

Abstract
BAK and BAX, the effectors of intrinsic apoptosis, each undergo major reconfiguration to an activated conformer that self‐associates to damage mitochondria and cause cell death. However, the dynamic structural mechanisms of this reconfiguration in the presence of a membrane have yet to be fully elucidated. To explore the metamorphosis of membrane‐bound BAK, we employed hydrogen‐deuterium exchange mass spectrometry (HDX‐MS). The HDX‐MS profile of BAK on liposomes comprising mitochondrial lipids was consistent with known solution structures of inactive BAK. Following activation, HDX‐MS resolved major reconfigurations in BAK. Mutagenesis guided by our HDX‐MS profiling revealed that the BCL‐2 homology (BH) 4 domain maintains the inactive conformation of BAK, and disrupting this domain is sufficient for constitutive BAK activation. Moreover, the entire N‐terminal region preceding the BAK oligomerisation domains became disordered post‐activation and remained disordered in the activated oligomer. Removal of the disordered N‐terminus did not impair, but rather slightly potentiated, BAK‐mediated membrane permeabilisation of liposomes and mitochondria. Together, our HDX‐MS analyses reveal new insights into the dynamic nature of BAK activation on a membrane, which may provide new opportunities for therapeutic targeting.
Keywords: apoptosis, BAK, BCL‐2, hydrogen‐deuterium exchange mass spectrometry, membrane
Subject Categories: Autophagy & Cell Death, Membranes & Trafficking, Structural Biology
Hydrogen‐deuterium exchange mass spectrometry of BAK on mitochondria‐like membranes identifies domains and conformations critically involved in activation.
Introduction
BAK and BAX are pro‐apoptotic members of the BCL‐2 family of proteins. Their expression and apoptotic activity are essential for cells to die in response to numerous apoptotic stimuli including anoikis, growth factor withdrawal and DNA damage (Wei et al, 2001; Czabotar et al, 2014). In a healthy cell, BAK and BAX are predominantly in an inactive conformation. Following an apoptotic stress, interaction with activated or upregulated BH3‐only proteins including BIM and caspase 8‐cleaved BID (cBID), BAK and BAX become activated undergoing a drastic change in their conformation. This reconfiguration includes exposure of N‐terminal and BH3 domain antibody epitopes, and dissociation of the α2‐5 (“core”) from α6‐8 (“latch”) domains (Hsu & Youle, 1998; Griffiths et al, 1999; Dewson et al, 2008; Oh et al, 2010; Czabotar et al, 2013; Brouwer et al, 2014). The activated forms then self‐associate by reciprocal interaction of the exposed BH3 domain with a hydrophobic groove of a partner molecule forming symmetric “BH3:groove” homodimers (Dewson et al, 2008, 2012; Czabotar et al, 2013; Brouwer et al, 2014; Subburaj et al, 2015; Zhang et al, 2016). These homodimers then multimerise presumably independent of both the BH3 domain and hydrophobic groove to form high molecular weight structures that permeabilise the mitochondrial outer membrane (Qian et al, 2008; Zhang et al, 2010; Bleicken et al, 2014; Subburaj et al, 2015). Ring‐like structures of BAX or BAK that may represent the elusive apoptotic pore have been reported in apoptotic cells (Grosse et al, 2016; Salvador‐Gallego et al, 2016), whilst recent studies have shown a role for mitochondrial outer membrane lipids in mediating higher order oligomerisation, consistent with previous work indicating that BAX and BAK form lipidic pores (Terrones et al, 2004; Qian et al, 2008; Salvador‐Gallego et al, 2016; Cowan et al, 2020).
X‐ray crystallography has provided important snapshots of BAK and BAX conformations in solution (Moldoveanu et al, 2006; Czabotar et al, 2013; Brouwer et al, 2014; Birkinshaw et al, 2021). In addition, biochemical approaches such as cross‐linking, limited proteolysis and spin labelling have provided insight into changes that occur on mitochondria (Annis et al, 2005; Dewson et al, 2008, 2009, 2012; Bleicken et al, 2010, 2014; Oh et al, 2010; Zhang et al, 2010). However, despite these advances, we lack complete understanding of the molecular changes in BAK/BAX conformation that occur upon activation to facilitate their homodimerisation and subsequent multimerisation to higher order structures. The events that lead to BAK and BAX pore formation have eluded characterisation potentially due to their highly dynamic nature and the requirement for a membrane environment. Indeed, lipid‐mediated association of dimers to form a lipidic pore or even random aggregation of dimers to cause membrane rupture are proposed mechanisms (Basanez et al, 2002; Terrones et al, 2004; Uren et al, 2017). Resolving the molecular details of these coordinated events in a membrane environment is key to understand their mechanism of action and will expedite efforts to unlock their emerging and clear potential as therapeutic targets (Niu et al, 2017; van Delft et al, 2019).
To overcome these barriers to conventional structural biology approaches and to provide new insight into the conformational dynamics of BAK activation as they occur in a membrane, we employed hydrogen‐deuterium exchange mass spectrometry (HDX‐MS). HDX‐MS exploits the exchange of hydrogen at the amide backbone, the rate of which is governed by hydrogen bonding and solvent accessibility and so is determined by protein conformation, protein‐protein and protein‐lipid interactions. HDX‐MS can be performed in a membrane (Wales & Engen, 2006) and has been used to determine the molecular interactions of large membrane complexes to almost single residue resolution (Sticht et al, 2005), whilst revealing subtle and dynamic changes in protein conformation. Thus, HDX‐MS represents a potentially powerful approach to characterise BAK activating conformation change in a membrane environment.
We found that activation of BAK in membranes in response to cBID involved exposure of the N‐terminus consistent with published findings on the exposure of cryptic antibody epitopes in BAK in apoptotic cells (Griffiths et al, 1999, 2001). HDX‐MS also revealed that a significant proportion of the BAK N‐terminus becomes highly disordered upon its dissociation from the core of the protein. However, the exposed N‐terminus of BAK does not contribute to apoptotic pore formation, but rather limits BAK‐mediated membrane damage. Furthermore, mapping the exchange data to existing structures of the BAK monomer and BH3:groove homodimer reveal that disruption of key intramolecular interactions involving the conserved BH4 domain residues is sufficient to trigger BAK activation and apoptotic activity.
Results
Characterisation of BAK oligomerisation on liposomes
BAK is normally integrated into the mitochondrial outer membrane in healthy cells via its C‐terminal transmembrane anchor (Iyer et al, 2015). Consequently, full‐length BAK is unstable as a recombinant protein due to the hydrophobicity of its C‐terminal transmembrane domain. To circumvent this problem, we used recombinant mouse BAK with a 6xHis at its C‐terminus in lieu of its transmembrane anchor (BAK‐6H). BAK‐6H was then targeted to liposomes reconstituted with mitochondrial lipids and a Ni‐NTA lipid (Fig 1A). Consistent with previous reports with this system, although BAK alone promoted limited liposome permeabilisation, permeabilising activity was promoted by an activating BID BH3 peptide (Fig 1A). BAK efficiently localised to these liposomes and was largely monomeric and inactive in the absence of an activating stimulus, but oligomerised following activation with a BH3 peptide or recombinant cBID to efficiently permeabilise liposomes (Figs 1B and EV1A and B).
Figure 1. BAK‐6H activation and oligomerisation on liposomes mirrors changes on mitochondria.
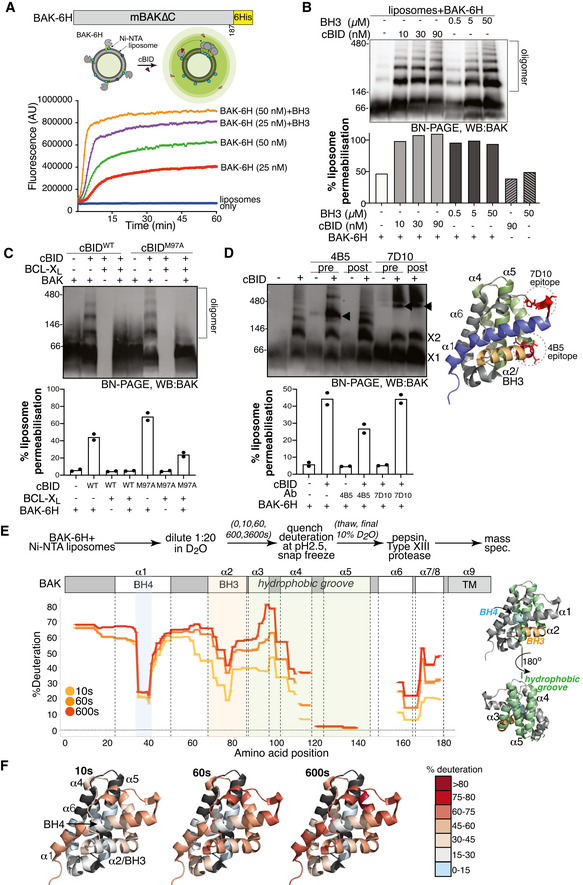
- BAK‐6H permeabilising activity is exacerbated by cBID. BAK‐6H (mBAK∆C21‐6H) was loaded at the indicated concentrations onto Ni‐NTA liposomes (5 µM) prior to the addition of cBID. The release of carboxyfluorescein was measured over time. Results representative of at least three independent experiments.
- BAK‐6H oligomerises on liposomes induced by cBID. Liposomes were incubated with BAK‐6H (150 nM) and the indicated concentration of cBID or a BID BH3 peptide for 60 min. Liposomes were solubilised in digitonin and BAK oligomers were analysed by BN‐PAGE. In parallel, samples were tested at endpoint for liposome permeabilisation based on the release of fluorescent dextran, normalised to total fluorescence of detergent permeabilised liposomes. Results representative of three independent experiments.
