

ABSTRACT
Multimeric cargo adaptors such as AP2 play central roles in intracellular membrane trafficking. We recently discovered that the assembly of the AP2 adaptor complex, a key player in clathrin-mediated endocytosis, is a highly organized process controlled by alpha- and gamma-adaptin-binding protein (AAGAB, also known as p34). In this study, we demonstrate that besides AP2, AAGAB also regulates the assembly of AP1, a cargo adaptor involved in clathrin-mediated transport between the trans-Golgi network and the endosome. However, AAGAB is not involved in the formation of other adaptor complexes, including AP3. AAGAB promotes AP1 assembly by binding and stabilizing the γ and σ subunits of AP1, and its mutation abolishes AP1 assembly and disrupts AP1-mediated cargo trafficking. Comparative proteomic analyses indicate that AAGAB mutation massively alters surface protein homeostasis, and its loss-of-function phenotypes reflect the synergistic effects of AP1 and AP2 deficiency. Taken together, these findings establish AAGAB as an assembly chaperone for both AP1 and AP2 adaptors and pave the way for understanding the pathogenesis of AAGAB-linked diseases.
KEY WORDS: Membrane trafficking, Endocytosis, Clathrin, Endocytosis, AP1, AP2
Summary: The assembly of the AP1 adaptor complex is a highly orchestrated process controlled by AAGAB.
INTRODUCTION
The coat protein clathrin drives vesicle budding at the plasma membrane, the trans-Golgi network and the endosome (Brodsky, 2012; Kirchhausen et al., 2014; Mettlen et al., 2018; Traub and Bonifacino, 2013). Since clathrin does not directly bind cargo proteins, it relies on adaptors to recruit cargo proteins to vesicle budding sites (Kaksonen and Roux, 2018; Mettlen et al., 2018; Page et al., 1999). A prominent clathrin adaptor is the heterotetrameric AP2 complex involved in clathrin-mediated endocytosis (CME), a major route for internalization of surface and extracellular molecules (Fotin et al., 2004; Kaksonen et al., 2006; Kirchhausen et al., 2014; McMahon and Boucrot, 2011; Traub and Bonifacino, 2013; Wang et al., 2016). AP2 adaptor is composed of two large subunits (α and β), one medium subunit (µ), and one small subunit (σ) (Fig. 1A) (Collins et al., 2002; Conner and Schmid, 2003; Hollopeter et al., 2014; Pearse and Robinson, 1984). After being recruited to the plasma membrane, AP2 recognizes sorting signals on cargo proteins and then recruits clathrin to drive vesicle budding (Blot and McGraw, 2008; Brodsky, 2012; Caceres et al., 2019; Fotin et al., 2004; Hirst et al., 2012; Kirchhausen et al., 2014; McMahon and Boucrot, 2011; Paczkowski et al., 2015; Park and Guo, 2014; Ramanan et al., 2011).
Fig. 1.

Downregulation of AP1 and AP2 subunits in AAGAB KO cells. (A) Diagrams of AP1 and AP2 adaptors. Despite their similarity in overall structure and configuration, subunits of adaptor proteins such as AP1, AP2 and AP3 are not functionally interchangeable, except for the β subunits of AP1 and AP2 (Traub and Bonifacino, 2013). (B) Representative immunoblots showing expression of the indicated proteins in WT and AAGAB KO HeLa cells. M.W., molecular weight. Blots shown are representative of three experiments. (C) Relative expression levels of AP1, AP2, AP3, COPI and COPII in AAGAB KO HeLa cells measured by mass spectrometry. Data are presented as percentage of total expression levels in WT cells. Average values of two biological replicates are shown. Error bars indicate s.d. The full dataset of the whole-cell proteomic analysis is shown in Table S1.
Recently, we discovered that AP2 assembly is a highly orchestrated process controlled by alpha- and gamma-adaptin-binding protein (AAGAB, also known as p34), a cytosolic factor identified in a genome-scale CRISPR screen of CME (Gulbranson et al., 2019). Heterozygous AAGAB mutations cause punctate palmoplantar keratoderma type 1 (PPKP1), a skin disease characterized by punctate hyperkeratosis of the palms and soles (Elhaji et al., 2020; Gerber et al., 2013; Giehl et al., 2012; Kono et al., 2017; Nomura et al., 2015; Pöhler et al., 2013, 2012). AAGAB first binds to the α subunit (α adaptin) to form an AAGAB–α binary complex, which then recruits a σ subunit to form an AAGAB–α–σ ternary complex. Subsequently, β and µ subunits displace AAGAB, leading to the formation of the AP2 complex (Gulbranson et al., 2019). Without the assistance of AAGAB, AP2 adaptor fails to form, leading to degradation of AP2 subunits and disruption of CME (Gulbranson et al., 2019). Together, these findings uncovered a previously unrecognized pathway in clathrin-mediated trafficking.
In this study, we demonstrate that, besides AP2, AAGAB also regulates the assembly of AP1, a clathrin cargo adaptor operating on the endosome and the trans-Golgi network (Hirst et al., 2012; Kaksonen et al., 2006; Park and Guo, 2014; Traub, 1997; Traub and Bonifacino, 2013). Although functionally distinct from AP2, AP1 is also a heterotetrameric complex composed of two large subunits (γ and β), one medium subunit (µ) and one small subunit (σ) (Fig. 1A) (Collins et al., 2002; Conner and Schmid, 2003; Heldwein et al., 2004; Hollopeter et al., 2014; Pearse and Robinson, 1984; Ren et al., 2013). AAGAB binds and stabilizes the γ and σ subunits of AP1, and its mutation abrogates AP1 assembly and disrupts AP1-mediated membrane trafficking. Our comparative proteomic analyses showed that AAGAB mutation strongly alters membrane protein homeostasis, and its loss-of-function phenotypes reflect the combinatorial effects of AP1 and AP2 deficiency. Taken together, these data demonstrate that AAGAB acts as an assembly chaperone for both AP1 and AP2 clathrin adaptors.
RESULTS
AP1 subunits are downregulated in AAGAB knockout cells
Without the assistance of AAGAB, AP2 subunits fail to assemble into the tetrameric AP2 complex and are degraded (Gulbranson et al., 2019). To determine whether AAGAB also regulates other substrates, we used mass spectrometry-based proteomics to detect and quantify proteins expressed in wild-type (WT) and AAGAB knockout (KO) HeLa cells (Fig. 1B,C). The proteomic analysis detected 7945 proteins in WT cells and 7722 proteins in AAGAB KO cells including AP1 and AP3, another two tetrameric cargo adaptors (Fig. 1C; Table S1). Despite their similarity in overall configurations and structures, AP1, AP2 and AP3 play distinct roles in membrane trafficking and their subunits are not functionally interchangeable, except for the β subunits of AP1 and AP2 (Folsch et al., 1999; Gravotta et al., 2012; Traub and Bonifacino, 2013; Wang et al., 2003). As expected, AP2 subunits were among the most depleted proteins in AAGAB KO cells (Fig. 1C; Table S1). All four subunits of AP1 adaptor (γ, β, µ and σ) were also strongly downregulated in AAGAB KO cells (Fig. 1C; Table S1), consistent with our previous observation that AP1 γ expression was diminished in AAGAB KO cells (Gulbranson et al., 2019). By contrast, expression levels of AP3 subunits (δ, β, µ and σ) remained unchanged in AAGAB KO cells (Fig. 1C; Table S1). The proteomic analysis also detected coat protein complexes I and II (COPI and COPII), which mediate vesicle budding in the early secretory pathway (Dell'Angelica and Bonifacino, 2019). We found that the expression of COPI and COPII did not change in AAGAB KO cells (Fig. 1C; Table S1). These proteomic data suggest that AAGAB selectively regulates AP1 and AP2 adaptors.
