

Abstract
During SARS-CoV-2 proliferation, the translation of viral RNAs is usually the rate-limiting step. Understanding the molecular details of this step is beneficial for uncovering the origin and evolution of SARS-CoV-2 and even for controlling the pandemic. To date, it is unclear how SARS-CoV-2 competes with host mRNAs for ribosome binding and efficient translation. We retrieved the coding sequences of all human genes and SARS-CoV-2 genes. We systematically profiled the GC content and folding energy of each CDS. Considering that some fixed or polymorphic mutations exist in SARS-CoV-2 and human genomes, all algorithms and analyses were applied to both pre-mutate and post-mutate versions. In SARS-CoV-2 but not human, the 5-prime end of CDS had lower GC content and less RNA structure than the 3-prime part, which was favorable for ribosome binding and efficient translation initiation. Globally, the fixed and polymorphic mutations in SARS-CoV-2 had created an even lower GC content at the 5-prime end of CDS. In contrast, no similar patterns were observed for the fixed and polymorphic mutations in human genome. Compared with human RNAs, the SARS-CoV-2 RNAs have less RNA structure in the 5-prime end and thus are more favorable of fast translation initiation. The fixed and polymorphic mutations in SARS-CoV-2 are further amplifying this advantage. This might serve as a strategy for SARS-CoV-2 to adapt to the human host.
Keywords: SARS-CoV-2, Human genome, CDS, Mutation, RNA structure, Translation
Introduction
Virus invades host cells and rapidly proliferates itself using the cellular resources. Understanding how SARS-CoV-2 efficiently utilizes the energies and resources from hosts is crucial for deciphering the molecular evolution of SARS-CoV-2 as well as controlling the pandemic (Hryhorowicz et al. 2021; MacLean et al. 2021). Despite many efforts on finding the molecular mechanisms of local adaptation of SARS-CoV-2 (Li et al. 2020a), a plausible explanation for why SARS-CoV-2 sequence is so perfect for invading human cells is still lacking.
Among the numerous biological processes, RNA translation is believed to be the most energy-consuming and rate-limiting step (Li et al. 2021; Zhao et al. 2021). For SARS-CoV-2, a successful invasion is measured by how many viral particles are produced, rather than how many viral RNAs are replicated. The rapid production of viral proteins is based on fast translation of viral RNAs, which further requires the proper binding and scanning of ribosomes at the 5-prime end of mRNA to allow efficient translation initiation. For hosts, there are multiple approaches (cis and trans) to enhance the translation initiation efficiency such as changing the ribosomes to a version with higher affinity to mRNAs, or improving the initiation factors (eIFs) to elevate their efficiency. However, for the virus, the RNAs are immersed in the same trans environment with host RNAs (where the availability of ribosomes is not determined by the virus), if the virus intends to compete with host mRNAs for tRNAs (Wang et al. 2020) or ribosomes, then the only way is to optimize the virus sequence (Taghinezhad et al. 2017).
It is known that a compact RNA structure at the 5-prime UTR (or at the 5-prime end of CDS) is not favorable for efficient ribosome scanning and initiation (Hall et al. 1982) (Fig. 1A). How complex an RNA structure is could be determined by the sequence features, mainly the GC content. As G:C base pair is biochemically more stable than A:T(U) base pair (Fig. 1B), an RNA sequence (or local sequence) is more likely to form a stable secondary structure with high GC content (Hofacker 2003; Yu et al. 2021) (Fig. 1C). Therefore, the competition for strong translation initiation becomes the competition for low GC content at the 5-prime end of CDS. For hosts, optimizing the mRNA sequence (cis) is not the only way to enhance translation initiation (because they can also optimize the trans factors). But for the virus, the “ability to express” is entirely determined by the cis features. This seems a limitation to the evolution and adaptation of viruses.
Fig. 1.