- BAK‐6H oligomerisation on liposomes is blocked by BCL‐XL. Liposomes were incubated with BAK‐6H (150 nM) together with cBID (WT, 90 nM) or cBID M97A (90 nM) that has reduced affinity for BCL‐XL, in the presence or absence of BCL‐XL for 60 min prior to analysis of BAK oligomerisation by BN‐PAGE. Results representative of two independent experiments. Liposome permeabilisation was monitored with the indicated combinations of cBID (90 nM), BAK‐6H (150 nM) and BCL‐XL (1 µM). Data are presented as mean of two independent experiments with percentage release relative to detergent‐lysed liposomes at a timepoint when permeabilisation with BAK and cBid was approximately 50% of detergent‐lysis maximum.
- BAK‐6H oligomerisation on liposomes involves its BH3 domain. Liposomes were incubated with BAK‐6H (150 nM) together with cBID (90 nM). Antibody recognising the BAK BH3 domain (4B5) or α1‐α2 loop (7D10), as shown on the structure of BAK (PDB:2IMS), were added (2.4 µg/µl) either prior to (pre) or after (post) incubation with cBID for 60 min. BAK:Ab complex is indicated (arrowhead). 7D10 which recognises the activated form of mBAK (Iyer et al, 2016) bound to and gel‐shifted BAK when added either before or after activation with cBID. In contrast, 4B5 failed to appreciably gel‐shift inactive BAK (lanes 3 and 5) or BAK that was already activated (lane 6), but gel‐shifted BAK when present during cBID activation (lane 4). Liposome permeabilisation was assessed after 20 min incubation with cBID (90 nM), BAK‐6H (150 nM) and antibodies 4B5 (2.5 µg) or 7D10 (2.5 µg). Data are presented as mean of two independent experiments with percentage release relative to detergent‐lysed liposomes at a timepoint when permeabilisation with BAK and cBid was approximately 50% of detergent‐lysis maximum.
- HDX‐MS reveals structural constraints of BAK on a membrane. BAK‐6H (150 nM) on liposomes was deuterated for 0, 10s, 60s or 600s and the incorporation of deuterium was assessed by mass spectrometry. Deuteration is expressed relative to a theoretical maximum. BH3 domain (orange), BH4 domain (blue) and hydrophobic surface groove comprising αhelices 3–5 (green) are shown on the structure of BAK (PDB:2IMS).
- Differential deuteration of BAK in a membrane. Relative deuteration is mapped onto the structure of inactive monomeric BAK (2IMS, Moldoveanu et al, 2006).
Source data are available online for this figure.
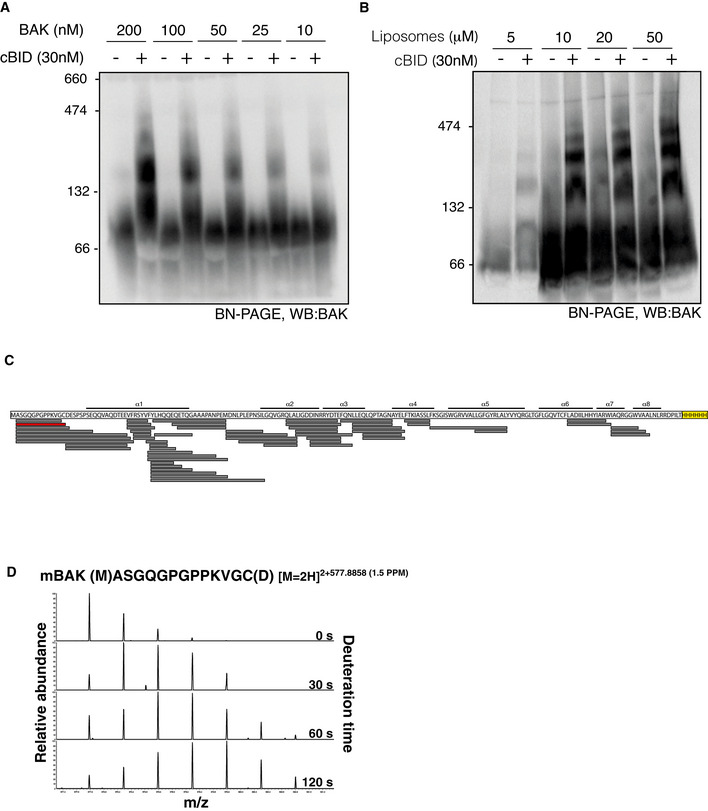
Figure EV1. HDX‐MS analysis of BAK‐6H during oligomerisation on liposomes.
-
A, BLiposomes were incubated with BAK‐6H and cBID at the indicated concentration prior to the analysis of BAK oligomerisation on BN‐PAGE.
-
CBAK peptide coverage following combined proteolysis with pepsin and Aspergillus Type XIII protease.
-
DExample of time‐dependent deuteration of BAK peptide. The increase in m/z of a BAK peptide (shown in red in C) due to incorporation with deuterium over time.
Source data are available online for this figure.
BAK oligomers forming on mitochondria of apoptotic cells are known to require interaction of an exposed BH3 domain with the hydrophobic groove of a partner BAK molecule (Dewson et al, 2008; Czabotar et al, 2014). To test if the pore‐forming oligomers observed in liposomes represent these BH3:groove homo‐oligomers, we tested for their ability to be blocked by pro‐survival protein BCL‐XL. Co‐incubation with recombinant BCL‐XL blocked BAK oligomerisation on liposomes driven by cBID as assessed by BN‐PAGE and liposome permeabilisation (Fig 1C). Blockade by BCL‐XL was not just due to its binding cBID to inhibit initial BAK activation as BCL‐XL could also inhibit oligomerisation of BAK and permeabilisation triggered by cBIDM97A that does not efficiently bind BCL‐XL (Lee et al, 2016). Following BAK activation on mitochondria, the BAK antibody 4B5 can bind the exposed BH3 domain to prevent BAK oligomerisation, but the antibody cannot recognise BAK prior to its activation or once it has oligomerised (Dewson et al, 2008). To confirm that the BAK oligomers induced on liposomes involved BH3:groove homodimers, we tested the ability of the 4B5 antibody to bind BAK either when added before or after BAK dimerisation. The 4B5 antibody failed to gel‐shift inactive BAK on liposomes (Fig 1D, lane 3), and likewise when added after BAK oligomerisation (Fig 1D). However, 4B5 antibody could bind when it was added during BAK activation and also impaired liposome permeabilisation (Fig 1D). In comparison, the 7D10 antibody, which binds residues in the BAK α1‐2 loop (Iyer et al, 2016), gel‐shifted BAK when added either during or after induction of BAK oligomerisation and did not block permeabilisation (Fig 1D). Together, these data support that during BAK activation on, and permeabilisation of, liposomes the BH3 domain is transiently exposed, but is then buried in the oligomer, thus mimicking BAK conformation changes that occur upon activation and oligomerisation on mitochondria.
BAK adopts an inactive conformation on liposomes revealed by HDX‐MS
To characterise deuterium uptake in membrane‐associated BAK, we first performed a time course of deuterium incorporation of recombinant BAK∆C21‐6H targeted to liposomes followed by quenching of exchange at pH 2.5 and 1°C and combined digestion with the acidic proteases pepsin and Type XIII from Aspergillus saitoi. We found that proteolysis by both proteases was necessary for efficient digestion of BAK resulting in near‐complete and overlapping peptide signature, although peptides C‐terminal of α4 were under‐represented (Fig EV1C). Mass spectrometry revealed a time‐dependent increase in the mass of peptides due to deuterium incorporation (Fig EV1D). The rate of exchange correlated well with the published structure of inactive human BAK (2IMS, Moldoveanu et al, 2006), with exposed termini and flexible loop regions exchanging more rapidly than structured regions in the hydrophobic core (Fig 1E and F). Interestingly, the BH4 domain was very resistant to exchange compared with flanking residues in the α1, suggesting a potentially important role for these residues in stabilising the inactive conformer of BAK. Additionally, residues in the α6/7/8 were relatively resistant to exchange despite their predicted solvent exposure in the structure of soluble BAK (Fig 1E and F) (Moldoveanu et al, 2006), suggesting that these residues may interface with lipids of the mitochondrial outer membrane.
HDX‐MS reveals reorganisation of BAK upon activation on membranes
Upon incubation with recombinant cBID to induce BAK activation and oligomerisation (Fig 1), HDX‐MS revealed significant changes in BAK conformation as indicated by changes in the rate of deuterium exchange (Fig 2A). Following incubation with cBID, there was a marked increase in deuteration of the BAK α1, particularly corresponding to residues in the conserved BH4 domain (Kvansakul et al, 2008), and in the first half of α2 (Fig 2A and B). This is consistent with the reported exposure of the N‐terminus and BH3 domain during BAK activation as indicated by antibody binding and limited proteolysis (Griffiths et al, 1999; Dewson et al, 2008).
Figure 2. HDX‐MS reveals dynamic changes in BAK activated on liposomes.