AAGAB directly interacts with AP1 subunits
The downregulation of AP1 in AAGAB KO cells could be because of a direct regulation of AP1 adaptor by AAGAB. Alternatively, AAGAB mutation might reduce AP1 levels indirectly through an AP2-dependent mechanism. To distinguish between these possibilities, we first examined whether AAGAB directly binds to AP1 subunits. Although AAGAB was previously found to associate with the γ subunit (γ adaptin) of AP1 in a yeast two-hybrid assay (Page et al., 1999), it was unknown whether the interaction was direct or biologically relevant. To address this question, we co-expressed AAGAB and AP1 γ subunit in Escherichia coli and purified the recombinant proteins using affinity chromatography and size-exclusion chromatography (SEC). We found that recombinant AAGAB and AP1 γ subunit formed a stoichiometric complex (Fig. 2A–C). Thus, AAGAB directly interacts with AP1 γ subunit to form a binary complex.
Fig. 2.

AAGAB directly interacts with AP1 adaptor. (A) SEC profiles of recombinant AAGAB–γ binary complexes and AAGAB itself from a Superdex 200 10/300 column. For Superdex 200, the excluded volume is 7 ml and the included volume is 19 ml. Elution positions of protein standards with known molecular weights (M.W.) are marked at the top. The binary complex consists of untagged human AAGAB and mouse AP1 γ subunit (trunk domain, residues 1–595). The proteins were co-expressed in E. coli, isolated using affinity chromatography and further purified using SEC. From its SEC profile, we estimate that AAGAB itself forms a homotetramer. (B) Coomassie Blue-stained gel showing the AAGAB–γ binary complex from SEC elution in A. (C) Coomassie Blue-stained gel showing AAGAB alone from SEC elution in A. Data shown in A–C are representative of three experiments. (D) Diagrams of HA-tagged full-length AP1 γ and σ subunits and 3xFLAG-tagged AAGAB used in IP experiments. (E) Representative immunoblots from three experiments showing the interaction of 3xFLAG-AAGAB with HA-tagged AP1 subunits. The 3xFLAG-AAGAB protein was transiently expressed in AAGAB KO HeLa cells with an empty vector (control) or plasmids encoding the indicated HA-tagged AP1 subunits. The 3xFLAG-AAGAB protein was immunoprecipitated from cell lysates using anti-FLAG antibodies, and the presence of 3xFLAG-AAGAB (bottom) and HA-tagged AP1 subunits (top) in the immunoprecipitates was detected using anti-FLAG and anti-HA antibodies, respectively. (F) Representative immunoblot from three experiments showing the expression of HA-tagged AP1 subunits in whole cell lysates prepared from the same cell samples used in E.
We then determined whether AAGAB interacts with AP1 subunits in the cell. AAGAB bearing an N-terminal 3xFLAG tag was co-expressed with hemagglutinin (HA)-tagged AP1 γ and σ subunits (Fig. 2D), which are analogous to AP2 α and σ subunits that interact with AAGAB during AP2 assembly (Gulbranson et al., 2019). Using co-immunoprecipitation (co-IP), we observed that AAGAB bound to AP1 γ subunit (Fig. 2D–F), in agreement with the biochemical data (Fig. 2A–C). Using antibodies recognizing endogenous AAGAB or AP1 γ subunit, we confirmed interactions of AAGAB and AP1 γ subunit in co-IP assays (Fig. S1). By contrast, AAGAB did not bind AP3 δ subunit (Fig. S1), which is the structural and functional equivalent within AP3 to the γ subunit within AP1 and α subunit within AP2 (Dell'Angelica and Bonifacino, 2019). These data are consistent with the observation that AP3 adaptor was not impacted in AAGAB KO cells (Fig. 1). AAGAB also interacted with the σ subunit of AP1 in the presence of the γ subunit (Fig. 2D–F), similar to the results of AAGAB–AP2 interactions (Gulbranson et al., 2019). Whereas AP2 σ subunit does not bind AAGAB in the absence of α subunit (Gulbranson et al., 2019), AP1 σ subunit interacted with AAGAB even without co-expression with the γ subunit (Fig. 2D–F). Taken together, these biochemical and co-IP data indicate that AAGAB directly recognizes AP1 subunits in the cell.
Surface proteomes of AP1-, AP2- and AAGAB-deficient cells
Next, we sought to determine whether AAGAB is required for AP1-mediated cargo trafficking in the cell. We began by measuring the surface proteome of AP1-deficient cells, in which the γ subunit-encoding gene AP1G1 was deleted using CRISPR-Cas9 genome editing (Fig. 3A). Clathrin-mediated transport plays key roles in determining surface levels of cargo proteins, which can be reliably quantified. However, it remained unclear how AP1 regulates surface protein homeostasis. Surface proteins of WT and AP1G1 KO cells were biotinylated, isolated using NeutrAvidin beads and analyzed using mass spectrometry (Fig. 3B). We observed that the surface proteome was strongly altered in AP1G1 KO cells with 695 proteins upregulated and 1031 proteins downregulated (Fig. 3C,D; Table S2). Thus, although AP1 is primarily involved in bidirectional trafficking between the Golgi and the endosome (Traub and Bonifacino, 2013), its mutation severely impairs surface protein homeostasis. Next, we determined the surface proteome of AP2-deficient cells, in which the σ subunit-encoding gene AP2S1 was deleted (Gulbranson et al., 2019). We observed that a large number of surface proteins were upregulated (220) or downregulated (228) in AP2-deficient cells (Fig. 3C,D; Table S3). The surface proteome of AAGAB KO cells was determined in a similar manner, revealing 379 upregulated surface proteins and 148 downregulated surface proteins (Fig. 3C,D; Table S4).
Fig. 3.

Surface proteomic analysis of WT and mutant cells. (A) Representative immunoblots from three experiments showing expression of the indicated proteins in WT and AP1G1 KO HeLa cells. M.W., molecular weight. (B) Procedure for mass spectrometry-based surface proteomics. (C,D) Comparative analysis of surface proteomes of WT, AP1G1 KO, AP2S1 KO and AAGAB KO HeLa cells. The Venn diagrams show the numbers of surface proteins upregulated (C) or downregulated (D) in a KO cell line (compared to WT cells). A protein is included if its surface level is increased or decreased by >10% in a KO cell line and the P value is <0.05. Full datasets of the surface proteomes are shown in Tables S2–S4. (E) Relative surface levels of the indicated proteins in AP1G1 KO, AP2S1 KO, and AAGAB KO HeLa cells based on surface proteomic data. Data are presented as percentage of surface levels in WT cells. Average values of two technical replicates are shown. Error bars indicate s.d. ***P<0.001 (one-way ANOVA and Tukey's multiple comparison test). Dashed line: WT levels.
By comparing the surface proteomes of the cell lines, we noted that a large group of surface proteins were moderately upregulated in AP1- or AP2-deficient cells, but greater increases were observed in AAGAB KO cells. For instance, surface levels of transferrin receptor (TfR, also known as TFRC), a classic cargo protein in membrane trafficking studies, were elevated in AP2-deficient cells because of impaired CME (Fig. 3E; Table S3) (Conner and Schmid, 2003; Gulbranson et al., 2019; Ricotta et al., 2002). AP1 mutation also moderately increased the surface levels of TfR (Fig. 3E; Table S2). However, increases in TfR surface levels were substantially greater in AAGAB KO cells (Fig. 3E; Table S4). Similar phenomena were observed for many other surface proteins including ECE1, TMEM206 (also known as PACC1), APLP2 and ITGA3 (Fig. 3E; Tables S2–S4). These data suggest that both AP1 and AP2 mutations contribute to the cargo trafficking defects observed in AAGAB KO cells. We also observed that AP1 and AP2 mutations had opposite effects on surface levels of particular cargo proteins such as folate receptor α (FOLR1) and CD166 (encoded by ALCAM) (Fig. 3E; Tables S2 and S3). However, surface levels of these cargo proteins in AAGAB KO cells remained largely unchanged (Fig. 3E; Table S4). These results further support the notion that the phenotypes of AAGAB KO cells reflect the combinatorial effects of AP1 and AP2 deficiency.
