

Abstract
The Golgi apparatus, the main glycosylation station of the cell, consists of a stack of discontinuous cisternae. Glycosylation enzymes are usually concentrated in one or two specific cisternae along the cis‐trans axis of the organelle. How such compartmentalized localization of enzymes is achieved and how it contributes to glycosylation are not clear. Here, we show that the Golgi matrix protein GRASP55 directs the compartmentalized localization of key enzymes involved in glycosphingolipid (GSL) biosynthesis. GRASP55 binds to these enzymes and prevents their entry into COPI‐based retrograde transport vesicles, thus concentrating them in the trans‐Golgi. In genome‐edited cells lacking GRASP55, or in cells expressing mutant enzymes without GRASP55 binding sites, these enzymes relocate to the cis‐Golgi, which affects glycosphingolipid biosynthesis by changing flux across metabolic branch points. These findings reveal a mechanism by which a matrix protein regulates polarized localization of glycosylation enzymes in the Golgi and controls competition in glycan biosynthesis.
Keywords: glucosylceramide synthase, glycosphingolipids, glycosylation, Golgi apparatus, GRASP55
Subject Categories: Membranes & Trafficking, Metabolism
Intra‐Golgi compartmentalization of glucosylceramide synthase (GCS) and lactosylceramide synthase 1 (LCS1) depends on their binding to matrix protein GRASP55, which prevents their sorting into retrograde COPI vesicles.
Introduction
Glycans are one of the fundamental building blocks of the cell and play key roles in development and physiology (Bishop et al, 2007; Kohyama‐Koganeya et al, 2011; Ryczko et al, 2016; Varki, 2017; Akintayo & Stanley, 2019). Cellular glycan profiles are sensitive to changes in cell state and/or differentiation and are also important contributors to the process (Russo et al, 2018b). Indeed, several developmental disorders are associated with impaired production of glycans (Chang et al, 2018). Thus, how the glycan biosynthesis is regulated to achieve specific cellular glycan profiles is an important biological problem. In eukaryotes, glycans are assembled mainly by the Golgi apparatus on cargo proteins and lipids that traverse the organelle (Stanley, 2011). Glycan biosynthesis happens in a template‐independent fashion (Varki & Kornfeld, 2015), yet the products are not random polymers of sugars but a defined distribution of glycans that is cell‐type and cargo‐specific (Rudd et al, 2015; Varki & Kornfeld, 2015). This suggests that their biosynthesis is guided by regulated program(s). Transcriptional programs have been identified that contribute to defining the glycome of a cell, but they only partially account for it (Nairn et al, 2008, 2012; Varki & Kornfeld, 2015). An obviously important but unexplored factor that influences glycosylation is the Golgi apparatus itself (Varki, 1998; Maccioni et al, 2002).
The Golgi apparatus is a central organelle of the secretory pathway that processes newly synthesized cargoes coming from the endoplasmic reticulum (ER), primarily by glycosylation, before sorting them toward their correct destination in the cell. It consists of a stack of 4–11 cisternae (Klumperman, 2011), populated by enzymes and accessory proteins that maintain a suitable milieu for the enzymes to act on biosynthetic cargoes. The stack is polarized with a cis‐side where cargoes arrive and a trans‐side from where they leave. The enzymes are not homogeneously distributed across the Golgi stack but are restricted or compartmentalized to 1–3 specific cisternae. The cisternal maturation model provides a conceptual framework for understanding Golgi enzyme compartmentalization (Nakano & Luini, 2010; Glick & Luini, 2011). According to the model, secretory cargoes are transported forward by the anterograde flux mediated by cisternal progression, which consists of constant formation and consumption of cis and trans cisternae, respectively. The retention of Golgi glycosylation enzymes in the face of this continuous forward flux is mediated by their retrograde transport that acts as counterbalance for the forward transport. The retrograde transport is promoted by coat protein complex I (COPI) machinery (Rabouille & Klumperman, 2005; Popoff et al, 2011; Papanikou et al, 2015; Ishii et al, 2016; Liu et al, 2018) and is assisted in this process by adaptor molecules like GOLPH3 (Tu et al, 2008, 2012; preprint: Rizzo et al, 2019), conserved oligomeric complex (COG) proteins, and Golgi matrix proteins especially Golgins (Eckert et al, 2014; Wong & Munro, 2014; Blackburn et al, 2019). However, the specific molecular mechanisms and processes by which the same retrograde transport pathway promotes localization of enzymes to distinct cisternae remain unknown.