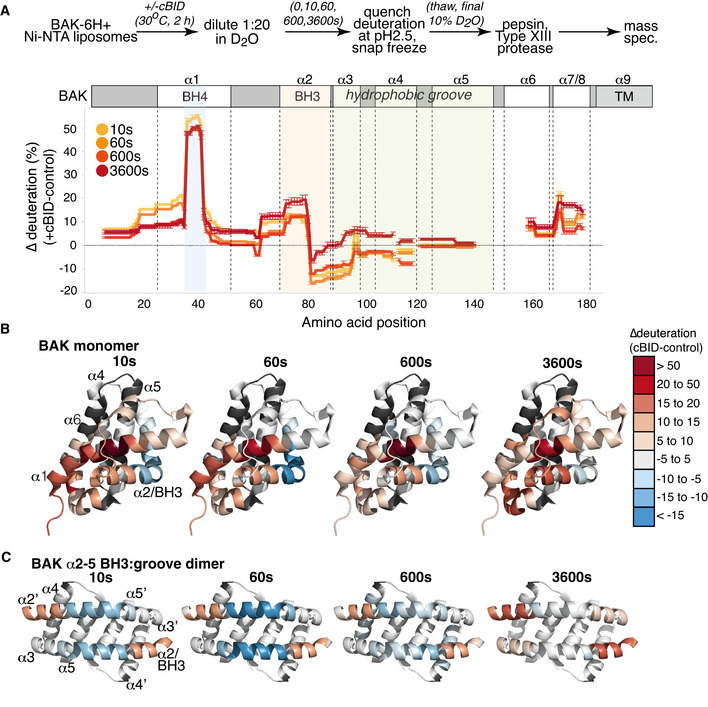
- BAK‐6H on liposomes was activated or not with cBID for 60 min prior to deuteration for 10s, 60s, 600s, or 3,600s. Incorporation of deuterium was assessed by mass spectrometry. Change in deuteration at each time point is expressed relative to BAK on liposomes without cBID.
- Deuteration of BAK in a membrane compared to inactive BAK is mapped onto the structure of inactive monomeric BAK (2IMS, Moldoveanu et al, 2006).
- Deuteration of BAK in a membrane compared to inactive BAK is mapped onto the structure of an activated BAK BH3:groove dimer (4U2V, Brouwer et al, 2014).
When changes in deuteration were mapped onto the structure of the hBAK α2‐5 homodimer (Brouwer et al, 2014; Birkinshaw et al, 2021), the hinge region between the α2 and α3 exhibited reduced exchange that might imply that the region becomes more structured‐ or is less solvent‐exposed due to protein:protein or protein:lipid interactions. However, we have previously shown that labelling of surface‐exposed residues (R88 and E92) in this α2/α3 region with 5 kDa PEG‐maleimide did not impair the ability of BAK to oligomerise or to mediate MOMP (Li et al, 2017). This argues against this region of the protein forming important interfaces with either protein or lipid. Comparisons of the X‐ray structures of inactive BAK with that of a truncated BAK α2‐5 homodimer reveals significant reconfiguration of the α2/α3 hinge region as the α2/BH3 domain becomes exposed and binds into the hydrophobic groove of an opposing BAK monomer, forming the activated BAK homodimer (Fig 2C) (Brouwer et al, 2014). The reduced rate of deuterium exchange in this α2/α3 hinge region in activated BAK is consistent with the straightening of the α2/α3 hinge region observed in dimeric BAK (Brouwer et al, 2014). The trend towards reduced exchange along the α4 suggests that this amphipathic helix is less exposed in the activated form of BAK. Although the peptide coverage in central hydrophobic α5 helix was relatively limited, there was no significant change in the deuterium exchange along this helix upon activation. That α4 becomes more buried whilst the α5 remains so is potentially consistent with the interaction of this lipophilic region with the mitochondrial outer membrane in activated and oligomerised BAK (Czabotar et al, 2013; Cowan et al, 2020).
Recently, a crystal structure of oligomeric BAK revealed a hexameric configuration of truncated BAK α2‐α5 homodimers in solution and identified lipids, including E. coli‐derived phosphoethanolamine (PE), that interacted with α5 residues to stabilise dimer:dimer interactions (Cowan et al, 2020). Whilst the hexamer itself was not considered to represent the precise orientation of BAK dimers within larger BAK oligomers on mitochondria, the structure revealed lipids cross‐linking between dimers and supported a model of lipid embedded dimers with a cytosol‐exposed α2α3α2′α3′ surface (equivalent to the surface on the outside of the hexamer) and a membrane buried α4α5α5′α4′ surface (equivalent to the surface facing the core of the hexamer) (Fig EV2). We mapped our HDX‐MS profiles to this hexameric structure (Fig EV2). Helices α2‐ α3 reside on the exterior of the hexamer consistent with their increased deuterium exchange following activation (Fig EV2). However, residues in helices α4 and α5 that face the core of the hexameric structure did not exhibit increased exchange following activation and oligomerisation of BAK on a membrane (Figs 2A and EV2), suggesting that α4‐α5 do not transition to a more solvent‐exposed environment and that they do not line a channel in oligomeric BAK. This aligns with the conclusions of Cowan et al that the protein‐protein interactions involved in the crystal do not support the biological relevance of the hexamer as an intermediate in a larger BAK pore‐forming oligomer (Cowan et al, 2020) and also reports that BAK forms disordered and heterogeneous complexes on mitochondria (Salvador‐Gallego et al, 2016; Uren et al, 2017). Whilst our HDX‐MS did not reveal an inhibition of exchange that might support discrete interactions of α5 residues with lipid, or an α4α5α5′α4′ interface that is promoted by lipid interaction, our approach of analysing the exchange profiles before or after full activation of BAK would not inform transient changes, including, α5 residues transitioning from being buried in the monomer, to transiently exposed, to being reburied in the oligomer with interfacing lipids. To interrogate such transient changes, continuous HDX‐MS (as opposed to pulse HDX‐MS employed here) involving sampling during BAK activation in the presence of deuterium would be informative to reveal transitional conformations.
Figure EV2. HDX‐MS mapped onto hexameric BAK.
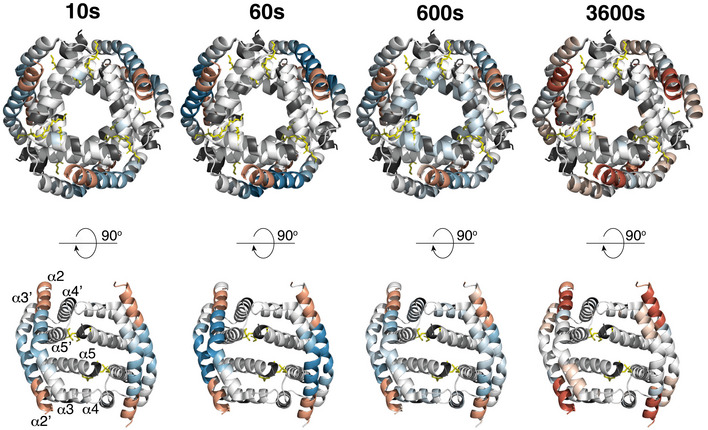
Change in deuteration of BAK compared to inactive BAK is mapped onto the structure of hexameric BAK core dimers (trimer of α2‐α5 homodimers) with bound E. coli lipids (yellow stick) (PDB:6UXM, Cowan et al, 2020). Colour coding as in Fig 2B. Lower panel is a 90° rotated view of the hexamer showing the uppermost 2 dimers only.
Crystal structures of inactive BAK lack the α9 C‐terminal transmembrane anchor and are in the absence of a membrane (Moldoveanu et al, 2006). Without the constraint of membrane anchoring via its α9 C‐terminus, the orientation of the preceding α7/8 helices in relation to the rest of the protein when it is tail‐anchored in the MOM is unknown. Our HDX‐MS identified a potential interaction between residues E24 at the N‐terminal end of α1 and R169 in α7 (Fig 3A) consistent with a salt‐bridge identified between these residues in a crystal structure of truncated BAK in solution. Whilst deuterium exchange of both residues was limited in the inactive conformer, it was significantly enhanced upon activation (Fig 3A). This confirms that the α1 and α7/α8 helices are indeed in close proximity in the inactive conformer of membrane‐anchored BAK and that they dissociate during BAK activation consistent with the structures of activated BAK dimers (Birkinshaw et al, 2021). To test this in cells, we engineered a variant of BAK with cysteines introduced at E24, R169 or both on an otherwise Cys‐null (C14S/C166S) background, stably expressed them in Bax −/− Bak −/− MEFs and confirmed expression and apoptotic activity (Fig 3B). Prior to BAK activation, oxidant treatment of enriched mitochondrial fractions caused wild‐type BAK to migrate faster on non‐reducing SDS–PAGE due to an induced intramolecular disulphide linkage between its two cysteines (C14, C166; Fig. 3C) (Cheng et al, 2003; Dewson et al, 2009). Whilst the migration of BAK single cysteine and ∆Cys control variants was not altered by oxidant as expected, BAK E24C/R169C likewise migrated faster on SDS–PAGE, indicating close proximity of E24 and R168 in the inactive conformer of BAK on the MOM (Fig 3C). This intramolecular disulphide linkage was reduced following activation with cBID in favour of intermolecular linkage, indicating that the α1 and α7 dissociate upon activation consistent with the HDX‐MS (Fig 3A and D).
Figure 3. BAK α1 and α7 dissociate upon activation on membranes.
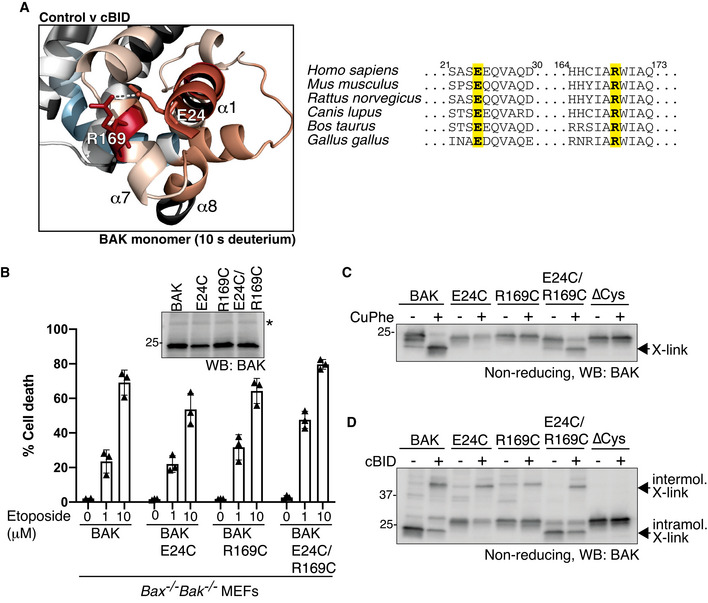
- HDX‐MS reveals a significant increase in deuteration at positions E24 (α1) and R169 (α7) following activation with cBID. These positions are conserved suggesting a potentially important hydrogen bonding interaction.
