

Summary
The retinal pigment epithelium-choriocapillaris (RPE-CC) complex in the eye is compromised in age-related macular degeneration (AMD) and related macular dystrophies (MDs). Yet, in vitro models of RPE-CC complex that enable investigation of AMD/MD pathophysiology are lacking. By incorporating iPSC-derived cells into a hydrogel-based extracellular matrix, we developed a 3D-RPE-CC model that recapitulates key features of both healthy and AMD/MD eyes and provides modular control over RPE and CC layers. Using this 3D-RPE-CC model, we demonstrated that RPE and mesenchyme secreted factors are both necessary for formation of fenestrated CC-like vasculature. Our data shows that choroidal neo-vascularization (CNV) and CC atrophy occur in the absence of endothelial cell dysfunction and are not necessarily secondary to drusen deposits underneath RPE cells, and CC atrophy and/or CNV can be initiated systemically by patient serum or locally by mutant RPE-secreted factors. Finally, we identify FGF2 and matrix-metalloproteinases as potential therapeutic targets for AMD/MDs.
eTOC
We developed a patient-derived in vitro model of the retinal-pigment epithelium-choriocapillaris (RPE-CC) complex to investigate local and systemic factors underlying age-related macular degeneration (AMD) and similar maculopathies. The model captures hallmarks of RPE-CC morphogenesis and AMD, provides insight into factors that induce dysfunction, and led to identification of therapeutic targets.
Graphical Abstract
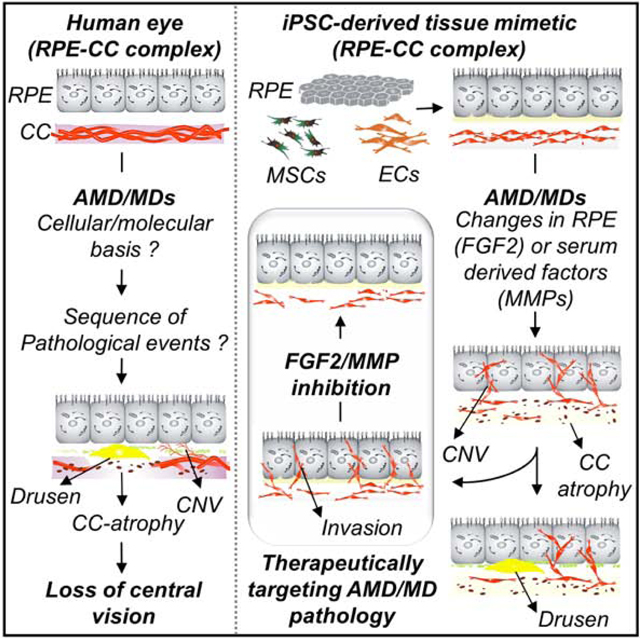
Introduction
The outer blood-retinal barrier (oBRB)-vascular complex is composed of retinal pigment epithelium (RPE) cells and the underlying fenestrated choriocapillaris (CC). The RPE-CC complex is the major conduit for transport of biological molecules between the retina and blood (Strauss, 2005). Disruptions in the RPE-CC complex are implicated in numerous macular degenerative diseases, including age-related macular degeneration (AMD), that is expected to impact ~288 million people worldwide by 2040 (Wong et al., 2014). Both AMD and related macular dystrophies (MDs) are characterized by pathology of the RPE-CC complex, including formation of extracellular deposits (drusen) beneath the RPE monolayer, RPE and CC atrophy, the loss of CC-specific EC fenestrations and choroidal neo-vascularization (CNV) into the RPE and sub-retinal space. Currently, there is conflicting evidence about the primary contribution of either RPE or CC in initiation of AMD/MD pathology. It has been postulated that primary RPE dysfunction leads to drusen formation, which initiates CC atrophy in AMD/MD (Hageman et al., 2001, Roth et al., 2004). Conversely, CC thinning at the onset of AMD development suggests that loss of CC precedes RPE cell changes in AMD/MD (Biesemeier et al., 2014).
Although animal models have greatly contributed to our understanding of RPE and CC physiology, separating the local tissue (RPE, CC) and systemic contributions to AMD/MD development has been challenging in vivo. Important insights into RPE-CC interactions have been gained using contact and non-contact co-cultures using primary and immortalized cells in vitro (Benedicto et al., 2017, Chen et al., 2017, Dardik et al., 2010, Hamilton et al., 2007, Komez et al., 2016, Kumar et al., 2011, Spencer et al., 2017). For example, primary EC and RPE coculture models suggest a role for crosstalk between EC and RPE in RPE barrier function and basement membrane maturation (Benedicto et al., 2017). Importantly, human induced pluripotent stem cells (iPSCs) can be consistently differentiated into RPE, endothelial cells (ECs), and other cell type(s) (e.g., mesenchymal stem cells, “MSC”) of the RPE-CC complex in vivo, which enables the investigation of RPE-CC pathologies in vitro using patient-derived cells. However, current RPE-EC culture models lack key features of the in vivo RPE-CC complex essential for modeling human AMD/MD progression, including 3-dimensional (3D) cellular organization and CC-specific phenotypes such as fenestrated vasculature.
Here, we integrated iPSC-derived RPE, EC, and MSC from healthy and AMD/MD-presenting human donors into a hydrogel-based engineered extracellular matrix (ECM) to generate an iPSCRPE-CC model that recapitulated key features of both healthy and disease phenotypes in the human eye. The 3D RPE-CC model was used to decipher independent roles of RPE and RPE secreted factors and serum in initiating endothelial cell/CC dysfunction in AMD/MD. Altogether, the human 3D model described here provides a platform for investigating RPE-CC biology, AMD/MD pathophysiology, and testing new therapeutic interventions.
Results
See Table S1 for a complete list of abbreviations for cell types used here. All references to the RPE-CC model refer to iPSC-derived cell/model unless otherwise specified.
Engineered iPSC-RPE-CC model recapitulates key features of in vivo physiology
The in vivo RPE-CC complex is comprised of the RPE monolayer overlying fenestrated CC vascular networks. RPE, EC, and mesenchyme from surrounding ocular stroma are crucial for development of the in vivo RPE-CC complex (Bhutto and Lutty, 2012, Gage et al., 2005). Therefore, iPSC-derived RPE, ECs, and MSCs (Figure S1a-c) were utilized for the in vitro RPE-CC model (Figure 1a, Figure S1d). The CC vasculature was modeled by encapsulating ECs into a poly(ethylene glycol) (PEG) hydrogels crosslinked with matrix metalloproteinase (MMP)-degradable peptides and functionalized with the ubiquitous RGD adhesive ligand, a composition previously shown to support 3D vascular network formation (Peters et al., 2016, Zanotelli et al., 2016). The hydrogel entrapping ECs was formed on top of a monolayer of MSCs (Figure 1a, Figure S1d) to mimic support of RPE-CC provided by ocular stroma (Lutty et al., 2010, Sellheyer, 1990). A day after initiating the EC-MSC co-culture, RPE cells were plated on the apical EC-containing hydrogel surface (Figure 1a, Figure S1d). In the RPE-CC model (RPE-EC-MSC co-culture), a pigmented RPE monolayer overlying cluster of differentiation 31-positive (CD31+) vascular networks (Figure 1b) was seen in the orientation that emulated the in vivo tissue. Of note, RPE monolayers and CC-like vascular networks were evident in the RPE-CC construct by day 7 and were stable up to day 60, the last time point for experiments reported here.
Figure 1. Generation of an iPSC-RPE-CC model.

(a) Schematic representation of 3D iPSC-RPE-CC complex development. A solution containing PEG-NB, MMP degradable crosslinker, and RGD were used to encapsulate iPSC-ECs using photopolymerization (365 nm, 5 mW/cm2) over iPSC-MSCs. RPE cells were seeded the next day on the vascularized gel surface.
(b) Merged immunofluorescence (CD31+ vascular networks, red) and brightfield (pigmented RPE monolayer) images for RPE-CC constructs after ~ 60 days in culture.
(c-f) Immunofluorescence images illustrating EZRIN (green) and MITF (grey) expression in the RPE monolayer, and VE-CADHERIN in the vascular layer (red) for day 15 RPE-CC in planar views (top panels) and XZ views (bottom panels). DAPI (cell nuclei, blue) shown in the XZ view.
(g-j) Immunofluorescence images illustrating expression of ZO1 (grey) in the RPE monolayer, and CD31 (green), and PLVAP (red) in the vascular layer for ~day 30 RPE-CC in planar views (panels g, h, j) and XZ views (panel i). (i) XZ cross-sections correlating to the region shown in (g-h) is ~43 μm thickness. (j) Enlarged view of the blue outlined region shown in (g) illustrates a hexagonal pattern for ZO1 at RPE cell-cell junctions.
(k) Immunofluorescence images showing ZO1-positive tight junctions (red) in the RPE and vascular layer and VWF in the vascular layer (green) for day 18 RPE-CC cryosection in planar view (top panel) and YZ views at two different planes corresponding to tight junctions in the RPE monolayer (middle panel, YZ: 14.5 μm and 13.4 μm) and tight junctions in the vascular layer (bottom panel, 23.8 μm and 13.4 μm).
(l) Immunofluorescence images illustrating VE-CADHERIN (green), PLVAP (red), and DAPI (blue) expression for cryosection of CC layer from day 67 RPE-CC. Planar XY view (top panel) and orthogonal XZ view (bottom panel) illustrates rudimentary lumen formation.
(m-o) Immunofluorescence images for cryosections of CC layer from RPE-CC illustrating CD31 (green) and either (m) CLAUDIN-5 (red, day 67), (n) ZO1 (red, day 18), or (o) PLVAP (red, day 67) relative to the RPE monolayer (toward top of images). DAPI shown in blue.
Scale bar = 20 μm for all images.
See also Figure S1.
Several pieces of evidence demonstrate that the RPE-CC model recapitulates important physiological aspects of the in vivo RPE-CC complex. Specifically, the RPE monolayer showed proper localization of EZRIN, an RPE signature protein that is present in apical microvilli and basal infoldings, and Microphthalmia-associated transcription factor (MITF), a transcription factor expressed by RPE cells (Figure 1c-e). Furthermore, RPE adopted hexagonal morphology with Zonula occludens-1 (ZO1)+ tight junctions between cells (Figure 1g, 1k). Vascular networks in the CC layer underlying the RPE monolayer were positive for vascular endothelial cadherin (VE-CADHERIN), an endothelial cell-specific adherens junction protein that contributes to tubule formation (Figure 1c, 1f, 1l), universal endothelial cell markers, CD31 (Figure 1g-j), and von Willebrand factor (VWF) (Figure 1k), and formed rudimentary vascular lumens (Figure 1l). Consistent with in vivo CC tissue, CD31+ endothelial cells in the CC layer expressed the fenestration-associated protein, plasmalemma vesicle associated protein (PLVAP) (Figure 1g-j, 1o, Movie S1) (Baba et al., 2009). Note that in addition to VE-CADHERIN, (Figure 1c, 1f, 1l), expression of tight junction proteins, CLAUDIN-5 (Figure 1m) and ZO1 (Figure 1n), with ZO1 present at cell-cell boundaries (Figure 1n) was also seen in the CC layer. Overall, pattern of VE-CADHERIN, CLAUDIN-5, and ZO1 expression together were indicative of immature EC junctions.
Taken together, the 3D RPE-CC co-culture model reported here i) maintains long-term spatial organization of RPE and CC layers and ii) shows protein expression and localization in the RPE monolayer (EZRIN, MITF, ZO1) and CC (CD31, PLVAP, VWF, VE-CADHERIN, ZO1, CLAUDIN-5) that is consistent with the polarized RPE monolayer and fenestrated CC endothelium in vivo.
RPE-secreted paracrine factors are sufficient to promote the development of CC-like fenestrated endothelium in the presence of MSCs.
The RPE is necessary for successful development of CC in vivo (Baba et al., 2009, Blaauwgeers et al., 1999, Marneros et al., 2005). While previous co-culture models have demonstrated that RPE-secreted factors can promote cord-like formation by ECs (Qi et al., 2019), the precise role of RPE (e.g., paracrine or paracrine and juxtacrine) in the development of CC-specific phenotypes, such as fenestrated vasculature has not been studied due to lack of suitable in vitro models. Here, we aimed to take advantage of the modular nature of the 3D culture model (Figure 1 to investigate the contribution of RPE secreted factors in the formation of CC vasculature. EC and MSC were co-cultured in PEG hydrogels in the absence of the RPE monolayer but apically supplemented with conditioned medium collected from RPE cultures (RPE-CM), which is denoted “(RPE-CM)-CC” hereafter (Figure 2a). Similar to CC in the RPE-CC tissue mimetic (Figure 1), immunocytochemical analyses of (RPE-CM)-CC showed abundant CD31+ and PLVAP+ vascular networks (Figure 2b). Furthermore, the overall thickness of the CC-like vascular tissue in the RPE-CM CC model was similar in three independent experiments (42.3 ± 1.83 μm), and in the range of in vivo CC layer measurements (Adhi et al., 2015). Notably, electron microscopy of (RPE-CM)-CC revealed membrane structure consistent with RPE-facing fenestrated endothelium (Figure 2c) (Kim et al., 2020). Similarly, confocal images (Figure 2d-e) showed the PLVAP-associated punctate staining pattern (Figure 2d) and robust co-localization of CD31 and PLVAP in juxtaposition to the RPE monolayer (Figure 2e). PLVAP-positive vascular networks also expressed VE-CADHERIN and formed primitive lumens in the (RPE-CM)-CC model (Figure 2f). These results demonstrate that secreted factors from RPE-CM alone were sufficient for development of CC-like fenestrated vascular networks in the 3D RPE-CC model.
Figure 2. RPE secreted trophic factors are sufficient for development of CC-like vascular networks in the iPSC model.

