

Abstract
Background
AVT02 (adalimumab) is a proposed biosimilar to Humira®. AVT02 is produced at a 100 mg/mL concentration with a citrate-free formulation.
Objectives
The aim of this study was to compare the efficacy, safety and immunogenicity of AVT02 versus Humira® in subjects with moderate to severe chronic plaque psoriasis.
Methods
This double-blind, randomised, parallel group, active control study of adult subjects compared (at a 1:1 ratio) AVT02 with originator adalimumab 80 mg subcutaneously in Week 1, then 40 mg every other week. At Week 16, subjects who had received originator adalimumab were re-randomised at a 1:1 ratio to continue receiving originator adalimumab, or to switch to AVT02, every other week until Week 48, with final efficacy endpoint at Week 50. Subjects who initially received AVT02 continued to receive AVT02 from Week 16 to Week 48. The primary endpoint was percentage improvement in Psoriasis Area and Severity Index (PASI) score at Week 16. Secondary efficacy endpoints included percentage improvement in PASI score at additional timepoints, change from baseline in Dermatology Life Quality Index (DLQI) score and number and percentage of subjects achieving static Physician’s Global Assessment (sPGA) responses of ‘clear’ or ‘almost clear’. Additional secondary endpoints included comparison of adverse event profiles, anti-drug antibodies and neutralising antibodies, and serum trough levels of adalimumab at steady state.
Results
A total of 413 subjects were randomised (205 to AVT02 and 208 to originator). The percentage improvement in PASI score at Week 16 was 91.6% for AVT02-treated subjects and 89.6% for originator adalimumab. The 90% confidence intervals for the primary endpoint were within the pre-defined equivalence margin of ±10% (90% CI − 0.76 to 5.29; 95% CI − 1.34 to 5.88), and a comparable pattern for DLQI score (11.4-point and 10.6-point improvement in AVT02-treated and originator adalimumab-treated groups, respectively) and sPGA (90.5% in both groups achieving ‘clear’ or ‘almost clear’) at Week 16 supported the assessment. Efficacy persisted through Week 50 of the study in all treatment groups, including those who switched from originator adalimumab to AVT02, for percent improvement in PASI score, quality-of-life assessment and sPGA. The safety, tolerability and immunogenicity profiles between AVT02 and originator adalimumab were similar at Week 16, and this persisted in the switched and continued groups through Week 50.
Conclusion
Objective and subjective measures of efficacy supported the evaluation of biosimilarity between AVT02 and originator adalimumab at Week 16 and until Week 50, in switched and continued treatment groups. AVT02 was safe and well tolerated, with a safety and immunogenicity profile similar to that observed in originator adalimumab with no clinically meaningful difference between the two.
Clinical Trial Registration
EudraCT: 2017-003367-35; ClinicalTrials.gov: NCT03849404.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40259-021-00502-w.
Key Points
| The results of this clinical study supported the assessment of biosimilarity of AVT02 in terms of efficacy, safety, tolerability and immunogenicity as compared to originator adalimumab. |
| The efficacy, safety, tolerability and immunogenicity profiles were similar for subjects who were treated with only the originator, only AVT02, or who switched from originator to AVT02, up to the primary endpoint at Week 16 and through Week 50. |
| This high-concentration, citrate-free presentation of AVT02 could be a new option for patients with moderate to severe chronic plaque psoriasis currently treated with adalimumab. |
Introduction
Adalimumab was originally approved by the US Food and Drug Administration (FDA) in December 2002 and by the European Medicines Agency (EMA) in September 2003 for the treatment of rheumatoid arthritis and is currently approved in a variety of inflammatory diseases, including moderate to severe rheumatoid arthritis, moderate to severe juvenile idiopathic arthritis, psoriatic arthritis (PsA), ankylosing spondylitis, moderate to severe Crohn’s disease, moderate to severe ulcerative colitis, moderate to severe chronic plaque psoriasis (PsO), moderate to severe hidradenitis suppurativa and uveitis [1, 2]. Adalimumab (ATC code: L04AB04) is a tumour necrosis factor blocker. Adalimumab is a recombinant human immunoglobulin 1 (IgG1) monoclonal antibody created using phage display technology resulting in an antibody with human-derived heavy and light chain variable regions and human IgG1:κ constant regions [3]. The introduction of adalimumab provided a powerful new targeted treatment option for patients whose needs were not being met with standard oral systemic therapies, as well as an additional tool to manage the rising incidence of these conditions [4].
Nevertheless, the high price point of biologics can be a barrier to adoption [5]. Development of biosimilars may help reduce cost and improve access [6]. To date, six adalimumab biosimilars have been licensed by the FDA and 12 approved by the EMA, of which four have subsequently been withdrawn [7, 8]. Since 2017, a high concentration (100 mg/mL) adalimumab has been available (EMEA/H/C/000481/II/138/G and EMEA/H/C/000481/II/145/G) and is currently approved in several countries. Compared with the original 50 mg/mL formulation, this allows for half the volume to be injected for the same dose and is formulated without a citrate buffer [9]. The higher concentration formulation has been better tolerated, with less pain with each injection, and has been associated with greater adherence, fewer discontinuations and longer time on therapy than the original adalimumab formulation [10, 11].
AVT02 is being developed as a proposed biosimilar to the high concentration adalimumab, and analytical and functional data, to date, support the assessment of biosimilarity with no clinically meaningful differences observed between AVT02 and originator adalimumab. AVT02 is produced at the 100-mg/mL concentration with a citrate-free formulation.
The purpose of this study is to contribute to the totality of evidence supporting the biosimilarity assessment of the proposed biosimilar AVT02 and the originator adalimumab product. The aim is to observe comparable efficacy, safety, immunogenicity and pharmacokinetics with no evidence of clinically meaningful differences.
Materials and Methods
Study Design and Participants
This was a double-blind, randomised, parallel group, active control study conducted at 20 sites in four countries (Estonia, Georgia, Poland and Ukraine) and was registered with ClinicalTrials.gov (NCT03849404) and EudraCT (2017-003367-35). Due to the coronavirus disease 2019 (COVID-19) pandemic, some study procedures were amended per FDA and EMA guidance, to prioritise subject safety and data validity [12, 13].
The active period of the study comprised two stages. From baseline to Week 16, the study measured efficacy in AVT02 and the originator adalimumab (Stage 1). Between Week 16 and end of study at Week 52, the study measured long-term efficacy and safety, including switching from the originator product (Stage 2).