- BAK, or BAK with cysteine introduced at the indicated positions (on a BAK∆Cys background) were stably expressed in Bax −/− Bak −/− MEFs and assessed for BAK expression by immunoblotting (inset panel; * indicates non‐specific band) and apoptotic activity in response to etoposide treatment. Data are mean ± SD of three independent experiments.
- E24 and R169 are proximal in inactive BAK on mitochondria. Mitochondria‐enriched membrane fractions from cells in (B) were incubated with oxidant (CuPhe) and induced intramolecular disulphide linkage of BAK was assessed on non‐reducing SDS–PAGE.
- E24 and R169 dissociate during BAK activation. Mitochondria‐enriched membrane fractions from cells in (B) were incubated with cBID (100 nM) prior to oxidant (CuPhe) and the induced intramolecular and intermolecular disulphide linkage of BAK was assessed on non‐reducing SDS–PAGE.
Source data are available online for this figure.
Overall, our HDX‐MS data of BAK tethered to a membrane support the events revealed by crystal structures of truncated recombinant BAK protein in solution and biochemical assays of BAK conformation on mitochondria, whilst further supporting the formation of BH3:groove homodimers as a key step in BAK apoptotic function.
BH4 domain mutation is sufficient to activate BAK
Residues corresponding to the BAK BH4 domain (Kvansakul et al, 2008) were relatively resistant to deuteration in the inactive conformer, but became rapidly deuterated following activation (Figs 1E, 2A and 4A). This suggests that the BH4 domain is key in restraining BAK in its inactive configuration. Hence, we hypothesised that mutation of the BH4 domain would drive intrinsic BAK apoptotic activity. Given the important role for the N‐terminus in stable BAK protein expression, we employed conserved cysteine mutations in the BH4 domain of BAK (on a ∆Cys background) based on our previous experience with stable expression of such mutants (Fig 4A) (Westphal et al, 2014; Alsop et al, 2015; Li et al, 2017). To assess their intrinsic apoptotic activity, we expressed each BH4 mutant in Bak −/− Bax −/− MEFs under the control of a doxycycline‐inducible promoter. We compared these mutants to wild‐type BAK or BAK∆Cys as controls. First, we tested the induced expression of each and confirmed that BAK V34C and S37C expressed similarly to wild‐type BAK and BAK∆Cys (Figs 4B and EV3). In contrast, mutants of the residues F35 and Y38 that orientate towards the core α5 helix did not appreciably express following their induction (Unpublished observations) (Alsop et al, 2015), suggesting that the BH4 domain forms interactions that are key for BAK to adopt a stable fold. As expected, BH3 mimetic drugs ABT‐737 and S63845 failed to induce apoptosis in cells where BAK expression was not induced (Fig 4C). Upon induction with doxycycline, wild‐type BAK, BAK∆Cys and the two BH4 mutants, V34C and S37C, all mediated cell death in response to BH3 mimetic drugs (Fig 4C). However, induced expression of the BH4 mutants prompted significantly more cell death than either wild‐type BAK or BAK∆Cys in the absence of exogenous apoptotic stimuli (Fig 4C). Consistent with this constitutive activity, the expressed BH4 mutants spontaneously adopted an activated conformation with exposed N‐terminal epitopes (Fig 4D). Both of the BH4 residues V34 and S37 orientate towards the α2/BH3 (Fig 4A), suggesting that the BH4 domain, as well as forming important interactions with the α5 for BAK to adopt a stable fold, also serves to stabilise the BH3 domain in its buried configuration. Destabilising this interaction was sufficient to promote BAK activation even without stimulus to engage the canonical hydrophobic groove, thereby confirming the role of the BH4 domain in restraining inactive BAK.
Figure 4. Mutation of the BAK BH4 promotes constitutive activation.
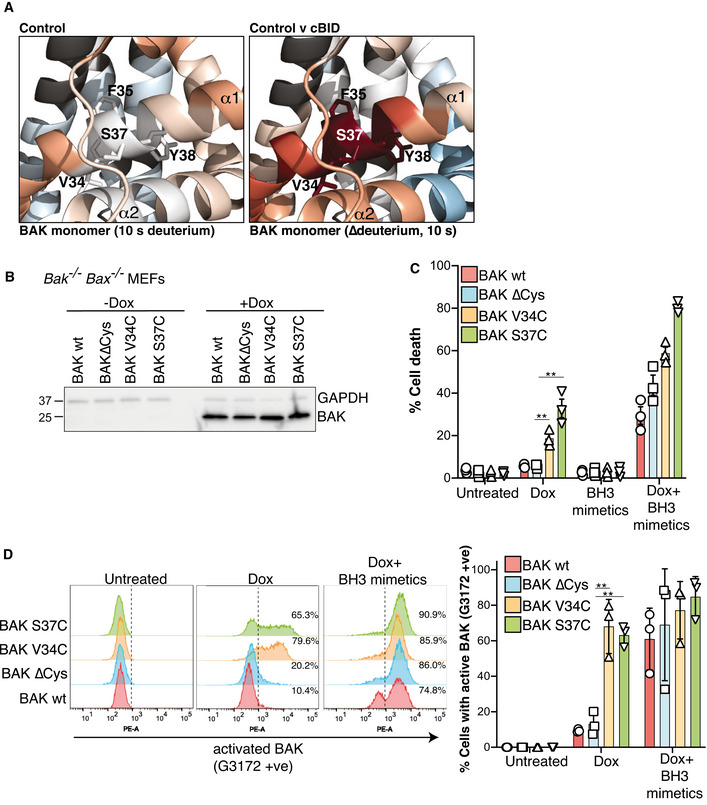
- HDX‐MS reveals a constrained BH4 domain in inactive BAK that is rapidly deuterated after activation. BAK BH4 residues in the α1 (V34, F35, S37 and Y38) corresponding to the BH4 motif Φ1Φ2XXΦ3Φ4 (Kvansakul et al, 2008) are indicated as sticks.
- BAK wild‐type or BAK engineered with the indicated mutations (on a BAK∆Cys background) were stably expressed in Bax −/− Bak −/− MEFs and expression was assessed by immunoblotting following 3 h doxycycline treatment.
- Bax −/− Bak −/− MEFs expressing the indicated BAK variants were treated with doxycycline (Dox, 3 h), incubated for a further 24 h in the presence or absence of BH3 mimetic compounds, and cell death was assessed by PI uptake. Data are expressed as mean ± SD of three independent experiments, **P < 0.01, unpaired Student’s t‐test.
- Mutation of BH4 promotes BAK activating conformation change. Cells in (B) were treated with doxycycline (3 h) to induce BAK expression followed by incubation with BH3 mimetics where indicated (for 2 h) and conformation change of BAK was assessed by intracellular flow cytometry with an antibody that recognises activated BAK (G3172). FACS profiles from one experiment are representative of the three independent experiments shown in Fig EV3. Collated data from three independent experiments are expressed as per cent G3172 positive cells (mean ± SD), **P < 0.01 unpaired Student’s t‐test.
Source data are available online for this figure.
Figure EV3. BAK BH4 mutants adopt an activated conformation without exogenous stimulus.
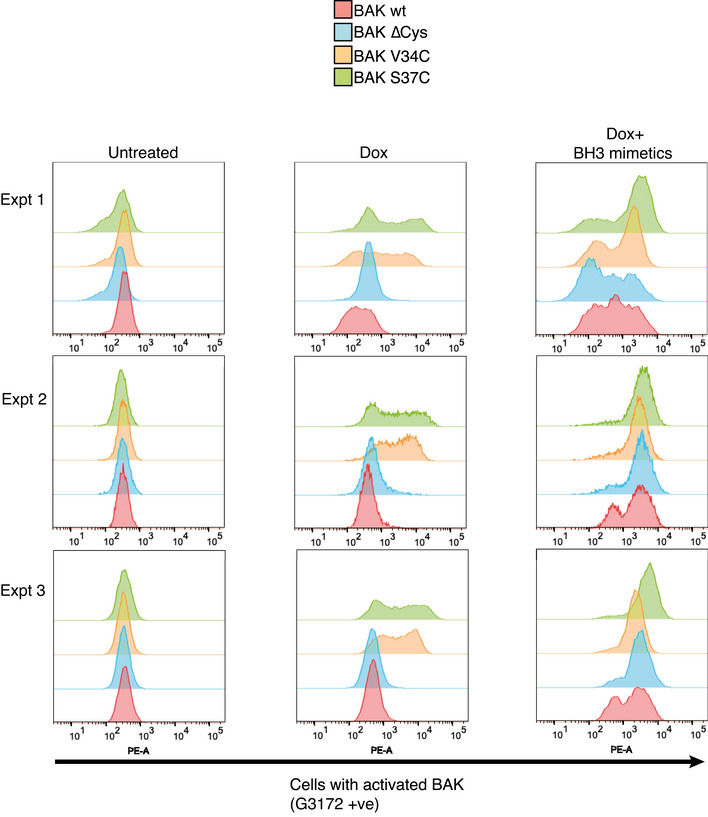
Bax −/− Bak −/− MEFs expressing BAK, BAK(∆Cys) or BAK(∆Cys) with mutations in the BH4 domain were incubated with doxycycline (3 h) to induce BAK expression followed by incubation with BH3 mimetics where indicated (for 2 h) and conformation change of BAK was assessed by intracellular flow cytometry with an antibody that recognises activated BAK (G3172). FACS profiles from three independent experiments are shown.