(a) Schematic representation of the experimental design for evaluating RPE-CM influence on CC-like vascular development in the absence of RPE-monolayer. Note that the CC layer in the presence of RPE-CM is referred to as “(RPE-CM)-CC”.
(b) Immunofluorescence images of (RPE-CM)-CC wholemount from day 18 culture in planar (top panel) and xz view (bottom panel) illustrating CD31 (green), PLVAP (red), and DAPI (blue) in the CC layer. Scale bar = 20 μm.
(c) Transmission Electron Micrograph (TEM) of day 12 (RPE-CM)-CC. Fenestrations run parallel to the membrane between the black arrowheads on the side facing the RPE-CM. Note that the side facing EC media and RPE-CM are indicated. Scale bar = 1 μm.
(d) Immunofluorescence images from day 14 (RPE-CM)-CC wholemount showing punctate PLVAP (red) in the CC layer. DAPI =blue. Scale bar = 20 μm.
(e) Immunofluorescence images from day 14 (RPE-CM)-CC cryosection illustrating localization of CD31 (green), PLVAP (red) relative to the RPE-CM. DAPI =blue. Scale bar = 20 μm.
(f) Top panel: Immunofluorescence images from day 18 (RPE-CM)-CC wholemount illustrating PLVAP (red), VE-CADHERIN (green), and DAPI (blue) in the CC layer. Bottom Panel: Immunofluorescence images from day 18 (RPE-CM)-CC wholemount showing localization of VE-CADHERIN (green) in planar and single plane xz cross-section consistent with primitive vascular lumen formation. Scale bar = 20 μm.
(g) Immunofluorescence images of CC layer showing CD31+ (green) vascular tubules and localization of Bruch’s membrane proteins, LAM (top image, red) or COL6 (second image from top, red) in day 67 cultures. Similarly, immunofluorescence images of vascular layer illustrating CD31+ (green) and COL6 in (RPE-CM)-CC (third image from top) and EC-MSC co-culture (bottom image). DAPI shown in blue. Scale bar = 20 μm.
See also Figure S2.
The combination of RPE and CC layers contributes to primitive Bruch’s formation in the RPE-CC model
RPE induced polarized expression of Bruch’s membrane proteins, COLLAGEN 6 (COL6) and LAMININ (LAM), towards the RPE monolayer (Figure 2g). Polarized expression of COL6 was not observed for the (RPE-CM)-CC model or EC-MSC co-cultures (Figure 2g), suggesting a role for juxtracrine signaling of RPE in the formation of Bruch’s membrane.
RPE-secreted VEGF-A and MSC-secreted TGF-ß1 are required for CC development in (RPE-CM)-CC mimetic.
To directly confirm the requirement of RPE released trophic factors and neighboring mesenchyme in the CC mimetic development (Figure 2), we next evaluated vascular network characteristics for EC encapsulated in PEG hydrogels (PEG-EC) and cultured in transwells under the following conditions i) EC medium (Figure S1e), ii) supplemented apically with RPE-CM and basally with EC medium (Figure S1f), and iii) overlying a confluent layer of MSCs on the transwell membrane surface and cultured apically with routine RPE media (retinal differentiation medium or RDM) and basally with EC medium (Figure S1g). Although ECs formed CD31-positive vascular networks in all conditions, consistent with requirement of both RPE and mesenchyme secreted factors in the CC development, expression of the fenestration marker PLVAP was not seen under any of the conditions (Figure S1e-g).
Vascular networks formed by EC cultured in PEG hydrogels (PEG-ECs) were typically stable for ~ 2 weeks in EC medium, which is consistent with previous studies (Chwalek et al., 2014, Zanotelli et al., 2016). Compared to PEG-ECs cultured in EC medium, PEG-ECs supplemented with RPE-CM showed increased area of vascular networks (Figure S2a, S2b; p ≤ 0.001), total lengths of tubules as measured by length of several cells in series that extend from one junction/node of tubule complexes (Figure S2a, S2c; p ≤ 0.01), numbers of tubes (Figure S2a, S2d; p ≤ 0.05). RPE-CM also induced EC proliferation during the first 24 hrs of culture (Figure S2e; p ≤ 0.01), which is consistent with prior in vitro and in vivo studies (Dardik et al., 2010, Marneros et al., 2005, Saint-Geniez et al., 2009). Co-cultures of PEG-ECs with MSCs resulted in dense vascular networks which were stable for at least 1.5 months both in the presence and absence of RPE or RPE-CM (Figure S2f), which is consistent with a role of mesenchymal cells in stabilizing vascular networks in vitro and in vivo (Lu et al., 2018, Moon et al., 2010, Stratman and Davis, 2012). Taken together, while RPE-CM and MSCs each play a pro-angiogenic role for ECs and improve vascular network formation and stability by PEG-ECs (Figures S1-S2), both were necessary to recapitulate fenestrated vasculature in the RPE-CC mimetic (Figures 1, 2).
To further investigate the role of RPE vs. MSC released paracrine factors in the development of CC-like vascular network in vitro, we compared the levels of specific cytokines in MSC vs. RPE-CM. Compared to MSCs, VEGF-A secretion was detected at higher levels for RPE cells both apically (Figure 3a, 3b, Table S2; p<0.05) and basally (Figure 3a, 3c, Table S2; p<0.001). Similar results were observed for cocultured EC-MSCs; basal VEGF-A secreted for (RPE-CM)-CC (EC+MSC+RPE-CM) culture was higher than EC-MSC cocultures (Figure 3a, 3d, Table S2; p<0.001). These results are in agreement with the requirement of basal VEGF-A secretion by RPE cells for CC development and maintenance in vivo. TGF-ß1 also plays an important role in the development of endothelial fenestration in vivo (Tosi et al., 2018, Wang et al., 2017). In contrast to VEGF-A secretion, TGF-ß1 secretion was higher for MSC-CM compared to RPE-CM (Figure 3a, 3e, 3f, Table S2; p<0.001). Note that, although secreted TGF-ß1 relative to total protein in EC+MSC cultures was higher than (RPE-CM)-CC cultures (Figure 3a, 3g), absolute levels of secreted TGF-ß1 were similar in EC+MSC and (RPE-CM)-CC cultures (Table S2).
Figure 3. Influence of cellular and secreted factors on formation of CC-like fenestrated endothelium.

(a) Schematic representation of experimental design and conditions. i) RPE monolayers (“RPE”) cultured on the transwell membrane in routine culture media (RDM) medium, ii) MSC monolayers (“MSC”) cultured on the transwell membrane in routine MSC culture medium, iii) EC-MSC co-cultures in RDM media apically and routine EC culture media of the transwell (“EC-MSC”) and iv) EC-MSC co-cultures treated apically with RPE-CM (“(RPE-CM)-CC” and basally with EC media.
(b-d) Quantitative ELISA analyses of VEGF-A for media collected from the (b) apical or (c, d) basal chambers of the transwell. Data normalized to (b, c) RPE and (d) (RPE-CM)-CC. n = 3 biological replicates. * p ≤ 0.05; *** p ≤ 0.001.
(e-g) Quantitative ELISA analyses of TGF-ß1 for medium collected from the (e) apical and (f, g) basal chambers of the transwell. Data normalized to (e, f) RPE and (g) (RPE-CM)-CC. n= 3 biological replicates. * p ≤ 0.05; *** p ≤ 0.001.
(h) Schematic representation of experimental set-up utilized to evaluate the impact of VEGF-A or TGF-ß1 inhibition using daily supplementation of neutralizing antibody αVEGF-A:10μg/ml; αTGF-ß1: 10μg/ml) on development of PLVAP-positive vascular networks in the (RPE-CM)-CC model.
(i, j) Immunofluorescence images (i) and quantitative analyses (j) showing CD31 (green), PLVAP (red), and DAPI (blue) expression in untreated (RPE-CM)-CC or (RPE-CM)-CC treated with neutralizing antibodies for VEGF-A (“αVEGF-A”; 10μg/ml) or TGF-ß1 (“αTGF-ß1”; 10μg/ml) at the day 7 time point. Of note, day 7 timepoint was chosen for this analysis as abundant amount of PLVAP-positive vascular networks were consistently observed by this timepoint in the CC model. The total number of PLVAP+ tubes were normalized to untreated (RPE-CM)-CC. n = 3 biological replicates. *** p ≤ 0.001. Scale bars = 20 μm.
See also Table S2.
Roles for TGF-ß1 and VEGF-A in the development of fenestrated CC-networks were further investigated using neutralizing antibodies (Figure 3h). The protocol described for producing fenestrated vascular networks in the (RPE-CM)-CC model (see Figure 2) was modified here by also incorporating anti-VEGF-A (10 μg/ml) or anti-TGF-ß1 (10 μg/ml) for the entire duration of the experiment, as shown in the schematic in Figure 3h. Immunocytochemical analyses demonstrated that anti-VEGF-A or anti-TGF-ß1 neutralizing antibodies reduced number of PLVAP-positive vascular networks at day 7 (Figure 3i, 3j), a time point at which PLVAP was consistently expressed in the (RPE-CM)-CC model. Altogether our data suggests that RPE-secreted VEGF-A and MSC-secreted TGF-ß1 contribute to the development of fenestrated CC-like vasculature in (RPE-CM)-CC model.
CNV and CC atrophy in the RPE-CC model can be initiated solely by alterations in RPE secreted factors due to RPE dysfunction.
We next utilized the modularity of the 3D RPE-CC model to investigate the singular role of RPE cell autonomous dysfunction in initiating vascular changes characteristic of AMD and related MDs. Notably, AMD and related MDs are characterized by pathologic changes spanning both the RPE and CC layers in the RPE-CC complex that include drusen deposits beneath the RPE cells, ECM accumulation, CNV, invasion of RPE and subretinal space by vessels of the CC/choroid, and CC atrophy (Bhutto and Lutty, 2012). In prior studies, we and others have utilized iPSC-based disease modeling to demonstrate that RPE-autonomous dysfunction in AMD/MDs is sufficient for drusen formation and ECM accumulation (Galloway et al., 2017). However, the specific role of cell autonomous dysfunction (RPE versus EC) in initiating CC atrophy and CNV-relevant pathologic changes has not yet been established. To address RPE cell autonomous dysfunction in initiating CNV and CC atrophy, iPSCs from patients with a monogenic MD, Sorsby’s Fundus Dystrophy (SFD), a disease characterized by both CNV and CC atrophy were used. SFD is caused by mutations within the Tissue Inhibitor Of Metalloproteinases 3 (TIMP3) gene (Weber et al., 1994). Of note, TIMP3 is a key regulator of matrix metalloproteinase (MMP) activity and is expressed by both human RPE (Ruiz et al., 1996) and ECs (Bugno et al., 1999), and consistent with this, TIMP3 protein was present within RPE and ECs used in the RPE-CC mimetic model. (Figure S3a-b).
RPE-Specific-TIMP3 mutation in SFD and altered composition of SFD-RPE-CM is sufficient to initiate CC atrophy in the RPE-CC and (RPE-CM)-CC model
Consistent with SFD pathology, immunocytochemical analyses showed reduced numbers of PLVAP+ vascular networks in both i) control vs. SFD RPE-CC cultures (Figure 4a, 4b) and ii) control vs. SFD (RPE-CM)-CC cultures (Figure 4c). Similarly, quantitative analysis of PLVAP showed reduced PLVAP-positive cells in SFD RPE-CC compared to control RPE-CC (Figure 4d, p <0.001). In agreement with immunocytochemistry data (Figure 4a-d), quantitative Western blot analyses showed reduced levels of CD31 protein in i) control vs. SFD RPE-CC cultures (Figure 4e, p <0.05) and ii) control vs. SFD (RPE-CM)-CC cultures (Figure 4f, p< 0.05).
Figure 4. SFD RPE or RPE-CM disrupts CC vascular networks.

(a-c) Immunofluorescence images illustrating CD31 (green), PLVAP (red), ZO1 (gray), and DAPI (blue) expression for parallel cultures of (a, b) RPE-CC at day 30 or (c) (RPE-CM)-CC at day 14. RPE-CC samples were formed using iPSC-derived RPE and ECs from control patients (“Control”) or patients with Sorsby’s Fundus Dystrophy (“SFD”). (RPE-CM)-CC samples were treated with CM from control or SFD iPSC-RPEs. Scale bar = 20 μm.
(d) Quantitative analysis of the number (#) of PLVAP+ cells (normalized to control) in parallel cultures of control RPE-CC vs. SFD RPE-CC. n = 3 biological replicates. *** p ≤ 0.001. (e, f) Western blot images and quantitative analysis of relative CD31 protein (~130 kDa) levels for parallel cultures of (e) Control vs SFD RPE-CC and (f) Control vs SFD (RPE-CM)-CC. Total CD31 protein is shown normalized to the whole protein level in the lysate. n = 3 biological replicates. * p ≤ 0.05.
(g) Top panel: Schematic representation showing the configuration of (RPE-CM)-CC or RPE-CC where individual components were derived using iPSCs from Control or SFD patients. Bottom panel: Immunofluorescence images from ~day 14 RPE-CC cultures illustrating density of PLVAP+ vascular networks (red) for each individual cellular configuration (schematic shown above the image). DAPI shown in blue. Scale bar = 20 μm.
To investigate the ability of RPE vs. EC-specific TIMP3 mutation to cause CC atrophy and CNV in SFD, we next exploited the modularity of the RPE-CC model. Specifically, in the RPE-CC or the (RPE-CM)-CC model, we utilized either i) control RPE/RPE-CM and control ECs, ii) SFD RPE/RPE-CM and control ECs iii) control RPE/RPE-CM and SFD ECs and iv) SFD RPE and SFD ECs (Figure 4g). Of note, MSCs utilized in all experiments were from a control iPSC line (Figure 4g), and therefore are consistent across all experiments.