Molecular basis of mRNA translation initiation. A No RNA structure near the start codon is favorable for efficient translation initiation. Strong RNA structure near the start codon usually leads to low initiation efficiency. B G:C base pair is biochemically more stable than A:U base pair. C Local RNA sequence with high GC content will lead to strong local structure, and vice versa
Nevertheless, viruses have developed multiple strategies to manage to survive in the host environment. Apart from translation initiation, another highly regulated step during translation is the elongation process, which is directly connected to codon usage bias (dos Reis et al. 2004; Sharp and Li 1986). Despite the fact that the codon usage of SARS-CoV-2 is poorly correlated with that of the human genome (human prefers G/C-ending codons while SARS-CoV-2 has over-represented A/U-ending codons) (Hou 2020; Roy et al. 2021), the virus does find a way to downregulate the host genes with similar codon usage by competing for rare-codon-interacting tRNAs (Alonso and Diambra 2020). Moreover, there is an interesting pattern about codon usage bias that the multiple-host viruses (like SARS-CoV-2 and related coronaviruses) are less adapted to hosts (lower correlation of codon usage with hosts) compared to the single-host viruses (Carmi et al. 2021). This is understandable because the multiple hosts may not have similar codon usage so that the virus could not “imitate” all the host genomes. However, these multiple-host viruses usually lack RNA degradation signals, thereby increasing the viral RNA expression before the translation step (Carmi et al. 2021). All the strategies mentioned here could help the virus adapt to hosts.
Despite the complexity of viral gene expression, the cis elements affecting translation initiation are the key feature that determines the production rate of viral proteins. Intuitively, there should be strong selection pressure acting on these cis features in viruses. For hosts, the selection on RNA structure might not be very strong as there are other ways to elevate translation (also see the “Discussion” section). Indeed, we observed that in SARS-CoV-2 but not human, the 5-prime end of CDS had lower GC content and less RNA structure than the 3-prime part, which was favorable for ribosome binding and efficient translation initiation. This observation on reference genome sequence could be the relic of purifying selection. For the ongoing natural selection, we also observed that the fixed and polymorphic mutations in SARS-CoV-2 had created an even lower GC content at the 5-prime end of CDS, but no similar patterns were observed for the fixed and polymorphic mutations in the human 1000-genome.
Our results demonstrate that the mutations in SARS-CoV-2 are further amplifying its advantage in efficient translation of viral RNAs. This might serve as a strategy for SARS-CoV-2 to adapt to the human host. The virus could only mutate with the aid of host cells (Li et al. 2020b, c), therefore, reducing the virus transmission, which cuts down the chance for virus mutation, might serve as an effective and simple approach to control the pandemic at this stage.
Materials and methods
Data collection
The SARS-CoV-2 and RaTG13 reference sequences were downloaded from Genebank (accession numbers NC_045512 and MN996532). The human (Homo sapiens), macaque (Macaca mulatta), and mouse (Mus musculus) reference genomes were downloaded from Ensembl (http://asia.ensembl.org/), versions hg38, Mmul_10, and mm10. The polymorphic sites of SARS-CoV-2 were retrieved from Li et al. (2020b). The human 1000-genome SNPs were downloaded from the official website (Kuehn 2008).
Inference of the direction of mutations based on outgroup species
The direction of fixed and polymorphic mutations was deduced from the outgroup sequence. For SARS-CoV-2, the outgroup was always RaTG13 under all circumstances. For human, the direction of the fixed mutations (from macaque to human) was inferred from the mouse sequence, which was an outgroup of all primates. The direction of the polymorphic mutations in human (SNPs in 1000-genome) could be judged from macaque sequence because macaque was the outgroup of all human populations. Briefly speaking, for all mutation sites investigated in humans, we required the nucleotide to be identical in the three reference genomes of human, macaque, and mouse. The SNPs of other conditions were not considered. This approach should be the widely accepted one in determining ancestral state (An et al. 2019; Jiang and Zhang 2019). We were fully aware that none of the extant species is the direct ancestor of human, but according to the maximum likelihood rule, we could surmise that the sites with identical nucleotides in human, macaque, and mouse reference genomes should be the ancestral state of human sequence.
Bins and GC contents
For a CDS of length L, we divided it into 10 equal bins so that each bin had a length of L/10 (or rounded to integer). The GC content with each bin = (number of G and C)/(L/10). Since the number of G and C usually increases with length L, the GC content had canceled the bias introduced by length. Moreover, in the “Discussion” section, we considered the length issue.
RNA structure and minimum free energy (MFE)
For each bin of CDS mentioned above, we calculated the folding energy MFE with software RNAfold (Hofacker 2003). Lower MFE reflected stronger RNA structure. However, MFE was highly correlated with the length of sequence, and could not be used as an indicator of “foldability.” Therefore, we compared the 10 bins within each CDS (with identical length), and ranked them from 1 to 10. The bin with the lowest MFE (strongest RNA structure) was labeled with 1, and likewise, the bin with the highest MFE (least structured) was labeled with 10. By this way, bins from different genes were comparable because the rank number only reflected the relative extent of RNA structure of each bin within a gene. The length effect was canceled.