Subjects eligible for inclusion were male and female adults, aged 18–75 years inclusive, with stable, moderate to severe chronic plaque psoriasis who had involved body surface area (BSA) ≥ 10% (Palm Method), score of ≥ 12 on the Psoriasis Area and Severity Index (PASI) and static Physicians Global Assessments (sPGA) ≥ 3 (moderate). Subjects that had a previous failure, inadequate response, intolerance or contraindication to at least one non-biologic systemic anti-psoriatic therapy were eligible for inclusion. If subjects had prior use of two or more biologics or had recently used certain topical or nonbiologic systemic therapies or phototherapy for treatment of psoriasis, they were excluded from the study.
Randomisation and Masking
Randomisation was performed by an Interactive Voice/Web Response System (IXRS) and subjects were randomised in a 1:1 ratio to receive either AVT02 or originator adalimumab in accordance with the randomisation schedule generated using permuted block randomisation. The randomisation code was prepared by a statistician not otherwise involved in the conduct of the study. Subject randomisation was stratified by presence or absence of PsA, and by prior use of a biologic therapy for the treatment of PsO or PsA.
After Stage 1 of the study at Week 16, responding subjects (subjects who had achieved at least a 50% reduction in PASI [PASI 50]) who were initially randomised to receive AVT02 continued to receive AVT02 in Stage 2. Responding subjects who were initially randomised to receive originator adalimumab were re-randomised in a 1:1 ratio to receive either AVT02 or originator adalimumab in Stage 2.
Subjects, investigators, site staff and the sponsor study team, inclusive of contract research organisation personnel, were unaware of treatment assignment until study closure and database lock. The pre-filled syringes (PFS) containing either AVT02 or originator adalimumab were masked by packaging. The double blind was maintained by each syringe being surrounded by a masking device. Subjects returned used syringes including the masking device and, while not all masking devices could be confirmed intact, no reports were received to the contrary.
Procedures
Subjects received an initial loading dose of either AVT02 or originator adalimumab 80 mg (2 × 40 mg) administered subcutaneously (SC), followed by 40 mg SC once every other week (EOW) starting 1 week after the loading dose, and continued to receive treatment until Week 14. At the end of Stage 1 at Week 16, those subjects still on study who had been receiving AVT02 continued to receive AVT02 40 mg SC EOW until Week 48. Those subjects still on study who had been receiving originator adalimumab were re-randomised for Stage 2 and received either AVT02 or originator adalimumab (40 mg EOW) until Week 48.
Study drug compliance was assessed based on study drug usage recorded by the subject or caregiver in the eDiary.
PASI was assessed at baseline and at Weeks 4, 8, 12, 16, 24, 32, 42 and 50. PASI 50, PASI 75, PASI 90 and PASI 100 are binary outcomes that indicate a 50%, 75%, 90% or greater, or 100% improvement, respectively, in PASI from baseline.
The sPGA of plaque psoriasis was assessed on a scale of 0–5. sPGA response was assessed at baseline and at Weeks 4, 8, 12, 16, 24, 32, 42 and 50. The percent of BSA (%BSA) affected was estimated by assuming that the subject’s hand represents roughly 1% of the body’s surface. The total %BSA was estimated as the number of hands necessary to cover the total affected area. Because of interobserver variability in estimated BSA, whenever possible, all assessments for a given subject were to be made by the same observer. Evaluation of the percentage of BSA affected by psoriasis was assessed at baseline and at Weeks 4, 8, 12, 16, 24, 32, 42 and 50.
The Dermatology Life Quality Index (DLQI) is a 10-question validated questionnaire with a maximum score of 30 and a minimum of 0. The higher the score, the more quality of life is impaired [14]. The change from baseline in quality of life as measured by DLQI scores was assessed at baseline and at Weeks 16, 24 and 50.
The Routine Assessment of Patient Index Data 3 (RAPID3) is a subset of the core variables found in the Multi-Dimensional Health Assessment Questionnaire, as a self-reported questionnaire that includes an assessment of physical function, PGA for pain and a PGA for global health. Change from baseline in RAPID3 at Week 12 was measured in the subset of subjects with PsA.
Routine safety parameters, including laboratory safety, vital sign measurements, 12-lead electrocardiogram (ECG) results, chest X-ray and physical examination findings were assessed throughout the study. Laboratory samples were analysed by a central laboratory. The frequency, type and severity of adverse events (AEs), including adverse drug reactions (ADRs), adverse events of special interest (AESI) and severity of injection-site reactions (ISRs), were assessed. All AEs from informed consent up to 6 weeks after the last dose of study drug were recorded.
Subjects described the characteristics of the ISR (pain/tenderness, erythema/redness, induration/swelling, pruritus/itching, hematoma/ecchymosis/bruising) at the site of injection in an eDiary. If ISR was graded as at least 1 then it was reported as an AE. Reported ISR characteristics were summarised separately by treatment group and stage.
The final safety follow-up visit was at Week 54, 6 weeks after receiving the final dose of study drug.
Serum samples were collected predose at Week 1/Day 1, Weeks 4, 8, 16, 24, 32, 50 (all predose as applicable) and at the follow-up visit at Week 54 (6 weeks after the final dose of study drug) or on early withdrawal for the detection of anti-drug antibodies (ADAs) and neutralising antibodies (nAbs) to AVT02 or originator adalimumab. The number and percentage of patients developing ADAs and nAbs was tabulated for each treatment arm. Only those subjects who were ADA-positive were then tested for the presence of nAbs.
A multi-tiered strategy consisting of screening assay, confirmation assay, titration assay and neutralising assay was performed for the evaluation of immunogenicity as recommended by the FDA and EMA [15, 16]. The ADA and nAb assays were each designed as a one-assay approach to detect anti-AVT02 and anti-Humira antibodies in one assay setup following current recommendations [17]. Serum samples were evaluated for ADA presence and ADA titre using a screening cut point (plate-specific floating cut point using a correction factor [CF] of 1.17; CF was calculated with a non-parametric approach using the 95% percentile), a confirmatory cut point (threshold at 33% inhibition) and a titration cut point (plate-specific floating cut point using a titration CF [TCF] of 1.250; TCF was calculated using the 99.9th percentile). Serum samples were evaluated for nAb presence using a neutralising cut point (threshold at 8.8% inhibition; threshold was calculated as mean of % inhibition plus 2.33 × SD as there was normal distribution after exclusion of statistical outliers).
The same serum samples were tested to establish trough levels of AVT02 and originator adalimumab measured at steady state.
Outcomes
The primary efficacy endpoint was the percent improvement in PASI from baseline to Week 16. Secondary efficacy endpoints were the percent improvement in PASI from baseline to Weeks 4, 8, 12, 24, 32, 42 and 50, PASI 50, PASI 75, PASI 90 and PASI 100 response, number and percentage of subjects achieving sPGA responses of clear (0) or almost clear (1), and the change from baseline in quality of life as measured by DLQI scores at Weeks 16, 24 and 50.
Additional secondary endpoints were the comparison of safety, tolerability, immunogenicity and steady-state pharmacokinetics between AVT02 and originator adalimumab.