The compartmentalized localization of enzymes has been suggested to influence both sequential as well as competing glycosylation reactions. The localization of enzymes along the cis‐trans axis reflecting their order of action (Dunphy & Rothman, 1985) has been suggested to influence the efficiency of sequential processing reactions (Fisher et al, 2019). On the other hand, the promiscuity of glycosylation enzymes (Biswas & Thattai, 2020) makes compartmentalized localization of competing enzymes a critical factor in determining the specificity in glycan output (i.e., the type and quantity of glycans produced) (Dunphy & Rothman, 1985; Pothukuchi et al, 2019; Jaiman & Thattai, 2020). When two or more enzymes compete for a substrate, the order in which they get access to it can substantially influence the glycans produced and subsequently the physiological outcomes. Competing reactions are frequent in glycosylation pathways, and all known glycosylation pathways have one or more competing glycosylation steps. Nevertheless, how the compartmentalized localization of competing enzymes is achieved, how it is regulated to influence glycosylation reactions, and what the physiological relevance of this regulation is remain unexplored.
To evaluate and understand the contribution of Golgi compartmentalization in regulating glycosylation, we have focused our study on sphingolipid (SL) glycosylation. We chose this model system for several reasons: a. It is well characterized from both biochemical and transcriptional perspectives (Halter et al, 2007; D'Angelo et al, 2013; Russo et al, 2018b); b. the glycosylation reaction is less influenced by the cargo structure in contrast to protein glycosylation and thus is a cleaner system to study effects of Golgi processes on glycosylation; c. there are simple biochemical methods available to analyze SL glycosylation (D'Angelo et al, 2013); and d. finally, SLs have important roles in physiology and development (Hannun & Obeid, 2018; Russo et al, 2018a). The SL glycosylation pathway exhibits the essential features of glycosylation pathways like localization of enzymes reflecting their order of action and also at least two competing reaction steps that are important in determining the metabolic outcome of the pathway (see below). Further, while enzymes of the pathway are well characterized, molecular players regulating their sub‐Golgi compartmentalization are unknown. By studying SL glycosylation, we identify GRASP55 as an important factor that compartmentalizes two enzymes catalyzing critical branch points of the SL glycosylation pathway. GRASP55 binds to and prevents the entry of these enzymes into retrograde transport carriers. This retaining action of GRASP55 is essential for dynamic compartmentalization of these enzymes in the Golgi stack. The competing enzymes thus positioned at appropriate levels in the Golgi stack regulate cargo flux across competing reactions of the pathway and determine the metabolic outcome viz. sphingolipid produced by the cell. These results delineate a molecular mechanism of enzyme compartmentalization and how it controls cell surface glycan profile.
Results
Disruption of Golgi organization alters SL biosynthesis
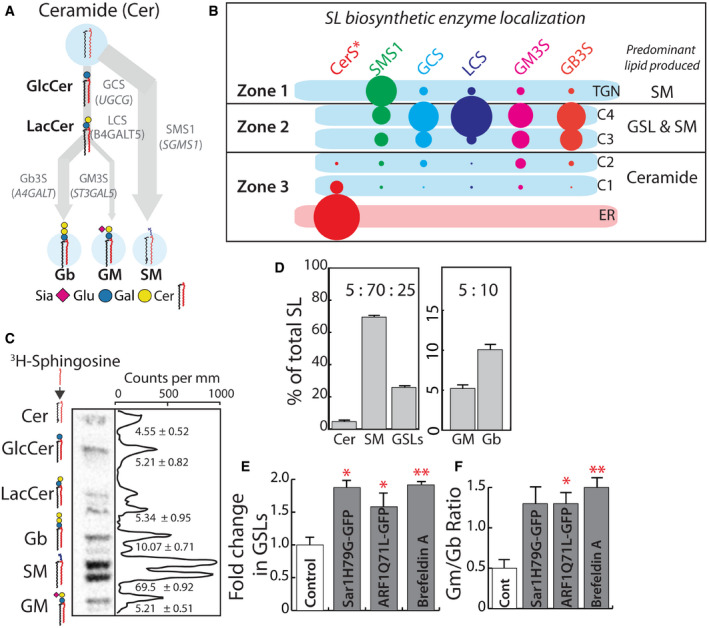
