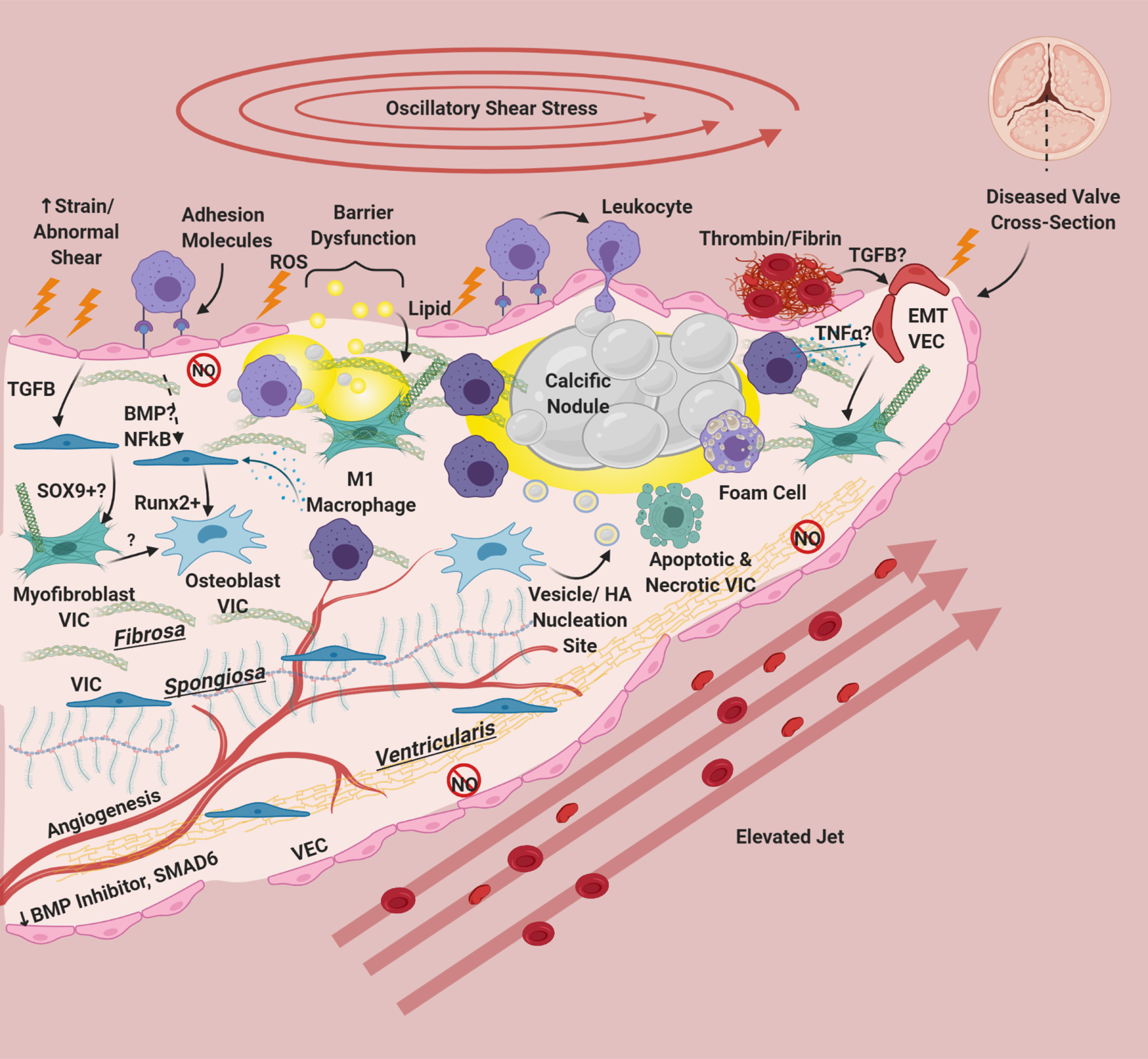
Risk factors cause damage, inflammatory adhesion expression, barrier disruption, and lipid deposition in/underneath the endothelium. Deposited lipids, specifically oxidized lipids can cause inflammation and damage in resident cells. Endothelial dysfunction results in eNOS down-regulation and dysfunction, abrogating protective eNOS signaling to VIC and in hemostasis. This is especially apparent on the ventricularis side of the valve. There is also a reduction in protective BMP inhibitors and SMAD6 on the ventricularis side of the valve. Leukocytes bind to VEC adhesion molecules and undergo extravasation into the tissue. Clotting factors including thrombin, tissue factor, and fibrin have been associated with calcified valves, and platelets recently have been implicated as sources of TGFB in valve disease. TGFB from VEC has been shown to induce Sox9 expression and to produce activated myofibroblast-like VIC. Paracrine signals from macrophages, specifically M1 macrophages has been shown to induce osteogenic differentiation of VIC. NFκB and BMP (possibly) from VEC can also modulate VIC osteoblastic differentiation. Osteoblastic VIC may excrete vesicles or undergo apoptosis/necrosis that deposit calcium and create HA nucleation sites, which grow over time to form large calcific nodules. Inflammatory infiltrate localizes around nodules, and areas of angiogenesis. Foam cells are observed in CAVD, but do not form dense necrotic cores. Inflammatory cytokines like TGFB and TNFα can cause VEC to undergo EndMT, creating fibrotic activated VIC-like cells. Thickening of the valve happens not only through lipid deposition/calcification but also fibrotic remodeling from activated VIC. CAVD induces angiogenesis in the normally avascular leaflet. For reasons unknown but hypothesized to be due to laminar shear stress, the ventricularis of the CAVD valve remains largely unaffected
VIC contribute to valve thickening by undergoing myofibroblastic differentiation and depositing fibrotic matrix90. They also play a role in the calcification process through apoptosis, creating calcific nucleation sites. This apoptotic process is commonly referred to as dystrophic calcification.90 VIC additionally undergo osteoblastic differentiation to directly modulate calcium deposition and bone formation90.
It has been hypothesized that fibrotic differentiation of VIC may precede osteoblastic differentiation in a linear trajectory of disease. This hypothesis is supported by Schloter et al., who demonstrated with transcriptomics that VIC isolated from fibrotic areas of CAVD valves exhibit intermediate gene profiles between non-diseased and calcific regions91. Additionally, Hjortnaes et al. have demonstrated that myofibroblastic aSMA silencing prevents Runx2+ osteogenic differentiation and calcification in VICs76 However, there is also evidence that VIC may directly undergo osteoblastic differentiation without a fibrotic intermediate93. Whether a fibrotic transition state is always necessary for osteoblastic VIC differentiation and why some myofibroblastic VIC further differentiate to an osteogenic state remains to be explored. Additionally, endothelial cells have been demonstrated to undergo transdifferentiation into myofibroblastic VIC, a process called EndMT, and subsequently invade the valve and promote remodeling81,94–96.
Inflammation and Disease initiation: Risk Factors Promote NO-dysfunction in VEC
Though much is unknown about the initiation of CAVD, it is thought to start with early endothelial inflammation and dysfunction. This makes sense as the endothelium serves as a barrier and first defense protecting the valve tissue from the blood. Endothelial dysfunction allows for the extravasation of inflammatory cells into the valve tissue and allows for deposition of pro-inflammatory molecules and lipid deposition. Drawing off of the atherosclerotic literature, it is likely that smoking, diabetes, and in addition to hyperlipidemia initiate CAVD at least in part through an endothelial NO-dysfunction/ROS damage related mechanism34,97. However, studies directly investigating causality of these risk factors in CAVD are somewhat lacking.
In an early study assessing the role of eNOS in CAVD, Rajamannan et al. found that atorvastatin was able to upregulate eNOS and protect against calcification, suggesting a role for eNOS in VIC calcification prevention98 Subsequently,, Rajamannan directly investigated the effects of adding an inflammatory lipid, low-density lipoprotein (LDL), on VEC eNOS activity. She found that addition of LDL significantly downregulated VEC eNOS enzyme activity, and increased nitrite and Caveolin I99. Caveolins are invaginations of the endothelial cell membrane that are involved in transcytosis of lipids and can directly inhibit eNOS, additionally reducing NO bioavailability100. It is also well known in the atherosclerotic literature that shear stress can modulate VEC nitric oxide production, and this phenomenon in valve disease is discussed further below. NO dysfunction in endothelial cells can occur through a phenomenon called eNOS uncoupling66,67,101. This often happens when there is not enough BH4 or L-arginine present in the cell66,67. In this scenario, eNOS directly transfers electrons to the oxygen reactant creating superoxide, a damaging reactive oxygen species66. The superoxide can then react with nitric oxide to create peroxynitrate, another type of reactive oxygen species66,67. This peroxynitrate can oxidize BH4 and remove it from the system, creating a destructive positive feedback loop67,102. Our lab has demonstrated that endothelial eNOS uncoupling likely contributes to oxidative damage in valve disease, as addition of BH4 was able to reduce Tumor Necrosis Factor-alpha (TNFα) induced reactive oxygen species in endothelial cells-vitro71. Interestingly, this effect of BH4 was only observed in the fibrosa, and not ventricularis side of the valves71 (See Figure 3). It follows that eNOS uncoupling reduces bio-available NO for VIC, abrogating the protective role VEC have been found exert on VIC and perpetuating disease (Figure 3).
Endothelial Sources of VIC Pathogenesis
There are many cytokine pathways that have been found to play a role in the fibrotic and calcific remodeling of CAVD, however, their cellular source and whether they act in an autocrine, juxtacrine, or paracrine manner largely remains to be elucidated.
Specifically, the signaling pathways BMP, Notch, Wnt, and TGFB have been found to play a role in chondrogenic and osteogenic processes of CAVD, and could be modalities through which dysfunctional VEC promote osteogenic differentiation in VIC in CAVD, although studies directly investigating these interactions are limited.. At the transcriptional level, many of these pathways appear to target Runx2 and/or Sox9 nuclear activity. Sox9 promotes chondrogenic differentiation of mesenchymal cells103. Runx2 inhibits chondrogenic differentiation and differentiates cells into an immature osteoblastic lineage103–105. In limb development, Sox9 acts upstream of Runx2 expression and osteoblastic differentiation, as knockdown of Sox9 prevents both cartilage and bone formation106. In endochondral ossification, where cartilage is gradually replaced by bone in limb development, Runx2 was required for ossification but its knock-out did not affect Sox9 expression, indicating a time-dependence for these two transcription factors in osteoblastic differentiation107. Furthermore, continued Sox9 expression can inhibit Runx2, indicating its upregulation and subsequent downregulation is necessary for bone formation through endochondral ossification104,108.
Pathways generally on the pro-osteogenic spectrum of VIC differentiation include BMP and Wnt signaling, while TGFB and Notch are generally considered anti-calcific, with caveats. BMP stands for bone morphogenic protein, and signals through down-stream SMAD 1/5/8 in the cell109. It is mainly found localized to the endothelium and sub-endothelial area of the valve as BMP2/4 and also evidenced by SMAD 1/5/8 expression75,110. BMP2/4 has also been noted in myofibroblasts/osteoblasts near inflammatory infiltrate and bone formation, and BMP-4 was found in the fibrosa and spongiosa in response to shear stress6,110. In VIC cells, BMP pathways are known to promote osteoblastic differentiation, through downstream expression of ALP, Runx2, and osteopontin111–113. It is possible that this endothelial-specific expression/activation of the endothelial BMP pathway could promote osteoblastic differentiation in VIC, and could be a promising area for future study.
TGFB is in the same superfamily as BMP, and signals downstream through SMAD2/3 in the cell114,115. It is known to induce Sox9 nuclear localization and aSMA+ fibrotic differentiation in VIC, and is also found to be upregulated in CAVD valves116–118. In response to shear stress, its expression is localized to the valvular endothelium, like BMP110. Huk et al. discovered that in co-culture and in mice, TGFB from VEC induces Sox9 nuclear localization in VIC which prevents VIC calcification117. However, many studies investigating calcification in VIC have also paradoxically shown that TGFB supplementation is necessary for nodule formation. This has been seen in-vitro culture and leaflet explants, where cells form nodules in a spatial pattern with necrosis and apoptosis, especially in a cyclic-strained environment28,118–120. One possible explanation is that TGFB supplementation initially upregulates Sox9, which inhibits nodule formation especially in the absence of a mechanical stimulus, but this initial upregulation of Sox9 primes the cells for a subsequent Runx2 upregulation terminal osteoblastic differentiation in a time- dependent endochondral ossification like process121,122. Further investigation is warranted to elucidate this mechanism.
The Wnt pathway is largely considered to induce calcification in the aortic valve. Usually B-catenin, a downstream signal of the Wnt pathway is continuously phosphorylated and degraded. However, upon Wnt binding B-catenin is not phosphorylated and translocates to the nucleus123. Upon activation, the Wnt/b-catenin pathway generally increases calcification and osteoblastic differentiation through upregulation through upregulation of Runx2124. Jenke et al. have demonstrated that TGFB, which has been found to be secreted by VEC to effect VIC, can inhibit both Wnt/B-catenin and BMP signaling116,123. They present a paradigm where TGFB induces myofibroblastic differentiation of VIC, while Wnt and BMP induce osteogenic/calcific differentiation, with TGFB an inhibitor of Wnt/BMP116.
Notch1 signaling usually requires cell-cell contact, and is activated when the protein is cleaved and the intracellular domain is translocated to the nucleus. From there, it upregulates Hairy repressors which are classically thought to induce an anti-calcific effect through repression of Runx2 and BMP15,125–128. The Notch1 pathway one of the first pathways whose mutation was directly shown to induce CAVD15. However, since this seminal paper, there has been debate on whether Notch-1 itself has pro- or anti- calcific effects on VIC cells. A follow up study by Nigam and Srivastava corroborated Garg et al.’s findings in-vitro and in-vivo, finding that Notch1 prevented calcification through inhibition of BMP-2128. Additionally, Acharya et al. have found that Notch1 inhibits calcification by upregulating Sox9 activity129. They also interestingly found increased total Notch1 expression in calcified valves, with reduced Notch1 locally in areas of calcification129. Furthermore, a study conducted by Hadji et. al. found increased expression of long non-coding RNA H19 from DNA hypomethylation to be inductive of osteogenic cell fate through the down-regulation of Notch1130. This indicates a role for not just genetics, but also epigenetics in modulating disease.
Conversely, Zeng et al. found in response to lipopolysaccharide (LPS) stimulus, Notch1 inhibition with DAPT actually decreased osteogenic BMP and alkaline phosphatase (ALP) levels in VIC, possibly through an inflammatory ERK1/2 / NFKB pathway131. Wang et al. further discovered that intercellular adhesion molecule (ICAM) and lymphocyte-function associated antigen 1 (LFA) signaling is also involved in this phenomenon, and another group found that Notch signaling in VEC promotes apoptosis in VIC132,133. Differences in the results of these papers may be due to the differences in osteogenic stimulus used. It is important to note that even though Notch1 is a cell-cell contact pathway, few studies have investigated whether VIC and VEC communicate using this pathway and possible cross-talk effects on calcification. Future studies investigating Notch1 signaling between VIC and VEC in a direct-contact co-culture system are warranted.
