

Abstract
With the arrival of the Covid-19 pandemic, anti-viral agents have regained center stage in the arena of medicine. Out of the various drug targets involved in managing RNA-viral infections, the one that dominates almost all RNA viruses is RdRp (RNA-dependent RNA polymerase). RdRp are proteins that are involved in the replication of RNA-based viruses. Inhibition of RdRps has been an integral approach for managing various viral infections such as dengue, influenza, HCV (Hepatitis), BVDV, etc. Inhibition of the coronavirus RdRp is currently rigorously explored for the treatment of Covid-19 related complications. So, keeping in view the importance and current relevance of this drug target, we have discussed the importance of RdRp in developing anti-viral agents against various viral diseases. Different reported inhibitors have also been discussed, and emphasis has been laid on highlighting the inhibitor's pharmacophoric features and SAR profile.
Keywords: RdRp, HCV (Hepatitis), Influenza, Dengue, BVDV, Covid 19, RdRp inhibitors
Graphical abstract
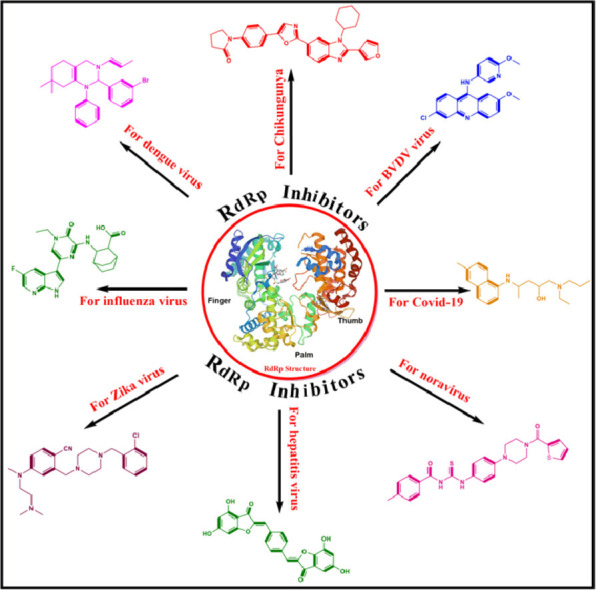
1. Introduction
Viruses are the most common infectious diseases causing agents. They typically contain RNA (Ribonucleic acid) or DNA (Deoxyribonucleic acid) as the core and source of genetic information encapsulated in a protein coat. One of the functions of the protein coats is to assist the virus in attaching to the host cell surface, followed by incursion and utilization of the host's bio-machinery for replication. Therefore, based on genetic material, the viruses are of two types, RNA and DNA viruses, out of which RNA-viruses result in critical affliction on healthcare systems throughout the globe [1, 2]. Further, depending on the type of genome, RNA viruses can be categorized into double-stranded (dsRNA), positive sense single-strand (RNA), and negative sense single-strand (ssRNA) [3]. The most recent RNA virus causing a burden on the global health care system is severe acute respiratory syndrome virus 2, the causative agent of COVID-19 [4], [5], [6], [7], [8]. Targeting RNA-viruses and their management is complicated due to an intrinsic viral property of integrating the viral genetic material with the host cell genome, which helps them in bypassing the human immune system [9]. Further utilizing the host machinery, RNA viruses increase their genetic content [10]. Also, RNA viruses maintain mutation rates, which further complicates the management of viral diseases [11,12].
With the advancements in current tools and techniques, all the essential proteins targeted to develop anti-virals are well-known. Still, RNA-dependent RNA polymerases (RdRps) remain among the most critical viral drug targets [13, 14]. It is involved in the genome replication process of an RNA from an RNA template, which is then involved in the encoding of some of the important proteins for the proper functioning and survival of viruses [15]. Following their functional parallel, RdRps also share structural resemblance with DNA polymerases and reverse transcriptases, which also require them to preserve conserved sequences and motifs through their span. Recently there are few reviews published on RdRps as potent anti-viral target [16], [17], [18] however they focus on either the structure of RdRp or on the chemical classes of the reported inhibitors, while this work provides the exploration of RdRp as target for different viral infection of positive-sense single-stranded RNA viruses along with the rationales and architecture on which different RdRp inhibitors are designed in the last five years. This work provides the binding mode analysis of different RdRp inhibitors which could be utilized for further designing of potent inhibitors.
2. Structure of viral RNA-dependent RNA polymerases
The core domain of RdRp, consisting of fingers, thumb and palm sub-domains, mainly performs the catalytic function involving binding of template, entry of nucleoside triphosphate (NTP), and polymerization (Fig. 1 ). The palm sub-domain, present at the intersection of the thumb and fingers sub-domains, possess the majority of the conserved structural elements which are essential for the catalytic activity. In the palm sub-domain, a RNA Recognizing Motif (RRM) comprising of three-strands and catalytic aspartates is present [19]. The sub-domain is responsible for the identification of NTPs over deoxy NTPs and catalysis of phosphorylation reaction via metal ion coordination [20]. The palm sub-domain is conformationally dynamic and following the binding of NTP, it undergoes significant restructuring to facilitate catalysis [21]. In some RdRp proteins, belonging to different RNA-viruses, shuffling in the sequence of the catalytic sub-domain results in an uncommon topology [22].
Fig. 1.

3-D structure of RdRp along with the motifs of palm, finger and thumb domains (PDB ID: 6M71).
The residues in the thumb sub-domain are responsible for padding alongside the template RNA, which stabilizes the starting NTPs on the template. Following polymerization, thumb sub-domain is involved in the migration of newly formed templates [23, 24]. The fingers sub-domain has a vital function of maintaining the architecture of the active site and holding the RNA template to facilitate polymerization [25]. This sub-domain aids in recognition of the template RNA via binding with the major groove. It consists of four intertwined fingers, which upon interacting with the thumb sub-domain, close the active site [26, 27].
Structurally, seven motifs (A-G) are present in viral RdRps which epitomizes the preserved structural constituents of RdRps [28]. The palm sub-domain contains motifs A to E and motifs G and F are present in the fingers sub-domain. In rare cases, RNA viruses possess an added motif H in the fingers sub-domains. A catalytic motif DX2–4D (an aspartate motif) having invariant first aspartate across numerous RdRps, is present in Motif A. Another aspartate, second in sequence, forms H-bond with 2′OH of the inbound NTP and therefore, along with a preserved asparagine of motif B, assists in the discernment of NTPs over deoxy NTPs [6]. In some RNA viruses, the concerned aspartate is replaced by a conserved lysine permitting them to utilize manganese as the cofactor, in place of magnesium [29]. While in Motif B, a threonine is present in the palm sub-domain, within the α-helix facing the active site, which contribute to the binding of the template RNA. It also links the N-terminal helix rising from the palm sub-domain to a strand of the fingers sub-domain. Acting as a pivot, it accommodates conformational deviations linked to the binding of the template and the substrate [30]. Whereas in Motif C, a preserved GDD (Gly-Asp-Asp) motif indispensable for complexation with the metal ions is present. Structurally, motifs A and C are juxtaposed and help in forming the RRM. Also, the aspartate from DX2–4D and the preserved aspartates of GDD align with each other at the tip of the RRM to aid in catalysis [31]. Further, motif D possess conserved glycine which participate in the adjustment of the conformation due to proper NTP binding. Detailed studies disclosed that motif D, responsible to select NTPs, is one of the most dynamic elements with variation of around 6 Å [32]. It is also reported to be responsible for the migration of the thumb sub-domain in the course of elongation. While a preserved lysine of motif D performs the key job of deprotonating the leaving pyrophosphate group [33]. Moreover, there is a primer grip formed by motif E, which maintains the proper orientation of 3′‑hydroxyl of the primer during catalysis. An aromatic residue present on the face of the N-terminal side pointing at motif C remains conserved in this motif [34]. Furthermore, there is a motif F which consists of positively charged amino acid residues that protect the negative charges of the incoming NTP's phosphate groups by positioning themselves right above the palm sub-domain. This motif also contains a key conserved arginine in its C-terminal [28]. Lastly, motif G, consisting of loop, forms a part of the entrance tunnel for the template and also interacts with the priming NTPs [35].
A lot of channels/tunnels which pass through in various directions are also part of RdRps. They connect the catalytic sites with the external surface of the protein [36]. These channels are also considered as key targets in the development of RdRp inhibitors. The template entry channels possess positively charged residues which favor the inward movement of template RNA and NTPs into the catalytic domain. Another channel eases the inward movement of the substrate and divalent cations into the catalytic domain and upon polymerization, also enables the discharge of the pyrophosphate moiety [37, 38]. Palm and thumb sub-domains both form an exit channel, which releases RNA template.
3. FDA approved rdrp inhibitors and their binding mode
There are several RdRp inhibitors approved by FDA for the management of different viral infections (Fig. 2 ) as listed in the Table 1 .
Fig. 2.