SL biosynthesis starts with the production of ceramide (Cer) in the ER, which is then processed in the Golgi to sphingomyelin (SM) or glycosphingolipids (GSLs). The model cell system we use, HeLa cells, produces two species of GSLs—globosides (Gb3) and gangliosides (GM1 and GM3) (Halter et al, 2007; D'Angelo et al, 2013; Russo et al, 2018b) (See Fig 1A for schematic of the SL system in HeLa cells). This SL pathway includes sequential processing of Cer to complex GSLs as well as two bifurcating steps where the substrates get differentially channeled. The first is the bifurcation between SM and glucosylceramide (GlcCer) biosynthesis, where the substrate Cer is channeled into either of the pathways. The second is the biosynthesis of Gb3 or GM3 from lactosylceramide (LacCer). These two critical steps determine the amount and type of SLs produced by the cell. We first examined the localization of SL biosynthetic enzymes and found that they localize to three distinct zones in the secretory pathway (Fig 1B, Appendix Fig S1): (i) the early secretory pathway including the ER and the cis/medial‐Golgi (C1, C2 cisternae), where Cer biosynthetic enzymes are localized (33), have little if any SL biosynthetic enzymes except for a slightly elevated amount of GM3S and GlcCer synthase (GCS) in the cis/medial‐Golgi compared with other GSL biosynthetic enzymes; (ii) medial/trans‐Golgi (C3, C4 cisternae) where most of the GSL biosynthetic enzymes are present alongside substantial amounts of Sphingomyelin synthase 1 (SMS1) and (iii) trans‐Golgi network (TGN), where SMS1 predominates. While all the GSL biosynthetic enzymes show a gradient of increasing concentration from cis‐ to trans‐Golgi, the gradient is much sharper in the case of GB3S and LacCer synthase (LCS) compared with GCS and GM3S (Appendix Fig S1). Thus, the SL biosynthetic enzymes are distributed reflecting their order of action with precursor (Cer) producing enzymes in the early secretory pathway and the Cer processing enzymes in late secretory pathway, which is in turn divided into two distinct zones where GSL and SM biosynthesis predominate. Of note, we expressed HA‐tagged enzymes (see Materials and Methods) for our studies since the endogenous enzymes were barely detectable and efficient antibodies for EM studies of endogenous enzymes were not available. Nevertheless, the localization mostly reflects expected localization based on enzyme activity and previously published evidence (Parashuraman & D'Angelo, 2019). A notable exception is the localization of GCS that was shown to be on the cis‐side of the Golgi (Halter et al, 2007) contrary to what we report here. This is because the earlier studies had used a construct with a tag that blocks the signal for intra‐Golgi localization that we identify and describe here. When this signal is blocked, localization of GCS is altered resulting in localization to cis‐Golgi (see below).
Figure 1. Disruption of SL biosynthetic machinery organization alters SL output.
-
ASchematic representation of GSL biosynthetic pathway in HeLa cells (Glu, glucose; Gal, galactose; Sia, N‐acetylneuraminic acid; Cer, ceramide). Products of biosynthesis are represented in bold and enzymes that catalyze the reactions in gray. The arrows represent the SL metabolic flux from ceramide.
-
BSchematic representation of GSL biosynthetic zones in HeLa, SM biosynthesis predominates in TGN, whereas GSL and SM productions happen in medial/trans‐Golgi (C3 and C4 cisternae). Cis‐Golgi/ER is where Ceramide biosynthesis happens with little, if any, SL production. CerS* refers to the group of Ceramide synthases localized to the ER. The size of the lipid label arbitrarily represents the proportion of the lipid expected to be synthesized in the compartment based on the localization of corresponding enzymes.
-
CHigh‐performance thin‐layer chromatography (HPTLC) profile of HeLa cells pulsed for 2 h with [3H]‐sphingosine and chased for 24 h. The peaks corresponding to each SL species are indicated, and numbers represent each SL species as percentage of total SL.
-
DThe total radioactivity associated with Cer, SM, and GSLs (GluCer, LacCer, Gb, and GM), or GM and Gb were quantified and presented as percentages relative to total. Data represented as means ± SD of three independent experiments.