Reactivation of EndMT by VEC in CAVD
Perhaps one of the largest direct interactions between valvular endothelial cells and interstitial cells in CAVD could occur during valvular endothelial to mesenchymal transition. For an in-depth review of VEC EndMT in CAVD, the reader is encouraged to reference Ma et al.94.Briefly, endothelial to mesenchymal transition (EndMT) is thought to be a re-activation of endothelial differentiation to an interstitial-cell phenotype and invasion into the valvular interstitial space. These transformed endothelial cells have been shown to have a myofibroblastic phenotype that can induce remodeling and could interact directly interact with valvular interstitial cells in the surrounding area95,96. Additionally, it can be imagined that widespread invasion of transformed endothelial cells into the interstitial space would compromise endothelial barrier function, allowing disease-promoting lipids, toxins cytokines, and inflammatory cells from the blood into the interstitial space.
Over the twenty years since the ability of aortic endothelial cells to undergo EndMT was discovered by Paranya et al., there has been much debate over the presence of EndMT in adult valves in both homeostasis and disease134. Our group has discovered through lineage tracing that adult aortic endothelial cells are able to undergo invasion into the interstitial space and that these cells are localized near calcific lesions in a CAVD mouse model but not in non-CAVD mice81. This indicates that this process occurs in adult disease but likely not homeostasis81. However, a study by Kim et al. came to the opposite conclusion. In their Tie2-CreER marfan-syndrome mice, they did not see aSMA+ CD31+ mice, and found that the lineage tracing they performed using g-gal+ Tie2 was actually marking CD45+ lymphocytes135. It is important to note that these two mouse models differed and the study by Kim et al. investigated the mitral valve, not aortic valve, which may reconcile some of these differences.
Hjortnaes et al. investigated the role of VIC paracrine signaling on VEC EndMT in-vitro using a transwell co-culture system96. They used two modes of promoting EndMT: TGFB media and osteogenic media. VIC were able to suppress aSMA, matrix metallo-proteinase (MMP)-2, and Slug expression in TGFB+ treated VEC, as well as osteocalcin, aSMA, osteopontin, Runx2, and calcium deposition in osteogenic-media treated VEC. This points to a protective paracrine role of VIC against EndMT in VEC cells, although the mechanism through which this signaling occurred was not investigated in this study. Interestingly, Hjortnaes et al. did not observe a reciprocal protective paracrine effect of VEC co-culture with VIC, and instead actually saw increased mineralization in the co-culture condition in osteogenic media vs. VIC alone96. This is in direct contrast to a large body of work indicating a protective role for VEC on VIC calcification25,65,69,117,136. Our group, as well as Hjortnaes et al. found evidence of increased remodeling ability of EndMT VEC, through upregulated MMP’s, and a notable propensity of EndMT transformed VIC to remodel collagen fibers96,137. Our group was able to attribute the EndMT process in response to TNFα to an AKT/NFKB pathway137. There has also been evidence of EndMT VEC present in calcified human explanted aortic valves, but not healthy control valves96,137. Gendron et al. have recently the ability of diseased VIC to undergo the reverse process back to endothelial cells. When challenged with vascular endothelial growth factor B (VEGF-B) and after 10 days of in-vivo implantation in matrigel, there was no evidence that VIC from diseased patients were able to differentiate to an endothelial phenotype, indicating that VEC EndMT plasticity is likely not reversible138.
IV. Inflammation in the Diseased Valve
Risk Factors Promote Adhesion Molecule Expression in VEC
Inflamed VEC express adhesion molecules and exert immunomodulatory functions, to interact with the immune system. Some of these markers include P/E selectins, ICAM, vascular adhesion protein 1(VCAM), and activated leukocyte adhesion molecule (ALCAM). Selectins facilitate rolling of leukocytes and attachment, and ICAM/VCAM bind leukocytes and assist with extravasation139. Interestingly, Guerraty et al. found that a unique adhesion molecule ALCAM was the only differentially expressed endothelial adhesion molecule between the fibrosa and ventricularis, indicating a potential role in CAVD and warranting further investigation140. Vandana et al. and Ciortan et al. investigated the effects of hyperglycemia on VIC/VEC co-cultures in a diabetes model35. They found that high glucose resulted in direct upregulation of the adhesion molecules VCAM-1 and E-selectin in VEC at 7 and 14 days35. Mechanical stimuli have also been found to directly cause increased inflammatory adhesion markers in VEC, and are discussed below. It is important to note that VIC have also been shown also express inflammatory adhesion molecules like VCAM, ICAM, and E-selectin, however the expression of adhesion molecules to facilitate leukocyte infiltration is usually physiologically attributed to endothelial cells36,141.
Inflamed VIC/VEC Recruit Inflammatory Cells to the Valve
As discussed above, pro-inflammatory VIC and VEC express adhesion molecules that allow inflammatory cells to the enter the valve. These inflammatory cells are generally thought to wreak more havoc on the valve, though destructive proteinases, and the release of additional pro-inflammatory cytokines, which promote pro-fibrotic and calcific phenotypes of resident valvular cells. Paradoxically, these infiltrating inflammatory cells may also serve a protective role against disease. Resident valvular inflammatory cells may also contribute to this process, but specifics regarding their role in disease has yet to be elucidated.
Initiation of Inflammatory cell infiltrate:
In CAVD, inflammation appears before valvular calcification. This has been demonstrated in-vivo by Abdelbaky et al., who used cancer surveillance fluorodeoxyglucose positron emission tomography (FDG-PET) images from patients without active cancer. They were able to demonstrate in images taken approximately two years apart that inflammation identified by FDG-PET independently predicted subsequent valvular calcification142. Though inflammation likely plays a role in the initiation of CAVD, there have been varying reports of its extent in late-stage disease. Coté et al. noted that only 28.4% of the 285 explanted human CAVD valves that they investigated contained inflammatory infiltrate, while Natorska et al. reported 95% inflammation in their CAVD specimens89,143. However, Coté et al. also noted that there was a correlation between leukocyte density and stenosis progression, and inflammatory cells tended to localize around calcific nodules and cluster in areas of bone deposition and angiogenesis, indicating an active role in disease progression89. Additionally, Hjortnaes et al. found that a macrophage-targeting fluorescent nanoparticles, as an indicator of inflammation, were positively correlated with valvular calcification, indicating that inflammation increases with disease severity144. This diverging evidence for the extent and presence of inflammation in late stage valve disease demonstrates the inherent variability in pathogenesis from patient to patient and prompts a nuanced understanding of inflammation in a wide range of CAVD disease etiologies.
Immune cells in Calcific Lesions
Pathophysiologically, inflammation in early CAVD is thought to be in many cases a response to lipid deposition through an ill-functioning endothelial barrier, similarly to atherosclerosis145. In diseased valves and in-vitro models, vascular endothelium has been reported to upregulate Lox1 which is involved in endocytosis of oxidized low-density lipoprotein (OxLDL), in addition to caveolins (discussed above), and CD73, although much remains unknown about this aspect of VEC barrier function99,146–148. A distinct cellular source for the conversion of LDL to OxLDL remains unclear, however it could be any ROS-producing cell149. ROS are discussed further in the section entitled “Reactive Oxygen Species”. However, key differences have been noted between the lipid deposition, lesions, and inflammatory infiltrate in CAVD and atherosclerosis. An early study noted that these lesions had lipid deposition and were primarily under the basement membrane of endothelial cells with some extension to the fibrosa7. Olsson et al. further reported that the lipids remained in the fibrosa subendothelium and did not move deeper, unlike in atherosclerotic plaques150. Further investigation into these lipids concluded that only small, dense LDL’s correlated with aortic stenosis progression, and not high-density lipoprotein-c (HDL-c), LDL-c, or triglycerides151. The presence specifically of oxidized low-density lipoprotein (OxLDL) in aortic valves correlated with T-cell, leukocyte, and macrophage presence as well as increased TNFα and tissue remodeling151. Otto et al. noted that lesions tend to accumulate these inflammatory cells including macrophages and T-cells on their surface, and Olsson et al. noted that unlike atherosclerotic lesions, valve lesions did not form necrotic cores with a high density of foam cells, although some foam cells were identified7,150. Widespread micromineralization has been noted in the aortic valve, and is also seen in atherosclerosis7,152. Olsson et al. postulated that deposited valvular OxLDL may aid in calcific deposit nucleation instead of inducing necrosis, and that differences in OxLDL turnover in the valve vs. vasculature may be in part responsible for the differing calcific pathology in CAVD vs. atherosclerosis, which typically exhibits plaque rupture and necrosis150. Studies have found that small LDL particles and lipoprotein (a) (Lp(a)) presence has been associated with oxLDL in CAVD, and a mutation in Lp(a) has been found to be associated with aortic valve stenosis and calcification24,126,151. However, the role of LDL-C in CAVD is still unclear14. This is in contrast to atherosclerosis, where LDLc is implicated in lesion development153 It is important to consider these differences between CAVD and atherosclerosis, as drugs such as statins that have shown incredible success in treating atherosclerosis have failed in the treatment of CAVD154. The role of lipid deposition in general CAVD pathogenesis has been reviewed extensively elsewhere154,155, and the remainder of this section will focus on inflammatory cellular mediators and pathways in CAVD.
Many inflammatory cell types have been discovered in the aortic valve, and for a more extensive review of these cell types the reader should refer to Raddaz et al.156. Briefly, cell types include macrophages, T-cells, B-cells, mast cells, dendritic cells, and platelets. These inflammatory cellular mediators have been shown to be sources of pro-inflammatory cytokines and matrix-remodeling proteases in CAVD. Macrophages are perhaps the most studied of the inflammatory cells in CAVD, largely because their polarization state is known to induce homeostatic and pathogenic inflammatory remodeling or resolution. Notably, Li et al. discovered that M1 macrophages are increased and M2 macrophages are decreased in calcified valves vs. controls, indicating a more pro-inflammatory state157. Macrophages have colocalized or associated with matrix-remodeling MMP’s EMMPRIN, MT1-MMP, MMP2/9, and Cathepsin B158,159. Macrophages have also been shown to express pro-inflammatory cytokine TNFα and leukocytes express IL-1B in stenotic valves160,161. It is assumed that macrophages would additionally express pro-inflammatory IL-6 in CAVD valves as they do in-vitro157. VIC cultured with conditioned media from undifferentiated macrophages resulted in an increase in osteogenic markers, and this was additionally increased by M1 differentiation157.
Despite this incriminating evidence against leukocytes and macrophages in the pathogenesis of CAVD, complete depletion of macrophages in hyperlipidemic hamsters resulted in larger valvular lesions with increased lipid and collagen deposition, suggesting that macrophages also serve a protective role against CAVD87. Mast cells, an inflammatory cell type infamous for its role in allergic reactions, have additionally been found in the aortic valve near microvessels and regions of calcification162,163. Interestingly, Milutinovic et al. found that mast cells may also provide protection against calcification in CAVD, as stenotic valves with mast cells had lower calcification than valves without them164. These findings indicate that inflammatory cells at least in part serve a protective role in disease, in addition to potentiating it.
Interestingly, the platelet to lymphocyte ratio has been found to be increased in severe aortic stenosis when compared to patients with mild/moderate stenosis165. Additionally, areas of thrombin, fibrin, and tissue factor have been found in calcified aortic valves implicating a role for coagulation in the disease166–168. Valves that stenosed had significantly higher tissue factor-positive areas compared to aortic insufficiency, and additionally patients with aortic stenosis had fewer large von-Willebrand factor (vWF) proteins, and lower rates of platelet retention indicating abnormal coagulation166,169. Recently, platelets have been implicated as potential sources of TGFB in valve disease, a pro-calcific and EndMT cytokine that is discussed earlier sections170. Investigation into valvular endothelium interactions with the blood is still somewhat lacking, and could provide important insights into disease initiation and progression.
Reactive oxygen species:
Reactive oxygen species (ROS) in many cases are produced to cause damage to invading pathogens, and can wreak havoc on native tissue171. The presence of reactive oxygen species and ROS-producing intermediates have been found around calcific nodules in human valves172,173. Though superoxide has been found in macrophages of calcified valves, this was more pronounced in areas of non-macrophage cells around the calcification172. These areas were found to contain osteoclastic and osteoblastic cells, and indicate that unlike atherosclerotic lesions, macrophages may not be the primary contributor of ROS in valves172. Miller et al. found that eNOS uncoupling likely plays a role in superoxide production, and this has been further validated in VEC, as discussed71,173. In-vitro studies by Branchetti et al. and Liu et al. have additionally investigated the role of ROS and intermediates in VIC174,175. Liu et al. found that Nox2, an enzyme that produces ROS, is elevated in diseased valves and osteogenic-media cultured VIC175. Inhibition of Nox2/ROS production with viral transduction and a drug called Celastrol was able to reduce calcific effects in-vitro and in-vivo through a GSK3B/B-catenin pathway175. Furthermore, Branchetti et al. discovered that treatment of VIC with H2O2 induced DNA damage repair proteins more so in sclerotic and stenotic valves than control valves174. Additionally, they found that H2O2 increased Runx2 signaling through an AKT pathway, and that catalase and SOD were able to mitigate some of the H2O2-driven DNA damage and osteogenic differentiation effects174. Given that both VIC and VEC appear to produce and react to ROS, and that studies have indicated that macrophages may not be the main cellular source of ROS in CAVD, it is imperative to test the response of these two cell types in co-culture to more clearly understand how ROS in disease valves initiates and manifests.
Neovascularization
One feature of diseased aortic valves is the presence of neovascularization in the normally avascular valvular tissue176. Steiner et al. postulated that one source of this neovascularization could come from the valvular endothelium176. A study by Arevalos et al. investigated the ability of VIC and VEC cells in co-culture to undergo pericyte differentiation and angiogenic sprouting on an angiogenic Matrigel scaffold177. VIC and VEC in co-culture were able to form angiogenic sprout networks which interestingly collapsed into VEC-VIC spheroids over prolonged culture. The VIC seemed to exhibit some pericyte-like behaviors, including wrapping around VEC tubules and invasive stalks. Cells from these spheroids notably invaded the matrix, with VIC cells paving the way for VEC cell migration, and this was found to be modulated by Ang/Tie and ROCK pathways. This pericytic differentiation of VIC has been further confirmed by Gendron et al., who discovered that conditioned media from VIC isolated from diseased patients were able to promote significantly more proliferation, migration, and sprouting in cord blood endothelial progenitor cells138.