SFD RPE/RPE-CM of both homogenous mutant (SFD RPE, SFD EC) and heterogenous mutant cells (SFD RPE, control EC) (Figure 4g) led to a reduction in PLVAP+ vascular networks of the RPE/ (RPE-CM)-CC cultures at ~day 14 (Figure 4g). In contrast, vascular changes were not seen in RPE/(RPE-CM)-CC cultures with homogenous (control RPE, control EC) and heterogenous (control RPE, SFD EC) cells that employed control RPE (Figure 4g).
To further confirm CC atrophy, as opposed to reduced EC proliferation and CC formation in the presence of SFD RPE/RPE-CM, a time course analyses was performed comparing control RPE/(RPE-CM)-CC vs. SFD RPE/(RPE-CM)-CC complex. Specifically, light and/or confocal microscopy analyses showed i) abundant PLVAP-positive vascular networks in SFD RPE-CC (Figure S3c) at day 8 and ii) similar numbers of vascular networks in control (RPE-CM)-CC vs. SFD (RPE-CM)-CC complex until day 8 (Figure S3d). As shown in Figure 4b and Figure S3d, reduced numbers of vascular networks in SFD RPE/(RPE-CM)-CC were observed by day 14. To substantiate these findings further, we switched the RPE-CM from control RPE-CM to SFD RPE-CM after formation of vascular networks (Figure S4). Similar to the time course analyses of control (RPE-CM)-CC vs. SFD (RPE-CM)-CC complex (Figure S3d), reduced vascular network areas (Figure S4a-h, S4i, S4m; p< 0.01), tube numbers, (Figure S4b-h, S4j, S4n; p< 0.001), tubules lengths (Figure S4a-h, S4k, S4o; p< 0.001), and branching points (nodes/junctions) (S4a-h, S4l, S4p; p< 0.001) were observed in CC cultures supplemented apically with SFD RPE-CM after the formation of extensive vascular networks. Together, these data clearly show that soluble cues present in SFD RPE-CM are independently sufficient to initiate CC atrophy in the 3D RPE-CC model.
Of note, at the time of CC atrophy, a uniform RPE monolayer was present in both control and SFD RPE-CC (Figure 4a, S5a). This result is consistent with human AMD/MD, whereby CC atrophy precedes RPE atrophy (Biesemeier et al., 2014). Furthermore, drusen deposits have been implicated in CC atrophy (Korte et al., 1984, Mullins et al., 2011). Therefore, in a subset experiments, the timecourse of drusen deposits vs. CC atrophy was evaluated in SFD cultures. Remarkably, CC atrophy was observed at a timepoint (~2 weeks) when no drusen deposits were observed, as characterized by the co-localization of common drusen components TIMP3 and APOE (Figure S5b) (Galloway et al., 2017). The absence of drusen at ~2 weeks is consistent with prior reports of aged SFD RPE cells (~day 60–90 in culture) developing drusen-like deposits in culture (Galloway et al., 2017) and suggests CC atrophy occurs prior to drusen formation in this RPE-CC model.
CNV-relevant invasion of the RPE monolayer in the presence of SFD-RPE/RPE-CM
In SFD and other similar maculopathies, CNV and CC atrophy co-exist and areas of CNV (abnormal vessel growth that invades into RPE-ECM) are found alongside overall CC atrophy. In the RPE-CC model, the presence of TIMP3 mutation containing SFD-RPE alone (with either control or mutant ECs) resulted in invasion of RPE monolayers by underlying vascular networks, as evident from CD31 positive cells above the RPE basement membrane positive for COLLAGEN 4 (COL4) (Figure 5a), as would occur clinically in CNV (Grossniklaus and Green, 2004). Of note, numerous mechanisms including impaired RPE barrier function, breakdown of RPE-ECM, and proangiogenic factors in RPE secreted media can contribute to RPE-ECM invasion by ECs/vessels and initiate CNV (Nowak, 2006). To corroborate a direct role SFD RPE secreted factors in initiating ECM invasion by ECs, we investigated whether SFD RPE-CM was independently sufficient to promote ECM invasion by ECs using ECM-invasion assays (Justus et al., 2014). Consistent with the CNV phenotype in the SFD RPE-CC model (Figure 5a), increased migration of ECs through collagen (COL1)-coated transwell toward SFD RPE-CM was observed (Figure 5b, 5c; p< 0.05). Importantly, invasion was not increased for RPE-CM versus routine media used for RPE cell culture (Figure S6a, S6b).
Figure 5. SFD RPE-CM is sufficient to instigate CNV-relevant changes in an iPSC model.

(a) Immunofluorescence images (XZ view) illustrating CD31 (green), COL4 (red), and DAPI (blue) for a day 18 SFD RPE-CC sample formed using SFD-derived RPE and SFD-derived ECs see Figure 4g.iv). CD31+ endothelial cells invade into the RPE basement membrane marked with COL4 and above the RPE monolayer (Position of the i) RPE monolayer nuclei and ii) COL4-containing RPE-basement membrane is shown by white arrow in the left and right panels respectively). Note that COL4 also colocalizes with CD31 in the CC layer due to basement membrane associated with endothelial tubules. Scale bar = 20 μm.
(b, c) Light microscopy images (b) and quantitative analyses (b) of crystal violet stained ECs invading through COL1-coated transwell towards control RPE-CM (left) or SFD RPE-CM (right) at the 24 h timepoint. Data is presented normalized to control. n =6 biological replicates. Scale bar = 20 μm. * p ≤ 0.05.
(d) Quantitative analyses of Alamar blue cell proliferation assay of EC cultures supplemented with control RPE-CM versus. SFD RPE-CM at the 24 h timepoint. Data is presented normalized to control. n = 7 biological replicates. ** p ≤ 0.01.
(e) Immunofluorescence images showing CD31 (green) and DAPI (blue) expression in parallel cultures of PEG-EC gels supplemented with either control RPE-CM or SFD RPE-CM for 14 days. Scale bar = 20 μm.
(f-i) Quantitative analyses of CD31+ vascular networks in day 14 PEG-EC gels exposed to either Control or SFD RPE-CM. Exposure to SFD-RPE-CM results in increased (f) vascular density or covered area, (g) total number of tubes, (h) total tube length and (i) total number of branching points compared to control RPE-CM. Data is presented normalized to control. n = 3 biological replicates. **p ≤ 0.01.
Given that pathologic neovascularization of remnant ECs as consequence of CC atrophy likely contributes to CNV (Grossniklaus and Green, 2004), we next evaluated the influence of SFD-RPE-CM on EC proliferation and EC-derived vascular network formation. Consistent with the role of RPE-secreted pro-angiogenic factors in CNV development (Bhutto et al., 2006), SFD RPE-CM increased EC proliferation (Figure 5d, p ≤ 0.01). Furthermore, compared to control RPE-CM, SFD RPE-CM increased the vascular network density of EC-derived networks (Figure 5e, 5f; p<0.01) by positively influencing the number (Figure 5e, 5g; p<0.01) length (Figure 5e, 5h; p<0.01) and branching (Figure 5e, 5i; p<0.01) of tubules.
Altogether, we leveraged modular control of the RPE-CC model and comparisons between healthy and SFD-derived patient cells to show that CNV and CC atrophy, which are pertinent to AMD/MD pathology, can be initiated solely by RPE autonomous dysfunction. Furthermore, our data suggests that CC atrophy in SFD and similar maculopathies is not a secondary consequence of drusen deposits underneath RPE cells but is caused by alterations in RPE secreted factors.
Serum-derived factors regulate development of CNV-relevant changes in ECs and EC-derived vascular networks
Apart from pathological changes impacting RPE and CC layers, systemic factors have been implicated in CNV development (Cheung and Wong, 2014). For example, it has been postulated that altered abundance of pro-angiogenic factors (e.g., VEGF, MMP2) in the serum of AMD patients contributes to CNV development (Campochiaro et al., 1999, Kvanta et al., 2000). Given that increased levels of pro-angiogenic factors in AMD patient serum could contribute to CNV development by promoting pathologic neovascularization in remnant ECs present consequent to CC atrophy, we next evaluated the influence of AMD patient serum on CNV-relevant vascular changes (EC proliferation, invasion, and vasculogenesis) observed in presence of SFD RPE-CM. Of note, wet-AMD (wAMD) serum was obtained from patients with clinically diagnosed CNV. Furthermore, single nucleotide polymorphisms (SNPs) common to wAMD (ARMS2, CFH, HTRA1) were not present in samples analyzed by PCR (Table S3).
To represent the orientation of RPE-secreted factors and serum in relationship to CC in vivo, control RPE-CM was applied apically with serum supplemented basally to the EC/EC-derived vascular networks. Similar to SFD RPE-CM, 25% wAMD serum increased ECM invasion by ECs compared to serum obtained from control subjects (Figure 6a & 6b, p ≤ 0.05). Of note, the extent of EC invasion ECM in the presence of 25% normal human serum was similar to the normal cell culture medium (Figure S6c, S6d). Given that AMD is an age-associated disease and CNV symptoms generally appear in adults > 60 years of age, we also verified the consequence of aging on ECM invasion by ECs in the absence of CNV/AMD. Parallel experiments utilizing serum from normal “young” subjects (age range of 26–29) versus aged control subjects (age range of 63–75) showed that EC invasion of ECM as a function of serum age was not statistically different (Figure S6e, S6f). Additionally, AMD serum did not significantly alter EC proliferation when compared to normal human serum (Figure 6c). However, CD31-positive tubes exhibited increased density (Figure 6d, 6e, p ≤ 0.001), number (Figure 6d, 6f, p ≤ 0.001), length (Figure 6d, 6g, p ≤ 0.001) and branching (Figure 6d, 6h, p ≤ 0.01) after two weeks for cultures with normal sera/serum versus AMD sera/serum.
Figure 6. wAMD serum derived factors are sufficient to instigate CNV-relevant changes in an iPSC model.

(a, b) Light microscopy images (b) and quantitative analyses (b) of crystal violet stained ECs invading through COL1-coated transwell towards 25% age-matched serum (“Control serum”, left) or 25% wAMD serum (“wAMD serum”, right) at the 24 h timepoint. Data is presented normalized to Control. n = 12 normal serum, 7 wAMD serum. * p ≤ 0.05. Scale bar = 20 μm.
(c) Quantitative analyses of Alamar blue cell proliferation assay in EC cultures supplemented with 25% age-matched normal (Control) versus. wAMD serum at the 24 h timepoint. Data is presented normalized to Control. n = 12 normal serum, 7 wAMD serum.
(d) Immunofluorescence images showing CD31 (green) and DAPI (blue) expression in parallel cultures of PEG-EC gels supplemented with either 25% age-matched normal serum (“Control”) or 25% wAMD serum (“wAMD”). for 14 days. Scale bar = 20 μm.
(e-h) Quantitative analysis of CD31+ vascular networks in day 14 PEG-EC gels exposed to either 25% age-matched serum (“Control”) or 25% wAMD serum (“wAMD”). Exposure to wAMD serum results in increased (e) vascular density or covered area, (f) total number of tubes (g) total tube length and (h) total number of branching points compared to control serum. Data is presented normalized to Control. n = 12 normal serum, 7 wAMD serum. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001.
Overall, these experiments show that exposure to serum constituents from AMD patients is independently sufficient to induce CNV-relevant cellular changes, invasion of ECM by ECs, and neovascularization by promoting development of EC-derived vascular networks.
Inhibition of pro-angiogenic signaling can rescue CNV-relevant changes in an iPSC-model.
Inhibition of pro-angiogenic signaling by targeting RPE-secreted factors is a treatment strategy for wAMD (Bhutto et al., 2006, Farnoodian et al., 2017) and SFD (Gourier and Chong, 2015). In CNV, the loss of CC may initiate RPE ischemia and increased production of pro-angiogenic factors such as VEGF-A and FGF2, thereby promoting new vessel development and CNV (Grossniklaus and Green, 2004). The addition of a FGF2 neutralizing antibody to SFD RPE-CM suppresses invasion of ECM by ECs (p ≤ 0.01) (Figure 7a, 7b), consistent with a recent study showing a role of FGF2-mediated pro-angiogenic signaling in CNV development in SFD (Qi et al., 2019). In contrast, anti-VEGF-A had no effect on EC ECM invasion in the presence of SFD RPE-CM (Figure 7a, 7b). Interestingly, VEGF-A or FGF2 neutralizing antibodies did not alter EC proliferation in SFD RPE-CM (Figure S7a).
Figure 7. Inhibition of pro-angiogenic signaling in an iPSC model can rescue CNV-relevant changes.

(a) Representative brightfield microscopy images illustrating ECs (crystal violet, purple) that have invaded COL1-coated transwell membranes towards SFD-RPE-CM (“Untreated”) versus SFD-RPE-CM supplemented with anti-FGF2 (“αFGF2”: 2.5 μg/ml), SFD-RPE-CM supplemented with anti-VEGF-A (αVEGF-A: 2.0 μg/ml) at the 24 h timepoint. Scale bars = 20 μm.
(b) Quantitative analysis of EC invasion for conditions described in (a). See Methods for additional details. Data was normalized to Untreated SFD-RPE-CM n = 5 biological replicates. ** p ≤ 0.01.
(c) Immunofluorescence images illustrating CD31+ vascular networks for day 14 PEG-EC gels culture in 25% wAMD serum-containing medium alone (“Untreated”) or supplemented with 15 μM doxycycline (“Dox”). PEG-ECs were cultured in wAMD serum-containing medium (±Dox) that was exchanged daily for the 14-day duration of the experiment.
(d) Quantitative analysis of CD31+ vascular networks in day 14 PEG-EC gels cultured using the same conditions described in (c). Data was normalized to Untreated. n = 12 normal serum, 7 wAMD serum. ** p ≤ 0.01, *** p ≤ 0.001.
See also Figure S7.