-
E, FBiosynthesis of SL in HeLa cells expressing GTP‐locked mutants of Sar1 or ARF1 or treated with Brefeldin A (BFA; 5 μg/ml) was measured by [3H]‐sphingosine pulse‐chase assay. Radioactivity associated with GSLs was quantified and represented as fold change with respect to control. (E) For BFA‐treated cells, the SL output was measured 8 h after pulse. Data represented as means ± SD of two independent experiments. *P < 0.05, **P < 0.01 (Student’s t‐test). (F) The ratio of GM/Gb is represented. Data represented as means ± SD of two independent experiments. *P < 0.05, **P < 0.01 (Student’s t‐test).
Next, SL output of this system was measured by metabolic labeling with 3H‐sphingosine, a precursor of ceramide. This revealed the following distribution of products at quasi steady state i.e., 24 h after labeling: SM (70%), globosides (10%), and gangliosides (5%) and rest remaining as precursors (Cer, GlcCer or LacCer; 15%) (Fig 1C and D). The GSLs (globosides, gangliosides, and GSL precursors GlcCer and LacCer) together constituted 25% of total SLs produced. We will refer to the ratio of GSL:SM::25:70 as SL output and the ratio of gangliosides (GM) to Globosides (Gb), GM:Gb::5:10 as GSL output (Fig 1D). For simplicity, the SL output will be represented as GSL fraction since a change in GSLs is always associated with a proportional change in SM in the opposite direction. For GSL output, the situation is complex since a substantial portion of signal remains as precursors (GlcCer and LacCer), and so GSL output will be represented as a GM/Gb ratio which under the control conditions corresponds to 0.5 (GM:Gb::5:10). To summarize, the SL machinery has a compartmentalized localization across the Golgi in HeLa cells and produces a SL output such that 70% of the Cer is directed toward the production of SM and 25% toward the production of GSLs. Within this 25, 5% is directed toward the production of gangliosides and 10% toward the production of globosides.
This distribution of glycoforms produced by the Golgi apparatus has largely been ascribed to the expression of the corresponding glycosylation enzymes (Maccioni et al, 2002; Nairn et al, 2008, 2012). To assess the contribution of enzyme compartmentalization to this, we monitored SL output after disrupting the spatial organization of SL biosynthetic enzymes by a) overexpressing GTP‐locked mutants of monomeric GTPases——secretion‐associated Ras‐related GTPase (Sar1 H79G) and ADP ribosylation factor 1 (ARF1 Q71L) that are well known to disorganize the secretory pathway (Zhang et al, 1994; Aridor et al, 1995) and b) by treating the cells with Brefeldin A, which causes relocation of Golgi enzymes back to the ER. Overexpression of Sar1 H79G led to collapse of the Golgi apparatus into the ER with SL biosynthetic enzymes showing a reticular ER pattern (Appendix Fig S2A). On the other hand, overexpression of ARF1 Q71L mutant led to disruption of stacked cisternal structure of the Golgi, which was replaced by tubulo‐vesicular clusters (Appendix Fig S2B), with no separation between cis‐ and trans‐Golgi markers (Appendix Fig S2C) (List of recombinant DNA used in this study are listed in Appendix Table S2). The treatment with Brefeldin A led to the translocation of the enzymes back into the ER as expected, apart from SMS1 which while present in the ER also displayed presence in some punctate structures (Appendix Fig S2A). The SL output was altered in these cells, and consistently, in all three conditions there was an increased production of GSLs over SM and gangliosides over globosides (Appendix Fig S2D and E). The SL output represented as fold change in GSL fraction showed that GSL production in these cells increased by 1.5–1.9 fold over control cells (Fig 1E). Similarly, GSL output measured as GM/Gb ratio changed from 0.5 in control cells to 1.3–1.5 in treated cells (Fig 1F). These data suggest that impaired spatial organization of enzymes correlates with altered SL output, and especially, the output from steps involving competing reactions is sensitive to disorganization of the Golgi. The contribution of enzyme expression to determination of glycosylation is well established (Nairn et al, 2012) but the contribution of the Golgi organization and its importance to this process was not clear. These results underscore a significant and substantive role played by the Golgi apparatus in determining the glycan output of a cell.