AAGAB is required for AP1- and AP2-mediated cargo trafficking
To validate the results of surface proteomics, we examined the surface levels of TfR using flow cytometry. We also found that surface TfR was moderately increased in AP2-deficient cells (Fig. 4A,B). Likewise, surface levels of TfR were mildly elevated in AP1-deficient cells (Fig. 4A,B). However, KO of AAGAB led to a greater increase in surface TfR (Fig. 4B). We also examined surface staining of TfR using confocal microscopy. Consistent with the flow cytometry data, surface staining of TfR was moderately elevated in AP1- or AP2-deficient cells but the increase was substantially greater in AAGAB KO cells (Fig. 4C). Next, we generated AP1G1 and AP2S1 double KO cells (Fig. 4A). We found that surface levels of TfR were strongly upregulated in the double KO cells, comparable to its levels in AAGAB KO cells (Fig. 4B,C). The total protein level of TfR was also upregulated in AAGAB KO cells (Fig. S2A–D), which was caused by increased expression of TfR mRNA (Fig. S2E), although a reduction in lysosomal degradation might also contribute to the upregulation. Thus, the upregulation of surface TfR results from increased total expression as well as a shift of localization from intracellular compartments to the cell surface. Together, these data confirmed the proteomic results and demonstrated that AAGAB is required for both AP1- and AP2-mediated cargo trafficking in the cell.
Fig. 4.

AAGAB is required for both AP1- and AP2-mediated cargo trafficking. (A) Representative immunoblots from three experiments showing the expression of the indicated proteins in WT and mutant HeLa cells. M.W., molecular weight. (B) Normalized surface levels of TfR in WT and mutant HeLa cells measured by flow cytometry. Cells were disassociated by Accutase and stained with monoclonal anti-TfR antibodies and APC-conjugated secondary antibodies. APC fluorescence measurements of ∼5000 cells were collected on a CyAn ADP analyzer. Mean APC fluorescence of mutant cells was normalized to that of WT cells. Data are presented as mean±s.d., n=3. ***P<0.001. P values were calculated using one-way ANOVA and Dunnett's multiple comparison test. (C) Representative confocal microscopy images from three experiments showing surface levels of TfR in WT and mutant HeLa cells. Surface TfR levels of non-permeabilized cells were labeled using anti-TfR antibodies and Alexa Fluor 568-conjugated secondary antibodies (red). Nuclei were stained with Hoechst 33342 (blue). Images were captured using a 100× oil immersion objective on a Nikon A1 Laser Scanning confocal microscope. Scale bars: 20 µm. DKO, double knockout.
Genes encoding AAGAB, AP1 and AP2 are dispensable for cell survival or proliferation
Next, we examined whether the genes encoding AAGAB, AP1 and AP2 are essential to cell survival or proliferation. Deletion of an essential gene results in cell death or growth arrest, which precludes reliable analysis of gene functions. To globally identify essential genes, a genome-wide CRISPR mutant library of HeLa cells was cultured continuously for four weeks. The abundance of gRNAs in the mutant cell population was then compared with that in the original CRISPR DNA library (Gulbranson et al., 2017; Li et al., 2014; Wang et al., 2019). A total of 1391 genes were predicted to be essential based on depletion of their gRNAs in the mutant cell population. We found that 13 of the 14 genes encoding AAGAB, AP1 or AP2 were clearly nonessential (Fig. 5; Table S5). AP2S1 falls into the essential gene category but its ranking is very close to the cutoff (Fig. 5; Table S5). As such, its mutation is not expected to significantly affect cell fitness. Indeed, AP2S1 KO HeLa cells exhibited no apparent growth defect in a range of cell-based assays (Figs 3 and 4). These results suggest that, although surface protein homeostasis is severely compromised in AP1-, AP2- and AAGAB-deficient cells, the survival and proliferation of these mutant cells remain largely intact.
Fig. 5.
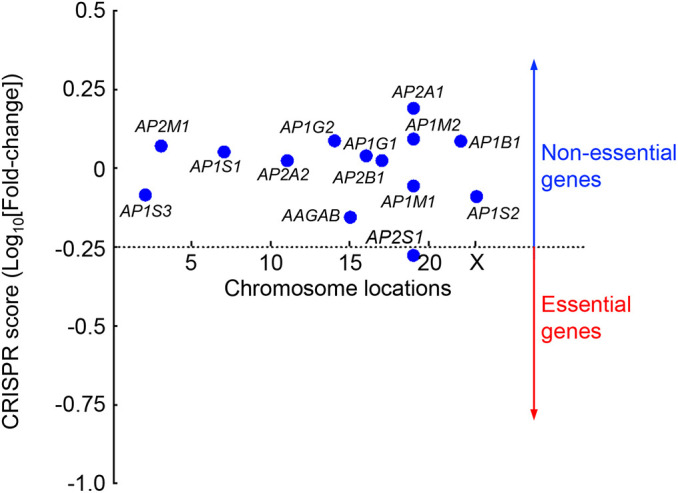
Genes encoding AAGAB, AP1 and AP2 are dispensable for cell survival or proliferation. To identify essential genes, we took advantage of a genome-wide HeLa mutant library generated using the GeCKO V2 CRISPR library (Gulbranson et al., 2017; Wang et al., 2019). The mutant cells were cultured continuously for four weeks before the abundance of gRNAs was analyzed by deep sequencing. Depletion of gRNAs was calculated by comparing the passage control population to the initial GeCKO V2 CRISPR DNA library. A CRISPR score was calculated for each gene based on log10(fold-changes) in the abundance of its corresponding gRNAs using an established algorithm (Li et al., 2014). The 1391 genes with CRISPR scores below the horizontal cutoff line of −0.25 are predicted to be essential to cell survival or growth. Only the 14 genes encoding AAGAB, AP1 and AP2 are shown. A full list of genes ranked according to their CRISPR scores is shown in Table S5.
AAGAB stabilizes AP1 γ and σ subunits
Finally, we sought to further characterize the molecular mechanism by which AAGAB regulates AP1 adaptor. GST-tagged AP1 γ subunit was expressed in E. coli with or without His6-tagged AAGAB. We found that GST-tagged γ subunit could not be obtained from the soluble fractions of E. coli lysates in the absence of AAGAB (Fig. 6A,B). Soluble AP1 γ subunit was isolated only when AAGAB was co-expressed (Fig. 6A,B). Similar results were observed when AAGAB and AP1 γ subunit were purified through the His6 tag on AAGAB (Fig. 6C,D). These results suggest that AP1 γ subunit is an intrinsically unstable protein prone to misfolding and must be stabilized by AAGAB. Next, we co-expressed GST-tagged AP1 γ subunit, His6-tagged AAGAB and untagged AP1 σ subunit in E. coli before GST-tagged γ subunit was isolated. We observed that recombinant AAGAB, γ subunit and σ subunit formed a ternary complex (Fig. 6A,B), in agreement with the co-IP data (Fig. 2D–F). The ternary complex was also obtained when proteins were isolated through the His6 tag on AAGAB (Fig. 6C,D). When γ and σ subunits were co-expressed in E. coli, soluble proteins could be obtained only when AAGAB was also expressed (Fig. S3). These biochemical data indicate that AAGAB promotes AP1 assembly by binding and stabilizing the γ and σ subunits.
Fig. 6.

AAGAB binds and stabilizes AP1 subunits. (A) Diagram of the GST pull-down assay measuring the interaction of AAGAB with the γ and σ subunits of AP1. His6-SUMO-tagged AAGAB was co-expressed with GST or GST-tagged γ subunit (trunk domain, residues 1–595) in E. coli with or without untagged σ subunit. Proteins were isolated from E. coli lysates using glutathione beads. (B) Coomassie Blue-stained gels showing the binding of GST-tagged γ subunit to His6-SUMO-tagged AAGAB and untagged σ subunit. Note that σ subunit intrinsically stains less because of its small size. Images are representative of three experiments. (C) Diagram of the nickel bead pull-down assay measuring the interaction of GST-tagged γ subunit (trunk domain, residues 1–595) with His6-SUMO-tagged AAGAB and untagged σ subunit. Proteins were co-expressed in E. coli as in A and isolated using nickel beads that recognized the His6 tag of AAGAB. (D) Coomassie Blue-stained gels showing the binding of His6-SUMO-tagged AAGAB to GST-tagged γ subunit and untagged σ subunit. Images are representative of three experiments. M.W., molecular weight.