Binding mode of FDA-approved drugs in the catalytic domain of different viral RdRps.
Table 1.
List of FDA approved RdRp inhibitors.
| S. No. | Name | Viral Infection | Mode of action | References |
|---|---|---|---|---|
| 1. | Remdesivir (Phase 3) | COVID-19, Ebola virus | It terminates the nascent viral RNA chains when interacted with them. | [39, 40] |
| 2. | PSI-7977 (Sofosbuvir) | Hepatitis C virus | Specifically inhibits the hepatitis C NS5B protein, undergoes metabolism intracellularly and forms uridine analog triphosphate (GS-461,203), a pharmacologically active compound. | [41] |
| 3. | Galidesivir | Flavivirus (broad spectrum) | Competes with natural nucleotide at its binding site and changes the conformation of the viral enzyme by altering electrostatic interactions. | [42] |
| 4. | Ribavirin | Hepatitis C virus | The metabolite ribavirin as ribavirin triphosphate (RTP) inhibits viral mRNA polymerase. | [43] |
| 5. | Favipiravir | Influenza | Phosphorylated favipiravir interferes with nascent RNA strand, and stops the elongation of RNA strand and prevents proliferation of virus. | [44] |
| 6. | Dasabuvir | Hepatitis C virus | Binds with NS5B polymerase in the palm domain and brings the structural changes which stops further extension of the viral genome. | [43] |
4. RdRp inhibitors for various viral infections
4.1. For BVDV (Bovine viral diarrhea virus)
In 2020, Ibba et al., designed and synthesized naphthoimidazole derivatives as an isosteric replacement of imidazoquinoline scaffold, earlier described by them as RdRp inhibitors in BVDV. They analysed the binding interaction to study the point of contacts of heteroatoms present in the scaffold with critical residues of the RdRp. Upon substitution of nitrogen in quinoline with a carbon prevented H-bond contact with Arg295 leading to a complete loss of inhibitory activity, as exhibited by their anti-viral assays. This analysis confirmed the relevance of interaction with Arg295. Although the top compound 1 did retain weak anti-viral activity, the isosteric replacement resulted in less active compounds (Fig. 3 ). The study finally established that the interaction between the aromatic ring and Ser411 residue of the RdRp in itself is not accountable for anti-BVDV activity [45].
Fig. 3.

Compound 1 as anti-BVDV agent with SAR analysis.
In 2020, Musiu et al., developed 104 quinolinecarboxamide analogues via optimizing two quinolinecarboxamide analogues, reported as selective inhibitors of RdRp of pestiviruses. They developed a 3D-QSAR model from these quinolinecarboxamide derivatives and concluded that introduction of aromatic rings with para substitutions of negatively charged groups at ring 1 enhances the activity. Similarly, addition of aromatic rings without para substitutions of negatively charged groups at ring 2 also enhances the activity (Fig. 4 ). Further they selected a BVDV variants resistant to numerous RdRp inhibitors and found that it was only partially inhibited by these derivatives. It was observed that although the derivatives inhibited the wild-type replication complexes, they could not inhibit mutant replication complexes. Interestingly, the derivative (2) did inhibit the activity of replication complexes but they could not inhibit the purified RdRp [46].
Fig. 4.

SAR profile of compound 2 as anti-BVDV agent.
In 2018, Loddo et al., described the synthesis of 9-aminoacridine derivatives as anti-viral agents via RdRp inhibition. The molecules were found able to selectively inhibit BVDV replication. Most potent compound 3 (Fig. 5 ) with EC50 value of 1.2 µM, showed favourable selectivity index in comparison to ribavirin (standard drug). All the derivatives had high CC50 values establishing the safety profile. Further the top molecules were subjected to biochemical assay for BVDV RdRp inhibitory potential. The analysis disclosed the compounds to have sub-micromolar level potency against RdRp. Further, ITC (Isothermal titration calorimetry) measurements were performed to determine their binding affinities, which were found to be in the range of 0.57–48 µM (Kd values). Detailed experimental analysis established that the 9-aminoacridine derivatives showed their action by inhibiting the replication of virus assembly via inhibition of RdRp of BVDV [47].
Fig. 5.

Substituted 9-aminoacridine as potent molecule against BVDV RdRp target.
In 2017, Santacruz et al., presented a study involving development of a series of thiosemicarbazones and N4-aryl substituted thiosemicarbazones as probable anti-BVDV compounds. The cell-based biological validation established compound 4 as active derivative, with EC50 value of 0.7 ± 0.1 µM. For N4- thiosemicarbazones, the SAR analysis disclosed that di‑methoxy substitution on the indanic ring is crucial for the activity. Additionally, N4-aryl substitution on the thiosemicarbazone moiety is also a critical feature for the anti-viral potential. Furthermore, an electron-withdrawing group on the aryl ring linked to N4 site on the thiosemicarbazone improved the anti-BVDV activity (Fig. 6 ) [48].
Fig. 6.

1-indanone and N4-aryl substituted thiosemicarbazones as potent anti-BVDV agent.
Previously in 2016, the same group reported the identification of 5 (3-(imidazo[1,2-a:5,4-b’]dipyridin-2-yl)aniline) for the preferential inhibition of CPE (cytopathic effect) produced by BVDV using virus-cell-based assay, with the EC50-values of 13.0 ± 0.6 mM. Surprisingly, this molecule showed no in vitro inhibitory potential against RdRp of recombinant BVDV, but exhibited inhibitory action against BVDV replication complexes in a dose-dependent manner. Through docking studies, it was recognized that the fingertip area in pestivirus RdRp is very vital for the inhibition of its function (Fig. 7 ) [49].
Fig. 7.

Imidazodipyridinylaniline derivative as inhibitor of the replication of pestiviruses.
In 2011, Carta et al., expanded the quinoline ring with 1,2,3-triazole/imidazole/pyrazine to explore the cytotoxicity and anti-viral potential of linear N-tricyclic ring systems. Upon testing against representatives of different RNA viruses, these quinoline-based molecules could only potently inhibit BVDV. Out of the three classes, imidazoquinolines and pyridoquinoxalines had more potent anti-BVDV activity than triazoloquinolines. Overall, imidazoquinoline 6 (Fig. 8 ), was more potent with selective activity against BVDV (EC50 =1.2 µM) and potency comparable to that of 20-C-methyl-guanosine, a known Flaviviridae RdRp inhibitor. In addition, compound 6 also showed anti-HCV potential with EC50 = 3.1 µM in a replicon assay [50].
Fig. 8.

Imidazoquinolines derivative as anti-BVDV agent with SAR analysis.
In continuation to the previous study, Carta et al., designed, synthesis and evaluated a plethora of molecules with linear aromatic N-tricyclic scaffold. Following their previous work, whereby derivatives of imidazo[4,5-g]quinoline were found to be potent anti-BVDV agents, they extended the SAR via rational alterations of lead molecule, 2-phenyl-3H-imidazo[4,5-g]quinoline, having EC50 value of 4 μM and no cellular toxicity. Various substitutions at multiple sites and replacing aryl ring with a furan ring resulted in 12 imidazo[4,5-g]quinoline based compounds. SAR analysis disclosed that chlorine/cyano at 4′ position of phenyl ring improves the anti-viral potential of the respective compound. Overall, 7 (EC50 = 0.3 μM) was found to be most potent amongst the whole series of compounds. Further in-silico experiments established the probable mechanism of action of the designed compounds. A 3D pharmacophore model was also developed, which disclosed that one aromatic hydrophobic feature and two H-bond acceptors were basic features required in a potent inhibitor. It was followed by MM/PBSA scoring, which disclosed the significance of a couple of RdRp amino acid residues, i.e., Arg295 and Tyr674, as the essential H-bond contacts (Fig. 9 ), on the basis of enthalpic component of binding Gibbs free energy change [51].
Fig. 9.

Imidazoquinoline derivative bearing cyano group as potent anti-BVDV agent.
4.2. For hepatitis C virus (HCV)
In 2016, Meguellatia et al., developed pseudodimeric aurones, considering both structure-based and ligand-based approaches of drug discovery, as inhibitors of NS5B (Nonstructural protein 5B) RdRp. Overall, they synthesized 14 compounds having a linker between the benzofuranone moieties and examined their HCV RdRp inhibitory potential using an in vitro assay. The most potent compound was found to be pseudodimeric aurone 8, with inhibitory activity of IC50 = 1.3 μM. The good inhibitory activity was attributed to the presence of phenyl ring as a spacer and -OH group at 4th and 6th position of benzofuran moiety (Fig. 10 ). The biological study also revealed that the distance between the two benzofuran moiety and shape of the molecule is also very important for its inhibitory action. Molecular docking analysis also highlight the significance of -OH groups as they form H-bond with Asn291. Hydrophobic interaction of benzofuran moiety with residues like Pro197, Met414, Tyr415, Ile447 and Tyr448 was also observed. Anti-HCV activity of pseudodimeric aurones was also evaluated using infectious model which signifies the correlation between the result of both HCV RdRp inhibitory activity and anti-HCV activity [52].
Fig. 10.