GRASP55 regulates SL output by controlling substrate flux between competing glycosylation pathways
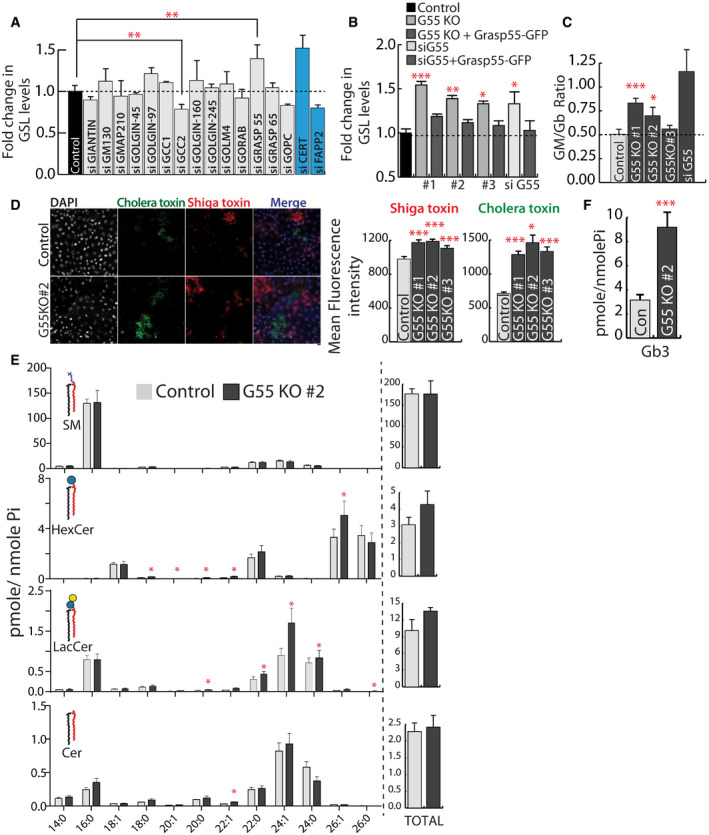
Given the importance of the organization of the Golgi apparatus, and likely of the SL biosynthetic machinery localized to the organelle, to determining SL output, we wanted to identify the molecular players involved in this process. Retention of enzymes in the Golgi depends on their COPI‐dependent retrograde transport. Golgi matrix proteins especially Golgins contribute to specificity in this process (Wong & Munro, 2014) and thus to compartmentalization of enzymes. So, to identify specific regulators of compartmentalization of SL biosynthetic enzymes, we systematically silenced Golgi matrix proteins and studied the effect on SL production. Among the 14 matrix proteins tested by depletion, downregulation of GRASP55 significantly increased the production of GSLs (a 40% increase in GSLs compared with control) while downregulation of GOPC and GCC2 led to a decrease in GSL levels (Fig 2A) (siRNA sequences used in this study to downregulate indicated human gene expression are listed in Appendix Table S3). We followed up on GRASP55 since its depletion altered SL output similar to that obtained by disorganization of the Golgi apparatus (Fig 1E).
Figure 2. GRASP55 regulates SL biosynthesis.
-
AHeLa cells were treated with control or indicated siRNA (pool of 4 or 2 as indicated in methods) for 72 h and SL biosynthesis measured by [3H]‐sphingosine pulse‐chase assay. GSL levels are expressed as fold changes with respect to control. CERT and FAPP2 knockdowns (blue bars) were used as controls. Data represented are mean ± SD of three independent experiments **P < 0.01 (Student’s t‐test).
-
B, CEffect of GRASP55 depletion on SL biosynthesis monitored by [3H]‐sphingosine pulse‐chase assay in GRASP55 KO cells or cells treated with GRASP55 siRNA or following expression of GRASP55‐GFP in GRASP55 depleted cells. GSL levels are expressed as fold changes with respect to control. Data represented are mean ± SD of three independent experiments *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t‐test). (C) The levels of GM and Gb were quantified and represented as GM/Gb ratio. Data represented are mean ± SD of three independent experiments *P < 0.05, ***P < 0.001 (Student’s t‐test).
-
DControl and GRASP55KO cells were processed for Cy3‐conjugated Shiga Toxin (ShTxB) and Alexa488‐conjugated Cholera Toxin (ChTxB) staining followed by flow cytometry analysis. Mean fluorescence intensity was measured and represented. Data represented are mean ± SD of three independent experiments *P < 0.05, ***P < 0.001 (Student’s t‐test). Scale bar: 10 μm.