N‐terminal α1 helix of BAK becomes highly disordered when exposed during activation and impairs MOMP
HDX‐MS revealed that the N‐terminal 58 amino acids of BAK were rapidly and completely deuterated with no time‐dependent increase in deuteration following activation, in contrast to α2‐8 that showed a time‐dependent increase in deuteration both before and after activation (Fig 5A). This indicates that the entire N‐terminus of BAK preceding the α2(BH3) including the α1 become disordered once dissociated. Disordered regions of proteins are known to mediate protein:protein and also protein:lipid interactions. Indeed, cross‐linking analysis reveals residues in the BAK N‐terminus are in close proximity in oligomerised BAK (Uren et al, 2017), and we have recently shown that residues in the N‐terminus of activated BAK can be cross‐linked with diazirine‐labelled lipids (Cowan et al, 2020). Additionally, disordered regions of proteins increase their hydrodynamic radius, which has been proposed to enhance molecular crowding on membranes and impart tension to promote membrane curvature (Busch et al, 2015). Membrane curvature has been implicated as an important feature of MOMP mediated by BAK or BAX (Basanez et al, 2002). Thus, we hypothesised that the released and disordered N‐terminus may contribute to BAK‐mediated MOMP either by participating in BAK oligomer formation or promoting membrane disruption.
Figure 5. Residues preceding the BAK BH3 domain do not promote, and are not required for, pore formation.
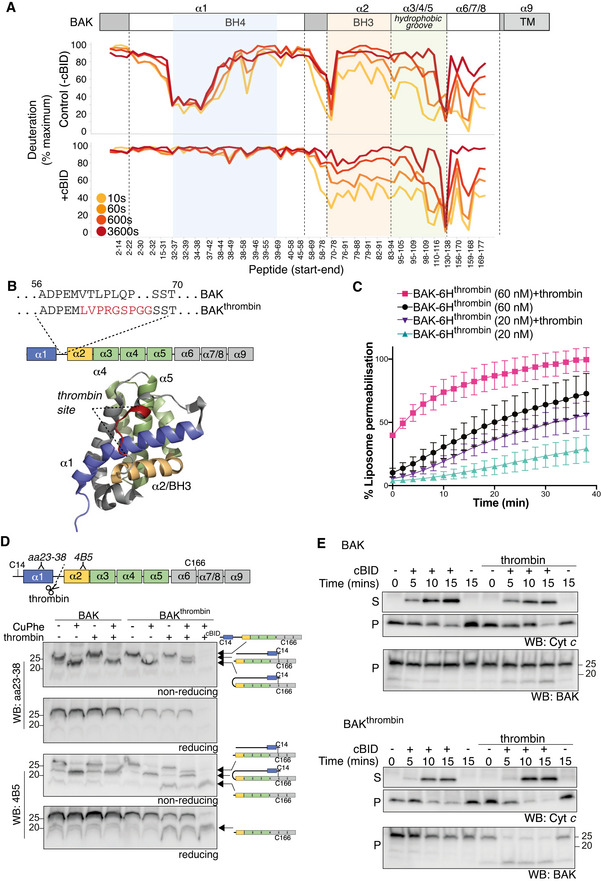
- BAK‐6H on liposomes was activated (+cBID, bottom) or not (Control, top) with cBID for 60 min prior to deuteration for 10 s, 60 s, 600 s, or 3,600 s. Incorporation of deuterium was assessed by mass spectrometry and is expressed relative to a non‐deuterated sample and a theoretical maximum. HDX‐MS reveals that following activation with cBID, maximal deuteration of the BAK N‐terminus (up to amino acid 58) occurred within 10 secs.
- BAK mutant with an engineered thrombin cleavage site in α1‐α2 loop of hBAK∆N22/∆C25/C166S with a 6xHis C‐terminal tag for recombinant protein expression or as full‐length protein for ectopic expression in MEFs (BAKthrombin).
- BAK cleaved within its α1‐2 loop by thrombin is able to mediate liposome permeabilisation. Recombinant BAK‐6Hthrombin pre‐cleaved or not with thrombin (see Fig EV4) was incubated with liposomes at either 20 or 60 nM and liposome permeabilisation induced by cBID was monitored over time. Data are presented as percentage liposome permeabilisation relative to detergent‐lysed liposomes, mean ± SEM of three independent experiments.
- BAK wild‐type or BAK with an engineered α1‐α2 loop thrombin cleavage site were stably expressed in Bax −/− Bak −/− MEFs. Membrane fractions were incubated with thrombin prior to incubation with copper phenanthroline (CuPhe) to induce disulphide linkage. Samples were run on SDS–PAGE under non‐reducing or reducing conditions and immunoblotted for BAK with an antibody recognising the BAK N‐terminus upstream of the thrombin cleavage site (aa23–38) or an antibody recognising the BH3 domain (4B5) downstream of the cleavage site. Schematic indicates the nature of the BAK protein detected by each antibody. Disulphide linkage indicated by a hashed line. Note that the N‐terminal portion of thrombin cleaved BAK could not be detected with the N‐terminal antibody unless it was intramolecularly disulphide‐linked. Where indicated cBID (100 nM) was added to activate BAK during thrombin cleavage. Data representative of two independent experiments.
- Cleavage of the BAK α1‐α2 loop potentiates MOMP. Membrane fractions from Bax −/− Bak −/− MEFs expressing BAK or BAKthrombin were incubated or not with thrombin in the presence or absence of cBID (10 nM) for the indicated times. Membrane (P) and supernatant (S) fractions were separated and immunoblotted for cytochrome c or BAK 4B5. Data representative of three independent experiments.
Source data are available online for this figure.
Exposure of the N‐terminus upon activation was one of the first characterised changes in the structure of BAK (and BAX) upon activation (Hsu & Youle, 1998; Griffiths et al, 1999). However, whether the N‐terminus contributes to MOMP when exposed is still unclear. A highly truncated BAK mutant comprising α2‐5/α9 that lacks the N‐terminus has been reported to kill cells upon transient over‐expression (George et al, 2007), although in a separate report a similar mutant failed to induce cell death when stably expressed due to a failure to associate with mitochondria (Ma et al, 2013). This suggests that the α1 is key in BAK adopting a stable and expressed fold. Indeed, our HDX‐MS data indicate that residues in the BH4 domain are highly constrained, suggesting that they play an important role in preserving the stability of the inactive BAK conformer, and potentially explain why BAK lacking its α1 cannot be stably expressed (Dewson et al, 2009). Hence, to test whether the disordered N‐terminus is important for membrane permeabilisation by BAK, we adopted a targeted proteolysis approach to specifically cleave the α1‐ α2 loop and allow the disordered α1 to dissociate from the folded protein following activation on membranes. We engineered a recombinant BAK variant with a thrombin cleavage site in the loop between the α1 and α2 helices, and a 6xHis tag at the C‐terminus to replace its C‐terminal transmembrane anchor (Fig 5B). Following treatment with thrombin, we purified the cleaved form of BAK by gel filtration revealing approximately 80% of BAK had its N‐terminus cleaved (Fig EV4). Cleaved BAK was then compared with uncleaved BAK in its ability to permeabilise liposomes. Cleavage of the α1‐2 loop prior to activation with cBID did not hinder the ability of BAK to permeabilise liposomes (Fig 5C), indicating that once exposed during BAK activation the N‐terminal amino acids are not required for either BAK oligomerisation or membrane permeabilisation. Rather, and contrary to our initial hypothesis that the BAK N‐terminus contributes to pore formation, cleavage in the α1‐2 loop actually potentiated BAK permeabilising activity (Fig 5C), suggesting that the exposed N‐terminus may impede BAK pore formation. Furthermore, loop cleavage potentially induced BAK activity even in the absence of BH3 activating stimulus (Fig 5C, see timepoint 0 for 60 nM BAK), which may be consistent with our previous findings that antibody binding to the α1‐2 loop can trigger BAK activation (Iyer et al, 2016).
Figure EV4. α1‐2 loop cleavage in recombinant BAK.
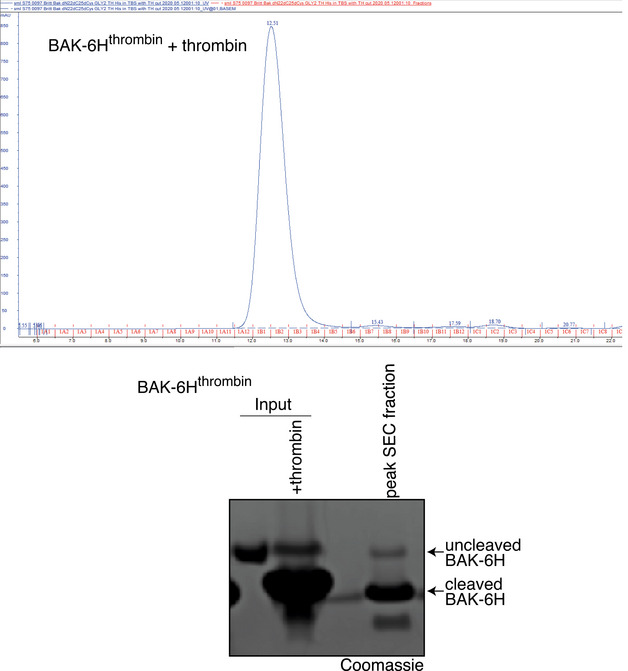
Recombinant BAK‐6Hthrombin was cleaved with thrombin prior to size exclusion chromatography. Coomassie‐stained SDS–PAGE of input and the peak fraction reveals efficient loop cleavage.
Source data are available online for this figure.