Matrix metalloproteinases (MMPs) regulate angiogenesis (Stetler-Stevenson, 2001) and increased levels of MMP2 and MMP9 in AMD patient serum have been linked to CNV (Lambert et al., 2002, Lambert et al., 2003). Therefore, a broad-spectrum MMP inhibitor, doxycycline, was investigated for its ability to rescue CNV-relevant changes in the iPSC tissue mimetic. Although doxycycline supplementation did not impact invasion of ECM by ECs (Figure S7b-c), PEG-EC gels supplemented with doxycycline-treated AMD serum exhibited decreased density, (Figure 7c, p<0.001), total tube length (Figure 7d, p< 0.01), total number of tubes (p= 0.07) and branching points (p<0.001) of CD31-positive networks after 2 weeks in culture compared to no doxycycline controls (untreated AMD serum).
Altogether, these experiments show that CNV relevant changes can be modulated by targeting pro-angiogenic signaling in a patient-derived human cell model.
Discussion
The RPE-CC interaction is critical not only for the development of choriocapillaris and synthesis of Bruch’s membrane but also for vision more generally (Strauss, 2005). In fact, dysfunction of either the RPE or CC initiate molecular events promoting pathogenic alterations, as observed by multimodal imaging studies in humans and animal models of macular degeneration (Fletcher et al., 2014, Forest et al., 2015). Manipulation of primary RPE and ECs or cell lines using overexpression and knockdown approaches or physiologically relevant stressors, such as hypoxia and aging, have been utilized to dissect the role of RPE versus ECs in the development of AMD and related MDs (Dardik et al., 2010, Forest et al., 2015, Kurihara et al., 2016, Samtani et al., 2009).
We have previously utilized patient-derived iPSC models of specific MDs, including SFD, to show that RPE-specific cell defects initiate drusen formation and basement membrane thickening (Galloway et al., 2017). With respect to vascular pathologies, the primary focus of in vitro cellular models has been on CNV. However, data on immortalized cell lines and primary mammalian cultures of RPE and ECs have not always agreed with the observed in vivo influence of RPE-EC crosstalk (Forest et al., 2015, Komez et al., 2016, Wang et al., 2011). Furthermore, the absence of pericytes/MSCs and subsequent lack of CC-like vasculature has limited their utility for disease modeling applications.
Here, we incorporated RPE, MSCs, and ECs in a spatially-controlled microenvironment using PEG hydrogels that were formed using MMP-degradable peptide crosslinkers and RGD adhesive motifs (Hoffman et al., 2013, Kraehenbuehl et al., 2009, Kraehenbuehl et al., 2008, Lutolf et al., 2003, Seliktar et al., 2004, Shubin et al., 2015, Van Hove et al., 2014, Zanotelli et al., 2016) to enable matrix remodeling that supports RPE-CC tissue formation and function. Notably, PEG-based hydrogels have been used in a variety of tissue mimetic approaches from fundamental studies to drug screening and discovery (Bhatia and Ingber, 2014, Bhise et al., 2014, Esch et al., 2015, Schwartz et al., 2015), including development of tissue organoids (Cruz-Acuna et al., 2018, Schwartz et al., 2015), neural tubes structures (Meinhardt et al., 2014, Ranga et al., 2016), and generation of the blood-brain barrier (Schwartz et al., 2015). Furthermore, PEG gels can be introduced step-wise (Aziz et al., 2017), providing spatiotemporal control over tissue evolution. Therefore, to mimic the organization of the RPE-CC complex in vivo, ECs were encapsulated in an MMP-degradable PEG scaffold to form CC-like vasculature and RPE were apically localized on the hydrogel surface. The modular function of our in vitro RPE-CC model was used to investigate parallels of RPE-CC development in vivo, specifically i) the contribution of RPE and MSC to the development of fenestrated CC-like endothelium in vitro and factors that support physiologically relevant features of the RPE-CC, ii) the contribution of cellular and exogenous factors in development of AMD/MD-relevant vascular changes.
Development of the RPE-CC complex in vivo requires synergistic interaction of the RPE, choroid, and mesenchyme from surrounding ocular stroma (Lutty et al., 2010). CC does not develop in the absence of the RPE monolayer (McLeod et al., 2009, Saint-Geniez and D’Amore, 2004) and surrounding pericytes and mesenchyme are essential for the stability of the choroidal vasculature (Lutty et al., 2010). Results using our RPE-CC model demonstrated that RPE and mesenchyme secreted factors were necessary for development of fenestrated CC-like endothelium (Figures 1–3). Similar to the in vivo CC, presence of primitive tubules in the RPECC (Figure 1) and the (RPE-CM)-CC (Figure 2) was concordant with mesenchymal presence in CC (Lutty et al., 2010). Additionally, MSCs were necessary to stabilize CC-like tubule networks, extending culture from ~2 weeks up to 2 months (Figures S2f), which is consistent with the role of pericyte/mesenchyme secreted factors in vivo (Park et al., 2017). Similar to the human eye, fenestrated vascular networks in the RPE-CC mimetic models i) required RPE-secreted VEGF-A (Figure 3) and ii) were apically localized towards the RPE monolayer (Figure 1, 2). TGF-ß1 is required for endothelial fenestration and vascular stability (Tosi et al., 2018, Wang et al., 2017), which was also found in our studies (Figure 3) and may be due to either MSC secretion or through EC-MSC activation of latent TGF-ß1 (Dickson et al., 2001). Thus, results using the RPE-CC model here were consistent with development of fenestrated endothelium and in vivo CC morphogenesis.
Also consistent with normal RPE-CC physiology, RPE-secreted factors from control subjects enhanced EC proliferation and vasculogenesis, but not invasion (Figure 2, S2a-e & S6a). We and others have previously shown that iPSC-RPE cells synthesize components of the RPE basal lamina, a constituent of the Bruch’s membrane in vivo (Galloway et al., 2017, Pilgrim et al., 2017). Here, COL6 and LAM are observed in the RPE-CC model in vitro (Figure 2g). Altogether, these results provide the first demonstration of an in vitro model that recapitulates physiological features of in vivo RPE-CC.
RPE-EC co-culture systems have been limited to culture durations ranging between 24 h and 2 weeks (Hamilton et al., 2007, Komez et al., 2016). Considering that diseases impacting the RPE-EC complex are age-associated and adult-onset (e.g., AMD, SFD), short-term RPE-EC interactions may not be sufficient for investigating disease pathophysiology. The tissue model described here extended stability of the RPE-CC model up to 2 months, which is within the time frame of development of major AMD/MDs pathological hallmarks (drusen deposits, ECM accumulation, CNV, CC atrophy) in culture (Galloway et al., 2017) (Figure 4).
With regard to the vascular pathologies in AMD, it is important to emphasize that other co-culture models utilizing RPE and EC have been utilized to study AMD pathophysiology (Hunt et al., 2017, Shokoohmand et al., 2017). For example, transmigration of non SFD iPSC-derived ECs across the RPE monolayer has been shown in RPE-EC co-cultures (Geisen et al., 2006). Using RPE-CM from SFD RPE cells in protocols (invasion and vascular network formation) that have previously been used to study the CNV pathology in culture (Geisen et al., 2006, Qi et al., 2019), we show that results obtained from these assays are consistent with the findings in our 3D RPE/(RPE-CM)-CC model (Figure 5). However, CC atrophy, a central pathologic feature that is among the first insults seen in both wet and dry-AMD (Bhutto and Lutty, 2012) has not been shown in cell models, as these models lacked CC-like vasculature. Importantly, the 3D SFD RPE-CC mimetic model presented here integrates CC atrophy jointly with pathologic neovascularization (Figure 4–6), providing a unique opportunity to study how loss of CC contributes to the instigation of CNV. In particular, our model provides a powerful tool to evaluate the temporal execution of molecular changes in the pathologic progression of the disease and the ability to link these cellular populations in a modular culture platform with patient-derived cells.
The RPE-CC model was specifically used to study cellular and locally-secreted soluble factors in the progression of AMD/MD relevant pathologies of CC atrophy and CNV. Our results provide evidence that CNV and CC atrophy can be initiated solely by alterations in RPE-secreted factors. It is noteworthy that an RPE-specific TIMP3 mutation in SFD led to CC atrophy and CNV-like behavior the RPE-CC model at a timepoint preceding overt pathologic changes, such as drusen formation and ECM thickening, which was not observed in the SFD RPE cell layer (Figure S5b) (Galloway et al., 2017). Therefore, atrophy of a specific cell layer (RPE vs. CC) layer might not be solely indicative of the cell type responsible for initiating disease pathology in AMD/MD. Furthermore, our data using EC/vascular networks show that both SFD RPE-CM and altered serum composition (wAMD serum) can independently initiate CNV-related changes, suggesting that AMD/SFD may be a consequence of multiple individual or combined local (e.g., RPE cell defect) and systemic factors.
In summary, we utilized a 3D RPE-CC tissue model to i) show the role of RPE and MSC secreted trophic factors in CC development, ii) develop CNV and CC atrophy in a patient-derived cell model of macular degeneration, iii) determine the unique contribution of local (RPE versus CC) and systemic influences (e.g., serum) to CNV and/or CC atrophy development in macular degeneration and iv) pharmacologically inhibit CNV-relevant cellular changes, EC invasion and vasculogenesis in an iPSC model system. Ultimately, the RPE-CC model described here provides a powerful platform to investigate and pharmacologically-target human disease pathophysiology using patient-derived cells.
Limitations of the Study
One limitation of this work is that we did not investigate the independent role of wet-AMD serum in initiating CC atrophy in the 3D iPSC-CC model. This is an important question and would require a large sample size of wet-AMD patients that would ideally be grouped by risk-factors (e.g., SNPs relevant to AMD) and should be pursued in future studies. Another limitation of the study is that the mechanism underlying CC loss and CNV development in the SFD RPE-CC model was not explored, although we did show that CNV-relevant cellular changes could be pharmacologically modulated by inhibition of FGF2. Also, the tissue mimetic described here does not include several complexities of the human eye that could be relevant for macular degeneration pathophysiology. For example, the photoreceptor layer or other cell type(s), including macrophages, melanocytes, and mast cells, present in the choroid., could be introduced. Similarly, we did not characterize the impact of RPE, EC, MSC co-culture on the gene/protein expression and functional characteristic of the individual RPE and CC layer. Lastly, current technical limitations with sample processing for immunocytochemical and electron microscopy resulted in the separation of transwell membrane/RPE monolayer from the CC layer and impacted consistent analysis of the tight junctions and/or the comprehensive RPE-CC complex.
Star Methods
RESOURCE AVAILABILITY
Lead Contact
Information and requests for reagents may be directed to and will be fulfilled by the corresponding author, Dr. Ruchira Singh (Ruchira_Singh@urmc.rochester.edu).
Materials availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human sera samples from 12 control subjects and 7 wAMD patients (equal representation of male and female subjects) were obtained with informed consent from the University of Rochester in accordance with an approved Institutional Regulatory Board (RSRB00056538) and conformed to the ethical norms and standards in the Declarations of Helsinki. Fibroblasts/human iPSC procurement and usage was also approved by the University of Rochester and fibroblasts from two SFD patients (S204C mutation in TIMP3) and three healthy subjects (sibling control, unrelated individual) were expanded and subsequently reprogrammed to obtain iPSCs.
METHOD DETAILS
Preparation of human sera
Peripheral blood samples from healthy volunteers (no history retinal diseases) and patients diagnosed with wAMD with clinically-documented CNV was collected in red top Vacutainer tubes. After allowing to clot at room temperature for 30 minutes, the samples were centrifuged at 1,500 x g for 10 minutes. The separated serum fraction was collected, aliquoted and frozen at −80 °C until used.
Cell culture
Fibroblasts were expanded from skin punch biopsies collected from Sorsby’s Fundus Dystrophy (SFD) patients and unaffected Controls and were maintained in DMEM containing 10% heat-inactivated FBS, 1% MEM nonessential amino acids (NEAAs), 1 mM Sodium pyruvate, 1%(v/v) Penicillin, and Streptomycin.
Reprogramming of fibroblasts into iPSCs
Human iPSC lines from SFD patients and Control were generated by reprogramming of patient fibroblasts, as previously described (Galloway et al., 2017, Okita et al., 2011). Briefly, fibroblasts were nucleofected with 1μg of pCXLE-hOCT4-shP53 (Addgene, 27077), pCXLE-hSK (Addgene, 27078) and pCXLE-hUL (Addgene, 27080) episomal plasmids using primary fibroblasts reprogramming kit (Lonza) and Nucleofector 2b device (Lonza, Program T-016). Nucleofected fibroblasts were maintained in fibroblast medium for 6 days and 1 × 105 cells were re-plated onto irradiated mouse embryonic fibroblast (MEF) feeders on a 100 mm dish. Media was replaced to iPSC basal medium (DMEM/F12 with 20% Knockout serum replacement or KSR, 1% MEM-NEAA, 1% Glutamax and 100 ng/mL FGF2; Thermo Fisher Scientific) the following day. iPSC colonies started appearing around day 17–30 and the colonies were manually picked up and expanded for further characterization and differentiation. Of note, presence of TIMP3 (S204C) mutation in SFD patient iPSCs and lack of S204C TIMP3 mutation in control iPSCs was confirmed using previously published primers and DNA sequencing (Galloway et al., 2017). Furthermore, standard G-banding karyotype analysis was performed, and all lines were demonstrated to be karyotypically normal.
Differentiation of iPSCs to MSCs
Differentiation of iPSCs into mesenchymal (MSCs) was conducted as previously described (Vodyanik et al., 2010). Briefly, iPSC cell lines grown initially on irradiated mouse embryonic fibroblasts (MEFs) were harvested with Collagenase IV and then co-cultured with OP9 cells and grown in MSC differentiation medium for 2.5 days. Using MACS, OP9 cells (CD29+ cells) were depleted and the remaining differentiated iPSC were grown in methylcellulose semi-solid medium (40% ES cult™, Stem Cell Technologies), 25% Stemspan™ serum-free expansion media (Stem Cell Technologies), 10% BIT 9500 supplement (Stem Cell Technologies), 1:100 Glutamax, 1:1000 Excyte supplement (Millipore), 100 μM monothioglycerol, 50 μg/mL ascorbic acid and 10 ng/mL hPDGF-BB for 12 days. On day 12, spherical colonies are picked and plated on collagen-coated plates to allow the proliferation of MSCs and were subsequently maintained in fibroblast media (DMEM with 20% heat-inactivated FBS, MEM nonessential amino acids (NEAAs), Sodium pyruvate, penicillin, and Streptomycin).