-
E, FSL levels as assessed by LC/MS or MALDI‐MS (Gb3) in control and GRASP55 KO (#2) cells. Data represented are mean ± SD of three independent experiments *P < 0.05, ***P < 0.001 (Student’s t‐test).
GRASP55 was originally identified as a Golgi stacking protein using in vitro studies (Shorter et al, 1999; Xiang & Wang, 2010), but recent studies with knockout (KO) animals and acute protein degradation have demonstrated that removing Grasp proteins does not affect stacking of Golgi (Grond et al, 2020; Zhang & Seemann, 2021). Other studies have shown that it plays a role in secretion (both conventional and unconventional) (D'Angelo et al, 2009; Kim et al, 2016; Chiritoiu et al, 2019) and glycosylation (Xiang et al, 2013; Jarvela & Linstedt, 2014). So, the function of GRASP55 at the Golgi apparatus and the mechanistic details remains unclear. We generated GRASP55 KO HeLa cells (3 independent clones) using clustered regularly interspaced short palindromic repeat (CRISPR)/ CRISPR‐associated protein 9 (Cas9) technique (Appendix Fig S3A) (gRNAs used in this study to generate GRASP55 knockout cell lines are listed in Appendix Table S1) (see Materials and Methods). Western blotting and immunofluorescence confirmed the complete abolishment of GRASP55 expression in these KO clones (Appendix Fig S3B and C) while expression levels of other Golgi matrix proteins were not altered (Appendix Fig S3D) (antibodies used in this study are listed in Appendix Table S4). Fragmentation of Golgi ribbon architecture was confirmed using both cis‐ and trans‐Golgi markers (Appendix Fig S4A–C). The Golgi stack itself did not reveal any obvious alterations with the length of the cisterna, the number of peri‐Golgi vesicles, or the fraction of membrane in vesicles and cisternae (Appendix Fig S4D–G). Metabolic labeling experiments showed that biosynthesis of GSLs significantly increased in all three clones (by 30–50%) with a corresponding decrease in SM (Fig 2B, Appendix Fig S5A and B) suggesting a bias toward GSL production in the absence of GRASP55. The total levels of complex GSLs (GM and Gb) also increased and GM/Gb ratio was altered favoring ganglioside production in 2 out of 3 clones, with a strong effect observed only in one clone (G55KO #1) and GRASP55 siRNA‐treated cells (Fig 2C). We also analyzed the kinetics of GSL production in GRASP55KO cells (G55 KO#2). SM was produced at similar but faster rate than GSLs in both control and GRASP55 KO cells (Appendix Fig S5A and B) likely because of faster CERT‐mediated transport of ceramide for SM biosynthesis (Hanada et al, 2003). We also observed differences in the rate of Gb and GM biosynthesis between control and GRASP55 KO cells (Appendix Fig S5A and B). While amounts of Gb were consistently more in GRASP55 KO cells across all time points, a large increase in GM in GRASP55 KO cells compared with control was observed only at the final time point (Appendix Fig S5A and B). Given the very low levels of GM in HeLa cells, the difference between control and GRASP55 KO cells is likely seen only after 24 h of chase. Next, we sought to confirm the increased levels of GSLs in GRASP55 KO cells by complementary methods. GRASP55 KO cells showed increased binding to bacterial toxins (Shiga and cholera toxins that bind Gb3 and GM1 respectively) compared with control cells (Fig 2D, Appendix Fig S5D) (all the Software used for analysis and figure preparation are listed in Appendix Table S8). Mass spectrometry analysis showed that there was an increase in the levels of several species of GlcCer, LacCer, and globosides in GRASP55 KO cell line (Fig 2E and F). A similar increase in GSLs was also observed in GRASP55 KO human fibroblast cell line (Wi‐26) (Appendix Figs S3E and S5C). Finally, the change in SL output associated with GRASP55 abolition was rescued by re‐expressing GRASP55‐GFP in these cell lines (Fig 2B, Appendix Fig S3C) suggesting that the observed changes in GSLs were specific. We conclude that GRASP55 acts as a regulator of substrate (Cer and LacCer) flux at the two steps that involve competing reactions—SM versus GSLs and gangliosides versus globosides in the SL biosynthetic pathway.