NFκB Signaling- Conserved Mode of VIC/VEC Inflammation inducing CAVD
Pro-inflammatory cytokines have been shown to contribute to CAVD, including TNFα, IL-1B, IL-6, etc178,179. Many of these cytokines, including TNFα, IL1B, and IL-6 converge in downstream activation of the pro-inflammatory nuclear factor kappa B (NFκB) pathway, which has been associated with atherosclerosis and CAVD137,180,181. Homeostatically, NFκB is inactivated through binding to inhibitory kappa B (IκB). Upon activation, a complex called IκB kinase (IKK) complex, which contains a subunit IKKβ (not to be confused with IκB), phosphorylates IκB, which releases NFκB and allows for translocation to the nucleus180. There, it induces inflammatory cytokines/chemokines, matrix degrading enzymes, and adhesion molecules, including VCAM-1, ICAM, Selectins, MMP’s, TNF, and IL1/6180. In innate immunity, toll-like receptors (TLR’s), and NOD-like receptors present in the cells of resident tissues, which sense molecular patterns associated with pathogens, can also activate NF-kB182. A large body of work has demonstrated that NFκB can be activated in VICs in response to TLR signaling, and that TLR signaling is associated with CAVD37. This has been reviewed extensively by Garcia-Rodriguez et al., and thus will not be discussed in depth here37.
Our group along with others have begun to study the role NFκB plays in CAVD. We have found that in the fibrosa of calcified valves, there is more NFκB nuclear signaling than the fibrosa of sclerotic and non-calcified valves81. As discussed above, Zeng et al. and Wang et al. have found that stimulation with an inflammatory LPS stimulus induces osteogenic differentiation of VIC through a downstream NFκB pathway, indicating an active role of NFκB in driving CAVD131,132.They found this pathway was activated through TLR receptor activation, Notch-1 and adhesion molecule ICAM-1 signaling. In VEC, our group found that the inflammatory cytokines TNFα and IL-6 can induce EndMT in adult endothelial cells through a downstream NFκB pathway137. In a follow up study, we additionally found that NFκB signaling can induce a calcific response in VEC in the presence of an osteogenic stimulus, evidenced by nodule formation, alizarin red S (ARS) deposition, and Runx2/osteocalcin (OCN) expression81. Knock out of NFκB in the entire valve of LDLR−/− mice had a protective role against CAVD, exhibited by decreased aortic peak velocity, peak gradient, mean gradient, leaflet thickness, and increased ejection fraction compared to LDLR−/− controls81. Further, knock out of NFκB exclusively in valvular endothelial cells was able to rescue hemodynamic function in LDLR−/− high-lipidemia mice, but not ejection fraction, indicating that NFκB-induced signaling from VEC to resident valvular cells plays a substantial but not all-encompassing role in the modulation of CAVD81. Notch signaling in VEC has been shown to promote apoptosis in VIC likely through a TNFα related pathway133. Thus, it would be interesting to investigate whether Notch 1 signaling is also involved with VEC NFκB signaling, and whether this signaling in VEC modulates VIC propensity towards calcification (Figure 5). The compounds EMD, curcumin, andrographolide, hydrogel sulfide, and caffeic acid have been discovered to have an anti-calcific effect on VIC by modulating at least in part the NFκB pathway, highlighting not only its importance in CAVD but also its promise for therapeutic targets183–185,186,187.
Figure 5. Inflammatory pathway NFκB Modulates CAVD intracellularly and through cell-cell interactions.
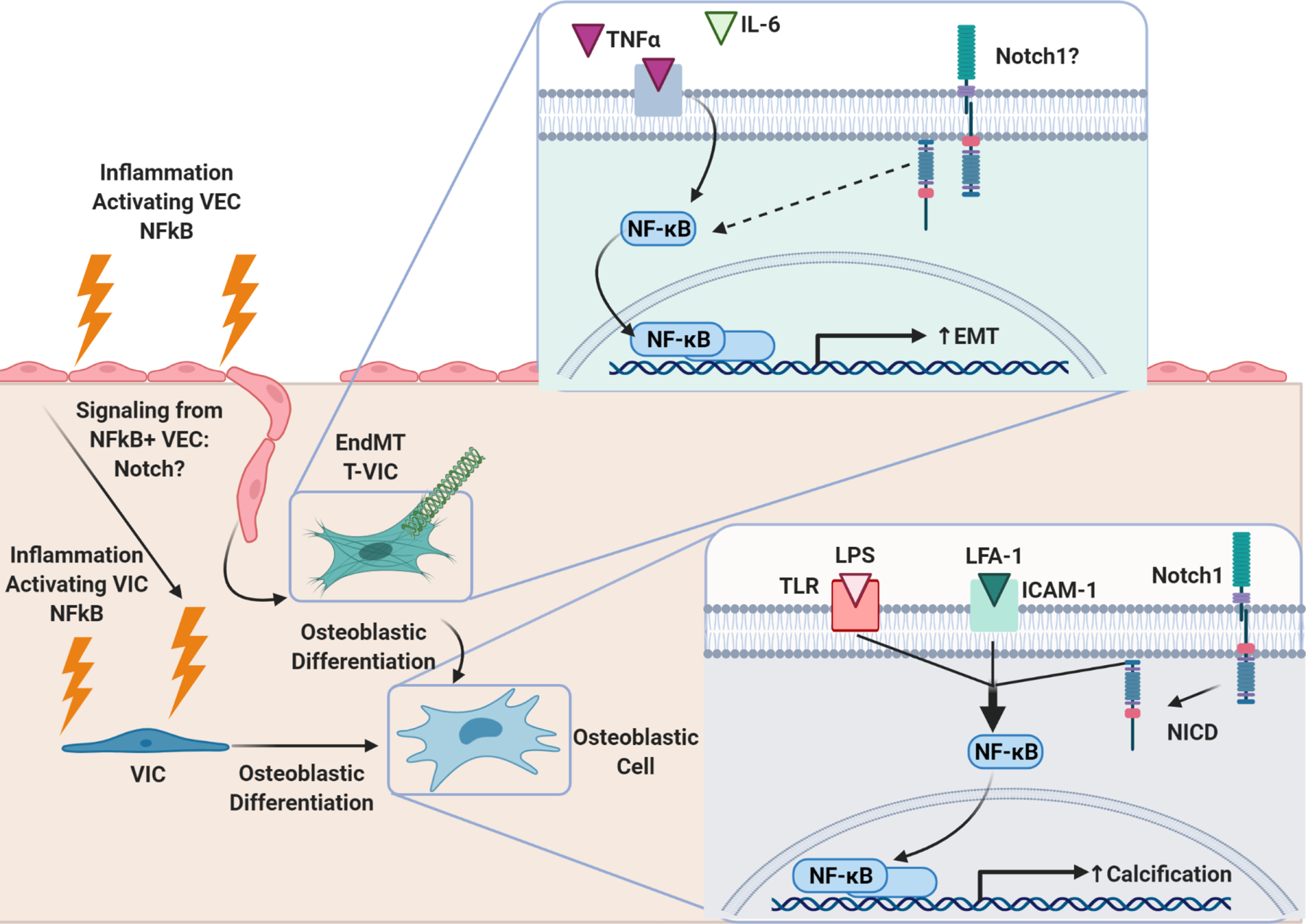
Bottom Right: NFκB activation in VIC through concerted efforts of inflammatory LPS/TLR signaling, LFA-1/ICAM-1 signaling, and Notch-1 signaling induces VIC osteoblastic differentiation and calcification. Left: Inflammation induces NFκB activation in VEC. NFκB+ VEC undergo EndMT, transforming into VIC (T-VIC). NFκB+ VEC have also been shown to exhibit evidence of osteoblastic differentiation. Knock-out of valve-specific NFκB in CAVD models prevents calcification and adverse valvular remodeling, consistent with the paradigm that NFκB activation in VEC promotes pathologic differentiation of VIC. Top right: Inflammatory cytokines TNFα and IL-6 activate NFκB signaling in VEC, which induces EndMT. It is unknown if Notch1 also plays a role in NFκB signaling in VEC. If so, Notch pathway could be a method through which VIC and VEC communicate in response to inflammation.
IV. Altered mechanics in CAVD perpetuate disease
Risk factors such as age as well as inflammation result in progressive stiffening of the aortic valve as resident cells differentiate and remodel the matrix, and this stiffness can be sensed by resident cells, modulating their phenotypes and contributing to disease in a positive feedback loop. As the valve stiffens, its mechanical properties alter, preventing its ability to efficiently open and close over the cardiac cycle. Thus, hemodynamic shear stress and cyclic strain in the valve is altered, and these mechanical stimuli can induce a pro-disease and pro-inflammatory state in valvular cells. Hypertension could also induce altered shear stresses on the valve, initiating and/or perpetuating disease. The following section describes current knowledge of VIC and VEC cellular response to these mechanical stimuli, in isolated mono-culture and dynamic co-culture studies.
VIC and VEC Responses to stiffness
The VIC response to stiffness under an activating stimulus has been studied extensively under many conditions generally ranges from a myofibroblastic and apoptotic state in stiff substrates to osteogenic in softer substrate54,188–190,191,192. Yip et al. investigated the formation of calcific nodules under stiff and soft substrates, and found interestingly that stiffer substrates tended to form aggregates that were apoptotic, while softer substrates formed more nodules overall that were osteoblastic and not apoptotic193. They attributed the formation stiffer apoptotic nodules to AKT signaling, and this was corroborated later by Wang et al., who found that inhibiting PI3K (upstream of AKT) was able to block nodule formation on stiff substrates191. Chen et al. found additionally that an increase in aSMA on increasingly stiff substrates was due to B-catenin signaling, while Santoro et al. found a role for YAP/TAZ signaling in this process54,190. It is likely that these pathways are acting either separately or in concert to modulate the VIC response to stiff substrates. Investigation into pathways involved in VIC nodule formation in softer substrates have found a role for RhoA in osteoblastic differentiation through demonstration that addition of Rock inhibitor decreased Runx2 and OCN at 1kPa188. Contrary to the studies mentioned above, in a 3D model Duan et al. reported that there was more aSMA activation of embedded VIC in softer vs. stiffermethacrylated hyaluronic acid/oxidized hyaluronic substrates194. However, high circularity indicative of poor cell spreading due to the methacrylated hyaluronic acid material may have affected cellular expression, as this trend was not seen in methacrylated hyaluronic acid/ gelatin combination gels194. It is important to consider 2D vs. 3D culture when interpreting stiffness results, as some stiffer substrates investigated in 2D may elicit different responses when embedded in a more physiologically relevant 3D model.
The role of matrix stiffness and composition has also been evaluated in endothelial cells195,196. Endothelial cells tend to increase EndMT evidenced by aSMA expression and matrix invasion with increasing stiffness of the substrate195,196. Dahal et al. found that this VEC transdifferentiation occurs through an ERK pathway, while Zhong et al. found a role for B-catenin, similarly to Chen et al’s VIC study190,195,196.
Gould et al. have investigated the role of stiffness on VIC activation in a VIC+VEC co-culture model136. They found similarly to previous reports that increased stiffness resulted in increased myofibroblastic activation and significantly more nodule formation. However, co-culture with VEC seeded on one end and VIC seeded on either side of a polyethelyne glycol (PEG) hydrogel was able to reduce the number of myofibroblastic VIC and nodule formation. As discussed above, VEC modulated this myofibroblastic and osteogenic activation in VIC through NO signaling, which likely modulates VIC ROCK activity136. As apoptotic VIC nodule formation through PI3K/AKT signaling has been found to dominate on substantially stiffer substrates, it would be interesting to see if VEC co-culture is also able to modulate these pathways through PI3K/AKT signaling. Additionally, as VEC have been shown to undergo increased EndMT in response to substrate stiffness, the effects of VIC-VEC co-culture on substrates of varying stiffnesses with characterization of VEC invasion could yield important insights in future studies.
VIC and VEC Responses to Pathologic Shear Stress
As the endothelium is the only cell type directly exposed to fluid flow from the valve, many studies have focused on the role it plays in mechanosensing and responding to shear in homeostasis and disease. Butcher et al. investigated transcriptional readouts of porcine aortic valvular endothelial cells (PAVEC) in comparison to porcine aortic endothelial cells (PAEC) under 20 dyne/cm^2 shear197. They found that both cell types down regulated BMP-4 with shear, but specifically cadherin-11 (CAD-11) was downregulated with shear in PAVEC, and that baseline levels of CAD-11 were much higher in PAVEC vs. PAEC197. This study indicates that though there are some similarities between the valvular and aortic endothelial response to shear, there are also marked differences. Our lab also investigated shear stress of different magnitudes on aortic endothelial cell transformation. We found that low laminar shear stress and low to moderate oscillatory shear stress induced EndMT markers aSMA, Snail, TGFB, NfKB, and ICAM-1 in endothelial cells198. These inflammatory markers and cytokines, notably TGFB, have been associated with VIC differentiation to a myofibroblastic and calcific phenotype. Holliday et al. interestingly found differences in mRNA due to applied shear stress but no significant differences in mRNA due to side origin of human aortic valvular endothelial cells199. This indicates that applied shear stimulus may supersede biological origin in driving shear responses of valvular endothelial cells. Holliday et al. also found that laminar shear reduces BMP4, and increases eNOS and IkB signaling. These eNOS results are corroborated by Richards et al.65. As discussed above, NO directly communicates with VIC, and IkB prevents the inflammatory transcription factor NFκB, which when activated could assist in recruitment of inflammatory cells and inflammatory activation of neighbors through cytokine expression200.
Studies have also investigated the role of shear stress on expression in the fibrosa and ventricularis endothelium in porcine leaflet explants110,201–203. In aortic explanted leaflets, elevated pulsatile shear stress resulted in increased VCAM-1, ICAM-1, BMP-4 and TGFB signaling on the aortic side of the leaflet110. A combination of BMP and TGFB-1 inhibitors was able to reduce VCAM-1, BMP-4, and TGFB-1 signaling compared to media control in supraphysiologic shear conditions on the aortic side of the leaflet201. It was concluded that there may be a potential synergistic role for BMP and TGFB signaling in promoting pro-inflammatory activation of endothelial cells under pathologic shear stress201. Sun et al. used same model to study the effects of both magnitude and frequency of shear203. They found that high magnitude normal frequency shear increased BMP-4 and TGFB (as expected) and also MMP’s 2/9 and Cathepsin L/S, and abnormal frequency at normal shear magnitude also increased degrative enzymes but not BMP’s/TGFB-1. Former studies in addition to this one noted that there was usually TGFB and BMP signaling localized along the endothelium and sub-endothelially110,203, while this study found MMP’s and cathepsins more uniformly spread throughout the tissue203. The effects of side-specific shear stress was also investigated in a similar model system, and it was found that inducing aortic shear flow pattern on the aortic side of the leaflet increased collagen and GAG content, while under ventricular flow increased elastin content compared to aortic flow202. This could indicate that shear stress type sensed by aortic or ventricular endothelium can influence ECM deposition type in VIC cells.