For the 100-bp MFE comparison, the 100-bp region around the focal mutation site was extracted. By default, the region covered from − 50 bp to + 50 bp of the mutation. If the site was within 50 bp of the mRNA ends, we extended the region to the opposite direction until 100 bp.
Results
The 5-prime end of CDS has lower GC content in SARS-CoV-2 but not in human
SARS-CoV-2 had 11 non-redundant CDSs while human (hg38) had ~ 20 thousand coding genes. For each CDS, we divided it into ten bins with equal length, and calculated the GC content (GC%) within each bin. By this way, bins from different genes were comparable because all GC contents ranged from 0 to 1 regardless of the absolute length (see “Materials and methods” section). In SARS-CoV-2, we found that the 5-prime-most bin has relatively low GC content compared with the rest part of CDS (Fig. 2A) and that this difference was significant (Fig. 2B). In sharp contrast, human genes exhibited an extraordinarily high GC content at the 5-prime end of CDS compared with the other part (Fig. 2C), the difference of which was also extremely significant (Fig. 2D). Indeed, the SARS-CoV-2 genes showed larger variance due to the fact that only 11 genes were available. Nevertheless, the global trend showed completely opposite patterns between SARS-CoV-2 and human.
Fig. 2.

GC content at the 5-prime end of CDS. Each CDS is divided into 10 equal bins. The GC content is calculated within each bin. A For SARS-CoV-2, the GC% in the first bin (the first 1/10 at the 5-prime of CDS) is relatively low. Error bar is the S.E.M. of all genes. B In SARS-CoV-2, the GC% in the first bin is significantly lower than the other parts of CDS. t-test is used to calculate the p-value. C For human, the GC% in the first bin is very high. Error bar is the S.E.M. of all human genes. D In human, the GC% in the first bin is significantly higher than the other parts of CDS. t-test is used to calculate the p-value
The 5-prime end of CDS has less RNA structure in SARS-CoV-2 but not in human
We continued to investigate whether the 5-prime end of CDS was likely to form RNA structures. We calculated the minimum free energy (MFE) of each bin of CDS and ranked the MFE within the CDS, assigning a rank number to each of the 10 bins (see “Materials and methods” section). “1” represented the bin with the lowest MFE (the strongest RNA structure) and “10” represented the bin with the highest MFE (the least RNA structure). By this operation, bins from different genes, although with different lengths, became comparable. In SARS-CoV-2, the 5-prime end of CDS had high MFE and thus low RNA structure (Fig. 3A), and the difference between the MFE rank of the first bin and the other parts was significant (Fig. 3B). In human, the 5-prime end of CDS had the lowest MFE (Fig. 3C), and significant difference was observed between the MFE of the first bin and the rest of bins of CDS (Fig. 3D). These results indicated that the RNA structure was depleted in the 5-prime of CDS in SARS-CoV-2, which was favorable for efficient translation initiation, whereas human genes tended to have complex RNA structure at the beginning of CDSs.
Fig. 3.

Rank of minimum free energy (MFE) of each bin of CDS. “1” represents the lowest MFE and “10” represents the highest MFE. A For SARS-CoV-2, the MFE rank of the first bin (the first 1/10 at the 5-prime of CDS) is relatively high. Error bar is the S.E.M. of all genes. B In SARS-CoV-2, the MFE rank of the first bin is significantly higher than those of other parts of CDS. t-test is used to calculate the p-value. C For human, the MFE rank of the first bin is very low. Error bar is the S.E.M. of all human genes. D In human, the MFE rank of the first bin is significantly lower than those of the other parts of CDS. t-test is used to calculate the p-value
Mutations and natural selection are continuously shaping the RNA structure
We have just found that the 5-prime end of CDS was less structured in SARS-CoV-2, which was potentially favorable for efficient translation initiation and provided the viral RNAs for an advantage over host mRNAs. However, in reality, evolutionary arms race is an ongoing process. Mutations are continuously changing the sequence features and shaping the fitness of both host and virus.
Generally, mutations that increase the GC content (like A > G, T > C mutations) are likely to increase the stability of an RNA structure. Inversely, mutations that decrease the GC content (like G > A, C > T mutations) could unravel or loosen an RNA structure. Since we already observed less RNA structure at the 5-prime CDS of SARS-CoV-2, if this feature really brought an advantage to SARS-CoV-2, then the virus sequence should continue to optimize. This long-lasting optimization process could be revealed by the mutation profile.