Exploratory efficacy endpoints were the same as the primary and secondary efficacy endpoints, as well as change from baseline in RAPID3, but measured at Week 12 in subjects with PsA.
Statistical Analysis
Based on analysis of a published reference study selected for its applicability to the present study, a sample size of 354 (177 per treatment group) was required for 90% power in a 5% level test for equivalence with equivalence margins ±10% based on a 90% two-sided confidence interval (CI) (as required by the FDA) [18]. A total of 400 (200 per treatment group) would provide 87.5% power in a 2.5% level test based on the same margins and a 95% two-sided CI (as required by EMA). Further details of the sample size calculation are given in the Electronic Supplementary Material (ESM).
The two-sided 90%/95% CIs of the difference between the AVT02 and originator adalimumab groups were calculated based on the analysis of covariance (ANCOVA) model, which included percent improvement as response variable, treatment, the two stratification factors and baseline PASI score as a covariate. Equivalence was achieved if the 90% CI/95% CI lay within (− 10% to 10%). Missing percent improvement was imputed using the last observation carry forward (LOCF) method for subjects with post-baseline assessment in Stage 1.
Methods as for the primary endpoint were also used for mean percent improvement in PASI from baseline to Weeks 8 and 12. For Weeks 24, 32, 42 and 50, there were no formal comparisons between the treatment groups. sPGA response rates at Weeks 16, 24 and 50 were calculated by treatment group based on observed data.
Change from baseline in DLQI at Weeks 16, 24 and 50 was summarised descriptively based on observed data. RAPID3 scores were summarised by treatment group in Stage 1 at baseline and Week 12.
Descriptive statistics for serum pharmacokinetic concentration were tabulated over time by visit up to Week 16 and were summarised descriptively by treatment sequence and by visit across the whole study. ADA and nAb impact on pharmacokinetics were additionally summarised.
The primary analysis was performed using the Full Analysis Set (FAS), which, consistent with the intention-to-treat principles, is defined as all randomised patients who received at least one dose of randomised study medication. For safety and pharmacokinetic endpoints, the Safety Analysis Set (SAF), defined as all patients who received at least one dose of the investigational product with treatment assignment based on actual treatment received, was analysed based on the actual treatment received.
Results
Subjects and Exposure
Screening of subjects began on 20 February 2019, with a total of 538 subjects screened and 413 randomised in a 1:1 ratio (AVT02: 205 subjects; originator adalimumab: 208 subjects; Fig. 1). One subject who had been randomised to the originator adalimumab group did not receive treatment as he withdrew consent to participate (Fig. 1). Last subject last visit for the whole study was on 20 July 2020.
Fig. 1.

Disposition of study subjects Stage 1 and Stage 2. aCOVID-19-related n = 2, Not COVID-19 related n = 5. bCOVID-19-related n = 2, Not COVID-19-related n = 7. cCOVID-19-related n = 1, Not COVID-19-related n = 1. PASI Psoriasis Area and Severity Index
Subject demographics and baseline characteristics were well balanced between all treatment groups and remained so after the transition and re-randomisation from Stage 1 to 2, including subjects who switched from originator adalimumab to AVT02 (Table 1). Subject disease characteristics at baseline, including DLQI, were also well balanced between treatment groups, in both Stage 1 and Stage 2 (Table 1).
Table 1.
Demographic and baseline characteristics (full analysis set)
| Stage 1 | Stage 2 | Overall N = 412 |
||||
|---|---|---|---|---|---|---|
| AVT02 N = 205 |
Originator N = 207 |
AVT02/ AVT02 N = 197 |
Originator/ AVT02 N = 97 |
Originator/ originator N = 98 |
||
| Gender, n (%) | ||||||
| Male | 125 (61.0) | 129 (62.3) | 122 (61.9) | 56 (57.7) | 67 (68.4) | 254 (61.7) |
| Female | 80 (39.0) | 78 (37.7) | 75 (38.1) | 41 (42.3) | 31 (31.6) | 158 (38.3) |
| Age (years) | ||||||
| Median (min, max) | 42.0 (20, 71) | 43.0 (18, 70) | 42.0 (20, 71) | 43.0 (22, 69) | 42.0 (18, 70) | 42.0 (18, 71) |
| Ethnicity, n (%) | ||||||
| Hispanic or Latino | 2 (1.0) | 2 (1.0) | 2 (1.0) | 1 (1.0) | 1 (1.0) | 4 (1.0) |
| Not Hispanic or Latino | 203 (99.0) | 205 (99.0) | 195 (99.0) | 96 (99.0) | 97 (99.0) | 408 (99.0) |
| Race, n (%) | ||||||
| White | 205 (100.0) | 207 (100.0) | 197 (100.0) | 97 (100.0) | 98 (100.0) | 412 (100.0) |
| Black or African American | 0 | 0 | 0 | 0 | 0 | 0 |
| Asian | 0 | 0 | 0 | 0 | 0 | 0 |
| PASI | ||||||
| Mean (SD) | 23.17 (8.538) | 22.98 (8.553) | 22.79 (8.062) | 22.74 (9.174) | 23.10 (7.915) | 23.08 (8.535) |
| Median (min, max) | 21.60 (12.1, 55.9) | 20.80 (12.0, 55.2) | 21.60 (12.1, 51.0) | 19.80 (12.1, 55.2) | 21.25 (12.0, 46.0) | 21.20 (12.0, 55.9) |
| sPGA, n (%) | ||||||
| Moderate | 112 (54.6) | 119 (57.5) | 108 (54.8) | 60 (61.9) | 51 (52.0) | 231 (56.1) |
| Severe | 76 (37.1) | 73 (35.3) | 74 (37.6) | 29 (29.9) | 40 (40.8) | 149 (36.2) |
| Very severe | 17 (8.3) | 15 (7.2) | 15 (7.6) | 8 (8.2) | 7 (7.1) | 32 (7.8) |
| BSA affected (%) | ||||||
| Mean (SD) | 32.3 (17.84) | 31.7 (17.88) | 31.9 (17.65) | 30.2 (18.03) | 32.3 (16.51) | 32.0 (17.84) |
| Median (min, max) | 28.0 (10, 86) | 26.0 (10, 84) | 28.0 (10, 83) | 25.0 (10, 84) | 28.0 (11, 82) | 28.0 (10, 86) |
| Months from PsO diagnosis to informed consent | ||||||
| Mean (SD) | 195.2 (131.43) | 198.6 (130.23) | 196.4 (131.80) | 193.0 (128.78) | 196.1 (127.20) | 196.9 (130.68) |
| Median (min, max) | 183.0 (6, 688) | 183.0 (7, 593) | 183.0 (6, 688) | 184.0 (20, 593) | 172.5 (20, 593) | 183.0 (6, 688) |
| PsA, n (%) | ||||||
| Presence | 43 (21.0) | 41 (19.8) | 40 (20.3) | 19 (19.6) | 18 (18.4) | 84 (20.4) |