Butcher and Nerem used a more simplified collagen gel co-culture system to study VIC alone in comparison to VIC+VEC co-culture under shear conditions204. They found that ECM remodeling and overall protein deposition and proliferation responses differed in response to shear in VIC only vs. VIC+VEC co-culture. Additionally, VIC+VEC had more GAG content than VIC alone under 20dynes shear at 4 days, less proliferation under shear at 4 days, and more protein content with shear204. In contrast, Wang et al. tested VIC alone in a PDMS microfluidic chip and found that above 0.78dyn/cm2 the VICs were elongated and aligned parallel to flow and that they had increased TGFB expression above 0.49dyn/cm2 205. This differs from the results found by Butcher et al., which did not largely find VIC alignment204. The 2D vs. 3D culture may play a role in differing results between these two studies. Wang et al. mentioned addition of VEC co-culture in their future work, and this will yield important information about VIC+VEC interactions under shear that could not be captured in their study204,205. For a timecourse summary of shear stress studies, please refer to figure 6.
Figure 6. Mechanical Stimulus: Shear Stress Time Point Observations.
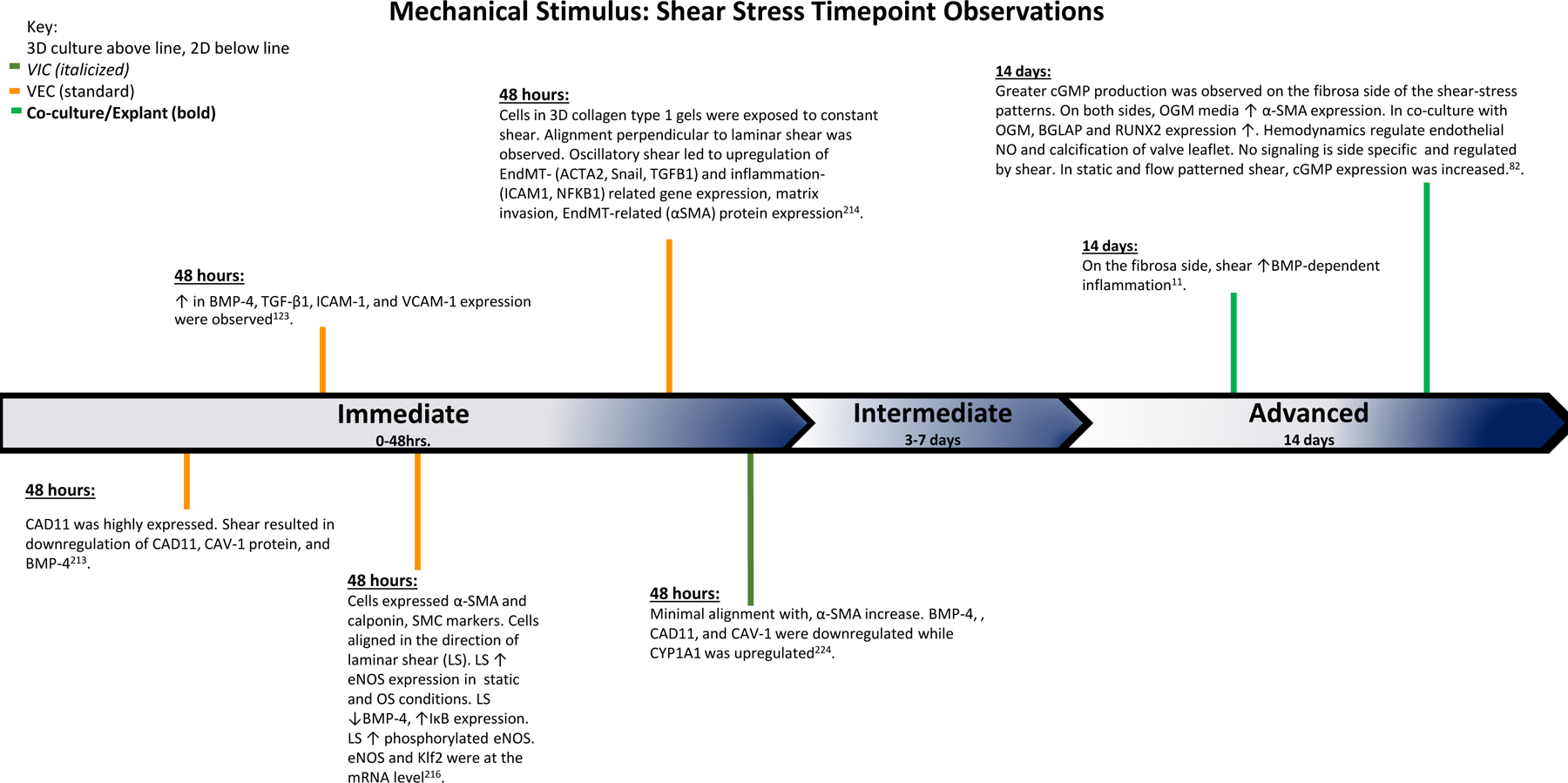
The findings of various studies observing the effects of shear stress over the course of the experiments. Based on the time points of experimental findings, the observations are classified as Immediate (0–48 hrs.), Intermediate (3–7 days), and Advanced (14 days).
VIC and VEC responses to Pathologic Static or Cyclic Strain
Studies investigating VEC only in response to homeostatic and pathologic cyclic strain have found that the least amount of inflammatory adhesion molecules ICAM-1, VCAM-1, and E-selectin are present at 10% strain in comparison to 20%206. Further investigation looking at differences between fibrosa and ventricularis VEC found that fibrosa VEC upregulate B1 integrin in comparison to the ventricularis, and ventricularis VEC alternatively upregulate VE-cadherin in comparison to the fibrosa at 20% strain207. These studies demonstrate that VEC alone are able to sense and respond to altered cyclic strain, specifically with changes in adhesion molecules that could promote inflammation. McIntosh and Warnock’s results also demonstrate that location matters when it comes to the response: i.e. VEC from the fibrosa will respond differently than VEC in the ventricularis to a cyclic-strain stimulus.
In contrast, many studies investigating VIC only and VIC+VEC concerted efforts on valve remodeling and calcification have focused on 14–15% strain. In VIC only, 15% strain yields the lowest expression of inflammatory markers, reduced IKKB, and increased pSMAD2/3141. A cyclic strain at 14–15% has also been reported to be pro-calcific and pro-remodeling in both VIC only120,208,209 and valve explant systems28,52,210. These results point to a complex role of inflammation on VIC remodeling and calcification, as the strain that induced the least number of inflammatory markers also exhibited notable calcific spheroids with hydroxyapatite (HA) deposits120,141,208. Our lab has investigated the role of VIC only culture in a 3D cyclically strained collagen gel. We have found that when VIC are cultured in 3D static-strained monoculture in ostogenic conditions, they exhibit a spike in RhoA signaling at day 1 that resolves at later timepoints, and that the addition of dominant negative (DN)-RhoA reduced the number of nodules at 14 days. Bouchareb et al. also found that 30 minutes after cyclic strain ROCK activity increased, returned back to baseline levels after one hour, and that addition of Rock inhibitor reduced calcification in a 2D VIC cyclic stretch model208. It appears that rapid RhoA/Rock signaling exerts a large influence over calcification and nodule formation in these VIC- only systems208,211. Interestingly, Gould et al. have discovered that endothelial NO signaling likely exerts an anti- myofibroblastic and calcific protective effect on VIC through ROCK signaling136. Investigation into the temporal regulation of calcification through RhoA/ROCK signaling in a co-culture cyclic-strain model could yield important insights into how these cells regulate each other in homeostatic and pathologic conditions.
Current methods to study VIC and VEC under a mechanical stimulus together in vitro include valvular explants mounted in a bioreactor, and mechanically-constrained gel co-culture. A series of studies by the Yoganathan group and others have tested explanted porcine aortic valves under cyclic stretch28,52,212. One notable finding was that the addition of cyclic stretch resulted in the upregulation of BMP2 specifically in the fibrosa side of leaflet explants, and that this co-localized with VWF, an endothelial marker28. Addition of noggin reduced the total calcification observed under mechanical stretch28. This indicates that the endothelial BMP side-specific response to cyclic stretch may be crucial to overall valve calcification, as BMP was co-localized with the endothelium and noggin, a BMP inhibitor, was able to reduce calcification17.
Our lab has also employed a collagen gel co-culture model containing aortic valvular endothelial cells seeded on top of interstitial cells to investigate the effects of co-culture on the cellular response to static mechanical constraint65. We discovered that addition of PAVEC to PAVIC prevents calcification and compaction in OGM conditions, and reduces aSMA, OCN, and Runx2 expression. With loss and gain of function experiments, we were able to attribute these effects to NO signaling from the valvular endothelial cells, indicating that NO signaling plays a role in modulating VIC phenotype not just under shear but also in static mechanically-constrained conditions. For a timecourse summary of the results discussed in this section, please refer to figure 7.
Figure 7. Mechanical Stimulus: Cyclic Stretch and Constant Tension Time Point Observations:
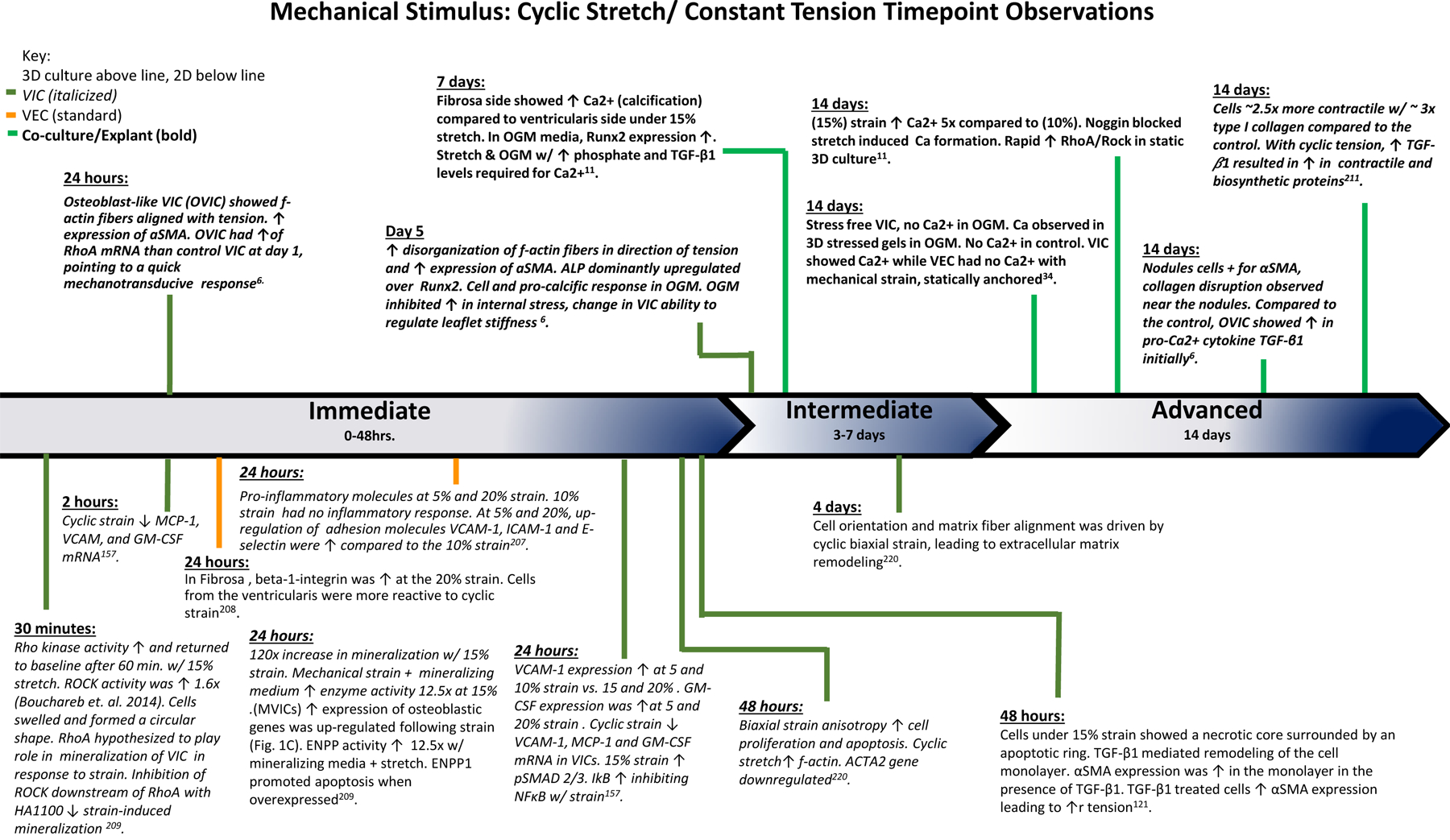
The findings of various studies observing the effects of cyclic stretch and constant tension over the course of the experiments. Based on the time points of experimental findings, the observations are classified as Immediate (0–48 hrs.), Intermediate (3–7 days), and Advanced (14 days).
V. Future Directions for CAVD Research
Temporal phases of CAVD progression
As discussed in the introduction, CAVD is usually not diagnosed in patients until they already have hemodynamic abnormalities due to widespread calcification and valve remodeling and left ventricular hypertrophy. In comparison, many in-vitro models test therapeutics on their ability to prevent valvular calcification. It is still unclear when a model investigating the prevention of calcification corresponds to in-vivo disease progression. Looking forward, as the field uncovers more therapeutics that show great promise in-vitro, it is crucial to gain a better understanding of when the effects of these therapeutics might provide the most benefit in-vivo.
For example, studies investigating the efficacy of preventative strategies against CAVD development have found that a combination of exercise and high cholesterol diet was able to prevent aortic valve disease in comparison to high cholesterol only. The endothelial lining was more intact in the exercise+cholesterol group than cholesterol alone, indicating the importance of maintaining the endothelial barrier in preventing CAVD213. However, this same group found in a follow up study that removing the high cholesterol diet or beginning exercise 16 weeks after mice were placed on a high cholesterol diet was not able to prevent valvular disease214. Additionally, in this study there was no longer preservation of the endothelial lining214. This indicates that there is likely a window for treatments that attempt to reduce the initiating factors, specifically endothelial damage, in CAVD. When that window begins and ends in humans and other animal models remains unclear.
Additionally, as discussed above, many studies investigating the interaction of mechanics and calcification have found that there is a time-dependent acute activation and subsequent return to baseline of the RhoA/Rock pathway208,211.This initial upregulation of the RhoA pathway had widespread effects on later calcification, although it was no longer upregulated during later timepoints. Though the Rho/Rock pathway played a significant role in calcification in these studies, targeting the pathway at later timepoints would not likely show efficacy. Thus, not only is it important to understand macroscopically when in-vitro models correspond to disease progression but it is also important to define when certain pathways are temporally modulated over disease progression.