Mutations could be classified into fixed mutations and polymorphic mutations (McDonald and Kreitman 1991). Take SARS-CoV-2 as an example, the fixed mutations were those diverged sites between RaTG13 and SARS-CoV-2 (Li et al. 2020c, d), which represented a relatively long timescale. The natural selection on fixed mutations took place in the past (Chang et al. 2021). The polymorphic mutations were those variant sites among the world-wide SARS-CoV-2 strains (Li et al. 2020b), which are now being shaped by recent natural selection events.
Mutations in SARS-CoV-2 tend to unravel the RNA structure at the beginning of CDS
We first looked at the fixed mutations in SARS-CoV-2 (Li et al. 2020c). For simplicity, the mutations were denoted as “from > to,” for instance, “G > A” meant guanosine-to-adenosine mutations from RaTG13 to SARS-CoV-2. Transitions took place more frequently than transversions; therefore, the following 4 types of mutations were the most abundant: A > G, G > A, T > C, and C > T. In CDS, from the 1st bin to the 10th bin, the total numbers of each mutation type were counted (Fig. 4A). G > A and C > T were the mutations that decreased the GC content, and they showed high abundance in the 1st bin of CDS, while A > G and T > C mutations that increased the GC content showed depletion in the 1st bin of CDS (Fig. 4A). This result indicated that the 5-prime end of CDS in SARS-CoV-2 was enriched with mutations that decreased the GC content, which were likely to unravel the RNA structure around the start codon, facilitating translation initiation. Naturally, we testified the folding MFE of the pre-mutation sequence and the post-mutation sequence. 100-bp region flanking each focal mutation site was used (see “Materials and methods” section). Each mutation site had two observation values. In the 1st bin of CDS, the post-mutation sequence showed significantly higher MFE than pre-mutation sequence, while the rest parts of CDS showed no such difference (Fig. 4B). This proved that the fixed mutations in SARS-CoV-2 tended to unravel the RNA structure at the beginning of CDS. These beneficial mutations in SARS-CoV-2 were maintained in its genome.
Fig. 4.

Fixed and polymorphic mutations in SARS-CoV-2 or human could change the folding MFE of RNAs. CDS is divided into 10 equal bins as previously described. Comparisons are made between the 1st bin and the other parts. MFE is folded by 100 bp around the focal mutation (from − 50 to + 50 bp). A For the fixed mutations from RaTG13 to SARS-CoV-2, four mutation types are the most abundant (denoted as “from > to”): A > G, G > A, T > C, and C > T. The total number of each type of mutation is counted within each bin. Red lines (G > A and C > T) are the mutations that decrease the GC content. They show high abundance in the 1st bin of CDS. Blue lines (A > G and T > C) are the mutations that increase the GC content. They show depletion in the 1st bin of CDS. B For each of the fixed mutations in SARS-CoV-2, we calculated the MFE before and after mutation. N mutations would have N pairs of observations. p-value was calculated by t-test. C For each of the polymorphic mutations in SARS-CoV-2, we calculated the MFE before and after mutation. N mutations would have N pairs of observations. The sites in the 1st bin of CDS and the remaining parts are compared separately. p-value was calculated by t-test. D For each of the fixed or polymorphic sites in human genome, the MFE before and after mutation were calculated. The sites in the 1st bin of CDS were shown and compared
We next investigated the polymorphic mutations in SARS-CoV-2 (Li et al. 2020b). Similarly, each mutation site had two observations for the MFE of pre- and post-mutation sequence. In the 1st bin of CDS, the post-mutation sequence showed significantly higher MFE than pre-mutation sequence, but the rest parts of CDS showed no difference (Fig. 4C). These results demonstrated that both fixed and polymorphic mutations in SARS-CoV-2 were continuously facilitating the translation initiation process (by allowing ribosomes to bind a less structured RNA), making the virus more likely to survive and proliferate in host cells.
SARS-CoV-2 is temporarily staying ahead in the evolutionary arms race
Nevertheless, evolution is an ongoing arms race between hosts and viruses, the “upgrading” of viral sequences does not ensure the success of virus invasion because the hosts’ mRNAs could also optimize their sequences (to compete for ribosomes and achieve high translation rate). We looked at the fixed and polymorphic mutations in human. Fixed sites were those mutations from monkey to human, which were inferred from outgroup species by requiring the nucleotide to be identical in the three reference genomes of human, macaque, and mouse (see “Materials and methods” for details), and polymorphic sites were retrieved from the 1000-genome project (and also requiring to be ancestral sites). For each mutation site, two MFE values of pre- and post-mutation sequence (100 bp around the focal mutation site) were calculated. It turned out that the human mutations did not change the global MFE at all (Fig. 4D). For a single mutation, it might change the MFE, but globally, the mutations that increased or decreased the MFE were of similar numbers, so that the global distribution did not show difference between the pre- and post-mutation sequences.