GRASP55 regulates the intra‐Golgi localization of GSL biosynthetic enzymes functioning at metabolic branch points
GRASP55 has been proposed to regulate glycosylation by regulating the kinetics of intra‐Golgi transport (Xiang et al, 2013) and/or ribbon formation (Jarvela & Linstedt, 2014). For GRASP55‐mediated regulation of GSL biosynthesis, neither is a likely explanation since the kinetics of GSL biosynthesis (Appendix Fig S5A) is very different from that of protein glycosylation (Xiang et al, 2013) and downregulation of several matrix proteins known to fragment Golgi ribbon does not affect GSL biosynthesis (Fig 2A). So, to understand how GRASP55 regulates GSL biosynthesis we first studied the consequences of GRASP55 deletion on the SM biosynthetic machinery (ceramide transfer protein—CERT and SMS1). GRASP55 deletion did not reduce the levels of CERT, and there was surprisingly a consistent increase in SMS1 transcript levels in all 3 clones (Appendix Fig S6A and B). GRASP55 deletion also did not alter their localization to Golgi (Appendix Fig S6C and D) or change the dynamics of CERT (Appendix Fig S6D and E). The kinetics of ceramide transport to the Golgi also remained unaltered (Appendix Fig S6F). These data suggest that SM biosynthesis is not directly affected by GRASP55 deletion.
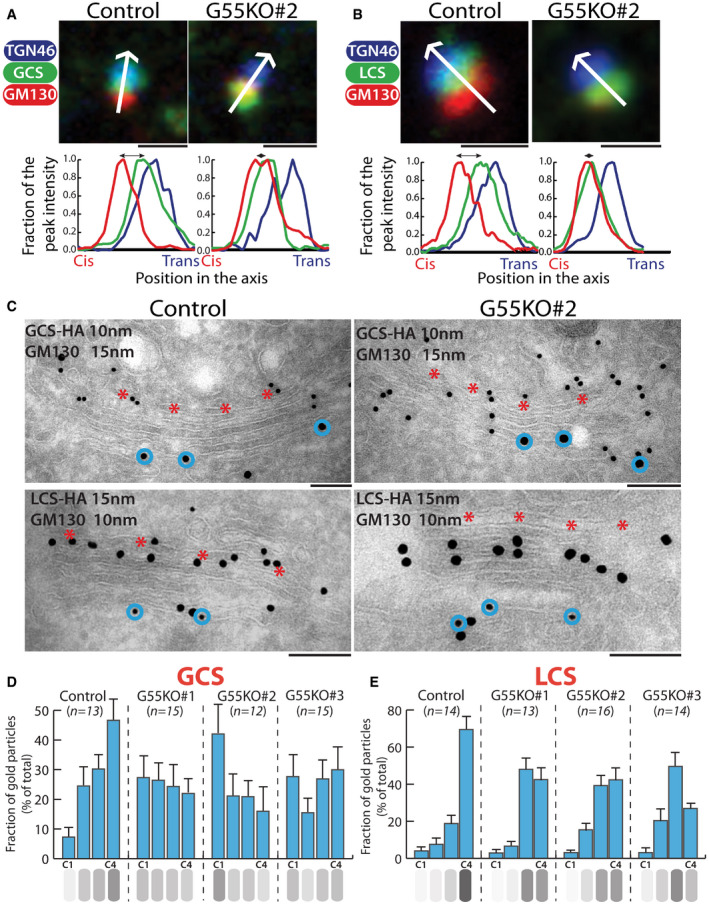
We next examined the effect of GRASP55 deletion on enzymes of the GSL biosynthetic branch (primers used in this study to determine mRNA levels of indicated gene are listed in Appendix Table S6). There were no consistent changes in their levels (Appendix Fig S7A) or their presence in the Golgi (Appendix Fig S7B) in GRASP55 KO cells (all the commercial kits used for cDNA extraction and mRNA analysis are listed in Appendix Table S7). Their intra‐Golgi localization was then examined in nocodazole‐induced ministacks with GM130 as a marker for cis‐Golgi/ cis‐Golgi network (CGN) compartment and TGN46 as a marker for TGN. Nocodazole‐induced ministacks were used since they show a clearer separation of cis‐ and trans‐Golgi markers and facilitate the intra‐Golgi localization of proteins (Rizzo et al, 2013; Beznoussenko et al, 2014). The peak localization of GSL biosynthetic enzymes was found in the medial/trans part of the Golgi in control cells (Appendix Fig S8). When GRASP55 was deleted, the intra‐Golgi localizations of GlcCer synthase (GCS) and LacCer synthase (LCS) were shifted in the direction of cis‐Golgi (Fig 3A and B, Appendix Fig S8A and B) while the localizations of SMS1, Gb3 synthase (Gb3S), and GM3 synthase (GM3S) were not altered (Appendix Fig S8C–E).