Work from our lab has attempted to model later-stage calcification in-vitro by introducing hydroxyapatite nanoparticles to our 3D cultures215. We have found that smaller and less-crystalline calcium deposits tend to promote more calcific-disease responses in VIC-only and VIC+VEC co-cultures than more crystalline structures. Additionally, there was no longer a protective effect against myofibroblastic/osteogenic differentiation and calcification from adding VEC in co-culture with VIC in the presence of HA nanoparticles. Interestingly, there was still an upregulation of eNOS in these cultures indicating that its effectiveness in promoting quiescence in VIC may be diminished when there is already calcification present. Further work modeling late-stage disease is warranted to identify VIC-VEC interactions that may offer protection in early disease progression but are no longer effective in later-stage disease.
Risk Factors in CAVD Progression
Table 1 presents many risk factors for CAVD, mechanisms for how they could cause the disease, as well as current experimental models to investigate them. Porras et al. has pointed out that unlike atherosclerosis, it appears that the risk factor for causing valve disease can directly influence how the disease progresses, as evidenced by sex-specific disease etiologies in CAVD216. Though CAVD patients in many cases present with many of these risk factors, understanding how each may play a role in the disease in-vitro could assist in identifying which populations promising therapeutic targets would benefit most. This could be accomplished by first developing representative models of each risk factor and then testing potential therapeutic targets across multiple models to determine whether there is a preferential efficacy in certain systems. For instance, a therapeutic that shows potential in preventing calcification in a diabetes-specific model with only male cells may not show the same effect with female cells, or in aged vs. young cells, or in a hyperlipidemia model. With the advent of personalized medicine and the invention of ever-increasing high-throughput experimental systems, the feasibility of performing multiplexed therapeutic experiments has shifted from dream to reality. Information gleamed from these types of experiments could help to explain failed clinical trials and prompt more targeted future clinical trials to bring therapeutics from bench to bedside.
3D Environmental Context of CAVD Progression
Although many in-vitro studies investigating the valve have focused on 2D culture systems, the unique 3D-structure of the valve positions VIC and VEC such that they may regularly communicate with each other during homeostasis and disease. This communication can influence calcific disease pathways, as evidenced by many of the articles discussed above. Specifically, it appears that VEC are able to exert a homeostatic influence on VIC, and serve as the gateway for infiltrating immune cells, lipids, cytokines, and other disease-initiating stimuli. Therapeutics that are able to rescue endothelial barrier function, therefore maintaining the semi-permeable barrier and restoring homeostasis, could prove fruitful.
It is also important to note that calcific lesions are not uniform over a diseased valve leaflet, but are often highly-organized structures spread heterogeneously over the diseased leaflet. This indicates that in addition to considering purely VIC and VEC interactions in CAVD, investigations into the spatial heterogeneity and differentiation spectrum in their relation to lesion formation could provide clues for how the disease progresses, and highlight nuanced complexity needed for therapeutic targets.
Though the interactions between VIC and VEC are critical to understand in the progression of CAVD, they are difficult to resolve using classical biological tools. Single cell gene sequencing and spatially-resolved proteomics are two novel techniques that will likely yield important information at a cell-resolved level to aid in the understanding of differentiation states and interactions between different cell types in CAVD, and how these might be exploited for future therapeutics. A novel study by Xu et al. is the first to employ single cell sequencing on calcified and healthy human aortic valves217. They were able to identify seven cellular sub-populations that were significantly upregulated in CAVD valves vs. healthy valves. They noted upon staining that these populations tended to be in the interstitial layer, and identified them as VDSC’s. However, there was still much unanswered about the identity and activity of these cell types.
Xu et al. additionally performed a differentiation trajectory, and determined that these VDSC’s likely originated from VIC, and a cellular cluster that likely was from a VEC-transitioning EndMT state217. Though this study yielded important insights at the single-cell level, there is much cellular heterogeneity from patient to patient, which may overshadow genes associated with disease progression. Thus, use of this technique in a more controlled experimental environment could yield more information about differentiation trajectories and expression at the single cell level.
Schlotter et al. also performed transcriptomics and proteomics on healthy, fibrotic, and calcific valve leaflets91. They were able to identify unique proteins and genes to each stage of disease progression. In studying pathways using network analysis of CAVD proteomic information, Schlotter et al. found MAPK signaling, lipid metabolism, coagulation, inflammation and ECM were the top subnetworks identified, further highlighting the important role of inflammation in disease progression. Additionally, they were able to find proteins specific to each layer of the valve in healthy and diseased valves, indicating that there is a baseline heterogeneity in these populations. They were able to identify a unique subpopulation of VIC that are GFAP+ and localize to the spongiosa, further highlighting the inherent heterogeneity of cells in the aortic valve based on location.
Additionally, it is important to remember that the valve is not a free-floating tissue, but rather is mechanically anchored and constantly exposed to mechanical stimuli. Abnormal mechanical stimuli appear to drive pro-inflammatory expression and promote CAVD myofibroblastic/osteogenic disease progression. Understanding how CAVD progresses under different mechanical stimuli can aid in elucidating risk factors as well as gain an understanding of which therapeutics might be most effective in which mechanical contexts.
Spatially, inflammation has been shown to be associated with calcific deposits, and is an important predictive factor for disease progression and severity. Modulating inflammatory cells themselves, specifically macrophages, has gained increasing interest in a wide range of diseases218. However, this proves to be a difficult strategy because the complexity through which the inflammatory system drives homeostasis and disease not only through the valve but in the body generally is still not fully understood. Modulating the macrophage response through blanketed removal was shown to promote, and not prevent disease, indicating that their presence in the valve is somewhat necessary87. Raddatz et al. found that is likely an already inflammatory valve that brings inflammatory infiltrate into the tissue in disease, and not the other way around, indicating that systems investigating isolated VIC and VEC interactions are likely a good model for disease initiation59. Information gathered using novel single-cell transcriptomics and spatially-resolved proteomics, as discussed above, could provide more detailed clues about the spatial distribution and identity of inflammatory cells that populate more advanced-stage CAVD valves.
Conclusions
In conclusion, as the aging population grows, incidence of calcific aortic valve disease will continue to rise. Though valve disease due to infection has been largely eradicated in western countries, worldwide this remains a major cause of heart disease and this should be considered when designing therapeutic strategies. As surgical treatment is not a long term solution for many or inaccessible due to resources and/or costs, there is a great opportunity to help patients globally through the design and implementation of medicinal therapeutics. Improved diagnostic tools that are biologically based could shed light to the elusive window of therapy where symptoms are not present, bolstering the odds of successful translation of therapeutics that modulate time-dependent CAVD processes. We have gained understanding of intricacies regarding the interplay of inflammation, VIC-VEC interactions, and mechanics in valve disease, which can pave the way for the development of effective biologically-based and targeted therapeutics. New proteomics and sequencing tools provide an opportunity for investigation of more nuanced biological processes, and will allow for further uncovering of unanswered questions that still remain regarding CAVD.
Acknowledgements:
We acknowledge Dr. Terence Gee, for his training, mentorship, expertise, and insight Figures 1–5 were created with BioRender.com
Sources of Funding:
This work was funded by the following NIH grants: R01 HL143247, R01 HL128745, R01 HL151190
Abbreviations
- CAVD
Calcific Aortic Valve Disease
- LVH
Left Ventricular Hypertrophy
- ECM
Extracellular Matrix
- GAG
Glycosaminoglycans
- NO
Nitric Oxide
- EBCT
Electron Beam Computed Tomography
- AS
Aortic Stenosis
- AFM
Atomic Force Microscopy
- RF
Rheumatic Fever
- RHD
Rheumatic Heart Disease
- TAVR
Transcatheter Aortic Valve Replacement
- SAVR
Surgical Aortic Valve Replacement
- LV
Left Ventricle
- VIC
Valvular Interstitial Cell
- VEC
Valvular Endothelial Cell
- eNOS
Nitric Oxide Synthase 3
- aSMA
alpha-Smooth Muscle Actin
- OGM
Osteogenic Media
- PAVIC
Porcine Aortic Valvular Interstitial Cells
- HUVEC
Human Umbilical Vein Endothelial Cell
- ROCK
Rho-Associated Protein Kinase
- BMP
Bone Morphogenic Protein
- CNP
C-type Natriuretic Peptide
- EndMT
Endothelial to Mesenchymal Transition
- LPS
Lipopolysaccharide
- ALP
Alkaline Phosphatase
- TGFB
Transforming growth factor beta
- LDL
Low-density lipoprotein
- TNFα
Tumor-Necrosis Factor Alpha
- ICAM
intercellular adhesion molecule-1
- LFA
Lymphocyte function-associated antigen 1
- VEGF
Vascular endothelial growth factor
- VCAM
Vascular cell adhesion protein 1
- ALCAM
Activated Leukocyte Adhesion Molecule
- FDG-PET
Fluorodeoxyglucose positron emission tomography
- HDL
High-density lipoprotein
- OxLDL
Oxidized low-density lipoprotein
- MMP
Matrix Metallo-Proteinase
- Lp(a)
Lipoprotein(a)
- vWF
Von-Willebrand Factor
- NFκB
Nuclear Factor Kappa B
- TLR
Toll-like Receptor
- ROS
Reactive Oxygen Species
- IkB
Inhibitory Kappa B
- IKK complex
IkB Kinase Complex
- IKKB
IKK-complex Beta subunit
- ARS
Alizarin Red S
- OCN
Osteocalcin
- EMD
Anthraquinone Emodin
- PEG
Polyethylene Glycol
- PAVEC
Porcine Aortic Valvular Endothelial Cells
- PAEC
Porcine Aortic Endothelial Cells
- CAD-11
Cadherin 11
- HA
Hydroxyapatite
- DN
Dominant-Negative
Footnotes
Disclosures:
None
References
- 1.Butcher JT, Mahler GJ, Hockaday LA. Aortic valve disease and treatment: the need for naturally engineered solutions. Adv Drug Deliv Rev 2011;63:242–268. doi: 10.1016/j.addr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Thubrikar M, Bosher LP, Nolan SP. The mechanism of opening of the aortic valve. J Thorac Cardiovasc Surg 1979;77:863–870. [PubMed] [Google Scholar]
- 3.Rader F, Sachdev E, Arsanjani R, Siegel RJ. Left ventricular hypertrophy in valvular aortic stenosis: mechanisms and clinical implications. Am J Med 2015;128:344–352. doi: 10.1016/j.amjmed.2014.10.054. [DOI] [PubMed] [Google Scholar]
- 4.Lorell BH, Carabello BA. Left Ventricular Hypertrophy. Circulation 2000;102:470–479. doi: 10.1161/01.CIR.102.4.470. [DOI] [PubMed] [Google Scholar]
- 5.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 7.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of “degenerative” valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 8.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yadgir S, Johnson CO, Aboyans V, Adebayo OM, Adedoyin RA, Afarideh M, et al. Global, Regional, and National Burden of Calcific Aortic Valve and Degenerative Mitral Valve Diseases, 1990–2017. Circulation 2020;141:1670–1680. doi: 10.1161/CIRCULATIONAHA.119.043391. [DOI] [PubMed] [Google Scholar]
- 10.Huygens SA, Goossens LMA, van Erkelens JA, Takkenberg JJM, Rutten-van Mölken MPMH. How much does a heart valve implantation cost and what are the health care costs afterwards? Open Hear 2018;5:e000672. doi: 10.1136/openhrt-2017-000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witberg G, Lador A, Yahav D, Kornowski R. Transcatheter versus surgical aortic valve replacement in patients at low surgical risk: A meta-analysis of randomized trials and propensity score matched observational studies. Catheter Cardiovasc Interv Off J Soc Card Angiogr Interv 2018;92:408–416. doi: 10.1002/ccd.27518. [DOI] [PubMed] [Google Scholar]
- 12.Antunes MJ. Repair of the rheumatic mitral valve: Is the controversy over? Asian Cardiovasc Thorac Ann 2020;28:374–376. doi: 10.1177/0218492320927316. [DOI] [PubMed] [Google Scholar]
- 13.Binotto M, Guilherme L, Tanaka A. Rheumatic Fever. Images Paediatr Cardiol 2002;4:12–31. [PMC free article] [PubMed] [Google Scholar]
- 14.Chen HY, Engert JC, Thanassoulis G. Risk factors for valvular calcification. Curr Opin Endocrinol Diabetes Obes 2019;26:96–102. doi: 10.1097/MED.0000000000000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 16.Marijon E, Mirabel M, Celermajer DS, Jouven X. Rheumatic heart disease. Lancet (London, England) 2012;379:953–964. doi: 10.1016/S0140-6736(11)61171-9. [DOI] [PubMed] [Google Scholar]
- 17.Frank S, Johnson A, Ross JJ. Natural history of valvular aortic stenosis. Br Heart J 1973;35:41–46. doi: 10.1136/hrt.35.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross JJ, Braunwald E. Aortic stenosis. Circulation 1968;38:61–67. doi: 10.1161/01.cir.38.1s5.v-61. [DOI] [PubMed] [Google Scholar]
- 19.Braunwald E Aortic Stenosis. Circulation 2018;137:2099–2100. doi: 10.1161/CIRCULATIONAHA.118.033408. [DOI] [PubMed] [Google Scholar]
- 20.Beckmann E, Grau JB, Sainger R, Poggio P, Ferrari G. Insights into the use of biomarkers in calcific aortic valve disease. J Heart Valve Dis 2010;19:441–452. [PMC free article] [PubMed] [Google Scholar]