Under this scenario, the mutations in SARS-CoV-2 had a global trend to elevate the translation initiation efficiency of viral RNAs, while in human, the mutations were globally “neutral” regarding host mRNA translation. In this evolutionary arms race of competing for ribosomes and rapid translation, SARS-CoV-2 is temporarily staying ahead (for now). This does not mean that novel mutations have any biases in SARS-CoV-2. Novel mutations should be random, but the virus replicates much faster than hosts and accumulates more mutations. Natural selection eliminates the viral sequences with unfavorable mutations and only maintains those viral sequences with advantageous ones. Considering that the evolutionary arms race is a complex process which may cause confusion to readers, the relevance and notes of this mutation pattern would be discussed in the following section.
Discussion
We have parsed the fixed and polymorphic mutations in SARS-CoV-2 and human genomes. Our general assumption is that the mutations that decrease the GC content, which unravel the RNA structure at the beginning of CDS, are beneficial for efficient translation. However, the net effect (or the net gain of fitness) of a novel mutation should be judged by multiple aspects. Affecting the RNA structure is only one feature of a mutation (Chamary and Hurst 2005). Codon usage bias is a typical feature that could affect mRNA translation rate so that the mutations in CDS that alter the codon usage could consequently affect translation (Li et al. 2021). Apart from the translation issue, the mutations in CDS could change amino acids and have more profound effects on the fitness of viral sequence (Becerra-Flores and Cardozo 2020). In addition, mutations that affect alternative splicing (Mroczek et al. 2017), stop codon read-through, or frameshift mutation could also cause strong consequences (Jiang et al. 2021a, b; Wang and Wang 2021). These aspects (changing amino acids, alternative splicing, frameshift etc.) are also worth studying. At this stage, without information of the other aspects and only focusing on the effect on RNA structure, one could draw the conclusion that SARS-CoV-2 is temporarily staying ahead in this competition because the viral mutations are continuously optimizing the viral sequence, while human mutations are globally “neutral” on RNA structures.
Regarding the translation process, our article only discusses translation initiation. What is also important is translation elongation, the speed of which is largely dependent on the codon composition (Li et al. 2021; Taghinezhad et al. 2017). Codon composition is again tightly connected to mutations in CDS. However, since the initiation efficiency is the major determinant of protein production rate and that elongation only fine-tunes translation, the focus on initiation is reasonable.
Moreover, regarding eukaryotic translation initiation, ribosomes usually bind mRNA through the interaction with the cap structure at the 5-prime end of mRNA, and then scan the mRNA sequence to find start codon ATG. Conceivably, both the two processes (binding mRNA and finding ATG) are affected by RNA structure. (1) Highly structured mRNA may have a steric effect that decrease the accessibility of 5-prime cap to the ribosomes; (2) highly folded mRNA makes ribosome movement difficult, which delays the ribosome to find an ATG and also slows down the loading of ribosomes to the CDS. Therefore, the global effect of highly structured mRNA is to decrease the translation initiation efficiency. Moreover, the absolute lengths of human CDSs and SARS-CoV-2 CDSs do not show global difference (Fig. 5). This makes sure that no bias has been introduced to the comparisons done in our study.
Fig. 5.