| Absence | 162 (79.0) | 166 (80.2) | 157 (79.7) | 78 (80.4) | 80 (81.6) | 328 (79.6) |
| Months from PsA diagnosis to informed consent | ||||||
| n | 43 | 41 | 40 | 19 | 18 | 84 |
| Mean (SD) | 65.9 (62.68) | 79.2 (67.68) | 70.5 (62.60) | 81.6 (74.56) | 81.0 (66.63) | 72.4 (65.12) |
| Median (min, max) | 45.0 (0, 243) | 62.0 (5, 276) | 58.0 (0, 243) | 64.0 (5, 276) | 57.0 (9, 255) | 56.5 (0, 276) |
| Prior use of biologic therapy, n (%) | ||||||
| Yes | 42 (20.5) | 36 (17.4) | 39 (19.8) | 20 (20.6) | 11 (11.2) | 78 (18.9) |
| No | 163 (79.5) | 171 (82.6) | 158 (80.2) | 77 (79.4) | 87 (88.8) | 334 (81.1) |
| DLQI score | ||||||
| Mean (SD) | 15.4 (6.44) | 15.6 (6.60) | 15.4 (6.44) | 15.0 (6.69) | 16.4 (6.29) | 15.5 (6.52) |
| Median (min, max) | 15.0 (1, 30) | 16.0 (1, 30) | 15.0 (1, 30) | 15.0 (1, 30) | 17.0 (2, 28) | 16.0 (1, 30) |
Percentages are based on the number of subjects in the full analysis set by treatment group. sPGA scores are as follows: ≥ 3 = moderate, ≥ 4 = severe and ≥ 5 = very severe
BSA body surface area, DLQI Dermatology Life Quality Index, n number of evaluable subjects, N number of subjects in treatment group, PASI Psoriasis Area and Severity Index, PsA psoriatic arthritis, PsO psoriasis, SD standard deviation, sPGA static Physician's Global Assessment
Among the 205 subjects receiving AVT02 in Stage 1, 97.6% of injections were administered in accordance with the protocol. Mean compliance for originator adalimumab (n = 207) in Stage 1 was 98.4%. Mean compliance remained high after transition into Stage 2. In subjects who continued with AVT02 (n = 197), 99.2% of study drug doses were administered per protocol. In subjects who continued with originator adalimumab (n = 98), mean compliance was 98.9% per protocol. In those subjects who switched from originator adalimumab to AVT02 (n = 97), mean compliance was 99.5%.
Efficacy
Primary Endpoint
At the conclusion of Stage 1 at Week 16, the percentage improvement in PASI score was 91.6% for AVT02-treated subjects and 89.6% for those treated with originator adalimumab. ANCOVA analysis of the difference in percent PASI improvement in the FAS from baseline between AVT02 and originator adalimumab (least squares means 89.2 [standard error 1.61] and 86.9 [standard error 1.65], respectively) was within the predefined equivalence margins of ±10% (90% CI [− 0.76 to 5.29]; 95% CI [− 1.34 to 5.88]; Table 2). The percent improvement in PASI from baseline to Week 16 was highly comparable between treatment groups (Fig. 2a). The robustness of the primary analysis result was supported by sensitivity analyses (ESM; Table S1 including a mixed model for repeated measures [MMRM] applied based on the FAS using the observed data up to Week 16, and ANCOVA applied based on per-protocol set). Also supporting the primary analysis, the %BSA change from baseline to all timepoints through Week 16 did not show notable difference across treatment groups (ESM; Table S2). Furthermore, the proportion of subjects achieving PASI 50, PASI 75, PASI 90 and PASI 100 at each timepoint was comparable across both treatment groups (ESM; Table S3).
Table 2.
ANCOVA of percent improvement in PASI in Stage 1 from baseline to Week 16 to assess therapeutic equivalence (LOCF; full analysis set)
| AVT02 N = 205 |
Originator N = 207 |
|
|---|---|---|
| LS mean (SE) | 89.2 (1.61) | 86.9 (1.65) |
| LS mean difference (SE; AVT02 vs originator) | 2.3 (1.84) | |
| 90% confidence interval | − 0.76 to 5.29 | |
| 95% confidence interval | − 1.34 to 5.88 | |
The two-sided 90% and 95% CIs of the differences of least squares means between the AVT02 and originator adalimumab groups are from the ANCOVA model including percent improvement as response variable, treatment and two stratification factors as factors and baseline PASI score as covariate
ANCOVA analysis of covariance, CI confidence interval, LOCF last observation carried forward, LS mean least–squares mean, N number of subjects in treatment group, PASI Psoriasis Area and Severity Index, SE standard error
Fig. 2.

Mean (± standard error) percent improvement from baseline in PASI by visit a through Stage 1/baseline to Week 16 and b Stage 2/Week 16–50 (observed data; full analysis set). Missing percent improvement in PASI is imputed using LOCF for subjects post-baseline assessment. Data in (b) relate to subjects who were PASI responders (PASI 50 response or higher) at Week 16. Baseline visit is included to show the magnitude of improvement and to provide a visual impression of efficacy profile over time. LOCF last observation carry-forward, LS mean least–squares mean, PASI Psoriasis Area and Severity Index, PASI 50 50% reduction in PASI, SE standard error
Secondary Efficacy Endpoints
In subjects who were PASI responders at the completion of Stage 1, the improvement in DLQI from baseline to Week 16 was likewise comparable across treatment groups (AVT02 mean 11.4 ± 6.63; originator adalimumab 10.6 ± 7.52). Additionally, the proportion of subjects achieving clear (0) or almost clear (1) sPGA rating was also consistent across treatment groups at the end of Stage 1 at Week 16 (ESM; Table S4, left panel).
At the conclusion of Stage 2, there was again a comparable pattern for the percent improvement in PASI from Week 16 to end of study between treatment groups (Fig. 2b). This long-term efficacy included subjects who remained on treatment for either AVT02 or originator adalimumab, as well as those who had switched from originator adalimumab in Stage 1 to AVT02 in Stage 2. The mean percent PASI improvement also remained similar at each timepoint in Stage 2 compared to results at Week 16 across treatment groups, supporting the assessment that efficacy was not compromised in subjects who switched from originator adalimumab to AVT02 (ESM; Table S5). The proportion of subjects achieving PASI 50, PASI 75, PASI 90 and PASI 100 was likewise comparable across treatment groups throughout Stage 2 of the study (ESM; Table S3).