We then tested if the BAK N‐terminus likewise inhibits BAK‐mediated cytochrome c release from mitochondria. The BAKthrombin mutant was expressed in Bax −/− Bak −/− MEFs and in its uncleaved form mediated apoptotic cell death (Fig EV5A and B). Additionally, induced intramolecular disulphide linkage of the two cysteines in BAK (C14, C166) that is diagnostic of the inactive conformer (Cheng et al, 2003; Dewson et al, 2009), supported the BAKthrombin variant adopted an inactive configuration (Fig 5D). In mitochondria‐enriched fractions, thrombin cleaved the inactive form of BAK with the α1‐α2 loop thrombin site, but not BAK with a wild‐type α1‐α2 loop (Fig 5D and E). To understand the molecular interactions in inactive BAK, we combined thrombin cleavage with disulphide linkage. After α1‐α2 loop cleavage, the N‐ and C‐termini could still be linked by an intramolecular disulphide bond, indicating that loop cleavage alone did not destabilise BAK from its inactive configuration and that important structural contacts in the α1 are retained (Fig 5D). Activation by cBID potentiated thrombin cleavage and the N‐terminus was no longer linkable to the C‐terminus (Fig 5D). These disulphide linkage data are consistent with the HDX‐MS profile of inactive BAK, in which constrained deuterium exchange in the BAK α1 indicated important intramolecular contacts, and the profile of activated BAK in which the N‐terminus exhibited markedly increased deuterium exchange.
Figure EV5. BAK α1‐2 loop cleavage does not inhibit cytochrome c release.
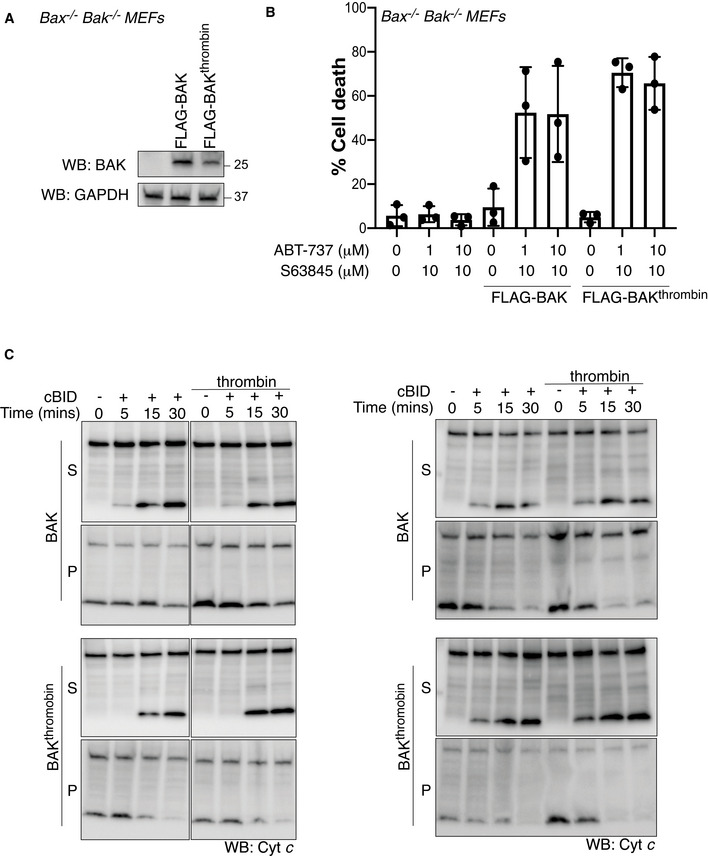
- FLAG‐BAK or FLAG‐BAKthrombin were stably expressed in Bax −/− Bak −/− MEFs and assessed by immunoblotting.
- Cells from A were treated with combined BH3 mimetic drugs for 24 h and cell death was analysed by PI uptake. Data are mean ± SD of three independent experiments.
- Mitochondria‐enriched fractions isolated from cells in A were incubated with cBID (10 nM) in the presence or absence of thrombin for the indicated times and membrane pellet (P) and supernatant (S) were separated and immunoblotted for cytochrome c or BAK (4B5). Data from two independent experiments are shown, third experiment is shown in Fig 5E.
Source data are available online for this figure.
To test the functional role of the disordered N‐terminus in BAK‐mediated MOMP, the effect of loop cleavage on cytochrome c release from isolated mitochondria induced by cBID was tested. As expected, co‐incubation with thrombin had no effect on the release of cytochrome c from mitochondria isolated from cells expressing wild‐type BAK (Fig 5E). Thrombin treatment efficiently cleaved the α1‐2 loop of BAKthrombin, but did not impair cBID‐induced cytochrome c release (Fig 5E). Instead, incubation with thrombin had a modest, but reproducible, potentiating effect on MOMP (Fig 5E), consistent with the liposome data (Figs 5C and EV5C).
Discussion
Recent studies have highlighted the potential of small molecules to influence the apoptotic activity of BAK and BAX (Gavathiotis et al, 2012; Niu et al, 2017; Garner et al, 2019; van Delft et al, 2019). Understanding the molecular details of how BAK and BAX undergo conformation change to oligomerise in the context of a membrane environment is key to the rational targeting of their apoptotic activity. Studies in cells and mitochondria using a variety of approaches including limited proteolysis, cross‐linking analysis and antibody epitope exposure have resolved gross events in BAK and BAX activation. These conformation changes include exposure of the N‐terminus and BH3 domain. Additionally, X‐ray crystal structures have identified atomic snapshots of different conformations of BAK and BAX in solution. A major transition in both BAK and BAX during their activation as revealed by such solution crystal structures was the dissociation of “core” (α2‐5) and “latch” (α6‐8) domains and the formation of BH3:groove symmetrical homodimers (Czabotar et al, 2013; Brouwer et al, 2014). However, we lack a clear understanding of the dynamic changes that occur in BAK and BAX in the context of a membrane environment at the residue level. To resolve this, we employed HDX‐MS of BAK resident in “mitochondria‐like” liposomes as a powerful approach to gain near residue resolution of the dynamics of BAK conformation change and interactions with protein or lipids.
Exposure of N‐terminal epitopes is an established early event in the activation of both BAK and BAX. Our HDX‐MS now reveals that once exposed the majority of the N‐terminus (amino acids 1–58) including α α1 helix of BAK becomes highly disordered. Studies have indicated that disordered regions of proteins can be important mediators of interactions with other proteins or with lipid (Jemth et al, 2018; Deryusheva et al, 2019). Additionally, disordered regions of membrane proteins can promote curvature of lipid bilayers (Busch et al, 2015). Oligomerisation and membrane curvature are thought to both be key features of BAK/BAX‐mediated mitochondrial permeabilisation (Basanez et al, 1999, 2002; Terrones et al, 2004; Dewson et al, 2008; Subburaj et al, 2015), suggesting that N‐terminal disorder could similarly enhance BAK permeabilising activity. The critical role that the α1 plays in the stable fold of inactive BAK, rationalised by its contacts with both the core and latch domains, has precluded the simple testing of an N‐terminal truncation variant, as we have shown previously that a BAK truncation mutant lacking its first 47 amino acids, which includes α1, does not express (Dewson et al, 2009). However, here we found that removal of the BAK N‐terminus from the activated conformer via targeted proteolysis did not inhibit permeabilisation of model liposomes or mitochondria, but potentially exacerbated it. These data indicate that residues preceding the α2/BH3 domain are not required for BAK to permeabilise mitochondria in agreement with recent data on detergent‐activated dimers of truncated BAK (Birkinshaw et al, 2021). The disordered N‐terminus, once released during activation, does not promote BAK, but may actually limit, BAK permeabilising activity. The mechanism of the potential inhibitory effect of the disordered N‐terminus is unclear, but it may involve inhibition of dimer or higher order oligomer formation. Alternatively, it is possible that loop cleavage potentiated steps in BAK conformation change, consistent with the α1/2 loop as a site for triggering BAK activation (Iyer et al, 2016). However, as thrombin cleavage did not provoke significant dissociation of the N‐terminus, this suggests that the stepwise changes in conformation are retained in BAK with a cleaved α1/2 loop.
X‐ray crystallographic studies of BAK and BAX in solution have revealed a major reconfiguration during activation involving dissociation of the “core” (α1‐α5) and “latch” (α6‐α9) domains (Czabotar et al, 2013; Brouwer et al, 2014). Whilst the relationship between the dissociated domains in a membrane remains unclear, disulphide constraints show that at the very least some separation of α5 and α6 is necessary for BAK and BAX apoptotic function on mitochondria (Czabotar et al, 2013; Brouwer et al, 2014). Although our HDX‐MS data indicate that peptides corresponding to the C‐terminal end of α6 do appear to become more dynamic or exposed, potentially consistent with core:latch dissociation upon activation of BAK on a membrane, the lack of peptide coverage in the α5‐α6 loop region unfortunately did not provide additional information on this major reconfiguration.
Our HDX‐MS data indicate that a major structural restraint on BAK activation is the interaction of the BH4 domain with the hydrophobic core α5 helix and with α2(BH3) that stabilises the inactive fold. Conservative mutations that would be predicted to disrupt the interaction with the core α5 abrogate folding and prevent stable expression. In contrast, mutations that would be predicted to disrupt interaction with the α2(BH3) are tolerated, but are prone to adopting an activated conformation. These data suggest that small molecules that target this specific region to destabilise the BH4: α2 interaction could be novel tool reagents or potential therapeutics to trigger or potentiate BAK activation. Importantly, this trigger site is independent of the canonical hydrophobic groove that is required for BAK activation by BH3‐only proteins and dimerisation.
BAK and BAX are emerging as potentially valuable therapeutic targets to treat diseases of dysregulated apoptosis including cancer and neurodegenerative disease (Gavathiotis et al, 2012; Niu et al, 2017; Garner et al, 2019; van Delft et al, 2019). Our studies using HDX‐MS to resolve the dynamics of BAK conformation change on membranes provides new insight into the molecular details of BAK activation to complement structural and biophysical studies. Such molecular insight is essential if BAK and BAX are to be successfully targeted with rationally designed small molecules as their pro‐survival counterparts have (Merino et al, 2018).