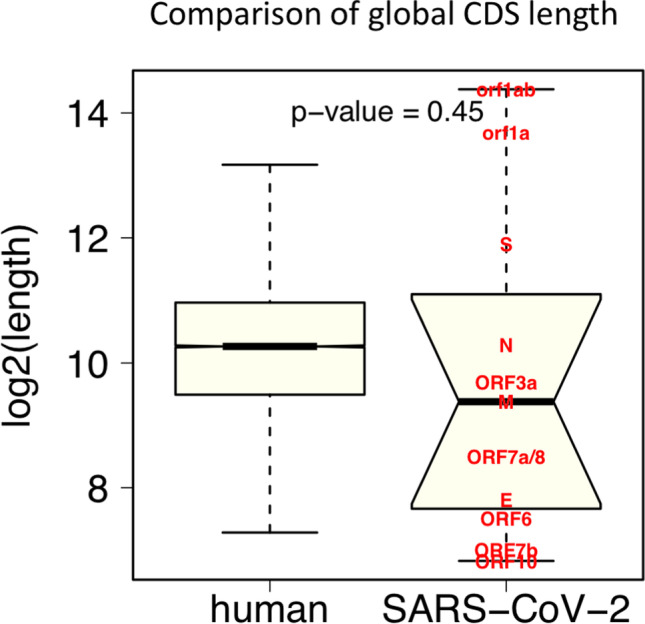
The comparison between CDS lengths of ~ 20,000 human coding genes versus 12 SARS-CoV-2 genes. t-test was used to get the p-value. The length of each SARS-CoV-2 gene is labeled in the graph
To further validate our assumption that SARS-CoV-2 has the advantage of fast translation initiation, one should first verify the association between RNA structure and translation initiation rate. An mRNA with high initiation efficiency is able to rapidly load ribosomes on CDS. Therefore, the ribosome density on an mRNA could reliably reflect its initiation efficiency. A technology termed ribosome profiling followed by next generation sequencing (Ribo-seq) accomplishes this aim (Ingolia et al. 2009). This technique allows researchers to profile the ribosome density of genes at genome-wide level. One could simply select the Ribo-seq data from two individuals or two cell lines, and calculate the ribosome density for each gene in each sample. Next, between the two samples, there are always many sample-specific mutations obtained from the sequencing data. For a same gene with (slightly) different sequences in two samples (understood as two versions), compare the MFE and ribosome density of the two versions. The version with higher MFE at the 5-prime CDS (less structure) is expected to have higher ribosome density on CDS (representing higher translation initiation rate). When the association between RNA structure and translation rate is proved, one might look further into the link between genotype and phenotype by investigating clinical data or in vitro experiments. Despite the fact that the virus has mRNA 5-prime favoring efficient translation, the translation rates of original viral sequences, including in vitro studies with isolated genes (non-optimized), result in low production of SARS-CoV-2 proteins. We propose the following possibilities. (1) No specific comparisons were made between the two sequence versions with different extent of RNA structure at the 5-prime end. One still did not know if the mutations leading to less RNA structure indeed contributed to translation rate. (2) Host cell systems had preference on endogenous RNAs compared to exogenous RNAs, which might affect the translatability of viral RNAs. For example, ADAR would label the endogenous RNAs as “self” so that the cellular defense system could deal with the “non-self” RNAs (Liddicoat et al. 2015). (3) Translation rate is determined by both initiation and elongation. Although high GC content was unfavorable for initiation, it might facilitate the elongation process due to codon optimality. The codon composition of endogenous mRNAs is highly optimal compared to that of virus. Therefore, the elongation process might serve as a compensation to the initiation stage to achieve high expression of endogenous genes.
The challenge in this current pandemic is that SARS-CoV-2 has the advantage of efficient translation initiation compared to host mRNAs, and that this advantage is continuously being amplified by the mutations in SARS-CoV-2. The virus is staying ahead in this evolutionary arms race. Note that generally, viruses co-evolve with their hosts, but SARS-CoV-2 has been introduced to the human population rather recently, and there is data that the virus mutates to adapt to the human host but we lack data on the co-evolution of the human host. There is no data on the changes already introduced to human genomes since 2020 as a result of the pandemic. Regarding this fact, we only claim that SARS-CoV-2 is “temporarily” staying ahead in the evolutionary arms race. Once we obtain the human mutation sites after a long period after the COVID-19, one may possibly find that human genome has caught up with the virus in the arms race.
To date, the patterns observed in the human mutation sites (in global population) do not indicate any optimal changes towards higher translation initiation rate. However, increasing the translation rate should be a general demand for virtually all viruses. Although human genome has only fought with SARS-CoV-2 for very short time, it has been fighting with other viruses for millions of years, and we still do not get any signals of increasing translation initiation. Maybe the mutation profile in human genome is dictated by other selection pressures so that we do not observe a pattern specific to translation. As mentioned in the “Introduction” section, the hosts have multiple ways to enhance the gene expression by cis and trans (but viruses do not), so that the selection pressure on a particular aspect might not be very strong in hosts.
Despite the many achievements in vaccine development, a simple but effective way to control the pandemic is to prevent the virus transmission. The mutations are introduced during virus infection. Without the aid of host cell machineries, the virus could not mutate by itself (Zhang et al. 2021). Even if a human individual is immune from severe symptom when infected by SARS-CoV-2, he/she could still spread the virus to others and the virus may mutate within this host. Therefore, blocking the virus transmission is an intuitive way to slow down virus mutation and control the pandemic.