DISCUSSION
In this study, we discovered that AAGAB regulates the assembly of AP1 adaptor, in addition to its role in AP2 formation. Overall, AAGAB-guided assembly of AP1 adaptor appears to be mechanistically similar to that of AP2 adaptor (Fig. 7) (Gulbranson et al., 2019). To assemble multimeric complexes such as AP1 and AP2, the initiation stage is particularly challenging because individual subunits are intrinsically unstable and prone to misfolding, aggregation and degradation. As an assembly chaperone, AAGAB helps overcome the challenge by binding and stabilizing the γ (AP1) or α (AP2) subunit in a binary complex. Subsequently, the binary complex recruits the σ subunit of AP1 or AP2 to form a ternary complex, which stabilizes the σ subunit and facilitates the formation of the γ–σ or α–σ hemicomplex. The hemicomplex then serves as an assembly platform for subsequent association with β and µ subunits. Once β and µ subunits join the γ–σ or α–σ hemicomplex, AAGAB is released, allowing the formation of the tetrameric AP1 or AP2 complex (Fig. 7) (Gulbranson et al., 2019). Without the assistance of AAGAB, AP1 and AP2 subunits fail to assemble into functional adaptors and are degraded. The consecutive binding of γ or α, and σ subunits to AAGAB probably represents a conserved route of AAGAB-guided AP1 or AP2 assembly. Interestingly, unlike the AP2 σ subunit, the AP1 σ subunit can bind to AAGAB in the absence of the γ subunit, suggesting that AAGAB–σ to AAGAB–σ–γ may represent an alternative pathway for AP1 assembly. These findings also suggest that AAGAB possesses independent binding sites for the γ and σ subunits. Additional research will be needed to determine whether the AAGAB–σ binary complex is competent for subsequent association with the γ subunit or constitutes an unproductive intermediate (i.e. a dead end in the assembly pathway).
Fig. 7.
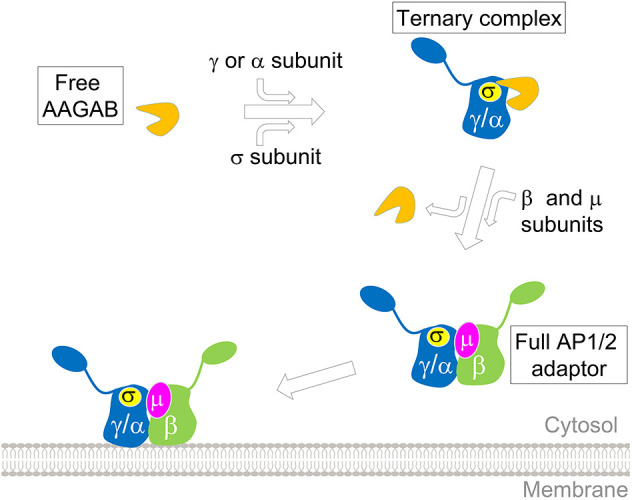
Model of AAGAB-guided assembly of AP1 and AP2 adaptors in the cytosol. AAGAB binds and stabilizes AP1 or AP2 subunits as well as their assembly intermediates, facilitating their assembly into tetrameric adaptor complexes.
Structural studies are required to define how AAGAB recognizes AP1 and AP2 subunits and whether differences exist between the binding modes of AAGAB–AP1 and AAGAB–AP2. The yeast protein Irc6p exhibits a similar domain configuration as AAGAB and interacts with AP1 (Gorynia et al., 2012). However, since Irc6p recognizes distinct substrates and does not regulate AP2 (Gorynia et al., 2012), it is unclear whether it represents a bona fide homologue of AAGAB. AAGAB selectively regulates AP1 and AP2 adaptors and is not involved in the assembly of other multimeric trafficking complexes such as AP3, COPI and COPII. However, since these complexes need to overcome similar challenges during assembly, we anticipate that their formation also requires specific assembly chaperones, analogous to the role of AAGAB in AP1 and AP2 assembly.
In the cell, AAGAB regulates both AP1- and AP2-dependent membrane trafficking. Since AP1 adaptor is involved in cargo transport from the trans-Golgi network to the endosome (Beacham et al., 2019; Brown et al., 2011; Dell'Angelica and Bonifacino, 2019; Gillingham et al., 1999; Li and Kandror, 2005), its mutation is expected to redirect such cargo to the plasma membrane (Lubben et al., 2007; Roeth et al., 2004; Shen et al., 2015). Defects in AP1-mediated retrograde endosome-to-Golgi transport, on the other hand, are expected to reroute cargo proteins to the lysosome for degradation (Hinners and Tooze, 2003). In an AP1 knocksideways experiment, acute mislocalization of AP1 adaptor led to substantial alterations in the composition of clathrin-coated vesicles (Hirst et al., 2012). Although it is difficult to directly compare the knocksideways results with our findings, a subset of cargo proteins, including FOLR1, SORT1 and ROR1, were identified in both studies (Hirst et al., 2012). In AP2-null cells, surface levels of a cargo protein could be upregulated due to CME defects. We note that a cargo protein might be indirectly controlled by AP1 and/or AP2 (i.e. through intermediate regulators), rather than being a direct target. For instance, mutations of AP2 may inhibit the retrieval and reuse of an exocytic regulator, resulting in downregulation of surface proteins dependent on the exocytic regulator for surface delivery. The overall consequence of AAGAB mutation is dictated by the dynamic balance of alterations in these AP1- and AP2-dependent trafficking pathways.
Our findings also set the stage for understanding the pathogenesis of AAGAB-linked PPKP1. Intriguingly, mutations in AP1-encoding genes such as AP1B1, AP1S1 and AP1S3 are also linked to skin disorders, including keratoderma (Alsaif et al., 2019; Boyden et al., 2019; Incecik et al., 2018; Mahil et al., 2016; Martinelli and Dionisi-Vici, 2014; Montpetit et al., 2008; Sanger et al., 2019; Setta-Kaffetzi et al., 2014). By contrast, AP2 has not been connected to skin conditions. Thus, we posit that AAGAB-linked PPKP1 might be caused mainly by imbalances in AP1-mediated cargo trafficking. Mice deficient in AP1 or AP2 die at an early stage of embryonic development (Meyer et al., 2000; Mitsunari et al., 2005; Zizioli et al., 1999). Thus, we anticipate that homozygous loss-of-function mutations of AAGAB also cause embryonic lethality. On the other hand, heterozygous AAGAB mutations found in PPKP1 are expected to subtly impact clathrin-mediated trafficking such that their effects are restricted to a small group of cargo proteins in selected tissues (e.g. the skin). AP1-linked skin diseases usually result from homozygous mutations in a β- or σ-encoding gene (Alsaif et al., 2019; Boyden et al., 2019; Favilli et al., 2009; Mahil et al., 2016; Montpetit et al., 2008; Setta-Kaffetzi et al., 2014). Since both β and σ subunits of AP1 are encoded by multiple paralogous genes, mutations in one gene are expected to impact a limited subset of membrane proteins in the skin that are particularly sensitive to AP1 perturbations. A major direction of future research is to identify the cargo protein(s) responsible for AAGAB- and AP1-linked diseases and to define how AAGAB and AP1 mutations selectively affect particular tissues such as the skin.
MATERIALS AND METHODS
Cell culture
HeLa and 293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FB Essence (FBE; Seradigm, #3100-500) and penicillin-streptomycin (Millipore-Sigma, #P4333). The cells were acquired from University of Colorado Cancer Center, which routinely authenticated cell lines.