Materials and Methods
Liposome permeabilisation assay
Liposomes were reconstituted with a combination of lipids that mimic the outer mitochondrial membrane (46% phosphatidylcholine (w/v), 25% phosphatidylethanolamine (w/v), 11% phosphatidylinositol (w/v), 10% phosphatidylserine (w/v) and 8% cardiolipin (w/v) and supplemented with 5% nickel chelating lipid (1,2‐Dioleoyl‐sn‐Glycero‐3‐[N‐(5‐amino‐1‐carboxypentyl)iminodiacetic‐acid)‐succinyl] (w/v)). Liposomes encapsulated self‐quenching 5(6)‐carboxyfluorescein prior to extrusion through a 100 nm pore size membrane and passed over a PD10 column to remove excess dye. Recombinant mouse BAKΔC21/6His, generated as previously described (Bernardini et al, 2019), was incubated with liposomes and where indicated was activated with recombinant human cBID (Uren et al, 2007) for the indicated times at room temperature in Falcon 96‐well U‐bottom tissue culture plates (Corning, NY, USA). Following incubation, fluorescence was measured (excitation 485 nm, emission 535 nm) in a Chameleon V plate reader (LabLogic Systems, Sheffield, UK). Baseline release values were subtracted and data were normalised to dye released by 1% CHAPS detergent. For the assays with targeted proteolysis, a hBAK‐6Hthrombin construct was generated by substituting the amino acids VTLPLQP in the α1‐2 loop region of hBAKΔN22ΔC25/C166S/6His (Bernardini et al, 2019) with the thrombin recognition sequence LVPRGSPGG starting at position 61. BAK cDNA was cloned into the bacterial expression vector pGEX‐6P3 and expression and purification was performed as previously described (Brouwer et al, 2014). To cleave the N‐terminus monomeric BAK‐6Hthrombin was incubated with thrombin protease (1 unit, Sigma, cat#10602400001) and calcium chloride (1 mM) and incubated overnight at 4°C, then purified by size exclusion chromatography using a superdex S200 10/300 in 20 mM tris pH 8, 150 mM NaCl. Cleaved purified samples were then added to liposomes as described above.
Cell culture, retroviral infection and cell death assays
Mouse embryonic fibroblasts (MEFs) generated from E14 Bax −/− Bak −/− C57BL/6 mice were immortalised and retroviral transduction performed as previously described (Dewson et al, 2008). Cells were maintained in Dulbecco’s Modified Eagles Medium supplemented with 8% FCS, 250 µM asparagine and 55 µM 2‐mercaptoethanol in 10% CO2 at 37°C and routinely passaged at < 80% confluency. Cells used were authenticated by STR profiling (https://www.garvan.org.au/research/capabilities/molecular‐genetics/cell‐line‐identification) and regularly tested to confirm Mycoplasma negativity (MycoAlert kit, Lonza, Basel).
To monitor cell death, MEFs were treated with combinations of BH3 mimetic drugs, BCL‐2/BCL‐XL/BCL‐W inhibitor (ABT‐737, 1 µM) with MCL1 inhibitor (S63485, 10 µM) (Oltersdorf et al, 2005; Kotschy et al, 2016) or etoposide (1, 10 µM). Alternatively, MEFs expressing doxycycline‐inducible constructs of BAK were treated with doxycycline for 12 h with or without co‐incubation with BH3 mimetic drugs. Dead cells were stained with propidium iodide uptake and quantified by flow cytometry. Statistical analysis was performed using an unpaired, two‐tailed Student’s t‐test.
SDS–PAGE, Blue Native PAGE and immunoblotting
For SDS–PAGE, protein lysates were analysed on Tris‐X gels (Bio‐Rad, CA, USA). For blue native PAGE, liposomes or enriched mitochondria were solubilised in 1% w/v digitonin and analysed essentially as described (Ma et al, 2013). Gels were transferred to PVDF membrane and the membrane was blocked overnight at 4°C in 5% skim milk TBS‐T (TBS buffer supplemented with 0.1% Tween‐20). Following blocking, the SDS–PAGE blots were washed with TBS‐T then incubated with rat anti‐BAK monoclonal antibody that recognises BAK amino acids 51–55 (7D10) or GAPDH as a loading control. For BN‐PAGE, blots were probed with anti‐BAK aa 23–38 (#B5897, Sigma). The membrane was washed three times in TBS‐T and then incubated with horseradish peroxidase‐conjugated secondary antibodies (#3010‐05, Southern Biotech). Membranes were washed three times in TBS‐T and proteins were detected with Enhanced Chemiluminescence reagent (Amersham Biosciences, Little Chalfont, UK) using a ChemiDoc XRS+ System (Bio‐Rad, CA, USA).
BAK activation by flow cytometry
The exposure of N‐terminal epitopes in BAK was assessed as described (Alsop et al, 2015). Briefly, MEFs were pre‐treated with the broad range caspase inhibitor Q‐VD.oph (50 µM, MP Biomedicals, LLC) followed by treatment with doxycycline and/or BH3 mimetic drugs (10 µM S63458 + 0.1 µM ABT‐737) for 3 h. MEFs were harvested, fixed and permeabilised using the eBioscience cell fixation and permeabilisation kit (Thermo Fischer, MA). Samples were then incubated with anti‐BAK G317‐2 monoclonal antibody (BD Pharmingen, CA) followed by phycoerythrin‐conjugated anti‐mouse antibody (Southern Biotech, Birmingham) and analysed on a FACS‐LSPIIW (Becton Dickinson). Statistical analysis was performed using an unpaired, two‐tailed Student’s t‐test.
Cytochrome c release and disulphide linkage on isolated mitochondria
Mitochondria‐enriched heavy membrane fractions were generated by permeabilising the plasma membrane with 0.025% w/v digitonin in permeabilisation buffer (20 mM Hepes (pH 7.5), 100 mM KCl, 2.5 mM MgCl2, and 100 mM sucrose). Membrane fractions were incubated with thrombin in the presence or absence of recombinant cBID at 30°C. Membranes were pelleted at 13,000 g for 5 min at 4°C and supernatant and membrane fractions assessed by immunoblotting for cytochrome c content. Alternatively, membrane fractions were treated with copper (II)(1,10‐phenanthroline)3 for 30 min on ice to induce disulphide linkage. Membrane fractions were then analysed under non‐reducing or reducing SDS–PAGE and immunoblotting with antibodies against the BAK N‐terminus (aa23–38, Sigma) or BH3 domain (4B5, (Dewson et al, 2009)).
Hydrogen‐deuterium exchange mass spectrometry (HDX‐MS)
HDX‐MS was performed essentially as previously described (Robin et al, 2018). Recombinant BAK‐6H (mBAKΔC21/6His) on liposomes was diluted 20‐fold in deuterated buffer containing PBS pH 7.5 at room temperature. Aliquots were taken at multiple time points (10 s, 60 s, 10 min, 30 min) with the hydrogen/deuterium exchange reaction suppressed by acidification of the sample to pH 2.5 using formic acid before snap‐freezing in liquid nitrogen. Digestion of the protein was carried out by thawing the sample in a 10‐fold dilution of H2O before addition of an equimolar concentration of pepsin (Sigma, USA) for 5 min on ice. Peptides were subjected to LC‐MS analysis using a dual pump (capillary for sample loading and nanoflow for analytical) 1100 series HPLC (Agilent, USA) coupled to an LTQ‐Orbitrap XL (Thermo, USA). Peptides were loaded onto an in‐house packed, reverse phase trap column (ReproSil‐Pur C18‐AQ (Dr. Maisch GmbH, Germany), 2 × 2 mm, 5 µm particle) before separation on an in‐house packed, reverse phase analytical column (ReproSil‐Pur C18‐AQ (Dr. Maisch GmbH), 200 mm × 150 µm, 3 µm particle) housed at 1°C. Peptides were loaded onto the trap column at 5% acetonitrile, 0.2% formic acid and elution performed using a gradient rising from 5 to 40% acetonitrile over 15 min, then 85% acetonitrile for 5 min before reconditioning the column at 5% acetonitrile for 15 min. Spectra were acquired in positive ion mode with m/z range from 250 to 1,850. Experiments were performed as experimental replicates (three independent exchange time courses with one batch of protein). Deuteration of peptides was determined by analysis of samples using HDX workbench, as previously described (Pascal et al, 2012). Additional analysis of HDX workbench output was performed using in‐house scripts written in R (version 3.3.1).
Author contributions
AIW, GI, JJS, RLN planned performed and analysed the HDX‐MS experiments. AZW, DD, DL, GD, IKLT, JPB, RB, RWB, ASH, SM, ZL planned, performed and analysed the cell and/or liposome experiments. GD, AIW and PEC conceptualised and coordinated the research. All authors contributed to drafting the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
The authors acknowledge Sweta Iyer, Ruth Kluck, Melissa Shi and Peter Colman for discussions and reagents. This research was supported by grants from the National Health and Medical Research Council Australia to PEC and GD (GNT1148174). DD and ASH are supported by Melbourne University Research Scholarships. GD is supported by a Fellowship from the Bodhi Education Fund. This work was supported by operational infrastructure grants through the Australian Government Independent Research Institute Infrastructure Support Scheme (9000587) and the Victorian State Government Operational Infrastructure Support, Australia.
The EMBO Journal (2021) 40: e107237.
See also: LE Sperl et al (October 2021) and AM Ojoawo & T Moldoveanu (October 2021)
Contributor Information
Andrew I Webb, Email: webb@wehi.edu.au.
Grant Dewson, Email: dewson@wehi.edu.au.
Data availability
All of the data generated and analysed during this study are included in this published article. Mass spectrometry data have been deposited to the ProteomeXchange Consortium (Deutsch et al, 2020) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD027288 (http://www.ebi.ac.uk/pride/archive/projects/PXD027288).