Differentiation of iPSCs to ECs
Differentiation of iPSCs into ECs was conducted as previously described (Vodyanik et al., 2010). iPSC maintained on Matrigel/mTeSR were adapted through E8BA (E8 media with BMP4 at 5 ng/mL and Activin at 25 ng/mL), E7i media (E6 media, 5 μM SB431542 (Miltenyi) and 10 ng/mL bFGF (PeproTech) prior to MACS purification of CD31+ cells using the MACS purification system, according to the manufacture’s protocol. The purified CD31+ population was maintained in E7v media (E6 media supplemented with 10 ng/mL bFGF (PeproTech) and 50 ng/mL VEGF165 (PeproTech) and eventually in Vasculife media (Lifeline Cell Technology, Frederick, MD) for subsequent passages.
Differentiation of iPSCs to RPE cells
Differentiation of iPSCs into RPE was conducted as previously described (Singh et al., 2013b, Singh et al., 2013a). iPSC colonies were dissociated and cultured in suspension to generate embryoid bodies (EBs). On day 6, EBs were transferred to the laminin-coated plate and maintained in neural induction medium (NIM, DMEM/F12 with 1% MEM-NEAA, 1% N2 Supplement and 2 μg/mL heparin and 1%(v/v) Penicillin, and Streptomycin). On day 14, the cell growth medium was switched from NIM to retinal differentiation medium (RDM, 70% DMEM / 30% F-12 with B-27 supplement and 1%(v/v) Penicillin, and Streptomycin). At ~day 20, optic vesicle-like structures were removed and the remaining cells were allowed to grow as adherent cultures. RPE were dissected and dissociated with 0.05% Trypsin EDTA and plated onto 24 well plates coated with laminin for 4–24h. RPE cells were thereafter cultured in RDM media containing 2% fetal bovine serum (FBS) until the cells had formed a confluent monolayer. After reaching confluence, FBS was removed from the cell culture media and cells were maintained in RDM. For tissue mimetic experiments, one-month old monolayers of RPE cells were trypsinized and re-plated on the apical side of hydrogels.
Hydrogel synthesis, characterization, and development as a tissue mimetic
Synthesis of 8-arm poly(ethylene glycol) (PEG)-Norbornene (PEG-NB)
N, N’-dicyclohexylcarbodiimide (DCC, Sigma) coupling was used to synthesize norbornene-functionalized 8-arm PEG, as previous described (Fairbanks et al., 2009). 20 g of 8-arm 20 kDa PEG (JenKem Technology), pyridine (5 mol/mol PEG) and 4-dimethylaminopyridine (Fisher Scientific) (0.5 mol/mol PEG) were dissolved in 25 mL dichloromethane (DCM, Sigma) for 30 min. Separately, DCC (5 mol/mol PEG) and norbornene-2-carboxylate (10 mol/mol PEG, Alfa Aesar) were dissolved in 100 mL DCM for 30 min. Then, the PEG solution was added to the carbodiimide-activated norbornene, which was stirred overnight at room temperature. After filtration, PEG-NB was precipitated in cold diethyl ether and dialyzed against distilled, deionized water (ddH2O) (1000 MWCO dialysis tubing, Spectrum Laboratories) for four days and then lyophilized for another three days. Norbornene-functionalization was measured by 1H-NMR [CDCl3] to be 90% (δ=3.5–3.8 of PEG ether protons compared to δ= 5.9–6.3 of norbornene vinyl protons).
Synthesis of RGD cell adhesive peptide
A Liberty1 automated peptide synthesizer with UV monitoring (CEM) was utilized to synthesize the cell adhesive ligand CGRGDSG (RGD) on FMOC-Gly-Wang resin (EMD), as previously described (Van Hove et al., 2014). All amino acids were dissolved at 0.2 M in N,N-Dimethylformamide (DMF, Fisher Scientific). 0.5 N, N’-Diisopropylcarbodiimide (DIC) in DMF was used as an activator, and 1 M Ethyl cyano(hydroxyimino)acetate (Oxyma Pure, Sigma-Aldrich) in DMF was used as activator base. 10 wt% piperazine (Alfa Aesar) in DMF with 10 vol% ethanol was used for deprotection. The resin was then cleaved from peptides using a cleavage cocktail comprised of 92.5 vol% trifluoroacetic acid (Alfa Aesar), 2.5 vol% each triisopropylsilane (Alfa Aesar), 2.5 vol% 3,6-dioxa-1,8-octanedithiol (Alfa Aesar), 2.5 vol% ddH2O and 2.5 vol% thioanisol for 4 h. After cleavage, the resin was removed by vacuum filtration, and the resulting solution was precipitated in ice-cold diethyl ether. The peptide was collected by centrifugation and molecular weight of 655 g/mol verified using a Bruker AutoflexTM III Smartbeam Matrix-Assisted Laser Desorption Ionization Time of Flight (MALDI-ToF) mass spectrometer using α-Cyano-4-hydroxycinnamic acid as matrix.
Hydrogel polymerization, characterization, and use as a tissue mimetic
PEG-NB (5 wt%), matrix metalloproteinase (MMP) sensitive crosslinker peptide (GKKCGPQGIWGQCKKG, Genscript, NJ, 10.7 mM, resulting in 62.5% crosslinking with PEG-NB) and 15 mM RGD were dissolved in phosphate buffered saline (PBS). 0.05 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), synthesized as described (Fairbanks et al., 2009), was then added to the hydrogel precursor solution. Acellular hydrogels (~ Ø6 × 2 mm, 40 μL/gel) were photopolymerized using 365 nm light at 5 mW/cm2 for 3 minutes. After incubation in PBS overnight, the compressive modulus of the hydrogels was measured to be 1.7 ± 0.4 kPa using an MTS QT/5 (5 N load cell).
To entrap ECs within hydrogels, ECs were dissociated using trypsin and re-suspended (1.5 × 105 cells/gel) in 50 μl pre-polymer solution, described above, which was subsequently photopolymerized in a 24-well PET Transwell® insert (6.5 mm, 0.4 μm Pore, Costar, Corning) and cultured using Vasculife media, which was exchanged every 3 days. For EC-MSC co-cultures, ~25,000 MSCs per well were first cultured on the PET transwell membrane surface overnight, and then EC-encapsulated PEG hydrogels were photopolymerized on top of the MSC monolayer. For EC-MSC-RPE hydrogels (“RPE-CC”), the apical surface of EC entrapped hydrogel cocultured with MSCs was coated with Laminin (20 μg/ml) for at least 3 hours at 37 °C and RPE post-dissociation was plated at a density of 2.5 × 105 cells in the presence of the ROCK inhibitor-Y-27632 (10 μM/ml, STEMCELL Technologies, Vancouver, BC). RPE-CC cultures were refreshed with Vasculife basally (“EC Medium”) and RDM apically (“RPE medium”) every other day.
For experiments determining the influence of RPE conditioned medium (“RPE-CM”) on EC, EC-MSC cocultures, RDM, RPE-CM from control RPE, and/or RPE-CM for RPE derived from SFD patients (“SFD-RPE-CM”) were added to the apical (top) transwell chamber and EC medium was added to the basal (bottom) chamber. RPM-CM was prepared by culturing a monolayer of RPE for at least 30 days. Of note, control and SFD RPE-CM was obtained from parallel cultures of RPE with the same EB date. EC-MSC samples treated with RPE-CM are referred to as “(RPE-CM)-CC” in the manuscript. For experiments determining the influence of serum, 10% normal serum (± 15 μM doxycycline) or 10% wAMD serum (in EC medium) were added to the basal chamber of the transwell and RPE-CM was added to the apical chamber. For RPE-CM and serum studies, culture media were refreshed every other day. At the conclusion of trials, hydrogel constructs were either fixed or lysed for further analysis.
EC Invasion Assay
Invasion assays were conducted as previously described (Justus et al., 2014). Briefly, EC were suspended in minimal medium (DMEM with 0.1% BSA and PSA) and seeded on the top (apical) surface of 3.0 μm PET transwell membranes (24-well transwell system, 6.5 mm, Costar, Corning) at a density of 50,000 cells per well. 500 μl of RDM, RPE-CM (from SFD-affected or control patients), or 25% serum (from AMD or control patients) was placed in the basal chamber of the transwell. To investigate how inhibition of pro-angiogenic factors influenced invasion of ECM by ECs, SFD-RPE-CM or control RPE-CM supplemented with FGF2 (2.5 μg/ml, Millipore) or VEGF-A (2.5 μg/ml, R&D system) neutralizing antibodies were added to the apical transwell chamber and samples were incubated for 24 hours at 37 °C. After 24 hours, the media from both the apical and basal chamber was removed and transwell membrane apically was swabbed with a Q-tip to remove residual cells. Following a gentle wash with 1X PBS the membranes were fixed in chilled 70% ethanol for 15 minutes and swabbed again to remove residual cells. Subsequently, transwell membrane were incubated for 15 minutes in a 0.2% crystal violet solution, rinsed in ddH2O and allowed to dry before imaging with 10x objective on a DMIRB Leica inverted microscope (Leica Microsystems, Buffalo Grove, IL) equipped with Moticam BTU 10 camera (Motic, Richmond, CA) and Moticam 2000 software. Of note, for quantification, in every experiment, ~15–20 images were taken for each sample in randomly selected areas and crystal-violet stained-ECs were counted manually. Data compilation and analyses was carried out using Microsoft Excel software.
EC Proliferation Assay
Alamar Blue was used to assess the proliferation of ECs in culture in accordance with the manufacturer’s recommendation (Bio-Rad, Hercules, CA). Briefly, 10% Alamar Blue was added to either cell plated in a 24 well (~50,000) or ECs encapsulated within hydrogel. Subsequently, aliquots of conditioned media were taken at either 2, 4, 6, and 24 hours post addition and the fluorescent intensity of reduced Alamar Blue was measured on a Tecan plate reader with excitation at 560 and emission at 590. The culture media served as the blank for fluorescence intensity measurements.
Immunofluorescence analysis
Whole mount staining of hydrogels
Hydrogels were fixed in 2% neutral buffered formalin for 2 hours. Subsequently, for wholemount analyses, fixed hydrogel samples were permeabilized and blocked in 0.25 % Triton X-100 with 1% BSA in 1X PBS. Next, the hydrogel samples were incubated with primary antibodies in 0.5X blocking buffer, rabbit anti-EZRIN (1:100 Cell Signaling, MA), sheep antiCD31 (1:50, R&D Minneapolis, MN), rabbit anti-COL4 (1:100, Abcam, UK), goat anti-MITF (1:100 Santa Cruz, TX), rabbit anti-PLVAP (1:100, Sigma-Aldrich, St. Louis, MO), mouse anti-VE-CADHERIN (1:100, Santa Cruz, TX), mouse anti-ZO1 (1:20, Thermo Fisher Scientific, MA),overnight at 4 °C. The next day, following 2 hours of washes in 1X PBS containing 0.25 % Triton X-100 and 1% BSA, hydrogel samples were incubated in host-specific Alexa-Fluor conjugated antibody (1:500, Thermo Fisher Scientific, MA) in 0.5X blocking buffer and Hoechst (1:2000, Thermo Fisher, MA) at 4°C overnight. Subsequently, the hydrogel samples were subjected to overnight washes and mounted with AQUA-Polymount media (Polysciences Warrington, PA) on depression slides (fisher scientific, NH). All images were captured on a confocal microscope (LSM 510 META, Zeiss, Thornwood, NY).
Embedding, sectioning, and staining of hydrogels
For preparing tissue sections, fixed hydrogels were infiltrated with 1% w/v poly-vinylalcohol (Sigma-Aldrich, St. Louis, MO) n PBS at 4 °C overnight. Next, the constructs were transferred into cryomolds filled with tissue freezing medium (General Data, Cincinnati, OH) and flash-frozen in liquid nitrogen. Cryosectioning was performed on the frozen blocks set at 16 μm and the sections were allowed to dry to slides (SuperFrost Plus slides, Fisher Scientific) and were either stored at −20 °C for future use or processed immediately as follows. Sections were rehydrated in chilled 1X PBS followed by incubation in ice-cold blocking buffer (1X PBS containing 10% normal donkey serum (NDS) and 0.1% Triton X-100 for 1 hour. Next, sections were incubated with primary antibodies in 0.5X blocking buffer, mouse anti-CD31 (1:50, Dako, Santa Clara, CA), sheep anti-CD31 (1:50, R&D Minneapolis, MN), rabbit anti-CLAUDIN-5 (1:50, Thermo Fisher Scientific, MA), rabbit anti-COL6 (1:200, Abcam, MA), rabbit anti-LAMININ (1:200, Abcam, UK), rabbit anti-PLVAP (Sigma, 1:100, Sigma-Aldrich, St. Louis, MO) sheep anti-VWF (1:100, Abcam, UK), mouse anti-VE-CADHERIN (1:100, Santa Cruz, TX) and mouse anti-ZO1 (1:20, Thermo Fisher Scientific, MA), After primary antibody incubation, sections were washed twice with 1X PBS containing 0.05% Triton X-100 and incubated in appropriate Alexa-fluor conjugated secondary antibodies (1:500, Thermo Fisher Scientific, MA) and Hoechst (1:2000, Thermo Fisher, MA) in 0.5X blocking buffer for 1 hour at room temperature. Following two more washes in 1X PBS containing 0.05% Triton X-100, the slides were mounted using ProLong™ gold anti-fade (Thermo Fisher, MA) solution. The images were captured and analyzed using confocal microscope (LSM 510 META, Zeiss, Thornwood, NY) equipped with Zen 2009 software (Zeiss). Of note, Table S4 briefly describes use of each staining marker used for immunocytochemical analyses.