- 21.Small A, Kiss D, Giri J, Anwaruddin S, Siddiqi H, Guerraty M, et al. Biomarkers of Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol 2017;37:623–632. doi: 10.1161/ATVBAHA.116.308615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kossar AP, Anselmo W, Grau JB, Liu Y, Small A, Carter SL, et al. Circulating and tissue matricellular RNA and protein expression in calcific aortic valve disease. Physiol Genomics 2020;52:191–199. doi: 10.1152/physiolgenomics.00104.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thériault S, Gaudreault N, Lamontagne M, Rosa M, Boulanger M-C, Messika-Zeitoun D, et al. A transcriptome-wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat Commun 2018;9:988. doi: 10.1038/s41467-018-03260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol 2013;60:27–35. doi: 10.1016/j.yjmcc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaura Y, Watanabe N, Shimaya M, Tomita Y, Fukaya T, Yoshida K. Impact of Cumulative Smoking Exposure on Subclinical Degenerative Aortic Valve Disease in Apparently Healthy Male Workers. Circ Cardiovasc Imaging 2019;12:e008901. doi: 10.1161/CIRCIMAGING.119.008901. [DOI] [PubMed] [Google Scholar]
- 27.Linefsky J, Katz R, Budoff M, Probstfield J, Owens D, Takasu J, et al. Stages of systemic hypertension and blood pressure as correlates of computed tomography-assessed aortic valve calcium (from the Multi-Ethnic Study of Atherosclerosis). Am J Cardiol 2011;107:47–51. doi: 10.1016/j.amjcard.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balachandran K, Sucosky P, Jo H, Yoganathan AP. Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner. Am J Pathol 2010;177:49–57. doi: 10.2353/ajpath.2010.090631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capoulade R, Clavel M-A, Mathieu P, Côté N, Dumesnil JG, Arsenault M, et al. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. Eur J Clin Invest 2013;43:1262–1272. doi: 10.1111/eci.12169. [DOI] [PubMed] [Google Scholar]
- 30.Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The Role of Lipids and Lipoproteins in Atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al., editors., South Dartmouth (MA): 2000. [Google Scholar]
- 31.Tintut Y, Hsu JJ, Demer LL. Lipoproteins in Cardiovascular Calcification: Potential Targets and Challenges. Front Cardiovasc Med 2018;5:172. doi: 10.3389/fcvm.2018.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflam 2011;2011:364310. doi: 10.4061/2011/364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F, Zhao Z, Cai Z, Dong N, Liu Y. Oxidized low-density lipoprotein promotes osteoblastic differentiation of valvular interstitial cells through RAGE/MAPK. Cardiology 2015;130:55–61. doi: 10.1159/000369126. [DOI] [PubMed] [Google Scholar]
- 34.Jensen HA, Mehta JL. Endothelial cell dysfunction as a novel therapeutic target in atherosclerosis. Expert Rev Cardiovasc Ther 2016;14:1021–1033. doi: 10.1080/14779072.2016.1207527. [DOI] [PubMed] [Google Scholar]
- 35.Vadana M, Cecoltan S, Ciortan L, Macarie RD, Tucureanu MM, Mihaila AC, et al. Molecular mechanisms involved in high glucose-induced valve calcification in a 3D valve model with human valvular cells. J Cell Mol Med 2020;24:6350–6361. doi: 10.1111/jcmm.15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciortan L, Macarie RD, Cecoltan S, Vadana M, Tucureanu MM, Mihaila AC, et al. Chronic High Glucose Concentration Induces Inflammatory and Remodeling Changes in Valvular Endothelial Cells and Valvular Interstitial Cells in a Gelatin Methacrylate 3D Model of the Human Aortic Valve. Polymers (Basel) 2020;12. doi: 10.3390/polym12122786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.García-Rodríguez C, Parra-Izquierdo I, Castaños-Mollor I, López J, San Román JA, Sánchez Crespo M. Toll-Like Receptors, Inflammation, and Calcific Aortic Valve Disease. Front Physiol 2018;9:201. doi: 10.3389/fphys.2018.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summerhill VI, Moschetta D, Orekhov AN, Poggio P, Myasoedova VA. Sex-Specific Features of Calcific Aortic Valve Disease. Int J Mol Sci 2020;21. doi: 10.3390/ijms21165620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, et al. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol 2014;34:2387–2393. doi: 10.1161/ATVBAHA.114.302523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephens EH, de Jonge N, McNeill MP, Durst CA, Grande-Allen KJ. Age-related changes in material behavior of porcine mitral and aortic valves and correlation to matrix composition. Tissue Eng Part A 2010;16:867–878. doi: 10.1089/ten.TEA.2009.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasan A, Saliba J, Pezeshgi Modarres H, Bakhaty A, Nasajpour A, Mofrad MRK, et al. Micro and nanotechnologies in heart valve tissue engineering. Biomaterials 2016;103:278–292. doi: 10.1016/j.biomaterials.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Tseng H, Grande-Allen KJ. Elastic fibers in the aortic valve spongiosa: a fresh perspective on its structure and role in overall tissue function. Acta Biomater 2011;7:2101–2108. doi: 10.1016/j.actbio.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine HJ. Rest heart rate and life expectancy. J Am Coll Cardiol 1997;30:1104–1106. doi: 10.1016/s0735-1097(97)00246-5. [DOI] [PubMed] [Google Scholar]
- 44.Arjunon S, Rathan S, Jo H, Yoganathan AP. Aortic valve: mechanical environment and mechanobiology. Ann Biomed Eng 2013;41:1331–1346. doi: 10.1007/s10439-013-0785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yap CH, Saikrishnan N, Tamilselvan G, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the aortic surface of the aortic valve leaflet. Biomech Model Mechanobiol 2012;11:171–182. doi: 10.1007/s10237-011-0301-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yap CH, Saikrishnan N, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the ventricular surface of the aortic valve leaflet. Biomech Model Mechanobiol 2012;11:231–244. doi: 10.1007/s10237-011-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernández Esmerats J, Heath J, Jo H. Shear-Sensitive Genes in Aortic Valve Endothelium. Antioxid Redox Signal 2016;25:401–414. doi: 10.1089/ars.2015.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest 2005;85:9–23. doi: 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- 49.Yip CYY, Simmons CA. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovasc Pathol Off J Soc Cardiovasc Pathol 2011;20:177–182. doi: 10.1016/j.carpath.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Pan S Molecular mechanisms responsible for the atheroprotective effects of laminar shear stress. Antioxid Redox Signal 2009;11:1669–1682. doi: 10.1089/ars.2009.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo D, Vesely I. Biaxial strain analysis of the porcine aortic valve. Ann Thorac Surg 1995;60:S374–8. doi: 10.1016/0003-4975(95)00249-k. [DOI] [PubMed] [Google Scholar]
- 52.Balachandran K, Sucosky P, Jo H, Yoganathan AP. Elevated cyclic stretch alters matrix remodeling in aortic valve cusps: implications for degenerative aortic valve disease. Am J Physiol Heart Circ Physiol 2009;296:H756–64. doi: 10.1152/ajpheart.00900.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vesely I, Noseworthy R. Micromechanics of the fibrosa and the ventricularis in aortic valve leaflets. J Biomech 1992;25:101–113. doi: 10.1016/0021-9290(92)90249-z. [DOI] [PubMed] [Google Scholar]
- 54.Santoro R, Scaini D, Severino LU, Amadeo F, Ferrari S, Bernava G, et al. Activation of human aortic valve interstitial cells by local stiffness involves YAP-dependent transcriptional signaling. Biomaterials 2018;181:268–279. doi: 10.1016/j.biomaterials.2018.07.033. [DOI] [PubMed] [Google Scholar]
- 55.Maleki H, Shahriari S, Durand L, Labrosse M, Kadem L. A metric for the stiffness of calcified aortic valves using a combined computational and experimental approach. Med Biol Eng Comput 2013;52. doi: 10.1007/s11517-013-1113-y. [DOI] [PubMed] [Google Scholar]
- 56.Shah NR, Coulter SA. An evidence-based guide for coronary calcium scoring in asymptomatic patients without coronary heart disease. Texas Hear Inst J 2012;39:240–242. [PMC free article] [PubMed] [Google Scholar]
- 57.Anstine LJ, Horne TE, Horwitz EM, Lincoln J. Contribution of Extra-Cardiac Cells in Murine Heart Valves is Age-Dependent. J Am Heart Assoc 2017;6. doi: 10.1161/JAHA.117.007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hulin A, Anstine LJ, Kim AJ, Potter SJ, DeFalco T, Lincoln J, et al. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arterioscler Thromb Vasc Biol 2018;38:636–644. doi: 10.1161/ATVBAHA.117.310667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raddatz MA, Huffstater T, Bersi MR, Reinfeld BI, Madden MZ, Booton SE, et al. Macrophages Promote Aortic Valve Cell Calcification and Alter STAT3 Splicing. Arterioscler Thromb Vasc Biol 2020;40:e153–e165. doi: 10.1161/ATVBAHA.120.314360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tompkins RG, Schnitzer JJ, Yarmush ML. Macromolecular transport within heart valves. Circ Res 1989;64:1213–1223. doi: 10.1161/01.RES.64.6.1213. [DOI] [PubMed] [Google Scholar]
- 61.Zeng Z, Yin Y, Huang A-L, Jan K-M, Rumschitzki DS. Macromolecular transport in heart valves. I. Studies of rat valves with horseradish peroxidase. Am J Physiol Heart Circ Physiol 2007;292:H2664–70. doi: 10.1152/ajpheart.01419.2006. [DOI] [PubMed] [Google Scholar]
- 62.Zeng Z, Yin Y, Jan K-M, Rumschitzki DS. Macromolecular transport in heart valves. II. Theoretical models. Am J Physiol Heart Circ Physiol 2007;292:H2671–86. doi: 10.1152/ajpheart.00608.2006. [DOI] [PubMed] [Google Scholar]
- 63.Mai J, Virtue A, Shen J, Wang H, Yang X-F. An evolving new paradigm: endothelial cells--conditional innate immune cells. J Hematol Oncol 2013;6:61. doi: 10.1186/1756-8722-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lei J, Vodovotz Y, Tzeng E, Billiar TR. Nitric oxide, a protective molecule in the cardiovascular system. Nitric Oxide Biol Chem 2013;35:175–185. doi: 10.1016/j.niox.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 65.Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, et al. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol 2013;182:1922–1931. doi: 10.1016/j.ajpath.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mónica FZ, Bian K, Murad F. The Endothelium-Dependent Nitric Oxide-cGMP Pathway. Adv Pharmacol 2016;77:1–27. doi: 10.1016/bs.apha.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 67.Kietadisorn R, Kietselaer BL, Schmidt HHHW, Moens AL. Role of tetrahydrobiopterin (BH4) in hyperhomocysteinemia-induced endothelial dysfuction: new indication for this orphan-drug? Am J Physiol Metab 2011;300:E1176–E1176. doi: 10.1152/ajpendo.00084.2011. [DOI] [PubMed] [Google Scholar]
- 68.Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation 2000;101:2345–2348. doi: 10.1161/01.cir.101.20.2345. [DOI] [PubMed] [Google Scholar]
- 69.El Accaoui RN, Gould ST, Hajj GP, Chu Y, Davis MK, Kraft DC, et al. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 2014;306:H1302–13. doi: 10.1152/ajpheart.00392.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol 2009;602:28–35. doi: 10.1016/j.ejphar.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 71.Farrar EJ, Huntley GD, Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS One 2015;10:e0123257. doi: 10.1371/journal.pone.0123257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gould ST, Matherly EE, Smith JN, Heistad DD, Anseth KS. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials 2014;35:3596–3606. doi: 10.1016/j.biomaterials.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res 2005;96:792–799. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yip CYY, Blaser MC, Mirzaei Z, Zhong X, Simmons CA. Inhibition of pathological differentiation of valvular interstitial cells by C-type natriuretic peptide. Arterioscler Thromb Vasc Biol 2011;31:1881–1889. doi: 10.1161/ATVBAHA.111.223974. [DOI] [PubMed] [Google Scholar]
- 75.Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, et al. Preferential Activation of SMAD1/5/8 on the Fibrosa Endothelium in Calcified Human Aortic Valves - Association with Low BMP Antagonists and SMAD6. PLoS One 2011;6:1–9. doi: 10.1371/journal.pone.0020969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li C, Xu S, Gotlieb AI. The response to valve injury. A paradigm to understand the pathogenesis of heart valve disease. Cardiovasc Pathol Off J Soc Cardiovasc Pathol 2011;20:183–190. doi: 10.1016/j.carpath.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 77.Bairati A, DeBiasi S. Presence of a smooth muscle system in aortic valve leaflets. Anat Embryol (Berl) 1981;161:329–340. doi: 10.1007/BF00301830. [DOI] [PubMed] [Google Scholar]
- 78.Chen J-H, Yip CYY, Sone ED, Simmons CA. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am J Pathol 2009;174:1109–1119. doi: 10.2353/ajpath.2009.080750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dutta P, Lincoln J. Calcific Aortic Valve Disease: a Developmental Biology Perspective. Curr Cardiol Rep 2018;20:21. doi: 10.1007/s11886-018-0968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hajdu Z, Romeo SJ, Fleming PA, Markwald RR, Visconti RP, Drake CJ. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J Mol Cell Cardiol 2011;51:955–965. doi: 10.1016/j.yjmcc.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gee T, Farrar E, Wang Y, Wu B, Hsu K, Zhou B, et al. NFκB (Nuclear Factor κ-Light-Chain Enhancer of Activated B Cells) Activity Regulates Cell-Type–Specific and Context-Specific Susceptibility to Calcification in the Aortic Valve. Arterioscler Thromb Vasc Biol 2020;40:638–655. doi: 10.1161/ATVBAHA.119.313248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis 2004;13:841–847. [PubMed] [Google Scholar]
- 83.Lester WM, Gotlieb AI. In vitro repair of the wounded porcine mitral valve. Circ Res 1988;62:833–845. doi: 10.1161/01.RES.62.4.833. [DOI] [PubMed] [Google Scholar]
- 84.Lester WM, Damji AA, Tanaka M, Gedeon I. Bovine mitral valve organ culture: Role of interstitial cells in repair of valvular injury. J Mol Cell Cardiol 1992;24:43–53. doi: 10.1016/0022-2828(92)91158-2. [DOI] [PubMed] [Google Scholar]
- 85.Deb A, Ubil E. Cardiac fibroblast in development and wound healing. J Mol Cell Cardiol 2014;70:47–55. doi: 10.1016/j.yjmcc.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. Int J Biochem Cell Biol 2003;35:113–118. doi: 10.1016/s1357-2725(02)00100-0. [DOI] [PubMed] [Google Scholar]
- 87.Calin MV, Manduteanu I, Dragomir E, Dragan E, Nicolae M, Gan AM, et al. Effect of depletion of monocytes/macrophages on early aortic valve lesion in experimental hyperlipidemia. Cell Tissue Res 2009;336:237–248. doi: 10.1007/s00441-009-0765-2. [DOI] [PubMed] [Google Scholar]
- 88.Lindman BR, Clavel M-A, Mathieu P, Iung B, Lancellotti P, Otto CM, et al. Calcific aortic stenosis. Nat Rev Dis Prim 2016;2:16006. doi: 10.1038/nrdp.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coté N, Mahmut A, Bosse Y, Couture C, Pagé S, Trahan S, et al. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation 2013;36:573–581. doi: 10.1007/s10753-012-9579-6. [DOI] [PubMed] [Google Scholar]