Conclusions
Compared with human RNAs, the SARS-CoV-2 RNAs have less RNA structure in the 5-prime end and thus are more favorable of fast translation initiation. The fixed and polymorphic mutations in SARS-CoV-2 are further amplifying this advantage. This might serve as a strategy for SARS-CoV-2 to adapt to the human host.
Acknowledgements
During this global pandemic, we would like to thank all the medical workers who fight against SARS-CoV-2 and make us safe and sound.
Abbreviations
- SNP
Single nucleotide polymorphism
- CDS
Coding sequence
- MFE
Minimum free energy
- S.E.M.
Standard error of mean
Author contribution
YZ, XJ, HW, YM, and XY analyzed the data and drafted this manuscript. WJ and BY supervised this work and revised the manuscript. All authors approved the submission and publication of this manuscript.
Availability of data and materials
The SARS-CoV-2 and RaTG13 reference sequences were downloaded from Genebank. The human (Homo sapiens), macaque (Macaca mulatta), and mouse (Mus musculus) reference genomes were downloaded from Ensembl (http://asia.ensembl.org/), versions hg38 and Mmul_10. The polymorphic sites of SARS-CoV-2 were retrieved from Li et al. (2020b). The human 1000-genome SNPs were downloaded from the official website (Kuehn 2008).
Declarations
Ethics approval and consent to participate
Not applicable. All data used in this study were public data and could be downloaded without restriction.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Key message
The mutations favorable for fast translation are maintained in the SARS-CoV-2 sequence so that the virus could compete with host RNAs for efficient translation.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Alonso AM, Diambra L. SARS-CoV-2 codon usage bias downregulates host expressed genes with similar codon usage. Front Cell Dev Biol. 2020;8:831. doi: 10.3389/fcell.2020.00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An NA, Ding W, Yang XZ, Peng J, He BZ, Shen QS, Lu F, He A, Zhang YE, Tan BC, et al. Evolutionarily significant A-to-I RNA editing events originated through G-to-A mutations in primates. Genome Biol. 2019;20:24. doi: 10.1186/s13059-019-1638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra-Flores M, Cardozo T (2020) SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract, e13525 [DOI] [PMC free article] [PubMed]
- Carmi G, Gorohovski A, Mukherjee S, Frenkel-Morgenstern M. Non-optimal codon usage preferences of coronaviruses determine their promiscuity for infecting multiple hosts. FEBS J. 2021;288:5201–5223. doi: 10.1111/febs.15835. [DOI] [PubMed] [Google Scholar]
- Chamary JV, Hurst LD. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 2005;6:R75. doi: 10.1186/gb-2005-6-9-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Li J, Li Q, Yu CP, Xie LL, Wang S (2021) Retrieving the deleterious mutations before extinction: genome-wide comparison of shared derived mutations in liver cancer and normal population. Postgrad Med J. [DOI] [PubMed]
- dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MN, Gabay J, Debarbouille M, Schwartz M. A role for mRNA secondary structure in the control of translation initiation. Nature. 1982;295:616–618. doi: 10.1038/295616a0. [DOI] [PubMed] [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W. Characterization of codon usage pattern in SARS-CoV-2. Virol J. 2020;17:138. doi: 10.1186/s12985-020-01395-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryhorowicz S, Ustaszewski A, Kaczmarek-Rys M, Lis E, Witt M, Plawski A, Zietkiewicz E. European context of the diversity and phylogenetic position of SARS-CoV-2 sequences from Polish COVID-19 patients. J Appl Genet. 2021;62:327–337. doi: 10.1007/s13353-020-00603-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Sci. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhang J. The preponderance of nonsynonymous A-to-I RNA editing in coleoids is nonadaptive. Nat Commun. 2019;10:5411. doi: 10.1038/s41467-019-13275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Cao X, Wang H. Comparative genomic analysis of a naturally born serpentized pig reveals putative mutations related to limb and bone development. BMC Genomics. 2021;22:629. doi: 10.1186/s12864-021-07925-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Cao X, Wang H (2021b) Mutation profiling of a limbless pig reveals genome-wide regulation of RNA processing related to bone development. J Appl Genet. [DOI] [PubMed]