Persistence of efficacy also remained comparable across all treatment groups in Stage 2 when measured by %BSA and sPGA at all timepoints from Week 24 to Week 50 (ESM; Table S6 and Table S4, right panel). These data were complemented by comparable improvement in DLQI across treatment groups in Stage 2 at Weeks 16, 24 and 50, including subjects who switched from originator adalimumab to AVT02 (Table 3).
Table 3.
Change from baseline in Dermatology Life Quality Index (DLQI) in Stage 2/Week 16 to Week 50 (full analysis set)
| AVT02/AVT02 | Originator/AVT02 | Originator/Originator | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Week 16 | Week 24 | Week 50 | Week 16 | Week 24 | Week 50 | Week 16 | Week 24 | Week 50 | |
| n | 196 | 193 | 180 | 97 | 95 | 90 | 98 | 96 | 85 |
| Mean (SD) | 11.7 (6.46) | 10.7 (7.00) | 10.7 (6.88) | 11.8 (7.06) | 10.7 (8.28) | 9.2 (9.28) | 10.2 (7.56) | 10.2 (7.46) | 9.3 (7.97) |
| Median (range) | 11.0 (−4, 29) | 11.0 (−21, 29) | 11.0 (−4, 28) | 11.0 (−1, 28) | 11.0 (−22, 28) | 9.0 (−24, 28) | 10.0 (−17, 28) | 10.0 (−20, 28) | 9.0 (−18, 28) |
n number of evaluable subjects, SD standard deviation
Exploratory Endpoints
Subgroup analysis in the subset of subjects with PsA also supported the evaluation of consistency between AVT02 (n = 43) and originator adalimumab (n = 41) treatment groups in the percentage improvement from baseline in PASI results (LOCF) through end of Stage 1 at Week 16 in the FAS (ESM; Table S7). Similar results in the PsA-positive population across groups, and compared to the overall population, in the data for sPGA and DLQI further supported the analysis (ESM; Tables S8 and S9). The change from baseline in RAPID3, moreover, supported the assessment of biosimilarity between AVT02 and originator adalimumab treatment groups at Week 12 (ESM; Table S10).
Efficacy as measured by percentage improvement from baseline in PASI results again persisted in the PsA subgroup through all Stage 2 timepoints to Week 50, including subjects who had been re-randomised at Stage 2 to switch from originator adalimumab to AVT02 (ESM; Table S11).
Safety
Safety Profile in Stage 1
The AE profile in the SAS was broadly similar across treatment groups in Stage 1 (Table 4) with both AVT02 and originator adalimumab being generally well tolerated. Most treatment emergent AEs (TEAEs) were not treatment related and a similar percentage of treatment-related TEAEs was reported across treatment groups. Of treatment-related TEAEs, the most commonly reported was ISR (Table 4).
Table 4.
Overview of treatment-emergent adverse events in Stage 1/through Week 16 and Stage 2/Week 16–54 of the study (safety set)
| Stage 1 | Stage 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AVT02 N = 205 |
Originator N = 207 |
AVT02/AVT02 N = 197 |
Originator/AVT02 N = 97 |
Originator/originator N = 98 |
||||||
| Subjects, n (%) | Events, n | Subjects, n (%) | Events, n | Subjects, n (%) | Events, n | Subjects, n (%) | Events, n | Subjects, n (%) | Events, n | |
| Adverse event category | ||||||||||
| Any TEAE | 92 (44.9) | 192 | 91 (44.0) | 245 | 116 (58.9) | 315 | 46 (47.4) | 153 | 49 (50.0) | 131 |
| Treatment-related TEAEs | 44 (21.5) | 84 | 41 (19.8) | 89 | 40 (20.3) | 135 | 17 (17.5) | 66 | 14 (14.3) | 39 |
| Maximum severity of TEAEs | ||||||||||
| Mild | 61 (29.8) | 117 | 51 (24.6) | 114 | 62 (31.5) | 175 | 28 (28.9) | 95 | 31 (31.6) | 76 |
| Moderate | 27 (13.2) | 37 | 37 (17.9) | 52 | 50 (25.4) | 79 | 18 (18.6) | 28 | 18 (18.4) | 34 |
| Severe | 4 (2.0) | 5 | 3 (1.4) | 3 | 4 (2.0) | 5 | 0 | 0 | 0 | 0 |
| TEAEs of special interestb | 38 (18.5) | 77 | 34 (16.4) | 76 | 45 (22.8) | 137 | 17 (17.5) | 64 | 13 (13.3) | 37 |
| TEAE leading to early withdrawalc | 3 (1.5) | 3 | 3 (1.4) | 3 | 6 (3.0) | 6 | 3 (3.1) | 3 | 1 (1.0) | 1 |
| Treatment-related TEAE leading to early withdrawal | 2 (1.0) | 2 | 3 (1.4) | 3 | 4 (2.0) | 4 | 3 (3.1) | 3 | 1 (1.0) | 1 |
| TESAEs leading to early withdrawal | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Treatment-related TESAEs leading to early withdrawal | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TESAEs | 2 (1.0) | 3 | 5 (2.4) | 5 | 4 (2.0) | 6 | 0 | 0 | 0 | 0 |
| Treatment-related TESAEs | 1 (0.5) | 1 | 0 | 0 | 1 (0.5) | 1 | 0 | 0 | 0 | 0 |
| Death | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| System organ classa MedDRA preferred term | ||||||||||
| Gastrointestinal disorders | 0 (0) | 0 | 0 (0) | 0 | 5 (2.5) | 5 | 2 (2.1) | 2 | 12 (12.2) | 16 |
| Diarrhoea | 0 (0) | 0 | 0 (0) | 0 | 0 | 0 | 1 (1.0) | 1 | 5 (5.1) | 5 |
| General disorders and administration site conditions | 40 (19.5) | 74 | 36 (17.4) | 78 | 34 (17.3) | 124 | 11 (11.3) | 56 | 12 (12.2) | 36 |
| Injection-site reaction | 34 (16.6) | 67 | 33 (15.9) | 75 | 31 (15.7) | 121 | 11 (11.3) | 56 | 10 (10.2) | 34 |
| Infections and infestations | 35 (17.1) | 42 | 39 (18.8) | 44 | 58 (29.4) | 81 | 23 (23.7) | 28 | 16 (16.3) | 21 |
| Nasopharyngitis | 11 (5.4) | 12 | 11 (5.3) | 13 | 23 (11.7) | 28 | 5 (5.2) | 6 | 4 (4.1) | 4 |
| Pharyngitis | 0 (0) | 0 | 0 (0) | 0 | 4 (2.0) | 4 | 5 (5.2) | 5 | 2 (2.0) | 2 |
| Investigations | 0 (0) | 0 | 0 (0) | 0 | 29 (14.7) | 44 | 12 (12.4) | 25 | 16 (16.3) | 21 |
| Alanine aminotransferase increased | 0 (0) | 0 | 0 (0) | 0 | 6 (3.0) | 7 | 5 (5.2) | 8 | 3 (3.1) | 4 |
AEs adverse events, BSA body surface area, DLQI Dermatology Life Quality Index, n number of evaluable subjects, N number of subjects in treatment group, PASI Psoriasis Area and Severity Index, PsA psoriatic arthritis, PsO psoriasis, SD standard deviation, sPGA static Physician's Global Assessment, TEAEs treatment-emergent adverse events, TESAEs treatment-emergent serious adverse events
aTEAE frequency ≥5%. Subjects are counted once within a system organ class and once for each unique preferred term. Adverse events are coded using MedDRA version 22.1
bAEs of special interest included injection-site reactions and liver enzyme abnormalities
cTEAEs leading to early withdrawal included liver enzyme abnormalities, mycobacterium tuberculosis complex test positive and psoriasis
Most TEAEs were mild in nature, and a similar percentage of subjects reported TEAEs classified as mild across both Stage 1 treatment groups (Table 4). Severe TEAEs were reported by 4 (2.0%) and 3 (1.4%) subjects in the AVT02 and originator adalimumab treatment groups, respectively, in Stage 1 of the study. For TEAEs of special interest (TEAESIs), there was no clinically meaningful difference between the treatment groups at the same time point.