- 90.Rutkovskiy A, Malashicheva A, Sullivan G, Bogdanova M, Kostareva A, Stensløkken K-O, et al. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J Am Heart Assoc 2017;6. doi: 10.1161/JAHA.117.006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schlotter F, Halu A, Goto S, Blaser MC, Body SC, Lee LH, et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation 2018;138:377–393. doi: 10.1161/CIRCULATIONAHA.117.032291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hjortnaes J, Goettsch C, Hutcheson JD, Camci-Unal G, Lax L, Scherer K, et al. Simulation of early calcific aortic valve disease in a 3D platform: A role for myofibroblast differentiation. J Mol Cell Cardiol 2016;94:13–20. doi: 10.1016/j.yjmcc.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Monzack EL, Masters KS. Can valvular interstitial cells become true osteoblasts? A side-by-side comparison. J Heart Valve Dis 2011;20:449–463. [PMC free article] [PubMed] [Google Scholar]
- 94.Ma X, Zhao D, Yuan P, Li J, Yun Y, Cui Y, et al. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiol Sin 2020;36:183–194. doi: 10.6515/ACS.202005_36(3).20200213A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Farrar EJ, Butcher JT. Heterogeneous susceptibility of valve endothelial cells to mesenchymal transformation in response to TNFα. Ann Biomed Eng 2014;42:149–161. doi: 10.1007/s10439-013-0894-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, et al. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 2015;242:251–260. doi: 10.1016/j.atherosclerosis.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zmysłowski A, Szterk A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis 2017;16:188. doi: 10.1186/s12944-017-0579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart 2005;91:806–810. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rajamannan NM. Oxidative-mechanical stress signals stem cell niche mediated Lrp5 osteogenesis in eNOS(−/−) null mice. J Cell Biochem 2012;113:1623–1634. doi: 10.1002/jcb.24031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol 2003;23:1161–1168. doi: 10.1161/01.ATV.0000070546.16946.3A. [DOI] [PubMed] [Google Scholar]
- 101.Gliozzi M, Scicchitano M, Bosco F, Musolino V, Carresi C, Scarano F, et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int J Mol Sci 2019;20. doi: 10.3390/ijms20133294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mónica FZ, Bian K, Murad F. The Endothelium-Dependent Nitric Oxide–cGMP Pathway. In: Khalil RA, editor. Endothelium, vol. 77, Academic Press; 2016, p. 1–27. doi: 10.1016/bs.apha.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 103.Komori T Regulation of osteoblast differentiation by transcription factors. J Cell Biochem 2006;99:1233–1239. doi: 10.1002/jcb.20958. [DOI] [PubMed] [Google Scholar]
- 104.Marie PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys 2008;473:98–105. doi: 10.1016/j.abb.2008.02.030. [DOI] [PubMed] [Google Scholar]
- 105.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 106.Akiyama H, Chaboissier M-C, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 2002;16:2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen H, Ghori-Javed FY, Rashid H, Adhami MD, Serra R, Gutierrez SE, et al. Runx2 Regulates Endochondral Ossification Through Control of Chondrocyte Proliferation and Differentiation. J Bone Miner Res 2014;29:2653–2665. doi: 10.1002/jbmr.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, et al. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lowery JW, Rosen V. The BMP Pathway and Its Inhibitors in the Skeleton. Physiol Rev 2018;98:2431–2452. doi: 10.1152/physrev.00028.2017. [DOI] [PubMed] [Google Scholar]
- 110.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler Thromb Vasc Biol 2009;29:254–260. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang X, Fullerton DA, Su X, Ao L, Cleveland JCJ, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 112.Yang X, Meng X, Su X, Mauchley DC, Ao L, Cleveland JC, et al. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: Role of Smad1 and extracellular signal-regulated kinase 1/2. J Thorac Cardiovasc Surg 2009;138:1008–1015.e1. doi: 10.1016/j.jtcvs.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 113.Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of Human Valve Interstitial Cells in Valve Calcification and Their Response to Atorvastatin. Circulation 2006;114:I-547–I–552. doi: 10.1161/CIRCULATIONAHA.105.001115. [DOI] [PubMed] [Google Scholar]
- 114.Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-β superfamily signalling. Genes to Cells 2002;7:1191–1204. doi: 10.1046/j.1365-2443.2002.00599.x. [DOI] [PubMed] [Google Scholar]
- 115.Sritharen Y, Enriquez-Sarano M, Schaff HV, Casaclang-Verzosa G, Miller JD. Pathophysiology of Aortic Valve Stenosis: Is It Both Fibrocalcific and Sex Specific? Physiology (Bethesda) 2017;32:182–196. doi: 10.1152/physiol.00025.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jenke A, Kistner J, Saradar S, Chekhoeva A, Yazdanyar M, Bergmann AK, et al. Transforming growth factor-β1 promotes fibrosis but attenuates calcification of valvular tissue applied as a three-dimensional calcific aortic valve disease model. Am J Physiol Circ Physiol 2020;319:H1123–H1141. doi: 10.1152/ajpheart.00651.2019. [DOI] [PubMed] [Google Scholar]
- 117.Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD, et al. Valve Endothelial Cell-Derived Tgfβ1 Signaling Promotes Nuclear Localization of Sox9 in Interstitial Cells Associated With Attenuated Calcification. Arterioscler Thromb Vasc Biol 2016;36:328–338. doi: 10.1161/ATVBAHA.115.306091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jian B, Narula N, Li Q, Mohler ER 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 2003;75:456–457. doi: 10.1016/s0003-4975(02)04312-6. [DOI] [PubMed] [Google Scholar]
- 119.Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER 3rd, et al. Transforming growth factor-beta1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Ann Thorac Surg 2007;83:946–953. doi: 10.1016/j.athoracsur.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 120.Fisher CI, Chen J, Merryman WD. Calcific nodule morphogenesis by heart valve interstitial cells is strain dependent. Biomech Model Mechanobiol 2013;12:5–17. doi: 10.1007/s10237-012-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 2013;5:a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mackie EJ, Ahmed YA, Tatarczuch L, Chen K-S, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 2008;40:46–62. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 123.Huelsken J, Behrens J. The Wnt signalling pathway. J Cell Sci 2002;115:3977–3978. doi: 10.1242/jcs.00089. [DOI] [PubMed] [Google Scholar]
- 124.Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PVN, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem 2005;280:33132–33140. doi: 10.1074/jbc.M500608200. [DOI] [PubMed] [Google Scholar]
- 125.Wang H, Zang C, Liu XS, Aster JC. The role of Notch receptors in transcriptional regulation. J Cell Physiol 2015;230:982–988. doi: 10.1002/jcp.24872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mathieu P, Boulanger M-C. Basic mechanisms of calcific aortic valve disease. Can J Cardiol 2014;30:982–993. doi: 10.1016/j.cjca.2014.03.029. [DOI] [PubMed] [Google Scholar]
- 127.Rutenberg JB, Fischer A, Jia H, Gessler M, Zhong TP, Mercola M. Developmental patterning of the cardiac atrioventricular canal by Notch and Hairy-related transcription factors. Development 2006;133:4381–4390. doi: 10.1242/dev.02607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, et al. Inhibitory Role of Notch1 in Calcific Aortic Valve Disease. PLoS One 2011;6:1–12. doi: 10.1371/journal.pone.0027743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fayez H, Marie-Chloé B, Simon-Pierre G, Nathalie G, Soumiya A, Guada M, et al. Altered DNA Methylation of Long Noncoding RNA H19 in Calcific Aortic Valve Disease Promotes Mineralization by Silencing NOTCH1. Circulation 2016;134:1848–1862. doi: 10.1161/CIRCULATIONAHA.116.023116. [DOI] [PubMed] [Google Scholar]
- 131.Zeng Q, Song R, Ao L, Weyant MJ, Lee J, Xu D, et al. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-κB activation. Arterioscler Thromb Vasc Biol 2013;33:1580–1590. doi: 10.1161/ATVBAHA.112.300912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang D, Zeng Q, Song R, Ao L, Fullerton DA, Meng X. Ligation of ICAM-1 on human aortic valve interstitial cells induces the osteogenic response: A critical role of the Notch1-NF-κB pathway in BMP-2 expression. Biochim Biophys Acta 2014;1843:2744–2753. doi: 10.1016/j.bbamcr.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang Y, Wu B, Farrar E, Lui W, Lu P, Zhang D, et al. Notch-Tnf signalling is required for development and homeostasis of arterial valves. Eur Heart J 2015;38:675–686. doi: 10.1093/eurheartj/ehv520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E, et al. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am J Pathol 2001;159:1335–1343. doi: 10.1016/s0002-9440(10)62520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim AJ, Alfieri CM, Yutzey KE. Endothelial Cell Lineage Analysis Does Not Provide Evidence for EMT in Adult Valve Homeostasis and Disease. Anat Rec (Hoboken) 2019;302:125–135. doi: 10.1002/ar.23916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gould ST, Matherly EE, Smith JN, Heistad DD, Anseth KS. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials 2014;35:3596–3606. doi: 10.1016/j.biomaterials.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol 2013;33:121–130. doi: 10.1161/ATVBAHA.112.300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gendron N, Rosa M, Blandinieres A, Sottejeau Y, Rossi E, Van Belle E, et al. Human Aortic Valve Interstitial Cells Display Proangiogenic Properties During Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol 2021;41:415–429. doi: 10.1161/ATVBAHA.120.314287. [DOI] [PubMed] [Google Scholar]
- 139.Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003;170:191–203. doi: 10.1016/s0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 140.Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Side-specific expression of activated leukocyte adhesion molecule (ALCAM; CD166) in pathosusceptible regions of swine aortic valve endothelium. J Heart Valve Dis 2011;20:165–167. [PMC free article] [PubMed] [Google Scholar]
- 141.Smith KE, Metzler SA, Warnock JN. Cyclic strain inhibits acute pro-inflammatory gene expression in aortic valve interstitial cells. Biomech Model Mechanobiol 2010;9:117–125. doi: 10.1007/s10237-009-0165-2. [DOI] [PubMed] [Google Scholar]
- 142.Abdelbaky A, Corsini E, Figueroa AL, Subramanian S, Fontanez S, Emami H, et al. Early aortic valve inflammation precedes calcification: a longitudinal FDG-PET/CT study. Atherosclerosis 2015;238:165–172. doi: 10.1016/j.atherosclerosis.2014.11.026. [DOI] [PubMed] [Google Scholar]
- 143.Natorska J, Marek G, Sadowski J, Undas A. Presence of B cells within aortic valves in patients with aortic stenosis: Relation to severity of the disease. J Cardiol 2016;67:80–85. doi: 10.1016/j.jjcc.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 144.Hjortnaes J, Butcher J, Figueiredo J-L, Riccio M, Kohler RH, Kozloff KM, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J 2010;31:1975–1984. doi: 10.1093/eurheartj/ehq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mathieu P, Arsenault BJ, Boulanger M-C, Bossé Y, Koschinsky ML. Pathobiology of Lp(a) in calcific aortic valve disease. Expert Rev Cardiovasc Ther 2017;15:797–807. doi: 10.1080/14779072.2017.1367286. [DOI] [PubMed] [Google Scholar]
- 146.Syväranta S, Alanne-Kinnunen M, Öörni K, Oksjoki R, Kupari M, Kovanen PT, et al. Potential pathological roles for oxidized low-density lipoprotein and scavenger receptors SR-AI, CD36, and LOX-1 in aortic valve stenosis. Atherosclerosis 2014;235:398–407. doi: 10.1016/j.atherosclerosis.2014.05.933. [DOI] [PubMed] [Google Scholar]
- 147.Twigg MW, Freestone K, Homer-Vanniasinkam S, Ponnambalam S. The LOX-1 Scavenger Receptor and Its Implications in the Treatment of Vascular Disease. Cardiol Res Pract 2012;2012:632408. doi: 10.1155/2012/632408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kaniewska E, Mielcarek M, Chester A, Slominska E, Yacoub M, Smolenski R. Oxidized low-density lipoproteins enhance expression and activity of CD39 and CD73 in the human aortic valve endothelium. Nucleosides, Nucleotides and Nucleic Acids 2016;35:713–719. doi: 10.1080/15257770.2016.1163377. [DOI] [PubMed] [Google Scholar]
- 149.Matsuura E, Hughes GR V, Khamashta MA. Oxidation of LDL and its clinical implication. Autoimmun Rev 2008;7:558–566. doi: 10.1016/j.autrev.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 150.Olsson M, Thyberg J, Nilsson J. Presence of Oxidized Low Density Lipoprotein in Nonrheumatic Stenotic Aortic Valves. Arterioscler Thromb Vasc Biol 1999;19:1218–1222. doi: 10.1161/01.ATV.19.5.1218. [DOI] [PubMed] [Google Scholar]
- 151.Mohty D, Pibarot P, Després J-P, Côté C, Arsenault B, Cartier A, et al. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler Thromb Vasc Biol 2008;28:187–193. doi: 10.1161/ATVBAHA.107.154989. [DOI] [PubMed] [Google Scholar]
- 152.Shi X, Gao J, Lv Q, Cai H, Wang F, Ye R, et al. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front Physiol 2020;11:56. doi: 10.3389/fphys.2020.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Crismaru I, Pantea Stoian A, Bratu OG, Gaman M-A, Stanescu AMA, Bacalbasa N, et al. Low-density lipoprotein cholesterol lowering treatment: the current approach. Lipids Health Dis 2020;19:85. doi: 10.1186/s12944-020-01275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhao Y, Nicoll R, He YH, Henein MY. The effect of statins on valve function and calcification in aortic stenosis: A meta-analysis. Atherosclerosis 2016;246:318–324. doi: 10.1016/j.atherosclerosis.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 155.Parisi V, Leosco D, Ferro G, Bevilacqua A, Pagano G, de Lucia C, et al. The lipid theory in the pathogenesis of calcific aortic stenosis. Nutr Metab Cardiovasc Dis 2015;25:519–525. doi: 10.1016/j.numecd.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 156.Raddatz MA, Madhur MS, Merryman WD. Adaptive immune cells in calcific aortic valve disease. Am J Physiol Heart Circ Physiol 2019;317:H141–H155. doi: 10.1152/ajpheart.00100.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li G, Qiao W, Zhang W, Li F, Shi J, Dong N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J Thorac Cardiovasc Surg 2017;153:1318–1327.e1. doi: 10.1016/j.jtcvs.2017.01.052. [DOI] [PubMed] [Google Scholar]
- 158.Joghetaei N, Akhyari P, Rauch BH, Cullen P, Lichtenberg A, Rudelius M, et al. Extracellular matrix metalloproteinase inducer (CD147) and membrane type 1-matrix metalloproteinase are expressed on tissue macrophages in calcific aortic stenosis and induce transmigration in an artificial valve model. J Thorac Cardiovasc Surg 2011;142:191–198. doi: 10.1016/j.jtcvs.2010.09.051. [DOI] [PubMed] [Google Scholar]