References
- Alsop AE, Fennell SC, Bartolo RC, Tan IK, Dewson G, Kluck RM (2015) Dissociation of Bak alpha1 helix from the core and latch domains is required for apoptosis. Nat Commun 6: 6841 [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz‐Aguado JA, Penn LZ, Leber B, Andrews DW (2005) Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J 24: 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanez G, Nechushtan A, Drozhinin O, Chanturiya A, Choe E, Tutt S, Wood KA, Hsu Y, Zimmerberg J, Youle RJ (1999) Bax, but not Bcl‐xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc Natl Acad Sci USA 96: 5492–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM, Zimmerberg J (2002) Bax‐type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem 277: 49360–49365 [DOI] [PubMed] [Google Scholar]
- Bernardini JP, Brouwer JM, Tan IKL, Sandow JJ, Huang S, Stafford CA, Bankovacki A, Riffkin CD, Wardak AZ, Czabotar PE et al (2019) Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J 38: e99916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkinshaw RW, Iyer S, Lio D, Luo CS, Brouwer JM, Miller MS, Robin AY, Uren RT, Dewson G, Kluck RM et al (2021) Structure of detergent‐activated BAK dimers derived from the inert monomer. Mol Cell 81: 2123–2134 e2125 [DOI] [PubMed] [Google Scholar]
- Bleicken S, Classen M, Padmavathi PV, Ishikawa T, Zeth K, Steinhoff HJ, Bordignon E (2010) Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem 285: 6636–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleicken S, Jeschke G, Stegmueller C, Salvador‐Gallego R, Garcia‐Saez AJ, Bordignon E (2014) Structural model of active bax at the membrane. Mol Cell 56: 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE (2014) Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell 55: 938–946 [DOI] [PubMed] [Google Scholar]
- Busch DJ, Houser JR, Hayden CC, Sherman MB, Lafer EM, Stachowiak JC (2015) Intrinsically disordered proteins drive membrane curvature. Nat Commun 6: 7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517 [DOI] [PubMed] [Google Scholar]
- Cowan AD, Smith NA, Sandow JJ, Kapp EA, Rustam YH, Murphy JM, Brouwer JM, Bernardini JP, Roy MJ, Wardak AZ et al (2020) BAK core dimers bind lipids and can be bridged by them. Nat Struct Mol Biol 27: 1024–1031 [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63 [DOI] [PubMed] [Google Scholar]
- Czabotar P, Westphal D, Dewson G, Ma S, Hockings C, Fairlie W, Lee E, Yao S, Robin A, Smith B et al (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- Deryusheva E, Nemashkalova E, Galloux M, Richard CA, Eleouet JF, Kovacs D, Van Belle K, Tompa P, Uversky V, Permyakov S (2019) Does intrinsic disorder in proteins favor their interaction with lipids? Proteomics 19: e1800098 [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez‐Riverol Y, Carver JJ, Kundu DJ, Garcia‐Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS et al (2020) The ProteomeXchange consortium in 2020: enabling “big data” approaches in proteomics. Nucleic Acids Res 48: D1145–D1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM (2008) To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell 30: 369–380 [DOI] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM (2009) Bak activation for apoptosis involves oligomerization of dimers via their alpha 6 helices. Mol Cell 36: 696–703 [DOI] [PubMed] [Google Scholar]
- Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, Kluck RM (2012) Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ 19: 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner TP, Amgalan D, Reyna DE, Li S, Kitsis RN, Gavathiotis E (2019) Small‐molecule allosteric inhibitors of BAX. Nat Chem Biol 15: 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD (2012) Direct and selective small‐molecule activation of proapoptotic BAX. Nat Chem Biol 8: 639–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George NM, Evans JJ, Luo X (2007) A three‐helix homo‐oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev 21: 1937–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths GJ, Corfe BM, Savory P, Leech S, Esposti MD, Hickman JA, Dive C (2001) Cellular damage signals promote sequential changes at the N‐terminus and BH‐1 domain of the pro‐apoptotic protein Bak. Oncogene 20: 7668–7676 [DOI] [PubMed] [Google Scholar]
- Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Corfe BM, Dive C, Hickman JA (1999) Cell damage‐induced conformational changes of the pro‐apoptotic protein Bak in vivo precede the onset of apoptosis. J Cell Biol 144: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S (2016) Bax assembles into large ring‐like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J 35: 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y‐T, Youle RJ (1998) Bax in murine thymus is a soluble monomeric protein that displays differential detergent‐induced conformations. J Biol Chem 273: 10777–10783 [DOI] [PubMed] [Google Scholar]
- Iyer S, Anwari K, Alsop AE, Yuen WS, Huang DCS, Carroll J, Smith NA, Smith BJ, Dewson G, Kluck RM (2016) Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nat Commun 7: 11734–11744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, Dewson G, Kluck RM (2015) Bak apoptotic pores involve a flexible C‐terminal region and juxtaposition of the C‐terminal transmembrane domains. Cell Death Differ 22: 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemth P, Karlsson E, Vogeli B, Guzovsky B, Andersson E, Hultqvist G, Dogan J, Guntert P, Riek R, Chi CN (2018) Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins. Sci Adv 4: eaau4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin‐Braizat G, Chanrion M, Kelly GL, Gong J‐N, Moujalled DM et al (2016) The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538: 477–482 [DOI] [PubMed] [Google Scholar]
- Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA, Huang DC, Colman PM (2008) Vaccinia virus anti‐apoptotic F1L is a novel Bcl‐2‐like domain‐swapped dimer that binds a highly selective subset of BH3‐containing death ligands. Cell Death Differ 15: 1564–1571 [DOI] [PubMed] [Google Scholar]
- Lee EF, Grabow S, Chappaz S, Dewson G, Hockings C, Kluck RM, Debrincat MA, Gray DH, Witkowski MT, Evangelista M et al (2016) Physiological restraint of Bak by Bcl‐xL is essential for cell survival. Genes Dev 30: 1240–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Tan IKL, Ma SB, Hockings C, Kratina T, Dengler MA, Alsop AE, Kluck RM, Dewson G (2017) BAK alpha 6 permits activation by BH3‐only proteins and homooligomerization via the canonical hydrophobic groove. Proc Natl Acad Sci USA 114: 7629–7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Hockings C, Anwari K, Kratina T, Fennell S, Lazarou M, Ryan MT, Kluck RM, Dewson G (2013) Assembly of the Bak apoptotic pore: A critical role for the Bak alpha6 helix in the multimerization of homodimers during apoptosis. J Biol Chem 288: 26027–26038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A (2018) BH3‐mimetic drugs: blazing the trail for new cancer medicines. Cancer Cell 34: 879–891 [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson MH, Shore G, Gehring K (2006) The x‐ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688 [DOI] [PubMed] [Google Scholar]
- Niu X, Brahmbhatt H, Mergenthaler P, Zhang Z, Sang J, Daude M, Ehlert FGR, Diederich WE, Wong E, Zhu W et al (2017) A small‐molecule inhibitor of Bax and Bak oligomerization prevents genotoxic cell death and promotes neuroprotection. Cell Chem Biol 24: 493–506.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh KJ, Singh P, Lee K, Foss K, Lee S, Park M, Lee S, Aluvila S, Park M, Singh P et al (2010) Conformational changes in BAK, a pore‐forming proapoptotic Bcl‐2 family member, upon membrane insertion and direct evidence for the existence of BH3‐BH3 contact interface in BAK homo‐oligomers. J Biol Chem 285: 28924–28937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ et al (2005) An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature 435: 677–681 [DOI] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR (2012) HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom 23: 1512–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S, Wang W, Yang L, Huang HW (2008) Structure of transmembrane pore induced by Bax‐derived peptide: evidence for lipidic pores. Proc Natl Acad Sci USA 105: 17379–17383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin AY, Iyer S, Birkinshaw RW, Sandow J, Wardak A, Luo CS, Shi M, Webb AI, Czabotar PE, Kluck RM et al (2018) Ensemble properties of Bax determine its function. Structure 26: 1346–1359.e5 [DOI] [PubMed] [Google Scholar]
- Salvador‐Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, Garcia‐Saez AJ (2016) Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J 35: 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, Werner J, Krausslich HG (2005) A peptide inhibitor of HIV‐1 assembly in vitro. Nat Struct Mol Biol 12: 671–677 [DOI] [PubMed] [Google Scholar]
- Subburaj Y, Cosentino K, Axmann M, Pedrueza‐Villalmanzo E, Hermann E, Bleicken S, Spatz J, Garcia‐Saez AJ (2015) Bax monomers form dimer units in the membrane that further self‐assemble into multiple oligomeric species. Nat Commun 6: 8042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A, Basanez G (2004) Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem 279: 30081–30091 [DOI] [PubMed] [Google Scholar]
- Uren RT, Dewson G, Chen L, Coyne SC, Huang DCS, Adams JM, Kluck RM (2007) Mitochondrial permeabilization relies on BH3 ligands engaging multiple pro‐survival Bcl‐2 relatives, not Bak. J Cell Biol 177: 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren RT, O'Hely M, Iyer S, Bartolo R, Shi MX, Brouwer JM, Alsop AE, Dewson G, Kluck RM (2017) Disordered clusters of Bak dimers rupture mitochondria during apoptosis. Elife 6: e19944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft MF, Chappaz S, Khakham Y, Bui CT, Debrincat MA, Lowes KN, Brouwer JM, Grohmann C, Sharp PP, Dagley LF et al (2019) A small molecule interacts with VDAC2 to block mouse BAK‐driven apoptosis. Nat Chem Biol 15: 1057–1066 [DOI] [PubMed] [Google Scholar]
- Wales TE, Engen JR (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25: 158–170 [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal D, Kluck RM, Dewson G (2014) Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ 21: 196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J et al (2016) BH3‐in‐groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J 35: 208–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M, Boreham D, McFarlane N, Ding J, Johnson AE et al (2010) Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem 285: 17614–17627 [DOI] [PMC free article] [PubMed] [Google Scholar]