TEAEs leading to early withdrawal in the study were reported by a similar percentage of subjects in all treatment groups in Stage 1 of the study (Table 4). There were no events leading to early withdrawal that were classified as serious, and none that were judged treatment-related (Table 4).
Of the eight TESAEs reported by seven subjects in Stage 1 of the study, one was considered treatment-related, in a subject treated with AVT02 (Table 4).
In the subset of subjects with PsA, the same percentage of subjects reported TEAEs in both treatment groups in Stage 1 of the study. The severity of TEAEs was also similar (data not shown). Consistent with the overall population, the most frequently reported TEAESIs in the PsA subset was ISRs.
Safety Profile Through Stage 2
The comparable AE profile in the SAS persisted across treatment groups in Stage 2 of the study, including those subjects who were re-randomised to switch from originator adalimumab to AVT02 (Table 4). Similar results for all TEAEs, treatment-related TEAEs and TEAEs leading to early withdrawal, as well as TEAESIs, were observed for all treatment groups, supporting the assessment that safety was not compromised when switching from originator adalimumab to AVT02.
In Stage 2 of the study, a slightly higher percentage of subjects in the AVT02/AVT02 group reported TEAEs of moderate severity compared with the other treatment groups (Table 4). Additionally, four subjects (2.0%) in the AVT02/AVT02 treatment group reported severe TEAEs, comprising one instance of meningococcal meningitis, one orchitis, one thermal burn and one psoriatic arthropathy, with none reported in the originator adalimumab group nor in subjects who switched from originator adalimumab to AVT02. Also in the AVT02/AVT02 treatment group in Stage 2 of the study, a slightly higher proportion of subjects reported TEAESIs compared with the other treatment groups (Table 4). This primarily comprised ISRs. ISRs were the only TEAESI reported in at least 5% of subjects across all groups, and all were reported as mild in severity except for one event that was classified as moderate. For those subjects who reported TEAESIs that were liver enzyme abnormalities, most had pre-existing conditions that predisposed them to such a possibility, with the majority of such TEAESIs being classified as mild (data not shown).
There were no deaths reported during the study (Table 4).
No clinically meaningful changes from baseline over time were observed for any treatment group in Stage 1 or Stage 2 of the study for laboratory, vital signs, physical examination and ECG findings (ESM).
Immunogenicity
The time of onset and frequencies of development of ADAs and nAbs were similar in both treatment groups in Stage 1; ADA (+) was 20.5% at baseline rising to 85.9% at Week 16 in AVT02-treated subjects, and 18.8% at baseline rising to 89.8% at Week 16 in those treated with originator adalimumab. The frequency of nAbs at baseline and Week 16 was 2.9% and 65.3% for AVT02-treated subjects, respectively, and 2.4% and 73.5% in those treated with originator adalimumab, respectively.
Frequencies of ADA and nAbs were also similar across treatment groups, including those subjects who had switched, through Stage 2 of the study up to Week 54. Total incidence of ADA (+) and nAb (+) was 93.4% and 84.3%, respectively, in AVT02-treated subjects, 95.9% and 80.6%, respectively, in originator adalimumab-treated subjects and 91.8% and 83.5 %, respectively, in those who switched from the originator to AVT02. Of the subjects developing ADA and nAbs wholly in Stage 2, the incidences were 43.5% and 53.7%, respectively, in AVT02-treated subjects, 42.9% and 34.5%, respectively, in originator adalimumab-treated subjects and 20.0% and 27.3%, respectively, in those who switched from the originator to AVT02.
ADA titres were similar across treatment groups in Stage 1 of the study (Fig. 3a), and remained similar across groups through Stage 2 (Fig. 3b). Figures exclude those subjects with pre-dose ADA-positive titre at baseline.
Fig. 3.

Box plot of titres for positive anti-drug antibody (ADA) results by visits a through Stage 1/Week 4–16 and (b) Stage 2/Week 24–54 (safety analysis set). No baseline visit is presented—subjects with pre-dose ADA positive are excluded. LS mean least -squares mean, PASI Psoriasis Area and Severity Index, SE standard error
The mean percent PASI improvement by ADA status at each timepoint from baseline to Week 16 was very similar between treatment groups in ADA-positive subjects in the LOCF analysis of the FAS. In ADA-positive subjects, improvement was 90.8% in AVT02-treated subjects and 90.2% in those treated with originator adalimumab. In ADA-negative subjects, the figures were 97.2% and 86.3%, respectively.
PASI improvement remained consistent across treatment groups during Stage 2, including those subjects who were re-randomised to switch from originator adalimumab to AVT02. PASI improvement by ADA status was likewise not affected in subjects switching from originator adalimumab to AVT02 in Stage 2, with improvement of 89.4% in such subjects at Week 50, compared with improvement of 90.8% and 89.6% in subjects treated only with AVT02 or originator adalimumab, respectively. Improvement was similar in all three treatment groups in Stage 2 in the ADA-negative subset, and for both ADA-positive and ADA-negative subjects, this efficacy persisted regardless of nAb status (data not shown).
In the subset of subjects who were ADA-positive, a similar percentage reported TEAEs in both treatment groups in Stage 1 of the study, and all three treatment groups, including those who switched, in Stage 2 of the study, and this was regardless of nAb status. There was no clinically meaningful difference between treatment groups in ADA-negative subjects reporting TEAEs in any stage of the study, which was again regardless of nAb status.