Pseudodimeric aurones with potential HCV RdRp inhibitory activity.
In 2016, Mohamed et al., synthesized series of pyrrole, pyrrolo[2,3-d]pyrimidine and pyrrolo[3,2-e][1,2,4]triazolo[4,3-c]pyrimidine derivatives and evaluated them for HCV inhibitory activity. Pyrrole and pyrrolo[3,2-e][1,2,4]triazolo[4,3-c]pyrimidine derivatives were found inactive against HCV genotype 4, while pyrrolo[2,3-d]pyrimidine derivatives exhibited substantial inhibitory potential. Compound 9 with IC50 value of 82.1 ± 10.3 µM, showed best potency amongst the pool, which indicated that pyrrolo[2,3-d]pyrimidine is crucial nucleus for anti-HCV activity. Incorporation of arylamino group at 4th position also increased the anti-viral activity (Fig. 11 ). Molecular docking studies revealed that compound 9 formed H-bond with Gln446 and exhibited a docking score of −24.92, kcal/mol. Overall findings concluded that this molecule can be used as lead compound for the designing of more potent anti-HCV agents [53].
Fig. 11.
Pyrrolo[2,3-d]pyrimidine derivative with potential HCV inhibitory activity.
In 2016, Wei et al., described the utilization of random forest based virtual screening (RB-VS) followed by e-pharmacophore (PB-VS), and molecular docking (DB-VS) based screening to filter HCV NS5B polymerase inhibitors. The protocol involved sequential use of the three methods (RB-VS, PB-VS and DB-VS) to filter InterBioScreen database. Finally, 5 compounds were selected for further validation via biological assays. Upon biochemical and cell-based evaluation, all 5 hits inhibited NS5B polymerase with the IC50 values in low micro molar range (2.01–23.84 μM) and exhibited anti-HCV activity with EC50 values of 1.61 to 21.88 μM, without any cytotoxicity (CC50 > 100 μM). Out of all, 10 showed top anti-viral potential for HCV inhibition (Fig. 12 ) [54].
Fig. 12.

Benzoxazole derivative as potent HCV NS5B polymerase inhibitor.
In 2015, Cakır et al., identified 5-arylidene-4-thiazolidinones as effective HCV NS5B polymerase inhibitors. A pool of 5-arylidene-4-thiazolidinones was synthesized and subjected to NS5B RdRp biochemical assay. The arylidene derivatives showed inhibition potential of 71–91% against NS5B RdRp while compounds without arylidene group showed only 6–31% inhibition. The screening result of NS5B inhibitory activity yielded 11 (IC50 = 25.3 µM) as most active. During the high throughput cell-based anti-HCV screening, 11 showed inhibition of ≥70% in HCV RNA replication process with cell survival of ≤45% at 100 mM concentration. Molecular docking studies disclosed the various binding interactions including cation–π contact of Arg501 and 3-fluorophenyl ring, and electrostatic interaction of 2‑chloro-6-fluorophenyl ring and Asn527/Arg422 residues in the thumb pocket-II of NS5B (Fig. 13 ). [55].
Fig. 13.

5-arylidene-4-thiazolidinone derivative as potent inhibitor of HCV NS5B polymerase.
In 2014, Therese et al., performed a comparative pharmacophore analysis studying the co-crystallized HCV NS5B structures to explore the active conformation of the ligands inside the allosteric pockets of palm and thumb sub-domains. In the next step, a high-throughput virtual screening along with molecular docking exercise was performed to screen varied non-peptide hits. The resulting top 10 hits, exhibiting different scaffolds, were then evaluated using NS5B and anti-HCV inhibition assays. Out of all, 12 was the most promising lead compound with benzimidazole scaffold (Fig. 14 ), with IC50 value of 28.8 μM in NS5B biochemical assay and inhibition of 97% at 50 μM in anti-HCV cell-based assay [56].
Fig. 14.

Benzimidazole derivative with HCV NS5B inhibitory activity.
In 2013, Peng et al., synthesized many unique derivatives of anilinobenzothiazole and evaluated them for their inhibitory potential on viral genome replication as HCV RdRp inhibitors. The compounds were in vitro evaluated for HCV RdRp inhibitory activity using Ava5 cells (cell with HCV replicon). They disclosed that compound 13 showed 76% inhibition of HCV RdRp activity in non-competitive manner and exhibited EC50 value of 8 ± 0.5 µM. SAR analysis revealed that substituents at 4th position of phenyl ring were important for inhibitory action. Molecular docking experiment performed considering Thumb II Pocket of HCV RdRp suggested that compound 13 well occupied the pocket and was found to be surrounded by four main residues Leu419, Leu497, Arg501, and Trp528 which are responsible for its good inhibitory activity (Fig. 15 ) [57].
Fig. 15.

Anilinobenzothiazole derivative with HCV RdRp inhibitory activity.
4.3. For zika virus (ZIKV)
In 2020, Wang et al. designed and synthesized novel derivatives of 1,4-dibenzylsubstituted piperazine on the basis of an in-silico screening of an in-built small molecule library. In the next step, screened hits were evaluated for their cytopathic protection effect (CPE) using a standard ZIKV-infected Vero E6 cellular assay. Initial examination disclosed five unique 4-amino-2-(4-benzylpiperazin-1-yl)methylbenzonitrile analogues having CPE lessening effects at micro-molar level. Furthermore, best hit 14 (Fig. 16 ), at low micro-molar concentrations, showed a substantial dose-dependent anti-viral activity by inhibiting both RNA replication of Zika and virus protein expression [58].
Fig. 16.
1, 4-bibenzylsubstituted piperazine derivatives as Zika inhibitors.
In 2017, Pattnaik et al., identified potent RdRp inhibitors for ZIKV via structure-based approach. The study involved evaluation of virus replication inhibitory potential of a library of small molecules via virtual screening, followed by validation of the screened hits using cell-based in vitro assays. Out of all, 15 was found to effectively inhibit ZIKV replication at nano molar concentrations with IC50 of 94 nM. Further, docking experiments were implemented to study the conformation and binding pose of 15 (Fig. 17 ), within the catalytic pocket of RdRp, which disclosed the allosteric mechanism involved in blocking the viral RNA synthesis. The two H-bond formed with residues Asp535 and Asp665 were key interactions as these coordinate with divalent metal ions (Mg2+) [59].
Fig. 17.

Binding pattern of 32 as potential drug candidate for ZIKV infections.
4.4. For Chikungunya virus (CHIKV)
In 2020, Kovacikova and Hemert discussed small molecule inhibitors for chikungunya virus, focussing on viral targets, mode of action, and mechanisms of antiviral drug resistance along with associated mutations [60]. In 2019, Yoon et al., designed and developed 6′-fluorinated analogues of aristeromycin, targeting viral RdRp along with SAH hydrolase of the host. The SAR analysis of the 6′-fluorinated aristeromycin analogues disclosed that incorporating fluorine on 6′-position upsurges the SAH hydrolase inhibitory activity along with inhibition of replication in certain +RNA viruses. It was also found that the 6′-fluorinated analogues more efficiently inhibited SAH hydrolase activity than the 6′-unsubstituted derivatives. Amongst all, 6′-β-fluoroaristeromycin, 16 (Fig. 18 ), proved to be better inhibitor for MERS-CoV replication, having an EC50 value of 0.20 μM. [61].
Fig. 18.

6′-fluorinated aristeromycin analogues with inhibitory effect against Chikungunya virus.
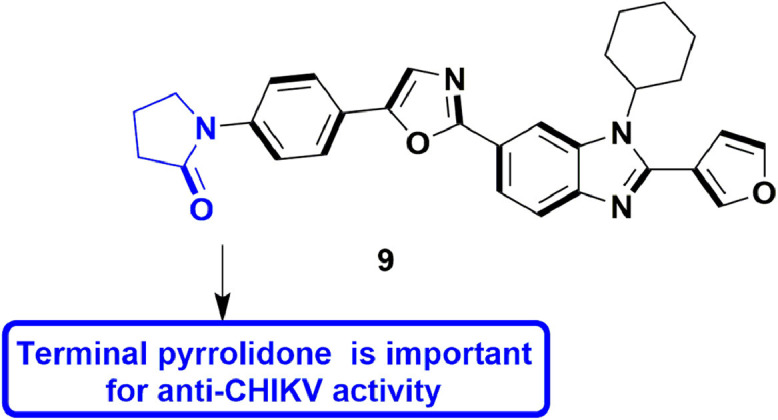
Wada et al., in 2017, screened chemical libraries virtually and found one candidate, a benzimidazole-based compound (17, Fig. 19 ), which at nano molar range, might potentially manage the infection of numerous CHIKV strains. Further to study the mechanism of inhibition, a resistant CHIKV (res-CHIKV) was isolated, which was then subjected to reverse-genetic recombinant CHIKVs to identify the key mutation associated with resistance. This detailed analysis disclosed that at the target site in the RdRp, the Met2295 residue was crucial for the binding of 9. They also confirmed that 9 used CHIKV replicons to inhibit the function of CHIKV RdRp [62].
Fig. 19.

Benzimidazole related compound as anti-CHIKV agent.
4.5. For covid-19
In 2020, Khan et al., screened NANPDB database for SARS-CoV-2 utilizing molecular docking approach. Out of all, 42 molecules showed improved docking score than Remdesivir. Top 5 hits along with top hit, 18, were subjected to MD simulation and free binding energy calculations, predicting better binding affinity than Remdesivir. Compound 18 (Diosmetin-7-O-Beta-d-apiofuranoside) exhibited high binding affinity score of −10.4 kcal/mol when compared to Remdesivir (−7.1 kcal/mol). It overall formed several H-bonds with different amino acid residues like Trp617, Tyr619, Lys621, Cys622, Asp623, Asp760, Asp761, Ala762, and Trp800, along with hydrophobic contacts with residues such as Asp618, Lys798, Glu811. Molinspiration Cheminformatics tool revealed the bioactivity score of 18 (Fig. 20 ) as 0.36 which is quite similar to Remdesivir (0.38). Finally, compound 18 was also found to comply pharmacokinetic rules such as Log P, hydrogen bond donor and acceptors [63].
Fig. 20.