Image analysis
3D reconstruction and orthogonal (xz, yz view analyses): Subsequent to confocal microscopy, image analyses for maximum intensity 2D projection, orthogonal (xz, yz view analyses and 3D reconstruction was carried out using Imaris (Bitplane, Switzerland) or Fiji-ImageJ (National Institute of Health, USA) software. Of note, in images where nonspecific binding of antibody aggregates in red and green channel in the vascular layer (likely due to nonspecific interactions with degrading hydrogel) interfered with image/data analyses, these punctate were removed in Fiji/Imaris or another imaging software. Of note, vasculature-were not removed from any images within the manuscript. Specifically, image smoothing was performed using background subtraction method and median filters for the channels corrupted by noise. Furthermore, to adjust the visualization of vasculature, localized aggregates were either manually deleted or 3D cropped. All movies were generated using windows Video Editor from the files exported from Zeiss or Olympus confocal microscope.
PLVAP cell count and PLVAP-vascular layer-thickness measurements: For PLVAP cell counts, maximum intensity 2D projections of DAPI and PLVAP staining within the vascular region were generated and merged into a composite image. The Cell Counter tool was used to quantify the number of nuclei within PLVAP-positive regions. For PLVAP thickness measurements, the width of PLVAP layer (based on PLVAP staining in xz view) at 5 different points per image were averaged to get the mean thickness of PLVAP-positive vascular networks in the CC layer. Of note, thickness of CC-tissue was based on PLVAP expression and excluded hydrogel and non-PLVAP containing vascular tissue when present.”
Evaluation of vascular network characteristics: Measurement of vascular density or area, number of tubes, total length of tubes, and branching points was carried out using Wimtube software (Wimasis, Cordoba Spain) that has been utilized by multiple studies to quantify vascular network characteristics both in vitro in 3D models (Khoo et al., 2011) and in vivo. With regard to individual vascular network characteristics, vascular density, or percent covered area is measured by computing the percentage of tubular structures in the image area. It is calculated by dividing the total number of pixels of the image by the pixels making up the tubular structure; number of tubules refers to the number tubular structure; total length of tubules is calculated as the complete length in pixels of the tubular structure; and number of branching points refers to nodes/junctions where three or more tubes converge. Of note, prior to image analyses, both confocal and light microscopy images were processed by ImageJ to reach an output image suitable for Wimtube (Khoo et al., 2011). Specifically, for confocal microscopy images maximum intensity z-stack projection were converted to a 32-bit image, inverted, and converted to a 16-bit image. Brightness and contrast of the image were adjusted to increase the signal to noise of vasculature. The 16-bit image was then converted to an 8-bit image a mask of the vascular skeleton was created using either a binary mask tool or the Auto Local Threshold based on the Mean. Subsequent to this step, the Image was despeckled and dark outliers were removed <6.0 pixels using the “Despeckle” and “Remove Outliers” tools in Fiji respectively. Remaining outliers (due to antibody aggregation) were manually removed using Fiji and uploaded to Wimtube software for automated analyses. Similarly, for light microscopy images, images were first converted to a 32-bit image, inverted and then converted to a 16-bit image. Next, brightness and contrast of the image were adjusted to make all vasculature brighter than the background and the image was smoothed and sharpened for creating an image output that could be evaluated by Wimtube software.
ELISA
VEGF-A and TGF-ß1 levels in cell culture supernatant was evaluated using commercially available Elisa kit (VEGF-A: DVE00, R&D systems, location; TGF-ß1: DB100B, R&D systems. Briefly, apical and basal media was collected from both apical and basal chamber of transwell containing i) RPE monolayer, ii) MSC monolayer, iii) EC-MSC co-cultures and iv) RPE-CC co-cultures and either processed directly or concentrated using Amicon Ultra-0.5 mL Centrifugal Filters (Millipore Sigma) with 3KDa cut-off following manufacturer’s instructions. Next media samples were processed following manufacturer’s instruction for ELISA analyses. ELISA data was reported as pg/ relative to total protein as measured by SYPRO Ruby Protein Blot Stain (ThermoFisher Scientific) on Western blots that utilized media samples used for ELISA assays.
Drusen analysis in RPE cultures
Control and SFD RPE cultures were grown in transwells (6.5 mm, 0.4 μm Pore, Costar, Corning) for 14 days. Next, in accordance with previous publication (Galloway et al., 2017), RPE was removed by using 0.05% Trypsin and the underlying transwell membrane was fixed in 4% paraformaldehyde for 30 min at room temperature. Subsequently, transwell membrane was blocked and permeabilized in10% normal donkey serum (ImmunoReagents Inc., Raleigh, NC) and 0.1% Triton-X-100 in 1X PBS (PBS-TX) for 1 hour at room temperature. Next, the transwell membrane was incubated in primary antibody for specific drusen-resident proteins, APOE (1:500, Millipore Sigma) and TIMP3 (1:100, Millipore Sigma) in 0.5X blocking buffer for overnight at 4°C. The next day, transwell membranes were washed twice in PBS-TX buffer and incubated in host-specific secondary antibodies, Alexa-fluor conjugated (1:500, Life Technologies) for 1h at room temperature. Following two washes in PBS-TX, the transwell membranes were incubated in Hoechst (Thermo Fisher, MA) for 15 minutes to stain nuclei, coverslipped and mounted on slides using Prolong™ gold (Life Technologies). The slides were subsequently imaged and analyzed using confocal microscope (LSM 510 META, Zeiss, Thornwood, NY) equipped with Zen 2009 software (Zeiss).
Transmission Electron Microscopy
PEG hydrogels were fixed with 4% paraformaldehyde/2.5 % glutaraldehyde in 0.1 M sodium cacodylate buffer for 24 hours prior to processing. Processing and imaging of the samples was carried out in the URMC Electron Microscope Shared Resource Laboratory. Briefly, hydrogels were post fixed in buffered 1% OsO4 /1.5% potassium ferrocyanide for 60 minutes at room temperature. Following post-fixation, dehydration utilized a graded series ethanol from 50% to 100% in 20 minutes steps prior to transition to propylene oxide and infiltration with EPON Araldite resin (Electron Microscopy Sciences (EMS), (Hatfield, PA). EPON Araldite resin infiltrated specimen were polymerized 48 hrs at 60ºC. Thin sections were cut at ~70 nm and placed onto formvar carbon coated nickel slot grids (EMS), sections were stained with 2% uranyl acetate and 0.3% lead citrate prior to imaging using a Hitachi 7650 TEM (Hitachi, Tokyo, Japan) and 11 MP Erlangshen ES1000W CCD camera (Gatan, Pleasanton, CA).
Quantitative real-time PCR analysis
RNA was extracted from ECs and MSCs using RNAeasy micro kit (Qiagen) in accordance with the manufacturer’s instructions. After 30 minutes of DNase I treatment, cDNA was prepared using iScript reverse transcriptase kit (Bio-Rad) and quantitative-real time PCR was performed with universal SYBR Green (Bio-Rad) and gene-specific primers pairs (Table S5, STAR resource Table) on CFX real-time PCR detection system (Bio-Rad). GAPDH served as a loading control and gene expression was normalized to the expression of GAPDH in all experiments. Quantitative PCR data were analyzed using the Bio-Rad CFX Manager 3.1 software and Microsoft Excel.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| CD31 | R&D | #AF806; RRID:AB_355617 |
| ZO1 | ThermoFisher | #33-9100; RRID:AB_2533147 |
| MITF | Santa Cruz Biotechnology | #sc-10999; RRID: AB_2142081 |
| Ezrin | Cell Signaling technology | #3145, RRID:AB_2100309 |
| VWF | Abcam | #ab11713; RRID:AB_298501 |
| PLVAP | Sigma | #HPA002279; RRID:AB_1079636 |
| APOE | Millipore, Sigma | #AB947; RRID:AB_2258475 |
| TIMP3 | Millipore, Sigma | #MAB3318; RRID:AB_94813 |
| COLVI | Abcam | #ab6588, RRID:AB_305585 |
| alpha-Smooth Muscle Actin | Abcam | #ab7187, RRID:AB_262054 |
| Claudin-5 | Thermo | # 34-1600, RRID:AB_2533157 |
| VE-Cadherin | Santa Cruz Biotechnology | #sc-9989, RRID:AB_2077957 |
| COLIV | Abcam | #ab19808, RRID:AB_445160 |
| Laminin | Abcam | # ab11575, RRID:AB_298179 |
| Alexa fluor 488 | ThermoFisher | #A-11015; RRID:AB_2534082 |
| Alexa fluor 546 | ThermoFisher | # A10040, RRID:AB_2534016 |
| Alexa fluor 633 | ThermoFisher | #A-21052; RRID:AB_2535719 |
| Secondary HRP | Abcam | #ab97125; RRID:AB_10680688 |
| Anti-FGF2 antibody | Millipore, Sigma | #05-117; RRID:AB_309632 |
| Anti-VEGF2 antibody | R&D system | #MAB293; RRID:AB_358222 |
| Anti-TGF-b antibody | R&D system | # MAB1835; RRID:AB_357931 |
|
| ||
| Episomal vectors | ||
|
| ||
| pCXLE-hOCT4-shP53 | Addgene | #27077 |
| pCXLE-hSK | Addgene | #27078 |
| pCXLE-hUL | Addgene | #27080 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| mTeSR1 medium | STEMCELL Technologies | #85850 |
| DMEM:F12 | GIBCO | #11765054 |
| DMEM | GIBCO | #11965118 |
| Matrigel | Corning | #354230 |
| E8 media | ThermoFisher | A1517001 |
| Non-essential amino acids | GIBCO | #11140050 |
| E6 media | ThermoFisher | #A1516401 |
| KOSR | GIBCO | #10828028 |
| Antibiotic-Antimycotic | GIBCO | #15240-062 |
| Sodium pyruvate | GIBCO | #11360070 |
| 2-Mercaptoenthanol | GIBCO | #21985023 |
| Collagenase IV | GIBCO | #17104019 |
| Collagen 1 | GIBCO | #A10483-01 |
| Collagen type I | GIBCO | #A1064401 |
| Glutamax | GIBCO | #35050061 |
| Laminin-521 | ThermoFisher | #23017015 |
| Phosphate Buffered Saline | Fisher Scientific | #BP39920 |
| b-FGF | Peprotech | #100-18B |
| BMP4 | Miltenyi | 130-111-164 |
| Activin | Miltenyi | 130-115-008 |
| human PDGF-BB | Peprotech | #100-14B |
| StemMACS SB431542 | Miltenyi | #130-105-336 |
| VEGF165 | Peprotech | #100-20 |
| ES cult Methylcellulose | STEMCELL Technologies | # M3120 |
| BIT 9500 supplement | STEMCELL Technologies | #09500 |
| Stemspan SFEM | STEMCELL Technologies | #09650 |
| 1-Thioglycerol | Sigma | #6145 |
| L-Ascorbic acid | Sigma | #A8960 |
| EX-CYTE supplement | Millipore | #811291 |
| Heat Inactivated FBS | GIBCO | #10437-028 |
| B-27 Supplement | GIBCO | #12587010B |
| N-2 Supplement | GIBCO | #17502001 |
| Heparin | Sigma Aldrich | #H-3149 |
| Trypsin 0.05% | GIBCO | #25300054 |
| Vasculife media | Lifeline Cell Technology | #LL-0002 |
| Doxycycline hyclate | Tocris | #4090 |
| ROCK inhibitor | STEMCELL Technologies | #Y-27632 |
| N,N-Dimethylformamide | Fisher Scientific | #D119-500 |
| Trifluoroacetic acid | Fisher Scientific, Alfa Aesar | #AA3177122 |
| Ethyl cyano(hydroxyimino)acetate | Oxyma Pure, Sigma-Aldrich | #233412 |
| Piperazine | Fisher Scientific, Alfa Aesar | #AAA1504922 |
| Triisopropylsilane | Fisher Scientific, Alfa Aesar | #AAL0958506 |
| SDL/MMP linker | Genscript | NA |
| Alamar Blue | Bio-Rad | #BUF012A |
| Crystal violet | Sigma | #3886 |
| AQUA-Polymount media | Polysciences | #18606-20 |
| Polyvinyl alcohol | Sigma | #341584 |
| SuperFrost Plus slides | Fisher Scientific | #12-550-15 |
| Hoechst 33342 | Thermo Fisher | #H3570 |
| ProLong gold | Thermo Fisher | #P36930 |
| Odyssey Blocking Buffer | LI-COR | #927-40100 |
| Tween-20 | Fisher Bioreagents | #BP337-100 |
| RIPA buffer | ThermoFisher | #89901 |
| Formaldehyde | Polysciences | #18814-10 |
| Tissue freezing medium | General Data | #TFM-5 |
| Triton-X | Sigma | #T9284 |
| Vacutainer tubes | BD | #367815 |
| 8-arm PEG-NB | Chemically synthesized | NA |
| RGD | Chemically synthesized | NA |
| Depression slides | Fisher Scientific | 12-560B |
| 7.5% Mini-PROTEAN® Precast Protein Gels | Biorad | #4561024 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Fibroblasts reprogramming kit | Lonza | #VPD-1001 |
| CD31+ Microbead kit | Miltenyi | #130-091-935 |
| RNeasy mini kit | QIAGEN | #74104 |
| iScript RT kit | Biorad | #1708840 |
| SYBR green mix | Biorad | #1725271 |
| TGF-beta 1 Quantikine ELISA | R&D | # DB100B |
| Human VEGF Quantikine ELISA Kit | R&D | #DVE00 |
| QIAamp DNA Blood Mini Kit | QIAGEN | #51104 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| Normal serum | Peripheral blood | n = 12 |
| Wet AMD serum | Peripheral blood | n = 8 |
| Control iPSC lines | Dermal fibroblasts | n = 3 |
| SFD iPSC lines | Dermal fibroblasts | n = 2 |
| Irradiated MEFs | ThermoFisher | #A34181 |
| OP9 stromal cells | ATCC | #CRL-2749 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Real-time PCR primers, see Table S5 | This paper | N/A |
| Genotyping primers, see Table S5 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Imaris | Bitplane, Oxford instruments | https://imaris.oxinst.com/ |
| Zeiss Zen (Black edition) | Carl Zeiss Microscopy GmbH | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| AngioQuant image analysis tool | MATLAB | The MathWorks |
| Wimtube | Onimagin Technologies | https://www.wimasis.com/en/WimTube |
| ImageJ (Fiji) | NIH | https://imagej.nih.gov/ij/docs/guide/146-2.html |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/p/excel/cfq7ttc0k7dx?activetab=pivot%3aoverviewtab |
| FluoView FV1000 | Olympus | https://www.olympus-lifescience.com/en/laser-scanning/ |
| ImageStudio | LI-COR | https://www.licor.com/bio/image-studio-lite/ |
| Infinity Analyze and Capture | Lumenera | https://www.lumenera.com/infinity-analyze-and-capture-for-windows.html |
| CorelDraw | Corel | https://www.coreldraw.com/en/ |
| Snapgene | GraphPad Software | https://www.snapgene.com/ |
|
| ||
| Others | ||
|
| ||
| Nucleofector 2b Device | Lonza | #AAB-1001 |
| 6.5 mm, 3.0 mm Pore, Membrane Insert | Corning | #3472 |
| 6.5 mm, 0.4 mm Pore, Membrane Insert | Corning | #3470 |
Genotyping of ARMS2, CFH, and HTRA1 genes
DNA was isolated from human peripheral blood samples collected from normal individuals and Wet AMD patients using the QIAamp DNA Blood Mini Kit (Qiagen) by following the manufacturer’s instructions. The OD260/280 of extracted genomic DNA was determined using Nanodrop (ThermoFisher). The purified DNA was then used as the template for amplification of ARMS2, CFH and HTRA1 gene using Emerald Mastermix (Takara), following the manufacturer’s instructions. Briefly, 100 ng of isolated DNA was added to PCR reaction mixture containing primer pair mix listed in Table S5 and STAR resource Table. PCR amplification for all the samples was carried out for 35 cycles; 98°C for 10 seconds for initial denaturation, annealing at 58°C for 30 seconds, 72°C for 1 minute for an extension (Thermocycler; Bio-Rad). Subsequently, PCR products were analyzed using agarose gel electrophoresis (2% agarose; ethidium bromide) on an Azure gel documentation system (Azure Inc) and submitted for processing by Sanger sequencing (Genewiz, South Plainfield, NJ). DNA sequence results were subsequently analyzed using SnapGene software (San Diego, CA).