Pharmacokinetics
There was no clinically meaningful difference in adalimumab serum concentration at steady state between AVT02 and originator adalimumab in Stage 1 of the study up to Week 16 (Fig. 4a), although AVT02 concentration was numerically slightly above the originator product at most time points.
Fig. 4.
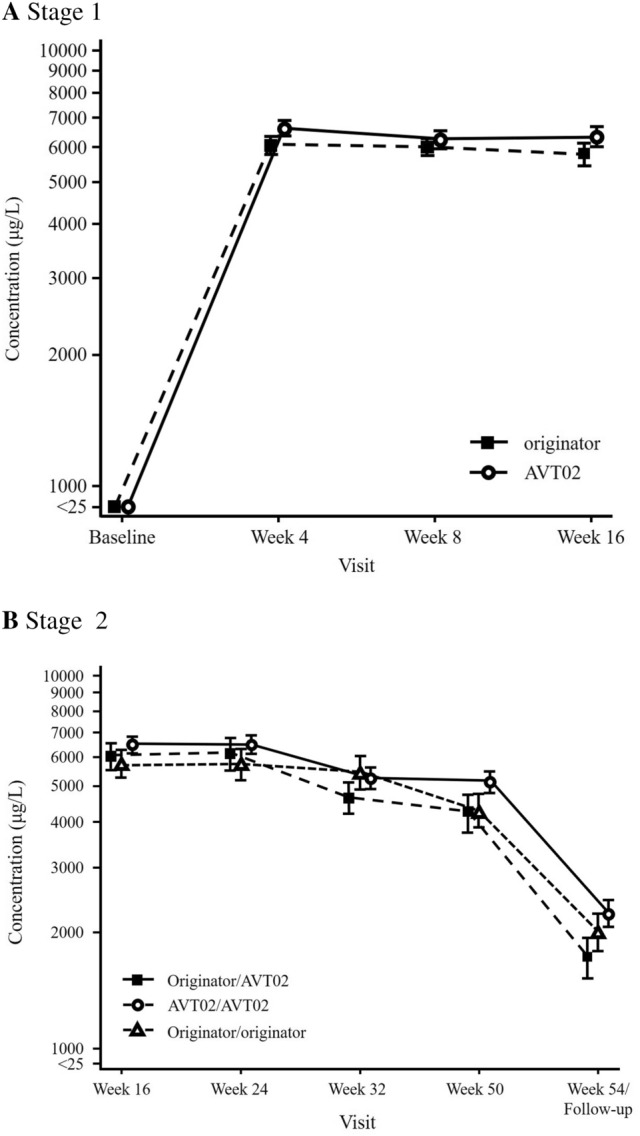
Mean (± standard error) adalimumab serum concentration vs time a through Stage 1/baseline to Week 16 and b Stage 2/Week 16–54 (safety analysis set). All baseline statistics are assigned a nominal value to enable plotting values of 0 on the log scale. LS mean least -squares mean, PASI Psoriasis Area and Severity Index, SE standard error
This degree of overlap was also observed in Stage 2 of the study up to Week 54, including in those subjects who switched from originator adalimumab to AVT02 (Fig. 4b).
Additionally, there was no clinically meaningful difference in the adalimumab serum concentration when comparing subjects treated with AVT02 or originator adalimumab in subgroups with or without ADAs, or with nAbs throughout the study period (data not shown).
Discussion
An originator biologic is a complex molecule with many post-translational modifications, resulting in multiple different forms and batch-to-batch variability [19]. Unlike traditional generic non-biologics, such originator products are too complicated to duplicate faithfully. Nevertheless, there is little variability between batches in originator biologic target binding and pharmacokinetics, leading to consistent efficacy and safety over time. A potential biosimilar must be shown to be comparable to the originator reference product in terms of physical and chemical properties, as well as demonstrating no clinically meaningful difference in efficacy, safety (including immunogenicity) and pharmacokinetic profiles.
In this study, we compared a 100-mg/mL, citrate-free formulation of proposed biosimilar AVT02 with recent batches of reference originator adalimumab product. The 100-mg/mL, citrate-free originator adalimumab is the most current formulation, one that augments the administration of originator adalimumab by reducing injection pain [10, 20].
The primary endpoint of the study was met, with the difference in percent PASI improvement from baseline to the end of Stage 1 of the study at Week 16 between AVT02 and originator adalimumab falling within the predefined equivalence margins, supporting the assessment of AVT02 having no clinically meaningful difference compared with originator adalimumab. The primary endpoint of therapeutic equivalence was supported not only by sensitivity analyses, but also with the assessment of sPGA score, where the proportion of subjects achieving a clear (0) or almost clear (1) sPGA rating was also consistent across treatment groups. Furthermore, the similarity in quality-of-life assessment, as measured by DLQI, mirrored the objective outcomes, with comparable improvement across treatment groups. AVT02 treatment also had a similar safety profile to that of originator adalimumab during Stage 1 of the study. Patients who were naïve to adalimumab were able to generally tolerate and effectively manage their disease condition with AVT02.
In real-life practice, patients may either start on a biosimilar or be switched to one. The study design included re-randomisation after completion of Stage 1, intended to mimic the potential real-life switching scenario that patients may experience. The mean percent PASI improvement achieved in Stage 1 in both treatment groups was similar and was maintained from Week 16 through Week 50 of the study across all treatment groups in Stage 2. Importantly, efficacy persisted for those subjects who switched from originator adalimumab treatment in Stage 1 to AVT02 treatment in Stage 2. This was true not only for percent PASI improvement, but for all efficacy assessments, and across all subgroups of subjects (with/without PsA, ADA and nAb status). Subjects who were re-randomised to switch treatments between Stage 1 and Stage 2 also reported comparable quality-of-life improvements to subjects who did not switch. Neither efficacy nor safety were compromised in subjects who switched treatments when compared with subjects who did not as no clinically meaningful differences in outcomes were observed. These results support that patients can expect no clinically meaningful differences in safety or efficacy when switching from originator adalimumab to AVT02.
Assessing both subjects who started on AVT02 and subjects who started on originator adalimumab and switched to AVT02 is a key strength of this study, combined with high compliance, with over 98% of subjects being dosed with study drug in accordance with the protocol from baseline to Week 50, leading to a robust data set.
Psoriatic arthritis is a common comorbidity of moderate to severe chronic plaque psoriasis, which typically responds well to tumour necrosis factor inhibitors like adalimumab. In the subset of subjects in this study who reported PsA, AVT02 had similar efficacy to originator adalimumab. However, the number of subjects in the PsA subset was small, precluding statistically meaningful conclusions. Nevertheless, the similar efficacy and safety in subjects with both moderate to severe chronic plaque psoriasis and psoriatic arthritis supports the use of AVT02 for all indications of originator adalimumab as part of a comprehensive programme to demonstrate the ‘totality of evidence’.