Docking interactions of Diosmetin-7-O-Beta-d-apiofuranoside.
In 2020, Athar and Beg elaborated the function of anti-HIV and anti-HCV drugs in inhibiting RdRp of SARS CoV 2. The study involved homology modeling followed by validation of the obtained protein structure and docking experiments to study the binding potential of HIV/HCV drugs with RdRp. Results suggested that Nelfinavir, Raltegravir and Delavirdine, out of the studied anti-HIV, and Paritaprevir, Beclabuvir and Ledipasvir, out of the studied anti-HCV, might has potential to inhibit RdRp of SARS CoV-2 [64].
Procacci et al., in the year 2020 disclosed that HCQ (hydroxychloroquine) might behave as a moderate inhibitor of some SARS-CoV2 replication functional proteins. They utilized both enhanced sampling MD techniques and non-equilibrium alchemical transformations together. The ∆G0 value for PLpro was found to be 7.7 ± 0.9 kcal/mol, while for 3CLpro, it was 8.5 ± 1.1 kcal/mol, and for RdRp, it was 9.1 ± 1.4 kcal/mol. Further by analysing the state in bound configurations, they could enhance the affinity for 3CLpro, leading to a HCQ-based, 19, with ∆G0 value = 9.8 ± 1.4 kcal/mol. The binding potential increased upon introducing methyl-naphtyl moiety which enhances the hydrophobic interactions (Fig. 21 ). Also, the -OH of 19 made H-bond with Ser46 residue [65].
Fig. 21.

A novel HCQ-inspired compound with docking interactions.
Gordon et al., described the mechanistic study of RdRp inhibition via remdesivir in MERS-CoV. Study involved expression of non-structural proteins including RdRp, as a part a polyprotein in insect cells. Further analysis demonstrated that an active complex involves both nsp8 and nsp12 (RdRp) along with remdesivir (triphosphate), which contests with ATP, an RdRp substrate. Importantly, it was observed that active complex had preference for remdesivir-triphosphate over ATP and two other nucleotide analogues which reflects that it is more proficiently assimilated. The mechanism of remdesivir was found to be delay in RNA chain termination. Overall, this study helped in explaining the affinity potential of remdesivir against RNA viruses [66].
4.6. For Dengue virus (DENV)
In 2020, Wan et al., utilized the five-component reaction to synthesize derivatives of octahydroquinazoline-5-one (OHQs). The compounds were tested for anti-dengue activity by lactate dehydrogenase release assay in DENV-2 infected BHK-21 cells. Results suggested that octahydroquinazoline-5-one bearing 4-BrPh at 2nd position exhibited maximum activity (EC50 = 2.85 µM) amongst all in the series. Further they performed SAR analysis by introducing different substituents at 2nd position. It was observed that at 2nd position of OHQs, phenyl substitutent with electron withdrawing group at its para position showed good activity (Fig. 22 ). In addition, OHQs with 3-BrPh resulted into a most potent compound 20 (EC50 = 1.31 ± 0.21 µM) [67].
Fig. 22.

Octahydroquinazoline-5-ones with potential anti-dengue activity.
In 2019, Shimizu et al., employed a HTS (high throughput screen) to filter a library of 16,240 molecules and identified a hit molecule 21. The molecule was tested against four types of dengue i.e. DENV1–4 and displayed EC50 values from 6.0 to 31.9 μM. Considering best for DENV2 (EC50 = 6.0 μM), 21 was docked in DENV2 RdRp protein. Docking analysis identified two binding sites (Site 1 on thumb sub-domain and site 2 on palm-subdomain) for 21 within the RdRp. The residues like Cys780, Tyr882, Met809 surrounds benzoxathiole ring of 21 and acetate group showed interactions with Trp833, Arg773 (Fig. 23 ). While in site 2 benzoxathiole ring is surrounded by Glu510, Gly511, Ser661, and Cys709. Cysteine residue was the common residue for site 1 and site 2 and have close binding contact with 21. Also, the mutation analysis of Cys-to Ala-revealed that interaction of compound with cysteine is important for its anti-viral action [68].
Fig. 23.

Compound 21 showing the interacting residues in the active domain of DENV2 RdRp.
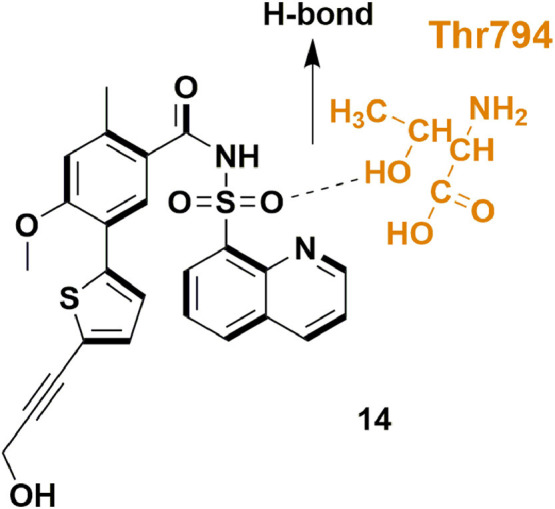
In 2018, Lim et al., identified a biphenyl acetic acid fragment targeting unique binding site, present at the thumb/palm boundary near to its N pocket, in the RdRp of dengue virus (DENV). They utilized a fragment-based screening via co-crystallized structure. Structure-directed modification resulted in a pool of nano molar range RdRp inhibitors (22; Fig. 24 ), which also showed sub-micro molar level EC50 values in cell-based assays [69].
Fig. 24.

Compound 14 with potential DENV RdRp inhibitory activity.
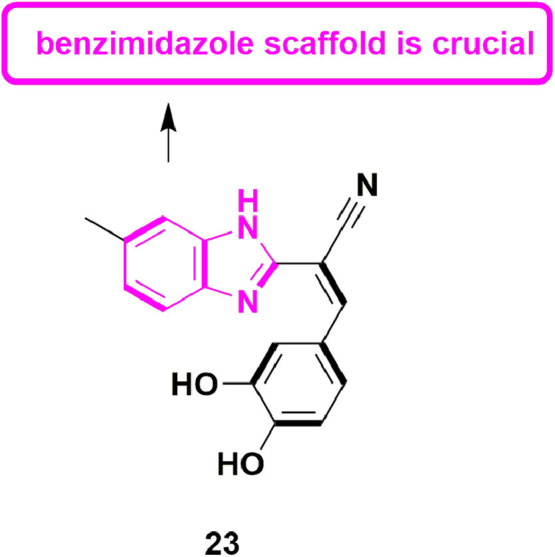
In 2017, Pelliccia et al., reported putative anti-DENV inhibitor which targets the NS5 RdRp. They recognized five lead molecules which showed potent anti-DENV activity. Out of five lead molecules, compound 23 (Fig. 25 ), EC50 = 5.99 ± 0.30 µM, exhibited most prominent anti-RdRp activity in cell-based assay. Further, enzymatic assays also confirmed the anti-RdRp activity of 23 with EC50 of 4.87 ± 0.24 µM. In-vivo DENV-infected ICR-suckling mouse model was used to evaluate the anti-DENV activity, which established that compound 23 was able to treat the DENV infection of mice [70].
Fig. 25.
Pyrazole derivative with potential RdRp inhibitory activity.
In 2016, Tarantino et al., identified a pyridobenzothiazole compound, 24, via an in silico docking approach. The compound was found to exhibit dengue RdRp inhibitory activity with IC50 = 1.5 ± 0.2 µM. Crystallographic analysis displayed that 24 was found to interact in the binding domain of fingers and priming loop of DENV 3 RdRp. Phenolic ring of 16 exhibited π-π stacking interaction with Trp795 in the priming loop which stabilized the molecule. Though, the bulky cyclohexane ring lead to loss of H-bonding with side chain Asn492 (Fig. 26 ). Further, mutational analyses identified a new binding cavity i.e. site 2, out of the primary loop. Interactions like H-bond with Cys753 was also observed, suggesting the importance of site 2 in RdRp inhibition [71].
Fig. 26.

Pyridobenzothiazole derivative with dengue RdRp inhibitory activity.
In 2016, Benmansour et al., reported various non-nucleoside based inhibitors of viral polymerase of dengue by modifying the N-phenyl-3- arylacrylamide scaffold of hit molecule ((2E)-N-(2- hydroxy‑4-nitrophenyl)−3-(5-nitrothiophen-2-yl)prop‑2-enamide). The 1,3,4-oxadiazoles and 1,2,4-oxadiazoles derivatives of the same hit compound were developed and evaluated for DENV-2 RdRp. It resulted in a potent compound 25 (bearing 1,2,4-oxadiazole moiety) with IC50 value of 2.2 ± 0.1 µM. The compound also exhibited anti-viral activity in micromolar concentration, when tested against DENV-infected Vero cells. SAR analysis indicated that presence of 4-chlorophenyl at 3rd position of oxadiazole moiety is important for DENV-2 RdRp activity (Fig. 27 ). In addition, bromo group at thiophene is also crucial for anti-viral activity [72].
Fig. 27.