Western blotting
Cells were lysed in RIPA buffer (Thermo Fisher Scientific, MA) containing protease inhibitor (Sigma-Aldrich, St. Louis, MO). 30 μg of protein lysates were resolved on 7.5% gels by SDS PAGE. Following SDS-PAGE, the proteins were transferred on to PVDF membrane and the membranes were blocked for one hour at room temperature in either 5% non-fat dry milk (NFDM) in PBS or commercially bought blocking solution (LI-COR Biosciences, NE). The membrane was then incubated overnight with sheep anti-CD31 antibody (1:200, R&D Minneapolis, MN) in blocking buffer at 4°C. Following washes with PBS-T (0.1% Tween-20), the membranes were incubated with HRP-conjugated rabbit anti-sheep secondary antibody (1:2000, Abcam, MA) at room temperature for 1 hour. The membrane was subsequently washed thrice in 1X PBS-T and analyzed using Azure™ C-series chemiluminescence documentation system (Azure Biosystems, CA). For quantification, ImageStudio (LI-COR Biosciences, NE) and Microsoft Excel software were utilized.
Experimental Set-up and Statistical Analyses
Each individual experiment was carried out at least in triplicates and in experiments comparing control versus SFD samples (RPE, ECs, RPE-CM, RPE-EC-MSC hydrogels), parallel age-matched cultures of control and SFD RPE and EC were utilized. In addition, three distinct control and two SFD lines including two different clones of each SFD line were utilized for experimental analysis. In experiments evaluating impact of serum composition, sera from eleven different control and eight distinct wAMD patients (equal representation of male and female in control versus wAMD group) were utilized. Of note, data throughout the manuscript is presented as mean ± SEM. Furthermore, even though the data in each individual experiment was carried on parallel cultures of patient vs. control and/or treated vs. untreated samples, given that our analysis was not comparing before treatment and after treatment consequences on the same sample, statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001 was determined using unpaired student’s t-test in Microsoft Excel.
Supplementary Material
Video constructed from z-stacks confocal images of the RPE-CC hydrogel-derived tissue mimetic at day 30 showing the presence of PLVAP+ (red) vascular networks beneath the RPE monolayer marked with ZO1(white). Nuclei were counterstained with DAPI (Blue).
Highlights.
First patient-derived 3D retinal pigment epithelium-choriocapillaris (RPE-CC) model
Captures hallmarks of RPE-CC morphogenesis, age-related macular degeneration (AMD)
Insight into cellular, soluble, systemic factors that initiate AMD pathology
Pharmacological targeting of AMD pathology in physiological iPSC model
Acknowledgments
This work was supported by grants from both private foundations and National Institutes of Health. Specifically private foundation grants that contributed to this work include BrightFocus Foundation Macular Degeneration Grant (to R.S.), David Bryant Trust (to R.S.), Foundation of Fighting Blindness Individual Investigator Award (to R.S), Knights Templar eye foundation grant (to R.S), Retina Research Foundation and Research to Prevent Blindness, RPB’s Career Development Award (to R.S.) and Unrestricted Challenge Grant to Department of Ophthalmology at the University of Rochester and the Department of Ophthalmic Research at Cleveland Clinic. National Institutes of Health grants that supported this work include R01EY028167 (to R.S), R01EY030183 (to R.S), R21EY027750 (to D.S.W.B. and R.S.), 5T32ES007026-43 (to JM), R01NS109427 (to W.L.M.) P30EY025585 (to Cleveland Clinic Foundation), EY027083, EY026181, P30EY025585, Cleveland and University of Rochester Department of Environmental Medicine Training Grant T32ES007026 (to J.A.M.).
Footnotes
Declaration of Interests
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ADHI M, FERRARA D, MULLINS RF, BAUMAL CR, MOHLER KJ, KRAUS MF, LIU J, BADARO E, ALASIL T, HORNEGGER J, FUJIMOTO JG, DUKER JS & WAHEED NK 2015. Characterization of Choroidal Layers in Normal Aging Eyes Using Enface Swept-Source Optical Coherence Tomography. PLoS One, 10, e0133080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZIZ AH, WAHLQUIST J, SOLLNER A, FERGUSON V, DELRIO FW & BRYANT SJ 2017. Mechanical characterization of sequentially layered photo-clickable thiol-ene hydrogels. J Mech Behav Biomed Mater, 65, 454–465. [DOI] [PubMed] [Google Scholar]
- BABA T, GREBE R, HASEGAWA T, BHUTTO I, MERGES C, MCLEOD DS & LUTTY GA 2009. Maturation of the fetal human choriocapillaris. Invest Ophthalmol Vis Sci, 50, 3503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENEDICTO I, LEHMANN GL, GINSBERG M, NOLAN DJ, BAREJA R, LEMENTO O, SALFATI Z, ALAM NM, PRUSKY GT, LLANOS P, RABBANY SY, AMINISHKIS A, MILLER SS, RAFII S. & RODRIGUEZ-BOULAN E. 2017. Concerted regulation of retinal pigment epithelium basement membrane and barrier function by angiocrine factors. Nat Commun, 8, 15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHATIA SN & INGBER DE 2014. Microfluidic organs-on-chips. Nat Biotechnol, 32, 760–72. [DOI] [PubMed] [Google Scholar]
- BHISE NS, RIBAS J, MANOHARAN V, ZHANG YS, POLINI A, MASSA S, DOKMECI MR & KHADEMHOSSEINI A. 2014. Organ-on-a-chip platforms for studying drug delivery systems. J Control Release, 190, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHUTTO I. & LUTTY G. 2012. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med, 33, 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHUTTO IA, MCLEOD DS, HASEGAWA T, KIM SY, MERGES C, TONG P. & LUTTY GA 2006. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res, 82, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIESEMEIER A, TAUBITZ T, JULIEN S, YOERUEK E. & SCHRAERMEYER U. 2014. Choriocapillaris breakdown precedes retinal degeneration in age-related macular degeneration. Neurobiol Aging, 35, 2562–2573. [DOI] [PubMed] [Google Scholar]
- BLAAUWGEERS HG, HOLTKAMP GM, RUTTEN H, WITMER AN, KOOLWIJK P, PARTANEN TA, ALITALO K, KROON ME, KIJLSTRA A, VAN HINSBERGH VW & SCHLINGEMANN RO 1999. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol, 155, 421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUGNO M, WITEK B, BERETA J, BERETA M, EDWARDS DR & KORDULA T. 1999. Reprogramming of TIMP-1 and TIMP-3 expression profiles in brain microvascular endothelial cells and astrocytes in response to proinflammatory cytokines. FEBS Lett, 448, 9–14. [DOI] [PubMed] [Google Scholar]
- CAMPOCHIARO PA, SOLOWAY P, RYAN SJ & MILLER JW 1999. The pathogenesis of choroidal neovascularization in patients with age-related macular degeneration. Mol Vis, 5, 34. [PubMed] [Google Scholar]
- CHEN LJ, ITO S, KAI H, NAGAMINE K, NAGAI N, NISHIZAWA M, ABE T. & KAJI H. 2017. Microfluidic co-cultures of retinal pigment epithelial cells and vascular endothelial cells to investigate choroidal angiogenesis. Sci Rep, 7, 3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEUNG CM & WONG TY 2014. Is age-related macular degeneration a manifestation of systemic disease? New prospects for early intervention and treatment. J Intern Med, 276, 140–53. [DOI] [PubMed] [Google Scholar]
- CHWALEK K, TSURKAN MV, FREUDENBERG U. & WERNER C. 2014. Glycosaminoglycan-based hydrogels to modulate heterocellular communication in in vitro angiogenesis models. Sci Rep, 4, 4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRUZ-ACUNA R, QUIROS M, HUANG S, SIUDA D, SPENCE JR, NUSRAT A. & GARCIA AJ 2018. PEG-4MAL hydrogels for human organoid generation, culture, and in vivo delivery. Nat Protoc, 13, 2102–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DARDIK R, LIVNAT T, NISGAV Y. & WEINBERGER D. 2010. Enhancement of angiogenic potential of endothelial cells by contact with retinal pigment epithelial cells in a model simulating pathological conditions. Invest Ophthalmol Vis Sci, 51, 6188–95. [DOI] [PubMed] [Google Scholar]
- DICKSON KM, BERGERON JJ, PHILIP A, O’CONNOR-MCCOURT M. & WARSHAWSKY H. 2001. Localization of specific binding sites for 125I-TGF-beta1 to fenestrated endothelium in bone and anastomosing capillary networks in enamel organ suggests a role for TGF-beta1 in angiogenesis. Calcif Tissue Int, 68, 304–15. [DOI] [PubMed] [Google Scholar]
- ESCH EW, BAHINSKI A. & HUH D. 2015. Organs-on-chips at the frontiers of drug discovery. Nat Rev Drug Discov, 14, 248–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAIRBANKS BD, SCHWARTZ MP, HALEVI AE, NUTTELMAN CR, BOWMAN CN & ANSETH KS 2009. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Adv Mater, 21, 5005–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARNOODIAN M, WANG S, DIETZ J, NICKELLS RW, SORENSON CM & SHEIBANI N. 2017. Negative regulators of angiogenesis: important targets for treatment of exudative AMD. Clin Sci (Lond), 131, 1763–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLETCHER EL, JOBLING AI, GREFERATH U, MILLS SA, WAUGH M, HO T, DE IONGH RU, PHIPPS JA & VESSEY KA 2014. Studying age-related macular degeneration using animal models. Optom Vis Sci, 91, 878–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOREST DL, JOHNSON LV & CLEGG DO 2015. Cellular models and therapies for age-related macular degeneration. Dis Model Mech, 8, 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAGE PJ, RHOADES W, PRUCKA SK & HJALT T. 2005. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci, 46, 4200–8. [DOI] [PubMed] [Google Scholar]
- GALLOWAY CA, DALVI S, HUNG SSC, MACDONALD LA, LATCHNEY LR, WONG RCB, GUYMER RH, MACKEY DA, WILLIAMS DS, CHUNG MM, GAMM DM, PEBAY A, HEWITT AW & SINGH R. 2017. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc Natl Acad Sci U S A, 114, E8214–E8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEISEN P, MCCOLM JR & HARTNETT ME 2006. Choroidal endothelial cells transmigrate across the retinal pigment epithelium but do not proliferate in response to soluble vascular endothelial growth factor. Exp Eye Res, 82, 608–19. [DOI] [PubMed] [Google Scholar]
- GOURIER HC & CHONG NV 2015. Can Novel Treatment of Age-Related Macular Degeneration Be Developed by Better Understanding of Sorsby’s Fundus Dystrophy. J Clin Med, 4, 874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROSSNIKLAUS HE & GREEN WR 2004. Choroidal neovascularization. Am J Ophthalmol, 137, 496–503. [DOI] [PubMed] [Google Scholar]
- HAGEMAN GS, LUTHERT PJ, VICTOR CHONG NH, JOHNSON LV, ANDERSON DH & MULLINS RF 2001. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res, 20, 705–32. [DOI] [PubMed] [Google Scholar]
- HAMILTON RD, FOSS AJ & LEACH L. 2007. Establishment of a human in vitro model of the outer blood-retinal barrier. J Anat, 211, 707–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFMAN MD, XIE C, ZHANG X. & BENOIT DS 2013. The effect of mesenchymal stem cells delivered via hydrogel-based tissue engineered periosteum on bone allograft healing. Biomaterials, 34, 8887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNT NC, HALLAM D, KARIMI A, MELLOUGH CB, CHEN J, STEEL DHW & LAKO M. 2017. 3D culture of human pluripotent stem cells in RGD-alginate hydrogel improves retinal tissue development. Acta Biomater, 49, 329–343. [DOI] [PubMed] [Google Scholar]
- JUSTUS CR, LEFFLER N, RUIZ-ECHEVARRIA M. & YANG LV 2014. In vitro cell migration and invasion assays. J Vis Exp. [DOI] [PMC free article] [PubMed]
- KHOO CP, MICKLEM K. & WATT SM 2011. A comparison of methods for quantifying angiogenesis in the Matrigel assay in vitro. Tissue Eng Part C Methods, 17, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM SA, KIM SJ, CHOI YA, YOON HJ, KIM A. & LEE J. 2020. Retinal VEGFA maintains the ultrastructure and function of choriocapillaris by preserving the endothelial PLVAP. Biochem Biophys Res Commun, 522, 240–246. [DOI] [PubMed] [Google Scholar]