- 159.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 160.Kaden JJ, Dempfle C-E, Grobholz R, Tran H-T, Kiliç R, Sarikoç A, et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 2003;170:205–211. doi: 10.1016/s0021-9150(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 161.Kaden JJ, Dempfle C-E, Grobholz R, Fischer CS, Vocke DC, Kılıç R, et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol 2005;14:80–87. doi: 10.1016/j.carpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 162.Wypasek E, Natorska J, Grudzień G, Filip G, Sadowski J, Undas A. Mast cells in human stenotic aortic valves are associated with the severity of stenosis. Inflammation 2013;36:449–456. doi: 10.1007/s10753-012-9565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Šteiner I, Stejskal V, Žáček P. Mast cells in calcific aortic stenosis. Pathol Res Pract 2018;214:163–168. doi: 10.1016/j.prp.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 164.Milutinovic A, Petrovič D, Zorc M, Vraspir Porenta O, Arko M, Pleskovič A, et al. Mast Cells Might Have a Protective Role against the Development of Calcification and Hyalinisation in Severe Aortic Valve Stenosis. Folia Biol (Praha) 2016;62:160–166. [DOI] [PubMed] [Google Scholar]
- 165.Akdag S, Akyol A, Asker M, Duz R, Gumrukcuoglu HA. Platelet-to-Lymphocyte Ratio May Predict the Severity of Calcific Aortic Stenosis. Med Sci Monit Int Med J Exp Clin Res 2015;21:3395–3400. doi: 10.12659/msm.894774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Natorska J, Marek G, Hlawaty M, Sobczyk D, Sadowski J, Tracz W, et al. Evidence for tissue factor expression in aortic valves in patients with aortic stenosis. Pol Arch Med Wewn 2009;119:636–643. [PubMed] [Google Scholar]
- 167.Natorska J, Marek G, Hlawaty M, Sadowski J, Tracz W, Undas A. Fibrin presence within aortic valves in patients with aortic stenosis: association with in vivo thrombin generation and fibrin clot properties. Thromb Haemost 2011;105:254–260. doi: 10.1160/TH10-09-0612. [DOI] [PubMed] [Google Scholar]
- 168.Breyne J, Juthier F, Corseaux D, Marechaux S, Zawadzki C, Jeanpierre E, et al. Atherosclerotic-like process in aortic stenosis: activation of the tissue factor-thrombin pathway and potential role through osteopontin alteration. Atherosclerosis 2010;213:369–376. doi: 10.1016/j.atherosclerosis.2010.07.047. [DOI] [PubMed] [Google Scholar]
- 169.Takahashi N, Tanabe K, Yoshitomi H, Adachi T, Ito S, Sugamori T, et al. Impairment of platelet retention rate in patients with severe aortic valve stenosis. J Cardiol 2013;62:171–175. doi: 10.1016/j.jjcc.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 170.Varshney R, Murphy B, Woolington S, Ghafoory S, Chen S, Robison T, et al. Inactivation of platelet-derived TGF-β1 attenuates aortic stenosis progression in a robust murine model. Blood Adv 2019;3:777–788. doi: 10.1182/bloodadvances.2018025817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bryan N, Ahswin H, Smart N, Bayon Y, Wohlert S, Hunt JA. Reactive oxygen species (ROS)--a family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur Cell Mater 2012;24:249–265. doi: 10.22203/ecm.v024a18. [DOI] [PubMed] [Google Scholar]
- 172.Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J, Pomerantzeff PMA, et al. Oxidant Generation Predominates Around Calcifying Foci and Enhances Progression of Aortic Valve Calcification. Arterioscler Thromb Vasc Biol 2008;28:463–470. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 173.Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Branchetti E, Sainger R, Poggio P, Grau JB, Patterson-Fortin J, Bavaria JE, et al. Antioxidant enzymes reduce DNA damage and early activation of valvular interstitial cells in aortic valve sclerosis. Arterioscler Thromb Vasc Biol 2013;33:e66–e74. doi: 10.1161/ATVBAHA.112.300177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu H, Wang L, Pan Y, Wang X, Ding Y, Zhou C, et al. Celastrol Alleviates Aortic Valve Calcification Via Inhibition of NADPH Oxidase 2 in Valvular Interstitial Cells. JACC Basic to Transl Sci 2019;5:35–49. doi: 10.1016/j.jacbts.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Steiner I, Krbal L, Dominik J. Blood vessels and lymphatics in calcific aortic stenosis--in support of its inflammatory pathogenesis. Cesk Patol 2010;46:33–36. [PubMed] [Google Scholar]
- 177.Arevalos CA, Berg JM, Nguyen JM V, Godfrey EL, Iriondo C, Grande-Allen KJ. Valve Interstitial Cells Act in a Pericyte Manner Promoting Angiogensis and Invasion by Valve Endothelial Cells. Ann Biomed Eng 2016;44:2707–2723. doi: 10.1007/s10439-016-1567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Parolari A, Loardi C, Mussoni L, Cavallotti L, Camera M, Biglioli P, et al. Nonrheumatic calcific aortic stenosis: an overview from basic science to pharmacological prevention. Eur J Cardio-Thoracic Surg Off J Eur Assoc Cardio-Thoracic Surg 2009;35:493–504. doi: 10.1016/j.ejcts.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 179.Mathieu P, Bouchareb R, Boulanger M-C. Innate and Adaptive Immunity in Calcific Aortic Valve Disease. J Immunol Res 2015;2015:851945. doi: 10.1155/2015/851945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pamukcu B, Lip GYH, Shantsila E. The nuclear factor--kappa B pathway in atherosclerosis: a potential therapeutic target for atherothrombotic vascular disease. Thromb Res 2011;128:117–123. doi: 10.1016/j.thromres.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 181.Alushi B, Curini L, Christopher MR, Grubitzch H, Landmesser U, Amedei A, et al. Calcific Aortic Valve Disease-Natural History and Future Therapeutic Strategies. Front Pharmacol 2020;11:685. doi: 10.3389/fphar.2020.00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Moynagh PN. The NF-kappaB pathway. J Cell Sci 2005;118:4589–4592. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 183.Éva Sikura K, Combi Z, Potor L, Szerafin T, Hendrik Z, Méhes G, et al. Hydrogen sulfide inhibits aortic valve calcification in heart via regulating RUNX2 by NF-κB, a link between inflammation and mineralization. J Adv Res 2021;27:165–176. doi: 10.1016/j.jare.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xu K, Zhou T, Huang Y, Chi Q, Shi J, Zhu P, et al. Anthraquinone Emodin Inhibits Tumor Necrosis Factor Alpha-Induced Calcification of Human Aortic Valve Interstitial Cells via the NF-κB Pathway. Front Pharmacol 2018;9:1328. doi: 10.3389/fphar.2018.01328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou T, Wang Y, Liu M, Huang Y, Shi J, Dong N, et al. Curcumin inhibits calcification of human aortic valve interstitial cells by interfering NF-κB, AKT, and ERK pathways. Phytother Res 2020;34:2074–2081. doi: 10.1002/ptr.6674. [DOI] [PubMed] [Google Scholar]
- 186.Liu M, Li F, Huang Y, Zhou T, Chen S, Li G, et al. Caffeic Acid Phenethyl Ester Ameliorates Calcification by Inhibiting Activation of the AKT/NF-κB/NLRP3 Inflammasome Pathway in Human Aortic Valve Interstitial Cells. Front Pharmacol 2020;11:826. doi: 10.3389/fphar.2020.00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang Y, Zhou X, Liu M, Zhou T, Shi J, Dong N, et al. The natural compound andrographolide inhibits human aortic valve interstitial cell calcification via the NF-kappa B/Akt/ERK pathway. Biomed Pharmacother 2020;125:109985. doi: 10.1016/j.biopha.2020.109985. [DOI] [PubMed] [Google Scholar]
- 188.Duan B, Yin Z, Hockaday Kang L, Magin RL, Butcher JT. Active tissue stiffness modulation controls valve interstitial cell phenotype and osteogenic potential in 3D culture. Acta Biomater 2016;36:42–54. doi: 10.1016/j.actbio.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kirschner CM, Alge DL, Gould ST, Anseth KS. Clickable, photodegradable hydrogels to dynamically modulate valvular interstitial cell phenotype. Adv Healthc Mater 2014;3:649–657. doi: 10.1002/adhm.201300288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chen J-H, Chen WLK, Sider KL, Yip CYY, Simmons CA. β-catenin mediates mechanically regulated, transforming growth factor-β1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol 2011;31:590–597. doi: 10.1161/ATVBAHA.110.220061. [DOI] [PubMed] [Google Scholar]
- 191.Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc Natl Acad Sci U S A 2013;110:19336–19341. doi: 10.1073/pnas.1306369110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Quinlan AMT, Billiar KL. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. J Biomed Mater Res A 2012;100:2474–2482. doi: 10.1002/jbm.a.34162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yip CYY, Chen J-H, Zhao R, Simmons CA. Calcification by Valve Interstitial Cells Is Regulated by the Stiffness of the Extracellular Matrix. Arterioscler Thromb Vasc Biol 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 194.Duan B, Hockaday LA, Kapetanovic E, Kang KH, Butcher JT. Stiffness and adhesivity control aortic valve interstitial cell behavior within hyaluronic acid based hydrogels. Acta Biomater 2013;9:7640–7650. doi: 10.1016/j.actbio.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dahal S, Huang P, Murray BT, Mahler GJ. Endothelial to mesenchymal transformation is induced by altered extracellular matrix in aortic valve endothelial cells. J Biomed Mater Res A 2017;105:2729–2741. doi: 10.1002/jbm.a.36133. [DOI] [PubMed] [Google Scholar]
- 196.Zhong A, Mirzaei Z, Simmons CA. The Roles of Matrix Stiffness and ß-Catenin Signaling in Endothelial-to-Mesenchymal Transition of Aortic Valve Endothelial Cells. Cardiovasc Eng Technol 2018;9:158–167. doi: 10.1007/s13239-018-0363-0. [DOI] [PubMed] [Google Scholar]
- 197.Butcher JT, Tressel S, Johnson T, Turner D, Sorescu G, Jo H, et al. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: influence of shear stress. Arterioscler Thromb Vasc Biol 2006;26:69–77. doi: 10.1161/01.ATV.0000196624.70507.0d. [DOI] [PubMed] [Google Scholar]
- 198.Mahler GJ, Frendl CM, Cao Q, Butcher JT. Effects of shear stress pattern and magnitude on mesenchymal transformation and invasion of aortic valve endothelial cells. Biotechnol Bioeng 2014;111:2326–2337. doi: 10.1002/bit.25291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. Am J Physiol Heart Circ Physiol 2011;301:H856–67. doi: 10.1152/ajpheart.00117.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol 2014;306:H317–25. doi: 10.1152/ajpheart.00182.2013. [DOI] [PubMed] [Google Scholar]
- 201.Hoehn D, Sun L, Sucosky P. Role of Pathologic Shear Stress Alterations in Aortic Valve Endothelial Activation. Cardiovasc Eng Technol 2010;1:165–178. doi: 10.1007/s13239-010-0015-5. [DOI] [Google Scholar]
- 202.Mongkoldhumrongkul N, Latif N, Yacoub MH, Chester AH. Effect of Side-Specific Valvular Shear Stress on the Content of Extracellular Matrix in Aortic Valves. Cardiovasc Eng Technol 2018;9:151–157. doi: 10.1007/s13239-016-0280-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sun L, Rajamannan NM, Sucosky P. Defining the role of fluid shear stress in the expression of early signaling markers for calcific aortic valve disease. PLoS One 2013;8:e84433. doi: 10.1371/journal.pone.0084433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng 2006;12:905–915. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- 205.Wang X, Lee J, Ali M, Kim J, Lacerda CMR. Phenotype Transformation of Aortic Valve Interstitial Cells Due to Applied Shear Stresses Within a Microfluidic Chip. Ann Biomed Eng 2017;45:2269–2280. doi: 10.1007/s10439-017-1871-z. [DOI] [PubMed] [Google Scholar]
- 206.Metzler SA, Pregonero CA, Butcher JT, Burgess SC, Warnock JN. Cyclic strain regulates pro-inflammatory protein expression in porcine aortic valve endothelial cells. J Heart Valve Dis 2008;17:571–577; discussion 578. [PubMed] [Google Scholar]
- 207.McIntosh CT, Warnock JN. Side-specific characterization of aortic valve endothelial cell adhesion molecules under cyclic strain. J Heart Valve Dis 2013;22:631–639. [PubMed] [Google Scholar]
- 208.Bouchareb R, Boulanger M-C, Fournier D, Pibarot P, Messaddeq Y, Mathieu P. Mechanical strain induces the production of spheroid mineralized microparticles in the aortic valve through a RhoA/ROCK-dependent mechanism. J Mol Cell Cardiol 2014;67:49–59. doi: 10.1016/j.yjmcc.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 209.Carrion K, Dyo J, Patel V, Sasik R, Mohamed SA, Hardiman G, et al. The long non-coding HOTAIR is modulated by cyclic stretch and WNT/β-CATENIN in human aortic valve cells and is a novel repressor of calcification genes. PLoS One 2014;9:e96577. doi: 10.1371/journal.pone.0096577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Merryman WD, Lukoff HD, Long RA, Engelmayr GCJ, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor-beta1 on the aortic valve myofibroblast. Cardiovasc Pathol Off J Soc Cardiovasc Pathol 2007;16:268–276. doi: 10.1016/j.carpath.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Farrar EJ, Pramil V, Richards JM, Mosher CZ, Butcher JT. Valve interstitial cell tensional homeostasis directs calcification and extracellular matrix remodeling processes via RhoA signaling. Biomaterials 2016;105:25–37. doi: 10.1016/j.biomaterials.2016.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Merryman WD, Lukoff HD, Long RA, Engelmayr GC, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor-β1 on the aortic valve myofibroblast. Cardiovasc Pathol 2007;16:268–276. doi: 10.1016/j.carpath.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Matsumoto Y, Adams V, Jacob S, Mangner N, Schuler G, Linke A. Regular Exercise Training Prevents Aortic Valve Disease in Low-Density Lipoprotein-Receptor-Deficient Mice. Circulation 2010;121:759–767. doi: 10.1161/CIRCULATIONAHA.109.892224. [DOI] [PubMed] [Google Scholar]
- 214.Schlotter F, Matsumoto Y, Mangner N, Schuler G, Linke A, Adams V. Regular Exercise or Changing Diet Does Not Influence Aortic Valve Disease Progression in LDLR Deficient Mice. PLoS One 2012;7:1–8. doi: 10.1371/journal.pone.0037298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Richards JM, Kunitake JAMR, Hunt HB, Wnorowski AN, Lin DW, Boskey AL, et al. Crystallinity of hydroxyapatite drives myofibroblastic activation and calcification in aortic valves. Acta Biomater 2018;71:24–36. doi: 10.1016/j.actbio.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Porras AM, McCoy CM, Masters KS. Calcific Aortic Valve Disease. Circ Res 2017;120:604–606. doi: 10.1161/CIRCRESAHA.117.310440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xu K, Xie S, Huang Y, Zhou T, Liu M, Zhu P, et al. Cell-Type Transcriptome Atlas of Human Aortic Valves Reveal Cell Heterogeneity and Endothelial to Mesenchymal Transition Involved in Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol 2020;40:2910–2921. doi: 10.1161/ATVBAHA.120.314789. [DOI] [PubMed] [Google Scholar]
- 218.Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-Targeted Therapeutics for Metabolic Disease. Trends Pharmacol Sci 2018;39:536–546. doi: 10.1016/j.tips.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]