1,2,4-oxadiazole derivative as potent DENV-2 RdRp inhibitor.
In 2015, Manvar et al., described the development of coupled thiazolidinone-thiadiazole molecules as novel inhibitors of DENV-2 NS5 RdRp. Around 40 molecules bearing different substituents on thiazolidinone and thiadiazole rings were synthesized. Out of them, 26 (IC50 = 2.1 µM) emerged as a potent compound. SAR study showed that 3-fluorobenzylidene at C5 of the thiazolidinone nucleus is important for activity (Fig. 28 ). Also, the existence of 2-chlorophenyl at C5 in the 1,3,4-thiadiazole ring is crucial. Molecular docking studies identified five binding sites (S1-S5). S1 is recognized as palm, S2 and S3 covers thumb pocket I and II, and S5 is located at fingers of protein. Binding energy score revealed site 3 with thumb pocket-II as desirable binding domain for compound 26 [73] The H-bond interactions were observed between NH of thiazolidinone ring and Pro884, and between imine group and Ser885. The fluorobenzylidene group showed aromatic interaction with Tyr882 whereas chloro group exhibited electrostatic interaction with Asn777 [73].
Fig. 28.

Hybrid thiazolidinone-thiadiazole derivative as DENV-2 NS5 RdRp inhibitor.
4.7. For influenza virus
In 2019, Yang et al., detailed the discovery of novel derivatives containing 5-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrazin-2(1H)-one skeleton as inhibitors of PB2 (a significant subunit of influenza RdRp). Biological evaluation results revealed compound 27 as one of the most potent compound among the series with sub-micro molar Kd values in surface plasmon resonance (SPR; 0.11 μM) and isothermal titration calorimetry (ITC; 0.19 μM) assays. Compound 27, when examined for in-vitro anti-viral activity, showed higher EC50 value (1.025 μM) than standard drug Ribavirin (9.116 μM). Molecular docking analysis disclosed that scaffold of compound 27 formed many important interactions like H-bond with Lys376 and Glu361 residues, electrostatic interaction as well as H-bond with Arg355 and π-π interactions with His357. SAR studies indicated the importance of ethyl group at N-1 of pyrazine ring of compound 27 (Fig. 29 ), if replaced by higher bulky group, a decrease in the anti-viral activity was observed [74].
Fig. 29.

5-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrazin-2(1H)-one as potent PB2 inhibitor for the treatment of influenza.
In 2019, Zhao et al., worked on identifying inhibitors of influenza A via screening strategy focussed on the transcription/replication stages by means of an RdRp-focussed filter. Followed by employing those same molecules in a replication-competent reporter virus-based approach. A library of natural product derived compounds (891 molecules) was subjected to the same approach to validate its utility, which established the primary screen to be rigorous for screening RdRp inhibitors for influenza A virus. In conclusion, out of the two screened hits, one was confirmed as a RdRp inhibitor [75].
In 2014, Lepri et al., designed various molecules as RdRp inhibitors aiming at PA−PB1 (polymerase acidic protein-basic protein 1) complex by modifying the chemical space of a molecule containing thiophene-3-carboxamide and pyrazolo[1,5-a]pyrimidine scaffolds [76], which was previously proved to possess RdRp inhibitory activity. Two approaches were utilized for the design of new compounds but thiophene-3-carboxamide moiety was preserved as an important moiety responsible for RdRp inhibition. Firstly, various new structural features were introduced to explore the interactions capable of interrupting of PA−PB1 complex. The second step utilized scaffold hopping to find new hits with different scaffolds as PA−PB1 complex inhibitor. When pyrazolo[1,5-a]pyrimidine was substituted with triazolo-pyrimidine ring, the PA−PB1 inhibitory activity increased three times. As a result, compound 28 containing triazolo-pyrimidine moiety with CF3 and cyclohexane substituents exhibited IC50 value of 7.5 ± 0.7 μM in ELISA PA−PB1 interaction assay. These results concluded that derivatives with linear shape are responsible for the good inhibitory activity. Scaffold hopping also resulted in a compound containing sulphonamide moiety with similar inhibitory activity as that of compound 28 (Fig. 30 ). Compounds 28 was also tested against FluA and FluB strains and displayed significant anti-viral potential. [77].
Fig. 30.

Compound 26 as potent PA−PB1 inhibitor for treatment of the influenza.
4.8. For norovirus
In 2020, Yi et al., explored the N-pocket in the RdRp of dengue virus (DENV) and its comparable B-site in the RdRp of human norovirus (hNV). They executed a structure-based virtual screening in parallel, considering both the binding sites, to identify hits which can hamper RdRps of hNV and DENV. They positively recognized Entrectinib (29) as an effective inhibitor of RdRps of both hNV and DENV. Explicitly, 29 displayed dose-responsive inhibition of both MNV (murine norovirus) and DENV2 with the IC50 values of 2.01 and 2.43 mM, respectively. It is reported to bind to respective RdRps and efficiently inhibit the catalytic function in the biological assays. The indazole ring of compound 29 exhibit cation-π interaction with Arg392 (Fig. 31 ). It also interacts with other residues like Val504, Glu506, and Asp507 in C-terminal of NV-RdRp. [78].
Fig. 31.

Entrectinib as potent compound for dual inhibition of hNV RdRp and DENV RdRp.
In 2019, Harmalkar et al., designed various stilbene analogs and tested their inhibitory effect on RNA replication in human norovirus (HNV) by means of HG23, a replicon-bearing viral cell line. Initially, authors screened a large number of natural products against NV which resulted into a hit containing stilbene and vinyl moiety. Taking this hit into consideration, various new compounds were synthesized by introducing substituted amides. Many amide derivatives possessed anti-NV activity with EC50 values within 1 to 2 μM. However, introduction of piperazine amide resulted in a potent compound 30 with 95.8% inhibition of NV replication. Moreover, compound 30 (Fig. 32 ) also showed good safety profile with therapeutic index of 41.2 and also showed less toxicity showing the maximum suppressive action on replication of virus [79].
Fig. 32.

Modified stilbene analog with piperazine moiety as inhibitor of NV replication.
In 2019, Giancotti et al., modified the skeleton of a HuNoV RdRp inhibitor and an inhibitor of Zika virus polymerase to design modified inhibitors for HuNoV RdRp. They synthesized 12 compounds and almost all the compounds showed partial inhibition of HuNoV (Human Noravirus) RdRp activity in in-vitro assays. Especially, out of 12, 5 compounds appeared to cause decrease in MNV-induced cytopathic effect with micro molar range EC50 values. Also, these compounds were not found substantially cytotoxic. These five hits were further subjected to human norovirus replicon assay possessing a geno-group GI. One of the 5 hits, 31 (Fig. 33 ), having a novel anti-viral scaffold, showed EC50 values in the low micro molar level in inhibiting HuNoV replication [80].
Fig. 33.