- KOMEZ A, BARAN ET, ERDEM U, HASIRCI N. & HASIRCI V. 2016. Construction of a patterned hydrogel-fibrous mat bilayer structure to mimic choroid and Bruch’s membrane layers of retina. J Biomed Mater Res A, 104, 2166–77. [DOI] [PubMed] [Google Scholar]
- KORTE GE, REPPUCCI V. & HENKIND P. 1984. RPE destruction causes choriocapillary atrophy. Invest Ophthalmol Vis Sci, 25, 1135–45. [PubMed] [Google Scholar]
- KRAEHENBUEHL TP, FERREIRA LS, ZAMMARETTI P, HUBBELL JA & LANGER R. 2009. Cell-responsive hydrogel for encapsulation of vascular cells. Biomaterials, 30, 4318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRAEHENBUEHL TP, ZAMMARETTI P, VAN DER VLIES AJ, SCHOENMAKERS RG, LUTOLF MP, JACONI ME & HUBBELL JA 2008. Three-dimensional extracellular matrix-directed cardioprogenitor differentiation: systematic modulation of a synthetic cell-responsive PEG-hydrogel. Biomaterials, 29, 2757–66. [DOI] [PubMed] [Google Scholar]
- KUMAR R, HARRIS-HOOKER S, KUMAR R. & SANFORD G. 2011. Co-culture of Retinal and Endothelial Cells Results in the Modulation of Genes Critical to Retinal Neovascularization. Vasc Cell, 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURIHARA T, WESTENSKOW PD, GANTNER ML, USUI Y, SCHULTZ A, BRAVO S, AGUILAR E, WITTGROVE C, FRIEDLANDER M, PARIS LP, CHEW E, SIUZDAK G. & FRIEDLANDER M. 2016. Hypoxia-induced metabolic stress in retinal pigment epithelial cells is sufficient to induce photoreceptor degeneration. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KVANTA A, SHEN WY, SARMAN S, SEREGARD S, STEEN B. & RAKOCZY E. 2000. Matrix metalloproteinase (MMP) expression in experimental choroidal neovascularization. Curr Eye Res, 21, 684–90. [PubMed] [Google Scholar]
- LAMBERT V, MUNAUT C, JOST M, NOEL A, WERB Z, FOIDART JM & RAKIC JM 2002. Matrix metalloproteinase-9 contributes to choroidal neovascularization. Am J Pathol, 161, 1247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMBERT V, WIELOCKX B, MUNAUT C, GALOPIN C, JOST M, ITOH T, WERB Z, BAKER A, LIBERT C, KRELL HW, FOIDART JM, NOEL A. & RAKIC JM 2003. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J, 17, 2290–2. [DOI] [PubMed] [Google Scholar]
- LU H, WANG F, MEI H, WANG S. & CHENG L. 2018. Human Adipose Mesenchymal Stem Cells Show More Efficient Angiogenesis Promotion on Endothelial Colony-Forming Cells than Umbilical Cord and Endometrium. Stem Cells Int, 2018, 7537589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUTOLF MP, LAUER-FIELDS JL, SCHMOEKEL HG, METTERS AT, WEBER FE, FIELDS GB & HUBBELL JA 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A, 100, 5413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUTTY GA, HASEGAWA T, BABA T, GREBE R, BHUTTO I. & MCLEOD DS 2010. Development of the human choriocapillaris. Eye (Lond), 24, 408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARNEROS AG, FAN J, YOKOYAMA Y, GERBER HP, FERRARA N, CROUCH RK & OLSEN BR 2005. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol, 167, 1451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLEOD DS, GREBE R, BHUTTO I, MERGES C, BABA T. & LUTTY GA 2009. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci, 50, 4982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEINHARDT A, EBERLE D, TAZAKI A, RANGA A, NIESCHE M, WILSCH-BRAUNINGER M, STEC A, SCHACKERT G, LUTOLF M. & TANAKA EM 2014. 3D reconstitution of the patterned neural tube from embryonic stem cells. Stem Cell Reports, 3, 987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOON JJ, SAIK JE, POCHE RA, LESLIE-BARBICK JE, LEE SH, SMITH AA, DICKINSON ME & WEST JL 2010. Biomimetic hydrogels with pro-angiogenic properties. Biomaterials, 31, 3840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLINS RF, JOHNSON MN, FAIDLEY EA, SKEIE JM & HUANG J. 2011. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest Ophthalmol Vis Sci, 52, 1606–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOWAK JZ 2006. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep, 58, 353–63. [PubMed] [Google Scholar]
- OKITA K, MATSUMURA Y, SATO Y, OKADA A, MORIZANE A, OKAMOTO S, HONG H, NAKAGAWA M, TANABE K, TEZUKA K, SHIBATA T, KUNISADA T, TAKAHASHI M, TAKAHASHI J, SAJI H. & YAMANAKA S. 2011. A more efficient method to generate integration-free human iPS cells. Nat Methods, 8, 409–12. [DOI] [PubMed] [Google Scholar]
- PARK DY, LEE J, KIM J, KIM K, HONG S, HAN S, KUBOTA Y, AUGUSTIN HG, DING L, KIM JW, KIM H, HE Y, ADAMS RH & KOH GY 2017. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun, 8, 15296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERS EB, CHRISTOFOROU N, LEONG KW, TRUSKEY GA & WEST JL 2016. Poly(ethylene glycol) Hydrogel Scaffolds Containing Cell-Adhesive and Protease-Sensitive Peptides Support Microvessel Formation by Endothelial Progenitor Cells. Cell Mol Bioeng, 9, 38–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PILGRIM MG, LENGYEL I, LANZIROTTI A, NEWVILLE M, FEARN S, EMRI E, KNOWLES JC, MESSINGER JD, READ RW, GUIDRY C. & CURCIO CA 2017. Subretinal Pigment Epithelial Deposition of Drusen Components Including Hydroxyapatite in a Primary Cell Culture Model. Invest Ophthalmol Vis Sci, 58, 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QI JH, BELL B, SINGH R, BATOKI J, WOLK A, CUTLER A, PRAYSON N, ALI M, STOEHR H. & ANAND-APTE B. 2019. Sorsby Fundus Dystrophy Mutation in Tissue Inhibitor of Metalloproteinase 3 (TIMP3) promotes Choroidal Neovascularization via a Fibroblast Growth Factor-dependent Mechanism. Sci Rep, 9, 17429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANGA A, GIRGIN M, MEINHARDT A, EBERLE D, CAIAZZO M, TANAKA EM & LUTOLF MP 2016. Neural tube morphogenesis in synthetic 3D microenvironments. Proc Natl Acad Sci U S A, 113, E6831–E6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTH F, BINDEWALD A. & HOLZ FG 2004. Keypathophysiologic pathways in age-related macular disease. Graefes Arch Clin Exp Ophthalmol, 242, 710–6. [DOI] [PubMed] [Google Scholar]
- RUIZ A, BRETT P. & BOK D. 1996. TIMP-3 is expressed in the human retinal pigment epithelium. Biochem Biophys Res Commun, 226, 467–74. [DOI] [PubMed] [Google Scholar]
- SAINT-GENIEZ M. & D’AMORE PA 2004. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int J Dev Biol, 48, 1045–58. [DOI] [PubMed] [Google Scholar]
- SAINT-GENIEZ M, KURIHARA T, SEKIYAMA E, MALDONADO AE & D’AMORE PA 2009. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A, 106, 18751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMTANI S, AMARAL J, CAMPOS MM, FARISS RN & BECERRA SP 2009. Doxycycline-mediated inhibition of choroidal neovascularization. Invest Ophthalmol Vis Sci, 50, 5098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWARTZ MP, HOU Z, PROPSON NE, ZHANG J, ENGSTROM CJ, SANTOS COSTA V, JIANG P, NGUYEN BK, BOLIN JM, DALY W, WANG Y, STEWART R, PAGE CD, MURPHY WL & THOMSON JA 2015. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc Natl Acad Sci U S A, 112, 12516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELIKTAR D, ZISCH AH, LUTOLF MP, WRANA JL & HUBBELL JA 2004. MMP-2 sensitive, VEGF-bearing bioactive hydrogels for promotion of vascular healing. J Biomed Mater Res A, 68, 704–16. [DOI] [PubMed] [Google Scholar]
- SELLHEYER K. 1990. Development of the choroid and related structures. Eye (Lond), 4 ( Pt 2), 255–61. [DOI] [PubMed] [Google Scholar]
- SHOKOOHMAND A, JEON JE, THEODOROPOULOS C, BALDWIN JG, HUTMACHER DW & FEIGL B. 2017. A Novel 3D Cultured Model for Studying Early Changes in Age-Related Macular Degeneration. Macromol Biosci, 17. [DOI] [PubMed] [Google Scholar]
- SHUBIN AD, FELONG TJ, GRAUNKE D, OVITT CE & BENOIT DS 2015. Development of poly(ethylene glycol) hydrogels for salivary gland tissue engineering applications. Tissue Eng Part A, 21, 1733–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH R, PHILLIPS MJ, KUAI D, MEYER J, MARTIN JM, SMITH MA, PEREZ ET, SHEN W, WALLACE KA, CAPOWSKI EE, WRIGHT LS & GAMM DM 2013a. Functional analysis of serially expanded human iPS cell-derived RPE cultures. Invest Ophthalmol Vis Sci, 54, 6767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINGH R, SHEN W, KUAI D, MARTIN JM, GUO X, SMITH MA, PEREZ ET, PHILLIPS MJ, SIMONETT JM, WALLACE KA, VERHOEVEN AD, CAPOWSKI EE, ZHANG X, YIN Y, HALBACH PJ, FISHMAN GA, WRIGHT LS, PATTNAIK BR & GAMM DM 2013b. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet, 22, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPENCER C, ABEND S, MCHUGH KJ & SAINT-GENIEZ M. 2017. Identification of a synergistic interaction between endothelial cells and retinal pigment epithelium. J Cell Mol Med, 21, 2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STETLER-STEVENSON WG 2001. The role of matrix metalloproteinases in tumor invasion, metastasis, and angiogenesis. Surg Oncol Clin N Am, 10, 383–92, x. [PubMed] [Google Scholar]
- STRATMAN AN & DAVIS GE 2012. Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. Microsc Microanal, 18, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAUSS O. 2005. The retinal pigment epithelium in visual function. Physiol Rev, 85, 845–81. [DOI] [PubMed] [Google Scholar]
- TOSI GM, ORLANDINI M. & GALVAGNI F. 2018. The Controversial Role of TGF-beta in Neovascular Age-Related Macular Degeneration Pathogenesis. Int J Mol Sci, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN HOVE AH, BELTEJAR MJ & BENOIT DS 2014. Development and in vitro assessment of enzymatically-responsive poly(ethylene glycol) hydrogels for the delivery of therapeutic peptides. Biomaterials, 35, 9719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VODYANIK MA, YU J, ZHANG X, TIAN S, STEWART R, THOMSON JA & SLUKVIN II 2010. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell, 7, 718–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, GEISEN P, WITTCHEN ES, KING B, BURRIDGE K, D’AMORE PA & HARTNETT ME 2011. The role of RPE cell-associated VEGF(1)(8)(9) in choroidal endothelial cell transmigration across the RPE. Invest Ophthalmol Vis Sci, 52, 570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X, MA W, HAN S, MENG Z, ZHAO L, YIN Y, WANG Y. & LI J. 2017. TGF-beta participates choroid neovascularization through Smad2/3-VEGF/TNF-alpha signaling in mice with Laser-induced wet age-related macular degeneration. Sci Rep, 7, 9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEBER BH, VOGT G, PRUETT RC, STOHR H. & FELBOR U. 1994. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet, 8, 352–6. [DOI] [PubMed] [Google Scholar]
- WONG WL, SU X, LI X, CHEUNG CM, KLEIN R, CHENG CY & WONG TY 2014. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health, 2, e106–16. [DOI] [PubMed] [Google Scholar]
- ZANOTELLI MR, ARDALANI H, ZHANG J, HOU Z, NGUYEN EH, SWANSON S, NGUYEN BK, BOLIN J, ELWELL A, BISCHEL LL, XIE AW, STEWART R, BEEBE DJ, THOMSON JA, SCHWARTZ MP & MURPHY WL 2016. Stable engineered vascular networks from human induced pluripotent stem cell-derived endothelial cells cultured in synthetic hydrogels. Acta Biomater, 35, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video constructed from z-stacks confocal images of the RPE-CC hydrogel-derived tissue mimetic at day 30 showing the presence of PLVAP+ (red) vascular networks beneath the RPE monolayer marked with ZO1(white). Nuclei were counterstained with DAPI (Blue).
Data Availability Statement
This study did not generate/analyze datasets/code.