Genome editing using CRISPR-Cas9
For genome editing experiments using CRISPR-Cas9, two gRNA sequences were selected to target early constitutive exons of a candidate gene. One of the gRNAs was subcloned into the pLenti-CRISPR-V2 vector (Addgene, #52961). The second gRNA was subcloned into a modified version of the pLentiGuide-Puro vector (Addgene, #52963), in which the puromycin selection marker was substituted with a hygromycin selection marker. CRISPR plasmids were transiently transfected into 293T cells together with pAdVAntage (Promega, #E1711), pCMV-VSV-G (Addgene, #8454) and psPAX2 (Addgene, #12260) using a previously established procedure (Gulbranson et al., 2017). The 293T culture media containing lentiviral particles were collected consecutively for four rounds and centrifuged in a Beckman SW28 rotor at 25,000 rpm (113,000 g) for 1.5 h at 4°C. Viral pellets were resuspended in PBS and used to infect target cells. Infected cells were sequentially selected using 1 µg/ml puromycin (Millipore-Sigma, #3101118) and 500 µg/ml hygromycin B (Thermo Fisher Scientific, #10687010). Sequences of gRNAs targeting the human AAGAB gene are 5′-CAGCTGGTCTCCTGAGAAGA-3′ and 5′-GCAGTAACAAGAAATTTGTT-3′. Sequences of gRNAs targeting the human AP2S1 gene are 5′-GCGTCTTGCCTGCCCGGTTC-3′ and 5′-GGTCCAGTTCACAGACATTG-3′. Sequences of gRNAs targeting the human AP1G1 gene are 5′-TGCCAGCCCCCATCAGATTG-3′ and 5′-ATTTTGCCACATTCCGACAT-3′.
To generate AP1G1 and AP2S1 double KO cells, the AP2S1-targeting gRNA sequence 5′-GCGTCTTGCCTGCCCGGTTC-3′ was subcloned into the lentiCRISPR-v2-Blast vector (Addgene, #83480). Lentiviruses generated using the CRISPR construct were used to infect AP1G1 KO HeLa cells before the cells were selected using 10 µg/ml of blasticidin (Millipore-Sigma, #15205).
Mass spectrometry
Stable isotope labeling with amino acids in cell culture (SILAC) and mass spectrometry were used for quantitative determination of whole-cell and surface proteomes. WT and mutant HeLa cells were grown in SILAC media (Thermo Fisher Scientific, #88364) supplemented with 10% dialyzed FBE. For whole-cell proteomics, WT HeLa cells were grown in the presence of light lysine and arginine (Millipore-Sigma, #L1262 and #A5131), while AAGAB KO cells were cultured in the presence of heavy lysine and arginine (Cambridge Isotope Laboratories, #CNLM-291 and #CNLM-539). After five days of labeling, cells at ∼60% confluence were harvested in a lysis buffer [100 mM triethylammonium bicarbonate (Millipore-Sigma, #18597), 5 mM Tris(2-carboxyethyl)phosphine (TCEP), 20 mM chloroacetamide (Millipore-Sigma, #C0267), and 1% sodium dodecyl sulfate (SDS)]. After incubation at 95°C for 15 min, equal amounts of lysates were mixed and used for mass spectrometry analysis (Gulbranson et al., 2017).
For surface proteomics, WT HeLa cells were grown in the presence of light lysine and arginine, while AP2S1 KO cells were grown in the presence of medium lysine and arginine (Thermo Fisher Scientific, #89988 and #88210). AAGAB KO cells were grown in the presence of heavy lysine and arginine. In a parallel experiment, WT HeLa cells were grown in the presence of light lysine and arginine, while AP1G1 KO cells were grown in the presence of heavy lysine and arginine. After five days of labeling, cells were labeled with Sulfo-NHS-Biotin (Thermo Fisher Scientific, #A39256) for one hour at 4°C. The reactions were quenched using 100 mM glycine in PBS before cells were lysed in a SILAC IP buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol, and a protease inhibitor cocktail). Equal amounts of cell extracts were mixed and biotinylated proteins were isolated using NeutrAvidin agarose resin (Thermo Fisher Scientific, #29201). Proteins were eluted using an SDS protein buffer (4% SDS and 50 mM Tris-HCl, pH 6.8).
Mass spectrometry samples were prepared according to the protein aggregation capture (PAC) method. Peptides were pre-fractionated using high-pH fractionation and concatenated into 10 fractions before they were resolved on a Thermo Ultimate 3000 RSLCnano system (Thermo Fisher Scientific) in a direct injection mode (Batth et al., 2019). Raw data files from mass spectrometry were processed using MaxQuant and Andromeda (v. 1.6.2.10) (Cox et al., 2011), and searched against the UniProt database of human protein sequences (downloaded September 2019, 74,468 entries). False discovery rates were set to 0.01 for both protein and peptide identifications with a minimum peptide length of four residues and a minimum peptide number of two. For SILAC ratio measurements, a minimum of two peptide ratios was used to calculate a protein ratio.
Flow cytometry
To stain surface TfR, HeLa cells were washed three times with KRH buffer (121 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, 0.33 mM CaCl2 and 12 mM HEPES, pH 7.0). Cells were then chilled on ice and stained with monoclonal anti-TfR antibodies (DSHB, #G1/221/12) at a final concentration of 0.1 µg/ml and allophycocyanin (APC)-conjugated secondary antibodies (Thermo Fisher Scientific, #17-4015-82) at a final concentration of 0.8 µg/ml. After dissociation from plates using Accutase (Innovative Cell Technologies, #AT 104), APC fluorescence of the cells was measured on a CyAn ADP Analyzer (Beckman Coulter). Mean APC fluorescence of mutant cells was normalized to that of WT cells. Data from populations of ∼5000 cells were analyzed using FlowJo software, v. 10 (FlowJo, LLC) based on experiments run in biological triplicates. To measure total TfR, cells were disassociated using Accutase, fixed using 2% paraformaldehyde, permeabilized using 0.1% saponin, and stained with monoclonal anti-TfR antibodies and APC-conjugated secondary antibodies.
Immunoblotting and immunoprecipitation
To detect proteins in whole cell lysates, cells grown in 24-well plates were lysed in the SDS protein buffer. To detect proteins on the cell surface, surface proteins (without SILAC labeling) were biotinylated and isolated as described in the Mass spectrometry section. Protein samples were resolved by 8% Bis-Tris SDS-PAGE and proteins were detected using primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. Primary antibodies used in this study included polyclonal anti-AAGAB antibodies (Bethyl Laboratories, #A305-593A) at a final concentration of 40 ng/ml, polyclonal anti-AP1-γ antibodies (Bethyl Laboratories, #A304-771A) at a final concentration of 1 µg/ml, monoclonal anti-AP2-µ antibodies (BD Biosciences, #611350) at a final concentration of 0.5 µg/ml, monoclonal anti-AP3-δ antibodies (DSHB, #SA4) at a final concentration of 0.5 µg/ml, monoclonal anti-α-tubulin antibodies (DSHB, #12G10) at a final concentration of 43 ng/ml, and monoclonal anti-FLAG antibodies (Millipore-Sigma, #F1804) at a final concentration of 1 µg/ml. HA-tagged proteins were directly detected using monoclonal anti-HA-HRP antibodies (Roche, #12013819001) at a final concentration of 25 ng/ml.
In immunoprecipitation (IP) experiments, cells grown in 6-well plates were lysed in an IP buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol and a protease inhibitor cocktail). Endogenous proteins were precipitated using Protein G agarose beads and polyclonal anti-AAGAB antibodies at a final concentration of 0.2 µg/ml or anti-AP1-γ antibodies (Millipore-Sigma, #A4200) at a final concentration of 1 µg/ml. Transiently expressed 3xFLAG-AAGAB was precipitated using Protein A agarose beads and anti-FLAG M2 antibodies (Millipore-Sigma, #F1804) at a final concentration of 0.5 µg/ml. Proteins in the immunoprecipitates were detected using immunoblotting.