Compound 31 as potent HuNoV RdRp inhibitor.
In 2014, Tarantino et al., evaluated naphthalene di-sulfonate (NAF2), a fragment-molecule derivative from suramin and NF023 (previously reported RdRp inhibitor of norovirus) [81] for their inhibitory action against Norovirus RdRp. This compound showed less RdRp inhibitory activity (IC50 = 14 µM) when tested in polymerase inhibition assays. However, this fragment molecule binds to thumb domain which is different binding site when compared to NF023 and suramin. Additionally, another structurally correlated molecule PPNDS (32; pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) tetrasodium salt) was tested for human Norovirus RdRp activity. The compound 32 exhibited IC50 value of 0.45 µM which is quite closer to suramin (0.24 ± 0.08 µM). Compound 32 also occupied well the thumb domain of hNV-RdRp protein. Various substituents of PPNDS showed many contacts including hydrophobic interactions of nitro group with Leu406, Ile411, Leu443 and Val504. The carbonyl group of pyridoxal ring showed H-bond with Glu168 residue. The sulfonate group at 4th and 8th position on naphthalene ring formed H-bond with Ser410 and Gln414 (Fig. 34 ). These findings highlighted the significance of binding site (thumb domain) for further development of Norovirus RdRp inhibitors [82].
Fig. 34.
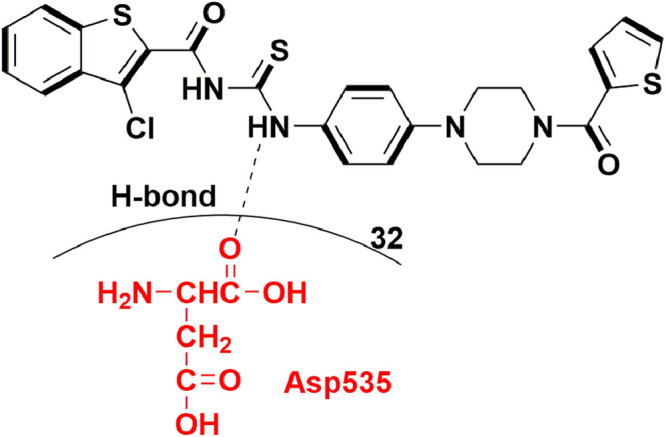
PPNDS as potent molecule with hNV-RdRp inhibitory activity.
5. Key observations for designing RdRp inhibitors against different viruses
Upon careful investigation of all the small molecule inhibitors reported as RdRp inhibitors for different viruses, some generalizations can be made. Therefore, here we will highlight the key structural features needed, in each virus type discussed above, to design potent RdRp inhibitors. Firstly, for RdRp inhibitors of BVDV, the most important requirements include H-bond interactions of the scaffold or substituents of the designed hits and Arg264, E265, Arg285, Arg295 and Y674 amino acid residues in the binding pocket. In case of HCV, the most important requirements include H-bond interactions with Asn291, Arg422, Gln446 and Ser476, and hydrophobic interactions with residues like Pro197, Met414, Tyr415, Ile447 and Tyr448 in the binding pocket. While to design RdRp inhibitors for Zika, attention should be given to incorporate pharmacophoric features which could form two H-bond interactions with D535 and D665 amino acid residues as these coordinates with divalent metal ions (Mg2+) in the pocket. In chikungunya, analysis of recent RdRp inhibitors suggests that interaction with M2295 at the target site in the RdRp is crucial for the binding and inhibition. In case of dengue, prospective RdRp inhibitors are expected to occupy two sites in the binding pocket. In one site interactions with Cys780, Tyr882 and Met809 are considered crucial while in the other site interactions with Glu510, Gly511, Ser661, and Cys709 are considered vital. To design the RdRp inhibitors for influenza, focus should be laid on the molecules which could form H-bond with Lys376, Glu361 and Arg355, along with π-π interactions with His357. In case of norovirus, RdRp inhibitors need to form H-bond interactions with Glu168, Ser410 and Gln414. However, a more important elucidation from these observations is the fact that different RdRp inhibitors are quite selective for the class of virus they are designed due to the different pharmacophoric requirements in each case and therefore, a more focussed and specific approach should be followed to design RdRp inhibitors.
6. Conclusion
Although anti-viral research has been continuous, COVID-19 has highlighted a need for a renewed focus on the management and treatment of viral infections. One of the key reasons for this alarming situation is incidence of the novel variants of previously manageable viral classes. The development of anti-viral drugs has become a key task for medicinal chemistry researchers in this past horrible year. And out of all the known protein targets from the viral genome, RdRp remains most sought-after target for the family of RNA-viruses. Expectedly, Molnupiravir, an RdRp inhibitor developed by Merck, has been approved by the FDA for the management of Covid-19 indicating the efficacy of RdRp targeted agents for the management of current pandemic. Consideration of the structure of viral RdRp is crucial for accurate computational as well as knowledge-based drug discovery protocols for pathological conditions like COVID-19. Therefore, the information provided in this compilation could be utilized to increase the library of pre-clinical molecules having potency and efficacy against Covid-19. The main composition of RdRps is quite similar to that of DNA and RNA polymerases, including motifs and sub-domains. Therefore, it is presumed that individually every RdRp inhibitor might possess common structural features or binding conformations that can explain a similar inhibitory effect throughout the RNA virus family. However, our compilation clearly suggests that RdRps belonging to different viruses have different crucial interaction points in their binding pocket and in each case, inhibitors require different pharmacophoric features to have significant affinity. This observation also hints at one of the reasons of why already reported RdRp inhibitors did not fare well against Covid-19. Accordingly, in the current review, the diverse RdRp inhibitors reported in the last few years have been analysed to draw conclusions about the designing of RdRp inhibitors. The structure activity relationship profile and binding conformations of the reported inhibitors are also discussed to elucidate some generalisations for the designing of further potent inhibitors. Overall, the information can be utilized to increase preclinical candidates targeting RdRp for current global pandemic.
Declaration of Competing Interest
Authors declare no conflict of interest.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
SP and RKR would like to acknowledge the faculty members of the ISF College of Pharmacy, Moga, Maharishi Markandeshwar (Deemed to University), Mullana, India and Director, CSIR-NEIST, Jorhat for their direction all the times. PKS would like to thank TCSMT for the Post-doctoral fellowship.
References
- 1.Cameron C.E., Götte M., Raney K.D. Springer; 2009. Viral Genome Replication. [Google Scholar]
- 2.Claverie J.-.M. Viruses take center stage in cellular evolution. Genome Biol. 2006;7(6):1–5. doi: 10.1186/gb-2006-7-6-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahlquist P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science. 2002;296(5571):1270–1273. doi: 10.1126/science.1069132. [DOI] [PubMed] [Google Scholar]
- 4.Ahlquist P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 2006;4(5):371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakrabarti S.S., Kaur U., Banerjee A., Ganguly U., Banerjee T., Saha S., Parashar G., Prasad S., Chakrabarti S., Mittal A. COVID-19 in India: are biological and environmental factors helping to stem the incidence and severity? Aging Dis. 2020;11(3):480–488. doi: 10.14336/AD.2020.0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia R., Ganti S.S., Narang R.K., Rawal R.K. Strategies and challenges to develop therapeutic candidates against COVID-19 pandemic. Open Virol J. 2020;14(1):16–21. doi: 10.2174/1874357902014010016. [DOI] [Google Scholar]
- 7.Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhary S., Sharma K., Singh P.K. Von Willebrand factor: a key glycoprotein involved in thrombo-inflammatory complications of COVID-19. Chem. Biol. Interact. 2021;348 doi: 10.1016/j.cbi.2021.109657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jensen S., Thomsen A.R. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012;86(6):2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Díez J., Ishikawa M., Kaido M., Ahlquist P. Identification and characterization of a host protein required for efficient template selection in viral RNA replication. Proc. Natl. Acad. Sci. 2000;97(8):3913–3918. doi: 10.1073/pnas.080072997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes E.C. What does virus evolution tell us about virus origins? J. Virol. 2011;85(11):5247–5251. doi: 10.1128/JVI.02203-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonelli M., Cichero E. Fight against H1N1 influenza A virus: recent insights towards the development of druggable compounds. Curr. Med. Chem. 2016;23(18):1802–1817. doi: 10.2174/0929867323666160210124930. [DOI] [PubMed] [Google Scholar]
- 13.Singh P., Pathania S., Rawal R. Exploring RdRp–remdesivir interactions to screen RdRp inhibitors for the management of novel coronavirus 2019-nCoV. SAR QSAR Environ. Res. 2020;31(11):857–867. doi: 10.1080/1062936X.2020.1825014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhatia R., Narang R.K., Rawal R.K. A summary of viral targets and recently released PDB IDs of SARS-CoV-2. Virology. 2015;478:75–85. [Google Scholar]