- Kuehn BM. 1000 Genomes Project promises closer look at variation in human genome. JAMA. 2008;300:2715. doi: 10.1001/jama.2008.823. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang X, Wang N, Wang H, Yin B, Yang X, Jiang W. GC usage of SARS-CoV-2 genes might adapt to the environment of human lung expressed genes. Mol Genet Genomics. 2020;295:1537–1546. doi: 10.1007/s00438-020-01719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yang X, Wang N, Wang H, Yin B, Yang X, Jiang W. Mutation profile of over 4500 SARS-CoV-2 isolations reveals prevalent cytosine-to-uridine deamination on viral RNAs. Future Microbiol. 2020;15:1343–1352. doi: 10.2217/fmb-2020-0149. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang XN, Wang N, Wang HY, Yin B, Yang XP, Jiang WQ. The divergence between SARS-CoV-2 and RaTG13 might be overestimated due to the extensive RNA modification. Future Virol. 2020;15:341–347. doi: 10.2217/fvl-2020-0066. [DOI] [Google Scholar]
- Li Y, Yang XN, Wang N, Wang HY, Yin B, Yang XP, Jiang WQ. Pros and cons of the application of evolutionary theories to the evolution of SARS-CoV-2. Future Virol. 2020;15:369–372. doi: 10.2217/fvl-2020-0048. [DOI] [Google Scholar]
- Li Q, Li J, Yu CP, Chang S, Xie LL, Wang S. Synonymous mutations that regulate translation speed might play a non-negligible role in liver cancer development. BMC Cancer. 2021;21:388. doi: 10.1186/s12885-021-08131-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Sci. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean OA, Lytras S, Weaver S, Singer JB, Boni MF, Lemey P, Kosakovsky Pond SL, Robertson DL. Natural selection in the evolution of SARS-CoV-2 in bats created a generalist virus and highly capable human pathogen. PLoS Biol. 2021;19:e3001115. doi: 10.1371/journal.pbio.3001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mroczek M, Kabzinska D, Chrzanowska KH, Pronicki M, Kochanski A. A novel TPM2 gene splice-site mutation causes severe congenital myopathy with arthrogryposis and dysmorphic features. J Appl Genet. 2017;58:199–203. doi: 10.1007/s13353-016-0368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Guo F, Singh B, Gupta S, Paul K, Chen X, Sharma NR, Jaishee N, Irwin DM, Shen Y. Base composition and host adaptation of the SARS-CoV-2: insight from the codon usage perspective. Front Microbiol. 2021;12:548275. doi: 10.3389/fmicb.2021.548275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghinezhad S, Razavilar V, Keyvani H, Razavi MR, Nejadsattari T. Codon optimization of Iranian human papillomavirus Type 16 E6 oncogene for Lactococcus lactis subsp cremoris MG1363. Future Virol. 2017;12:499–511. doi: 10.2217/fvl-2017-0032. [DOI] [Google Scholar]
- Wang N, Wang D (2021) Genome-wide transcriptome and translatome analyses reveal the role of protein extension and domestication in liver cancer oncogenesis. Mol Genet Genomics [DOI] [PMC free article] [PubMed]
- Wang Y, Gai Y, Li Y, Li C, Li Z, Wang X (2020) SARS-CoV-2 has the advantage of competing the iMet-tRNAs with human hosts to allow efficient translation. Mol Genet Genomics [DOI] [PMC free article] [PubMed]
- Yu YY, Li Y, Dong Y, Wang XK, Li CX, Jiang WQ (2021) Natural selection on synonymous mutations in SARS-CoV-2 and the impact on estimating divergence time. Future Virol
- Zhang YP, Jiang W, Li Y, Jin XJ, Yang XP, Zhang PR, Jiang WQ, Yin B (2021. Fast evolution of SARS-CoV-2 driven by deamination systems in hosts. Future Virol [DOI] [PMC free article] [PubMed]
- Zhao S, Song S, Qi Q, Lei W. Cost-efficiency tradeoff is optimized in various cancer types revealed by genome-wide analysis. Mol Genet Genomics. 2021;296:369–378. doi: 10.1007/s00438-020-01747-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The SARS-CoV-2 and RaTG13 reference sequences were downloaded from Genebank. The human (Homo sapiens), macaque (Macaca mulatta), and mouse (Mus musculus) reference genomes were downloaded from Ensembl (http://asia.ensembl.org/), versions hg38 and Mmul_10. The polymorphic sites of SARS-CoV-2 were retrieved from Li et al. (2020b). The human 1000-genome SNPs were downloaded from the official website (Kuehn 2008).