Immunofluorescence
Cells grown on microscope cover glasses (VWR, #89015-725) were washed three times with KRH buffer and fixed using prewarmed 2% paraformaldehyde. Surface TfR was stained using monoclonal anti-TfR antibodies (DSHB, #G1/221/12) at a final concentration of 0.1 µg/ml and Alexa Fluor 568-conjugated secondary antibodies (Thermo Fisher Scientific, #A11004) at a final concentration of 1 µg/ml. The nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific, #H3570) at a final concentration of 10 µg/ml. Images were captured using a 100× oil immersion objective on a Nikon A1 Laser Scanning confocal microscope and processed using FIJI software (Schindelin et al., 2012).
Purification of recombinant AAGAB–γ binary complexes using SEC
Ap1g1 and AAGAB were cloned into the first and second multiple cloning sites, respectively, of a modified pRSFDuet-1 vector (Millipore-Sigma, #71341). The resultant plasmid encodes the trunk domain of mouse AP1 γ subunit (residues 1–595) with an N-terminal His6-SUMO tag and untagged full-length human AAGAB. The plasmid was transformed into Rosetta (DE3) E. coli (Millipore-Sigma, #70954) grown at 37°C in LB medium containing 30 μg/ml chloramphenicol and 50 μg/ml kanamycin. After OD600 reached 0.5–0.8, 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added. After 16 h of incubation at 18°C, cells were pelleted, resuspended in a lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 25 mM imidazole and 5% glycerol) and lysed by sonication. After centrifugation (14,000 g, 1 h, 4°C), the supernatant was sonicated again to fragment nucleic acids. The supernatant was filtered through a 0.45 μm filter and then loaded onto a 5-ml HisTrap column (GE Healthcare). The column was washed with lysis buffer and eluted with a linear gradient of 25–500 mM imidazole in lysis buffer. Peak fractions were incubated with Ulp1 protease overnight at 4°C to remove the His6-SUMO tag. The proteins were then loaded onto a 5-ml HiTrap Q HP column (GE Healthcare) pre-equilibrated with Buffer A (50 mM Tris-HCl, pH 8.0, and 150 mM NaCl). Proteins were eluted with a linear gradient of 0–100% Buffer B (50 mM Tris-HCl, pH 8.0, and 1 M NaCl). Fractions containing target proteins were pooled and further purified by SEC using a Superdex 200 10/300 column (GE Healthcare) in a buffer containing 10 mM Tris-HCl, pH 8.0, 150 mM NaCl and 1 mM DTT. The plasmid encoding human AAGAB with an N-terminal His6-SUMO tag was generated in a previous study (Gulbranson et al., 2019), and the protein was purified using HisTrap and HiTrap Q HP columns as described above before loading onto a Superdex 200 10/300 column.
Pull-down assays using recombinant proteins
Ap1g1 encoding mouse AP1 γ subunit (trunk domain, residues 1–595) was subcloned into the pGEX–4T–3 vector (GE Healthcare). AP1S3 encoding human AP1 σ subunit (full-length) was subcloned into the pACYCDuet-1 vector (Millipore-Sigma, #71147). The plasmid encoding His6-SUMO-tagged human AAGAB was developed in a previous study (Gulbranson et al., 2019). The empty pGEX-4T-3 vector was used to express the GST control protein. Recombinant proteins were expressed in BL21 (DE3) E. coli as described previously (Gulbranson et al., 2019; Yu et al., 2019). When OD600 of E. coli cultured in 2xYT media reached ∼0.6, 1 mM IPTG was added to induce protein expression. After three hours of incubation at 37°C, cells were harvested and lysed. After centrifugation (3,600 g, 0.5 h, 4°C), proteins were isolated using glutathione beads (Thermo Fisher Scientific, #PI16101) or nickel beads (Thermo Fisher Scientific, #PI-88222), resolved by SDS-PAGE and stained with Coomassie Blue.
qPCR assay
An RNeasy Mini Kit (Qiagen, #74104) was used to purify total RNAs from HeLa cells. After treatment with ezDNAse (Thermo Fisher Scientific, #8091150), first-strand complementary DNA was synthesized using a SuperScript IV kit (Thermo Fisher Scientific, #18091050). Gene expression levels were measured by quantitative reverse transcription PCR on an Applied Biosystems™ 7500 Fast Real-Time PCR System using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, #172-5272) with gene-specific primer sets. The cycle threshold values of a gene were normalized to those of GAPDH, a reference gene, and the Δcycle threshold values were calculated. The results were plotted as fold changes relative to the WT sample. PCR primers for TFRC were as follows: 5′-CTCGGCAAGTAGATGGCGATA-3′ (forward) and 5′-ACGATCACAGCAATAGTCCCA-3′ (reverse). PCR primers for GAPDH were as follows: 5′-GACAGTCAGCCGCATCTTCT-3′ (forward) and 5′-GCGCCCAATACGACCAAATC-3′ (reverse).
Supplementary Material
Acknowledgements
We thank Drs James Hurley, Juan Bonifacino, Daniel Gulbranson, Gus Lienhard and Margaret Robinson for reagents or advice. We thank Yan Ouyang and Harrison Puscher for technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.S.; Methodology: C.W., Y.T., R.Y., I.D.; Software: S.W.; Validation: C.W., L.C., S.W., J.S.; Formal analysis: L.C., I.D., S.L., H.Y., Q.Y., J.S.; Investigation: L.C., B.W., Y.T., Q.Y., J.S.; Resources: C.W., B.W., T.N., S.L., Q.Y.; Data curation: C.W., L.C., H.Y.; Writing - original draft: C.W., L.C., J.S.; Writing - review & editing: C.W., L.C., T.N., S.L., H.Y., Q.Y.; Supervision: J.S.; Project administration: C.W., J.S.; Funding acquisition: J.S., Q.Y., S.L., S.W.