- 15.Poltronieri P., Sun B., Mallardo M. RNA viruses: RNA roles in pathogenesis, coreplication and viral load. Curr. Genomics. 2015;16(5):327–335. doi: 10.2174/1389202916666150707160613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar R., Mishra S., Maurya S.K. Recent advances in the discovery of potent RNA-dependent RNA-polymerase (RdRp) inhibitors targeting viruses. RSC Med. Chem. 2021;12(3):306–320. doi: 10.1039/D0MD00318B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian L., Qiang T., Liang C., Ren X., Jia M., Zhang J., Li J., Wan M., YuWen X., Li H. RNA-dependent RNA polymerase (RdRp) inhibitors: the current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021;213 doi: 10.1016/j.ejmech.2021.113201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Machitani M., Yasukawa M., Nakashima J., Furuichi Y., Masutomi K. RNA-dependent RNA polymerase, RdRP, a promising therapeutic target for cancer and potentially COVID-19. Cancer Sci. 2020;111(11):3976–3984. doi: 10.1111/cas.14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng K.K.-S., Arnold J.J., Cameron C.E. Springer; 2008. Structure-function Relationships Among RNA-dependent RNA polymerases, RNA Interference; pp. 137–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Černý J., Bolfíková B.Č., Valdes J.J., Grubhoffer L., Růžek D. Evolution of tertiary structure of viral RNA dependent polymerases. PLoS ONE. 2014;9(5):e96070. doi: 10.1371/journal.pone.0096070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J.-.H., Chung M.S., Kim K.H. Structure and function of caliciviral RNA polymerases. Viruses. 2017;9(11):329. doi: 10.3390/v9110329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graham S.C., Sarin L.P., Bahar M.W., Myers R.A., Stuart D.I., Bamford D.H., Grimes J.M. The N-terminus of the RNA polymerase from infectious pancreatic necrosis virus is the determinant of genome attachment. PLoS Pathog. 2011;7(6) doi: 10.1371/journal.ppat.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.te Velthuis A.J. Common and unique features of viral RNA-dependent polymerases. Cell. Mol. Life Sci. 2014;71(22):4403–4420. doi: 10.1007/s00018-014-1695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu G., Gong P. A structural view of the RNA-dependent RNA polymerases from the Flavivirus genus. Virus Res. 2017;234:34–43. doi: 10.1016/j.virusres.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Ferrer-Orta C., Ferrero D., Verdaguer N. RNA-dependent RNA polymerases of picornaviruses: from the structure to regulatory mechanisms. Viruses. 2015;7(8):4438–4460. doi: 10.3390/v7082829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Surana P., Satchidanandam V., Nair D.T. RNA-dependent RNA polymerase of Japanese encephalitis virus binds the initiator nucleotide GTP to form a mechanistically important pre-initiation state. Nucleic Acids Res. 2014;42(4):2758–2773. doi: 10.1093/nar/gkt1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collier A.M., Lyytinen O.L., Guo Y.R., Toh Y., Poranen M.M., Tao Y.J. Initiation of RNA polymerization and polymerase encapsidation by a small dsRNA virus. PLoS Pathog. 2016;12(4) doi: 10.1371/journal.ppat.1005523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jácome R., Becerra A., Ponce de León S., Lazcano A. Structural analysis of monomeric RNA-dependent polymerases: evolutionary and therapeutic implications. PLoS ONE. 2015;10(9) doi: 10.1371/journal.pone.0139001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukarska M., Fournier G., Pflug A., Resa-Infante P., Reich S., Naffakh N., Cusack S. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature. 2017;541(7635):117–121. doi: 10.1038/nature20594. [DOI] [PubMed] [Google Scholar]
- 30.Garriga D., Ferrer-Orta C., Querol-Audí J., Oliva B., Verdaguer N. Role of motif B loop in allosteric regulation of RNA-dependent RNA polymerization activity. J. Mol. Biol. 2013;425(13):2279–2287. doi: 10.1016/j.jmb.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y., Xiao M., Chen J., Zhang W., Luo J., Bao K., Nie M., Chen J., Li B. Mutational analysis of the GDD sequence motif of classical swine fever virus RNA-dependent RNA polymerases. Virus Genes. 2007;34(1):63–65. doi: 10.1007/s11262-006-0001-z. [DOI] [PubMed] [Google Scholar]
- 32.Yang X., Smidansky E.D., Maksimchuk K.R., Lum D., Welch J.L., Arnold J.J., Cameron C.E., Boehr D.D. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure. 2012;20(9):1519–1527. doi: 10.1016/j.str.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castro C., Smidansky E.D., Arnold J.J., Maksimchuk K.R., Moustafa I., Uchida A., Götte M., Konigsberg W., Cameron C.E. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat. Struct. Mol. Biol. 2009;16(2):212–218. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kidmose R.T., Vasiliev N.N., Chetverin A.B., Andersen G.R., Knudsen C.R. Structure of the Qβ replicase, an RNA-dependent RNA polymerase consisting of viral and host proteins. Proc. Natl. Acad. Sci. 2010;107(24):10884–10889. doi: 10.1073/pnas.1003015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu C., Fan S., Li C., Macken C., Kim J.H., Hatta M., Neumann G., Kawaoka Y. Functional analysis of conserved motifs in influenza virus PB1 protein. PLoS ONE. 2012;7(5):e36113. doi: 10.1371/journal.pone.0036113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrer-Orta C., Arias A., Escarmís C., Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2006;16(1):27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 37.van der Linden L., Vives-Adrián L., Selisko B., Ferrer-Orta C., Liu X., Lanke K., Ulferts R., De Palma A.M., Tanchis F., Goris N. The RNA template channel of the RNA-dependent RNA polymerase as a target for development of antiviral therapy of multiple genera within a virus family. PLoS Pathog. 2015;11(3) doi: 10.1371/journal.ppat.1004733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald S.M., Tao Y.J., Patton J.T. The ins and outs of four-tunneled Reoviridae RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2009;19(6):775–782. doi: 10.1016/j.sbi.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang C., Tian L., Liu Y., Hui N., Qiao G., Li H., Shi Z., Tang Y., Zhang D., Xie X. A promising antiviral candidate drug for the COVID-19 pandemic: a mini-review of remdesivir. Eur. J. Med. Chem. 2020;201 doi: 10.1016/j.ejmech.2020.112527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullard A. 2017 FDA drug approvals. Nat. Rev. Drug Discov. 2018;17(2):81–86. doi: 10.1038/nrd.2018.4. [DOI] [PubMed] [Google Scholar]
- 42.Westover J.B., Mathis A., Taylor R., Wandersee L., Bailey K.W., Sefing E.J., Hickerson B.T., Jung K.-.H., Sheridan W.P., Gowen B.B. Galidesivir limits Rift Valley fever virus infection and disease in Syrian golden hamsters. Antiviral Res. 2018;156:38–45. doi: 10.1016/j.antiviral.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers R.P., Shah H., Burak K.W., Cooper C., Feld J.J. An update on the management of chronic hepatitis C: 2015 Consensus guidelines from the Canadian Association for the Study of the Liver. Can. J. Gastroenterol. Hepatol. 2015;29(1):19–34. doi: 10.1155/2015/692408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayden F.G., Shindo N. Influenza virus polymerase inhibitors in clinical development. Curr. Opin. Infect. Dis. 2019;32(2):176–186. doi: 10.1097/QCO.0000000000000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ibba R., Piras S., Delogu I., Loddo R., Carta A. Anti-BVDV Activity Evaluation of Naphthoimidazole Derivatives Compared with Parental Imidazoquinoline Compounds. Open Medicinal Chem. J. 2020;14(1):65–70. doi: 10.2174/1874104502014010065. [DOI] [Google Scholar]
- 46.Musiu S., Castillo Y.P., Muigg A., Pürstinger G., Leyssen P., Froeyen M., Neyts J., Paeshuyse J. Quinolinecarboxamides Inhibit the Replication of the Bovine Viral Diarrhea Virus by Targeting a Hot Spot for the Inhibition of Pestivirus Replication in the RNA-Dependent RNA Polymerase. Molecules. 2020;25(6):1283. doi: 10.3390/molecules25061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loddo R., Francesconi V., Laurini E., Boccardo S., Aulic S., Fermeglia M., Pricl S., Tonelli M. 9-Aminoacridine-based agents impair the bovine viral diarrhea virus (BVDV) replication targeting the RNA-dependent RNA polymerase (RdRp) Bioorg. Med. Chem. 2018;26(4):855–868. doi: 10.1016/j.bmc.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Santacruz M.C.S., Fabiani M., Castro E.F., Cavallaro L.V., Finkielsztein L.M. Synthesis, antiviral evaluation and molecular docking studies of N4-aryl substituted/unsubstituted thiosemicarbazones derived from 1-indanones as potent anti-bovine viral diarrhea virus agents. Bioorg. Med. Chem. 2017;25(15):4055–4063. doi: 10.1016/j.bmc.2017.05.056. [DOI] [PubMed] [Google Scholar]
- 49.Musiu S., Leyssen P., Froeyen M., Chezal J.-.M., Neyts J., Paeshuyse J. 3-(imidazo [1, 2-a: 5, 4-b′] dipyridin-2-yl) aniline inhibits pestivirus replication by targeting a hot spot drug binding pocket in the RNA-dependent RNA polymerase. Antiviral Res. 2016;129:99–103. doi: 10.1016/j.antiviral.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Carta A., Briguglio I., Piras S., Corona P., Boatto G., Nieddu M., Giunchedi P., Marongiu M.E., Giliberti G., Iuliano F. Quinoline tricyclic derivatives. Design, synthesis and evaluation of the antiviral activity of three new classes of RNA-dependent RNA polymerase inhibitors. Bioorg. Med. Chem. 2011;19(23):7070–7084. doi: 10.1016/j.bmc.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 51.Carta A., Briguglio I., Piras S., Corona P., Ibba R., Laurini E., Fermeglia M., Pricl S., Desideri N., Atzori E. A combined in silico/in vitro approach unveils common molecular requirements for efficient BVDV RdRp binding of linear aromatic N-polycyclic systems. Eur. J. Med. Chem. 2016;117:321–334. doi: 10.1016/j.ejmech.2016.03.080. [DOI] [PubMed] [Google Scholar]