The nature of biologic therapeutics results in different batches and different products having slight variations in form, even as they show consistent target binding and pharmacokinetics. This leads to the possibility of altered immunogenicity, which is a particular concern for a biosimilar product. In this study there was no clinically meaningful difference between AVT02 and originator adalimumab as measured by time onset and frequency of ADA and nAbs, and ADA titre. This was true for subjects who received AVT02 throughout the study and for those subjects who switched to AVT02 from originator adalimumab.
Conclusion
The data presented here confirm the therapeutic equivalence of AVT02 to its reference product. Based on these findings, we anticipate that AVT02 can offer patients both comparable efficacy and improvements in subjective quality of life that originator adalimumab has provided.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank the subjects who participated in the study and all the investigators who contributed. The authors additionally thank Joseph McClellan of Alvotech for strategic guidance, and Lorna Rettig of Alvotech for medical writing assistance.
Declarations
Funding
The study was sponsored by Alvotech Swiss AG. Employees of the sponsor had a role in study design, data analysis and manuscript preparation. Employees of the funder had no role in data collection.
Competing interests
FB, JS, RD, EG, HO, HNH and HS are employees at Alvotech. SF has received research grants from Abbvie, Janssen, Lilly and Novartis and speaker honoraria from Alvotech, Abbvie, Amgen, Lilly, Novartis and Janssen. RK's company has received consultancy fees in relation to this study and in other studies conducted by Alvotech, but no consultancy fees have been received in relation to the writing of this manuscript. NR, GP, KK, and GG declare that they have no conflicts of interest that might be relevant to this work.
Availability of data
The datasets generated and/or analysed during the current study are available from the sponsor on reasonable request.
Code availability
Not applicable.
Ethics approval
The study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation Good Clinical Practice (ICH-GCP) guidelines and the appropriate regulatory requirements in the countries in which the study was conducted. The protocol, its amendments and informed consent documentation were reviewed and approved by the institutional review board(s) or independent ethics committee(s) at each study site.
Consent to participate
All patients signed an Institutional Review Board/Independent Ethics Committee-approved informed consent form before any study-specific procedures were performed.
Consent for publication
Not applicable.
Authors’ contributions
SRF, FB, JS, EG, HO, HNH, RK and HS were involved in the conception and design of the study. NR, GP, KK, GG, JS and RD were involved in the provision of study materials and patients and acquisition of the data. SRF, FB, JS, HO, HNH and HS did the analysis and/or the interpretation of the data. All authors revised the report critically. All authors approved the final version.
References
- 1.EMA. Humira (adalimumab). Summary of Product Characteristics 2021. https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf. Accessed 12 Aug 2021.
- 2.FDA. Humira (adalimumab) US Package Insert. AbbVie, Inc., 2021. https://www.rxabbvie.com/pdf/humira.pdf. Accessed 12 Aug 2021.
- 3.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008; 117(2): 244-279. Doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed]
- 4.Safiri S, Kolahi AA, Hoy D, et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the Global Burden of Disease study 2017. Ann Rheum Dis. 2019;78(11):1463–1471. doi: 10.1136/annrheumdis-2019-215920. [DOI] [PubMed] [Google Scholar]
- 5.Chen BK, Yang YT, Bennett CL. Why biologics and biosimilars remain so expensive: despite two wins for biosimilars, the supreme court’s recent rulings do not solve fundamental barriers to competition. Drugs. 2018;78(17):1777–1781. doi: 10.1007/s40265-018-1009-0. [DOI] [PubMed] [Google Scholar]
- 6.McCamish M, Yoon W, McKay J. Biosimilars: biologics that meet patients' needs and healthcare economics. Am J Manag Care. 2016;22(13 Suppl):S439–S442. [PubMed] [Google Scholar]
- 7.FDA. Biosimilar Product Information. https://www.fda.gov/drugs/biosimilars/biosimilar-product-information. Accessed 12 Aug 2021.
- 8.EMA. https://www.ema.europa.eu/en/medicines. Accessed 12 Aug 2021.
- 9.AbbVie Corporation. https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf. Accessed 24 Aug 2021.
- 10.Nash P, Vanhoof J, Hall S, et al. Randomized crossover comparison of injection site pain with 40 mg/0.4 or 0.8 ml formulations of adalimumab in patients with rheumatoid arthritis. RheumatolTher. 2016;3(2):257–270. doi: 10.1007/s40744-016-0041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergman M, Patel P, Chen N, et al. Evaluation of adherence and persistence differences between adalimumab citrate-free and citrate formulations for patients with immune-mediated diseases in the United States. Rheumatol Ther. 2021;8(1):109–118. doi: 10.1007/s40744-020-00256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FDA. Conduct of Clinical Trials of Medical Products During the COVID-19 Public Health Emergency: Guidance for Industry, Investigators, and Institutional Review Boards. https://www.fda.gov/media/136238/download. Accessed 12 Aug 2021.
- 13.EMA. Guidance on the Management of Clinical Trials During the COVID-19 (Corona Virus) Pandemic. https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-10/guidanceclinicaltrials_covid19_en.pdf. Accessed 12 Aug 2021.
- 14.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)–a simple practical measure for routine clinical use. ClinExp Dermatol. 1994;19(3):210–216. doi: 10.1111/j.1365-2230.1994.tb01167.x. [DOI] [PubMed] [Google Scholar]
- 15.U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER). Immunogenicity Testing of Therapeutic Protein Products—Developing and Validating Assays for Anti-Drug Antibody Detection. 2019.
- 16.European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) EMEA/CHMP/BMWP/14327/2006 Rev1. Guideline on Immunogenicity assessment of therapeutic proteins. May 2017.
- 17.Civoli F, Kasinath A, Cai XY, et al. Recommendations for the development and validation of immunogenicity assays in support of biosimilar programs. AAPS J. 2020;22:7. doi: 10.1208/s12248-019-0386-y. [DOI] [PubMed] [Google Scholar]
- 18.Gooderham M, Kaur P, Narbutt J, Philipp S, Spelman L, Weglowska J, et al. Clinical similarity of biosimilar ABP 501 to adalimumab in the treatment of patients with moderate to severe plaque psoriasis: a randomized, double-blind, multicenter, phase III study. J Am Acad Dermatol. 2017;76(6):1093–1102. doi: 10.1016/j.jaad.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 19.de Mora F, Balsa A, Cornide-Santos M, et al. Biosimilar and interchangeable: Inseparable scientific concepts? BrJ Clin Pharmacol. 2019;85(11):2460–2463. doi: 10.1111/bcp.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida T, Otaki Y, Katsuyama N, Seki M, Kubota J. New adalimumab formulation associated with less injection site pain and improved motivation for treatment. Mod Rheumatol. 2019;29(6):949–953. doi: 10.1080/14397595.2018.1520426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.