Funding
This work was supported by National Institutes of Health grants GM126960 (to J.S.), DK124431 (to J.S.), AG061829 (to J.S.), GM138685 (to Q.Y.) and AI156560 (to S.L.); an American Diabetes Association Basic Science Award (to J.S.); an AB Nexus Seed Grant from University of Colorado (to J.S.); a National Institutes of Health institutional predoctoral training grant GM088759 (to L.C.); and a Fellowship from the Postdoctoral Overseas Training Program at Beijing University of Chinese Medicine (to S.W.). Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/article-lookup/doi/10.1242/jcs.258587
References
- Alsaif, H. S., Al-Owain, M., Barrios-Llerena, M. E., Gosadi, G., Binamer, Y., Devadason, D., Ravenscroft, J., Suri, M. and Alkuraya, F. S. (2019). Homozygous loss-of-function mutations in AP1B1, encoding Beta-1 subunit of adaptor-related protein complex 1, cause MEDNIK-like syndrome. Am. J. Hum. Genet. 105, 1016-1022. 10.1016/j.ajhg.2019.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batth, T. S., Tollenaere, M. A. X., Rüther, P., Gonzalez-Franquesa, A., Prabhakar, B. S., Bekker-Jensen, S., Deshmukh, A. S. and Olsen, J. V. (2019). Protein aggregation capture on microparticles enables multipurpose proteomics sample preparation. Mol. Cell Proteomics 18, 1027-1035. 10.1074/mcp.TIR118.001270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beacham, G. M., Partlow, E. A. and Hollopeter, G. (2019). Conformational regulation of AP1 and AP2 clathrin adaptor complexes. Traffic 20, 741-751. 10.1111/tra.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot, V. and McGraw, T. E. (2008). Molecular mechanisms controlling GLUT4 intracellular retention. Mol. Biol. Cell 19, 3477-3487. 10.1091/mbc.e08-03-0236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden, L. M., Atzmony, L., Hamilton, C., Zhou, J., Lim, Y. H., Hu, R., Pappas, J., Rabin, R., Ekstien, J., Hirsch, Y.et al. (2019). Recessive mutations in AP1B1 cause ichthyosis, deafness, and photophobia. Am. J. Hum. Genet. 105, 1023-1029. 10.1016/j.ajhg.2019.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, F. M. (2012). Diversity of clathrin function: new tricks for an old protein. Annu. Rev. Cell Dev. Biol. 28, 309-336. 10.1146/annurev-cellbio-101011-155716 [DOI] [PubMed] [Google Scholar]
- Brown, F. C., Schindelhaim, C. H. and Pfeffer, S. R. (2011). GCC185 plays independent roles in Golgi structure maintenance and AP-1-mediated vesicle tethering. J. Cell Biol. 194, 779-787. 10.1083/jcb.201104019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres, P. S., Gravotta, D., Zager, P. J., Dephoure, N. and Rodriguez-Boulan, E. (2019). Quantitative proteomics of MDCK cells identify unrecognized roles of clathrin adaptor AP-1 in polarized distribution of surface proteins. Proc. Natl. Acad. Sci. USA 116, 11796-11805. 10.1073/pnas.1821076116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, B. M., McCoy, A. J., Kent, H. M., Evans, P. R. and Owen, D. J. (2002). Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523-535. 10.1016/S0092-8674(02)00735-3 [DOI] [PubMed] [Google Scholar]
- Conner, S. D. and Schmid, S. L. (2003). Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 162, 773-780. 10.1083/jcb.200304069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V. and Mann, M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794-1805. 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- Dell'Angelica, E. C. and Bonifacino, J. S. (2019). Coatopathies: genetic disorders of protein coats. Annu. Rev. Cell Dev. Biol. 35, 131-168. 10.1146/annurev-cellbio-100818-125234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhaji, Y., Hedlin, C., Nath, A., Price, E. L., Gallant, C., Northgrave, S. and Hull, P. R. (2020). AAGAB mutations in 18 Canadian families with punctate palmoplantar keratoderma and a possible link to cancer. J. Cutan. Med. Surg. 24, 28-32. 10.1177/1203475419878161 [DOI] [PubMed] [Google Scholar]
- Favilli, F., Anzilotti, C., Martinelli, L., Quattroni, P., De Martino, S., Pratesi, F., Neumann, D., Beermann, S., Novick, D., Dinarello, C. A.et al. (2009). IL-18 activity in systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 1173, 301-309. 10.1111/j.1749-6632.2009.04742.x [DOI] [PubMed] [Google Scholar]
- Fölsch, H., Ohno, H., Bonifacino, J. S. and Mellman, I. (1999). A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell 99, 189-198. 10.1016/S0092-8674(00)81650-5 [DOI] [PubMed] [Google Scholar]
- Fotin, A., Cheng, Y., Sliz, P., Grigorieff, N., Harrison, S. C., Kirchhausen, T. and Walz, T. (2004). Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature 432, 573-579. 10.1038/nature03079 [DOI] [PubMed] [Google Scholar]
- Gerber, P. A., Hevezi, P., Buhren, B. A., Martinez, C., Schrumpf, H., Gasis, M., Grether-Beck, S., Krutmann, J., Homey, B. and Zlotnik, A. (2013). Systematic identification and characterization of novel human skin-associated genes encoding membrane and secreted proteins. PLoS ONE 8, e63949. 10.1371/journal.pone.0063949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl, K. A., Eckstein, G. N., Pasternack, S. M., Praetzel-Wunder, S., Ruzicka, T., Lichtner, P., Seidl, K., Rogers, M., Graf, E., Langbein, L.et al. (2012). Nonsense mutations in AAGAB cause punctate palmoplantar keratoderma type buschke-fischer-brauer. Am. J. Hum. Genet. 91, 754-759. 10.1016/j.ajhg.2012.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham, A. K., Koumanov, F., Pryor, P. R., Reaves, B. J. and Holman, G. D. (1999). Association of AP1 adaptor complexes with GLUT4 vesicles. J. Cell Sci. 112, 4793-4800. 10.1242/jcs.112.24.4793 [DOI] [PubMed] [Google Scholar]
- Gorynia, S., Lorenz, T. C., Costaguta, G., Daboussi, L., Cascio, D. and Payne, G. S. (2012). Yeast Irc6p is a novel type of conserved clathrin coat accessory factor related to small G proteins. Mol. Biol. Cell 23, 4416-4429. 10.1091/mbc.e12-07-0507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravotta, D., Carvajal-Gonzalez, J. M., Mattera, R., Deborde, S., Banfelder, J. R., Bonifacino, J. S. and Rodriguez-Boulan, E. (2012). The clathrin adaptor AP-1A mediates basolateral polarity. Dev. Cell 22, 811-823. 10.1016/j.devcel.2012.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbranson, D. R., Davis, E. M., Demmitt, B. A., Ouyang, Y., Ye, Y., Yu, H. and Shen, J. (2017). RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis. Proc. Natl. Acad. Sci. USA 114, E8224-E8233. 10.1073/pnas.1712176114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbranson, D. R., Crisman, L., Lee, M., Ouyang, Y., Menasche, B. L., Demmitt, B. A., Wan, C., Nomura, T., Ye, Y., Yu, H.et al. (2019). AAGAB controls AP2 adaptor assembly in clathrin-mediated endocytosis. Dev. Cell 50, 436-446.e5. 10.1016/j.devcel.2019.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein, E. E., Macia, E., Wang, J., Yin, H. L., Kirchhausen, T. and Harrison, S. C. (2004). Crystal structure of the clathrin adaptor protein 1 core. Proc. Natl. Acad. Sci. USA 101, 14108-14113. 10.1073/pnas.0406102101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinners, I. and Tooze, S. A. (2003). Changing directions: clathrin-mediated transport between the Golgi and endosomes. J. Cell Sci. 116, 763-771. 10.1242/jcs.00270 [DOI] [PubMed] [Google Scholar]
- Hirst, J., Borner, G. H. H., Antrobus, R., Peden, A. A., Hodson, N. A., Sahlender, D. A. and Robinson, M. S. (2012). Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Curr. Biol. 22, 1711-1716. 10.1016/j.cub.2012.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollopeter, G., Lange, J. J., Zhang, Y., Vu, T. N., Gu, M., Ailion, M., Lambie, E. J., Slaughter, B. D., Unruh, J. R., Florens, L.et al. (2014). The membrane-associated proteins FCHo and SGIP are allosteric activators of the AP2 clathrin adaptor complex. eLife 3, e03648. 10.7554/eLife.03648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incecik, F., Bisgin, A. and Yılmaz, M. (2018). MEDNIK syndrome with a frame shift causing mutation in AP1S1 gene and literature review of the clinical features. Metab. Brain Dis. 33, 2065-2068. 10.1007/s11011-018-0313-4 [DOI] [PubMed] [Google Scholar]
- Kaksonen, M. and Roux, A. (2018). Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19, 313-326. 10.1038/nrm.2017.132 [DOI] [PubMed] [Google Scholar]
- Kaksonen, M., Toret, C. P. and Drubin, D. G. (2006). Harnessing actin dynamics for clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 7, 404-414. 10.1038/nrm1940 [DOI] [PubMed] [Google Scholar]