- 52.Meguellati A., Ahmed-Belkacem A., Nurisso A., Yi W., Brillet R., Berqouch N., Chavoutier L., Fortuné A., Pawlotsky J.-.M., Boumendjel A. New pseudodimeric aurones as palm pocket inhibitors of Hepatitis C virus RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2016;115:217–229. doi: 10.1016/j.ejmech.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Mohamed M.S., Sayed A.I., Khedr M.A., Soror S.H. Design, synthesis, assessment, and molecular docking of novel pyrrolopyrimidine (7-deazapurine) derivatives as non-nucleoside hepatitis C virus NS5B polymerase inhibitors. Bioorg. Med. Chem. 2016;24(9):2146–2157. doi: 10.1016/j.bmc.2016.03.046. [DOI] [PubMed] [Google Scholar]
- 54.Wei Y., Li J., Qing J., Huang M., Wu M., Gao F., Li D., Hong Z., Kong L., Huang W. Discovery of novel hepatitis C virus NS5B polymerase inhibitors by combining random forest, multiple e-pharmacophore modeling and docking. PLoS ONE. 2016;11(2) doi: 10.1371/journal.pone.0148181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Çakır G., Küçükgüzel İ., Guhamazumder R., Tatar E., Manvar D., Basu A., Patel B.A., Zia J., Talele T.T., Kaushik-Basu N. Novel 4-thiazolidinones as non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA polymerase. Arch. Pharm. 2015;348(1) doi: 10.1002/ardp.201400247. doi:10-22 10.1002/ardp.201400247. [DOI] [PubMed] [Google Scholar]
- 56.Therese P.J., Manvar D., Kondepudi S., Battu M.B., Sriram D., Basu A., Yogeeswari P., Kaushik-Basu N. Multiple e-pharmacophore modeling, 3D-QSAR, and high-throughput virtual screening of hepatitis C virus NS5B polymerase inhibitors. J. Chem. Inf. Model. 2014;54(2):539–552. doi: 10.1021/ci400644r. [DOI] [PubMed] [Google Scholar]
- 57.Peng H.-.K., Chen W.-.C., Lin Y.-.T., Tseng C.-.K., Yang S.-.Y., Tzeng C.-.C., Lee J.-.C., Yang S.-.C. Anti-hepatitis C virus RdRp activity and replication of novel anilinobenzothiazole derivatives. Antiviral Res. 2013;100(1):269–275. doi: 10.1016/j.antiviral.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y., Zhou R., Quan Y., Chen S., Shi X., Li Y., Cen S. Design, synthesis, and evaluation of novel 4-amino-2-(4-benzylpiperazin-1-yl) methylbenzonitrile compounds as Zika inhibitors. Bioorg. Med. Chem. Lett. 2020;30(4) doi: 10.1016/j.bmcl.2019.126906. [DOI] [PubMed] [Google Scholar]
- 59.Pattnaik A., Palermo N., Sahoo B.R., Yuan Z., Hu D., Annamalai A.S., Vu H.L., Correas I., Prathipati P.K., Destache C.J. Discovery of a non-nucleoside RNA polymerase inhibitor for blocking Zika virus replication through in silico screening. Antiviral Res. 2018;151:78–86. doi: 10.1016/j.antiviral.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Kovacikova K., van Hemert M.J. Small-molecule inhibitors of chikungunya virus: mechanisms of action and antiviral drug resistance. Antimicrob. Agents Chemother. 2020;64(12):e01788. doi: 10.1128/AAC.01788-20. -20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoon J.-s., Kim G., Jarhad D.B., Kim H.-.R., Shin Y.-.S., Qu S., Sahu P.K., Kim H.O., Lee H.W., Wang S.B. Design, synthesis, and anti-RNA virus activity of 6′-fluorinated-aristeromycin analogues. J. Med. Chem. 2019;62(13):6346–6362. doi: 10.1021/acs.jmedchem.9b00781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wada Y., Orba Y., Sasaki M., Kobayashi S., Carr M.J., Nobori H., Sato A., Hall W.W., Sawa H. Discovery of a novel antiviral agent targeting the nonstructural protein 4 (nsP4) of chikungunya virus. Virology. 2017;505:102–112. doi: 10.1016/j.virol.2017.02.014. [DOI] [PubMed] [Google Scholar]
- 63.Khan A., Khan M., Saleem S., Babar Z., Ali A., Khan A.A., Sardar Z., Hamayun F., Ali S.S., Wei D.-.Q. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARs-CoV-2 with natural products. Interdiscip. Sci. Comput. Life Sci. 2020;12(3):335–348. doi: 10.1039/D0CC03558K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beg M., Athar F. Anti-HIV and Anti-HCV drugs are the putative inhibitors of RNA-dependent-RNA polymerase activity of NSP12 of the SARS CoV-2 (COVID-19) Pharm. Pharmacol. Int. J. 2020;8(3):163–172. [Google Scholar]
- 65.Procacci P., Macchiagodena M., Pagliai M., Guarnieri G., Iannone F. Interaction of hydroxychloroquine with SARS-CoV2 functional proteins using all-atoms non-equilibrium alchemical simulations. Chem. Comm. 2020;56(62):8854–8856. doi: 10.1039/d0cc03558k. [DOI] [PubMed] [Google Scholar]
- 66.Gordon C.J., Tchesnokov E.P., Feng J.Y., Porter D.P., Götte M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020;295(15):4773–4779. doi: 10.1074/jbc.AC120.013056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wan Y., Wu S., Zheng S., Liang E., Liu S., Yao X., Zhu Q. A series of octahydroquinazoline-5-ones as novel inhibitors against dengue virus. Eur. J. Med. Chem. 2020;200 doi: 10.1016/j.ejmech.2020.112318. [DOI] [PubMed] [Google Scholar]
- 68.Shimizu H., Saito A., Mikuni J., Nakayama E.E., Koyama H., Honma T., Shirouzu M., Sekine S.-i., Shioda T. Discovery of a small molecule inhibitor targeting dengue virus NS5 RNA-dependent RNA polymerase. PLoS Negl. Trop. Dis. 2019;13(11) doi: 10.1371/journal.pntd.0007894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lim S.P., Noble C.G., Nilar S., Shi P.-.Y., Yokokawa F. Dengue and Zika: Control and Antiviral Treatment Strategies. Springer; 2018. Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from fragment screening and structure-guided design; pp. 187–198. [DOI] [PubMed] [Google Scholar]
- 70.Pelliccia S., Wu Y.-.H., Coluccia A., La Regina G., Tseng C.-.K., Famiglini V., Masci D., Hiscott J., Lee J.-.C., Silvestri R. Inhibition of dengue virus replication by novel inhibitors of RNA-dependent RNA polymerase and protease activities. J. Enzyme Inhib. Med. Chem. 2017;32(1):1091–1101. doi: 10.1080/14756366.2017.1355791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tarantino D., Cannalire R., Mastrangelo E., Croci R., Querat G., Barreca M.L., Bolognesi M., Manfroni G., Cecchetti V., Milani M. Targeting flavivirus RNA dependent RNA polymerase through a pyridobenzothiazole inhibitor. Antiviral Res. 2016;134:226–235. doi: 10.1016/j.antiviral.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 72.Benmansour F., Eydoux C., Querat G., de Lamballerie X., Canard B., Alvarez K., Guillemot J.-.C., Barral K. Novel 2-phenyl-5-[(E)-2-(thiophen-2-yl) ethenyl]-1, 3, 4-oxadiazole and 3-phenyl-5-[(E)-2-(thiophen-2-yl) ethenyl]-1, 2, 4-oxadiazole derivatives as dengue virus inhibitors targeting NS5 polymerase. Eur. J. Med. Chem. 2016;109:146–156. doi: 10.1016/j.ejmech.2015.12.046. [DOI] [PubMed] [Google Scholar]
- 73.Manvar D., Küçükgüzel İ., Erensoy G., Tatar E., Deryabaşoğulları G., Reddy H., Talele T.T., Cevik O., Kaushik-Basu N. Discovery of conjugated thiazolidinone-thiadiazole scaffold as anti-dengue virus polymerase inhibitors. Biochem. Biophys. Res. Commun. 2016;469(3):743–747. doi: 10.1016/j.bbrc.2015.12.042. [DOI] [PubMed] [Google Scholar]
- 74.Yang J., Du J., Huang C., Wang T., Huang L., Yang S., Li L. Discovery of 5-(5-fluoro-1H-pyrrolo [2, 3-b] pyridin-3-yl) pyrazin-2 (1H)-one derivatives as new potent PB2 inhibitors. Bioorg. Med. Chem. Lett. 2019;29(13):1609–1613. doi: 10.1016/j.bmcl.2019.04.042. [DOI] [PubMed] [Google Scholar]
- 75.Zhao X., Wang Y., Cui Q., Li P., Wang L., Chen Z., Rong L., Du R. A parallel phenotypic versus target-based screening strategy for RNA-dependent RNA polymerase inhibitors of the influenza a virus. Viruses. 2019;11(9):826. doi: 10.3390/v11090826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamada K., Koyama H., Hagiwara K., Ueda A., Sasaki Y., Kanesashi S.-n., Ueno R., Nakamura H.K., Kuwata K., Shimizu K. Identification of a novel compound with antiviral activity against influenza A virus depending on PA subunit of viral RNA polymerase. Microbes Infect. 2012;14(9):740–747. doi: 10.1016/j.micinf.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 77.Lepri S., Nannetti G., Muratore G., Cruciani G., Ruzziconi R., Mercorelli B., Palù G., Loregian A., Goracci L. Optimization of small-molecule inhibitors of influenza virus polymerase: from thiophene-3-carboxamide to polyamido scaffolds. J. Med. Chem. 2014;57(10):4337–4350. doi: 10.1021/jm500300r. [DOI] [PubMed] [Google Scholar]
- 78.Yi D., Li Q., Pang L., Wang Y., Zhang Y., Duan Z., Liang C., Cen S. Identification of a broad-spectrum viral inhibitor targeting a novel allosteric site in the RNA-dependent RNA polymerases of dengue virus and norovirus. Front. Microbiol. 2020;11:1440. doi: 10.3389/fmicb.2020.01440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harmalkar D.S., Lee S.-.J., Lu Q., Kim M.I., Park J., Lee H., Park M., Lee A., Lee C., Lee K. Identification of novel non-nucleoside vinyl-stilbene analogs as potent norovirus replication inhibitors with a potential host-targeting mechanism. Eur. J. Med. Chem. 2019;184 doi: 10.1016/j.ejmech.2019.111733. [DOI] [PubMed] [Google Scholar]
- 80.Giancotti G., Rigo I., Pasqualetto G., Young M.T., Neyts J., Rocha-Pereira J., Brancale A., Ferla S., Bassetto M. A new antiviral scaffold for human norovirus identified with computer-aided approaches on the viral polymerase. Sci. Rep. 2019;9(1):1–20. doi: 10.1038/s41598-019-54903-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mastrangelo E., Pezzullo M., Tarantino D., Petazzi R., Germani F., Kramer D., Robel I., Rohayem J., Bolognesi M., Milani M. Structure-based inhibition of Norovirus RNA-dependent RNA polymerases. J. Mol. Biol. 2012;419(3–4):198–210. doi: 10.1016/j.jmb.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 82.Tarantino D., Pezzullo M., Mastrangelo E., Croci R., Rohayem J., Robel I., Bolognesi M., Milani M. Naphthalene-sulfonate inhibitors of human norovirus RNA-dependent RNA-polymerase. Antiviral Res. 2014;102:23–28. doi: 10.1016/j.antiviral.2013.11.016. [DOI] [PubMed] [Google Scholar]