- Kirchhausen, T., Owen, D. and Harrison, S. C. (2014). Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 6, a016725. 10.1101/cshperspect.a016725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, M., Fukai, K., Shimizu, N., Nagao, J., Takeichi, T., Tsuruta, D., Sugiura, K. and Akiyama, M. (2017). Punctate palmoplantar keratoderma type 1 with a novel AAGAB frameshift mutation: intrafamilial phenotype variation due to ageing. J. Eur. Acad. Dermatol. 31, e175-e176. 10.1111/jdv.13906 [DOI] [PubMed] [Google Scholar]
- Li, L. V. and Kandror, K. V. (2005). Golgi-localized, γ-ear-containing, Arf-binding protein adaptors mediate insulin-responsive trafficking of glucose transporter 4 in 3T3-L1 adipocytes. Mol. Endocrinol. 19, 2145-2153. 10.1210/me.2005-0032 [DOI] [PubMed] [Google Scholar]
- Li, W., Xu, H., Xiao, T., Cong, L., Love, M. I., Zhang, F., Irizarry, R. A., Liu, J. S., Brown, M. and Liu, X. S. (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. 10.1186/s13059-014-0554-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubben, N. B., Sahlender, D. A., Motley, A. M., Lehner, P. J., Benaroch, P. and Robinson, M. S. (2007). HIV-1 Nef-induced down-regulation of MHC class I requires AP-1 and clathrin but not PACS-1 and is impeded by AP-2. Mol. Biol. Cell 18, 3351-3365. 10.1091/mbc.e07-03-0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahil, S. K., Twelves, S., Farkas, K., Setta-Kaffetzi, N., Burden, A. D., Gach, J. E., Irvine, A. D., Képíró, L., Mockenhaupt, M., Oon, H. H.et al. (2016). AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J. Invest. Dermatol. 136, 2251-2259. 10.1016/j.jid.2016.06.618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli, D. and Dionisi-Vici, C. (2014). AP1S1 defect causing MEDNIK syndrome: a new adaptinopathy associated with defective copper metabolism. Ann. N. Y. Acad. Sci. 1314, 55-63. 10.1111/nyas.12426 [DOI] [PubMed] [Google Scholar]
- McMahon, H. T. and Boucrot, E. (2011). Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12, 517-533. 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- Mettlen, M., Chen, P.-H., Srinivasan, S., Danuser, G. and Schmid, S. L. (2018). Regulation of clathrin-mediated endocytosis. Annu. Rev. Biochem. 87, 871-896. 10.1146/annurev-biochem-062917-012644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C., Zizioli, D., Lausmann, S., Eskelinen, E.-L., Hamann, J., Saftig, P., von Figura, K. and Schu, P. (2000). μ1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 19, 2193-2203. 10.1093/emboj/19.10.2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsunari, T., Nakatsu, F., Shioda, N., Love, P. E., Grinberg, A., Bonifacino, J. S. and Ohno, H. (2005). Clathrin adaptor AP-2 is essential for early embryonal development. Mol. Cell. Biol. 25, 9318-9323. 10.1128/MCB.25.21.9318-9323.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit, A., Côté, S., Brustein, E., Drouin, C. A., Lapointe, L., Boudreau, M., Meloche, C., Drouin, R., Hudson, T. J., Drapeau, P.et al. (2008). Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord. PLoS Genet. 4, e1000296. 10.1371/journal.pgen.1000296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura, T., Yoneta, A., Pohler, E., Suzuki, S., Osawa, R., Mizuno, O., Ohguchi, Y., Nomura, Y., Yamashita, T., McLean, W. H. I.et al. (2015). Punctate palmoplantar keratoderma type 1: a novel aagab mutation and efficacy of etretinate. Acta Derm. Venereol. 95, 110-111. 10.2340/00015555-1832 [DOI] [PubMed] [Google Scholar]
- Paczkowski, J. E., Richardson, B. C. and Fromme, J. C. (2015). Cargo adaptors: structures illuminate mechanisms regulating vesicle biogenesis. Trends Cell Biol. 25, 408-416. 10.1016/j.tcb.2015.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, L. J., Sowerby, P. J., Lui, W. W. Y. and Robinson, M. S. (1999). γ-synergin: an EH domain-containing protein that interacts with γ-adaptin. J. Cell Biol. 146, 993-1004. 10.1083/jcb.146.5.993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. Y. and Guo, X. (2014). Adaptor protein complexes and intracellular transport. Biosci. Rep. 34, e00123. 10.1042/BSR20140069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse, B. M. and Robinson, M. S. (1984). Purification and properties of 100-kd proteins from coated vesicles and their reconstitution with clathrin. EMBO J. 3, 1951-1957. 10.1002/j.1460-2075.1984.tb02075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohler, E., Mamai, O., Hirst, J., Zamiri, M., Horn, H., Nomura, T., Irvine, A. D., Moran, B., Wilson, N. J., Smith, F. J. D.et al. (2012). Haploinsufficiency for AAGAB causes clinically heterogeneous forms of punctate palmoplantar keratoderma. Nat. Genet. 44, 1272-1276. 10.1038/ng.2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöhler, E., Zamiri, M., Harkins, C. P., Salas-Alanis, J. C., Perkins, W., Smith, F. J. D., Irwin McLean, W. H. and Brown, S. J. (2013). Heterozygous mutations in AAGAB cause type 1 punctate palmoplantar keratoderma with evidence for increased growth factor signaling. J. Invest. Dermatol. 133, 2805-2808. 10.1038/jid.2013.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan, V., Agrawal, N. J., Liu, J., Engles, S., Toy, R. and Radhakrishnan, R. (2011). Systems biology and physical biology of clathrin-mediated endocytosis. Integr. Biol. 3, 803-815. 10.1039/c1ib00036e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X., Farías, G. G., Canagarajah, B. J., Bonifacino, J. S. and Hurley, J. H. (2013). Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755-767. 10.1016/j.cell.2012.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricotta, D., Conner, S. D., Schmid, S. L., von Figura, K. and Höning, S. (2002). Phosphorylation of the AP2 μ subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J. Cell Biol. 156, 791-795. 10.1083/jcb.200111068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeth, J. F., Williams, M., Kasper, M. R., Filzen, T. M. and Collins, K. L. (2004). HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 167, 903-913. 10.1083/jcb.200407031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, A., Hirst, J., Davies, A. K. and Robinson, M. S. (2019). Adaptor protein complexes and disease at a glance. J. Cell Sci. 132, jcs222992. 10.1242/jcs.222992 [DOI] [PubMed] [Google Scholar]
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B.et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setta-Kaffetzi, N., Simpson, M. A., Navarini, A. A., Patel, V. M., Lu, H.-C., Allen, M. H., Duckworth, M., Bachelez, H., Burden, A. D., Choon, S.-E.et al. (2014). AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 94, 790-797. 10.1016/j.ajhg.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Q.-T., Ren, X., Zhang, R., Lee, I.-H. and Hurley, J. H. (2015). HIV-1 Nef hijacks clathrin coats by stabilizing AP-1:Arf1 polygons. Science 350, aac5137. 10.1126/science.aac5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub, L. M. (1997). Clathrin-associated adaptor proteins — putting it all together. Trends Cell Biol. 7, 43-46. 10.1016/S0962-8924(96)20042-X [DOI] [PubMed] [Google Scholar]
- Traub, L. M. and Bonifacino, J. S. (2013). Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 5, a016790. 10.1101/cshperspect.a016790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. J., Wang, J., Sun, H. Q., Martinez, M., Sun, Y. X., Macia, E., Kirchhausen, T., Albanesi, J. P., Roth, M. G. and Yin, H. L. (2003). Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 114, 299-310. 10.1016/S0092-8674(03)00603-2 [DOI] [PubMed] [Google Scholar]
- Wang, L., Johnson, A., Hanna, M. and Audhya, A. (2016). Eps15 membrane-binding and -bending activity acts redundantly with Fcho1 during clathrin-mediated endocytosis. Mol. Biol. Cell 27, 2675-2687. 10.1091/mbc.e16-03-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S., Crisman, L., Miller, J., Datta, I., Gulbranson, D. R., Tian, Y., Yin, Q., Yu, H. and Shen, J. (2019). Inducible Exoc7/Exo70 knockout reveals a critical role of the exocyst in insulin-regulated GLUT4 exocytosis. J. Biol. Chem. 294, 19988-19996. 10.1074/jbc.RA119.010821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H., Crisman, L., Stowell, M. H. B. and Shen, J. (2019). Functional reconstitution of intracellular vesicle fusion using purified SNAREs and Sec1/Munc18 (SM) proteins. Methods Mol. Biol. 1860, 237-249. 10.1007/978-1-4939-8760-3_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizioli, D., Meyer, C., Guhde, G., Saftig, P., von Figura, K. and Schu, P. (1999). Early embryonic death of mice deficient in γ-adaptin. J. Biol. Chem. 274, 5385-5390. 10.1074/jbc.274.9.5385 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.