Figure 3. GRASP55 regulates the intra‐Golgi localization of GSL biosynthetic enzymes.
-
A, BControl and GRASP55 KO clones were transfected with HA‐tagged GSL biosynthetic enzymes, treated with nocodazole (33 µM) for 3 h, and processed for immunofluorescence with anti‐HA (green), anti‐GM130 (red), and anti‐TGN46 (blue) antibodies. The relative position of HA‐tagged enzymes with respect to GM130 and TGN46 was measured by line scanning and expressed as normalized positions of the peak intensity with the start of GM130 peak indicated as Cis and the end of TGN46 peak indicated as trans in the graph. The double‐headed arrows show the distance between the peak localization of GM130 and the enzymes. Scale bar 1 μm.
-
CControl and GRASP55 KO cells were transfected for 16 h with GCS‐HA or LCS‐HA and processed for cryoimmunolabeling with anti‐HA antibody (10‐nm gold particles) and anti‐GM130 antibody (15‐nm gold particles) in case of GCS‐HA and anti‐HA antibody (15‐nm gold particles) and anti‐GM130 antibody (10‐nm gold particles) in case of LCS‐HA. Representative images of the distribution of GCS‐HA and LCS‐HA are shown. Scale bar 200 nm. Red asterisk marks C4 cisterna and blue circles indicate GM130 labeling.
-
D, EDistribution of indicated enzymes across the Golgi stack was quantified and represented as fraction of Gold particles in each cisternae for GCS‐HA (D) and LCS‐ HA (E) (n indicated in the graph; data are Mean ± SEM).
The shift toward the cis‐Golgi was also confirmed by electron microscopy. GCS which is localized mostly to medial/trans‐Golgi (C3, C4 cisterna) with peak localization in C4 cisterna became evenly distributed across the stack in GRASP55 KO conditions (Fig 3C and D). The cis‐most cisterna (C1) which had minimal amount of GCS in control cells showed increased levels of GCS in GRASP55 KO cells, where it reached almost the same level as in other cisternae and in one clone (#2) even resulted in having the peak amount of enzyme (Fig 3D).
In case of LCS, there was a redistribution of the enzyme from trans‐Golgi (C4 cisterna) to medial‐Golgi (C2‐C3 cisternae) in GRASP55 KO cells (Fig 3C and E). Unlike GCS, LCS levels in the C1 cisterna did not change significantly (Fig 3E), and thus, the shift in intra‐Golgi localization was less pronounced in case of LCS. As a control, the localization of SMS1 was not altered under the same conditions (Appendix Fig S8F). To conclude, GRASP55 deletion changed the intra‐Golgi localization of two core enzymes of the GSL biosynthetic pathway involved in metabolic branching steps viz. GCS and LCS, shifting them from their mainly trans‐Golgi localization to more cis/medial‐Golgi localization. These observations raise the following questions: a. What is the mechanism by which the depletion of GRASP55 causes the shift in localization of GCS and LCS? and b. Is the displacement of enzymes responsible for metabolic effects observed after GRASP55 depletion?
GRASP55 interacts directly with GCS to promote its intra‐Golgi localization
To understand how GRASP55 regulates the localization of enzymes, we first studied whether GRASP55 interacts with enzymes of the GSL biosynthetic pathway. We expressed GRASP55‐GFP and HA‐tagged versions of GSL biosynthetic enzymes in HeLa cells, immunoprecipitated GRASP55‐GFP and analyzed for co‐immunoprecipitation of HA‐tagged enzymes by Western blotting. We found that both GCS and LCS co‐immunoprecipitated with GRASP55‐GFP while GM3S and Gb3S do not interact (Fig 4A). The interaction of GCS and LCS with endogenous GRASP55 was also observed in human fibroblast cells (Fig 4B). Surprisingly, in spite of significant homology between GRASP55 and GRASP65 proteins (Shorter et al, 1999), the enzymes do not interact with GRASP65, underscoring the specificity of the interaction (Fig 4B).
Figure 4. GRASP55 interacts with GCS and LCS.
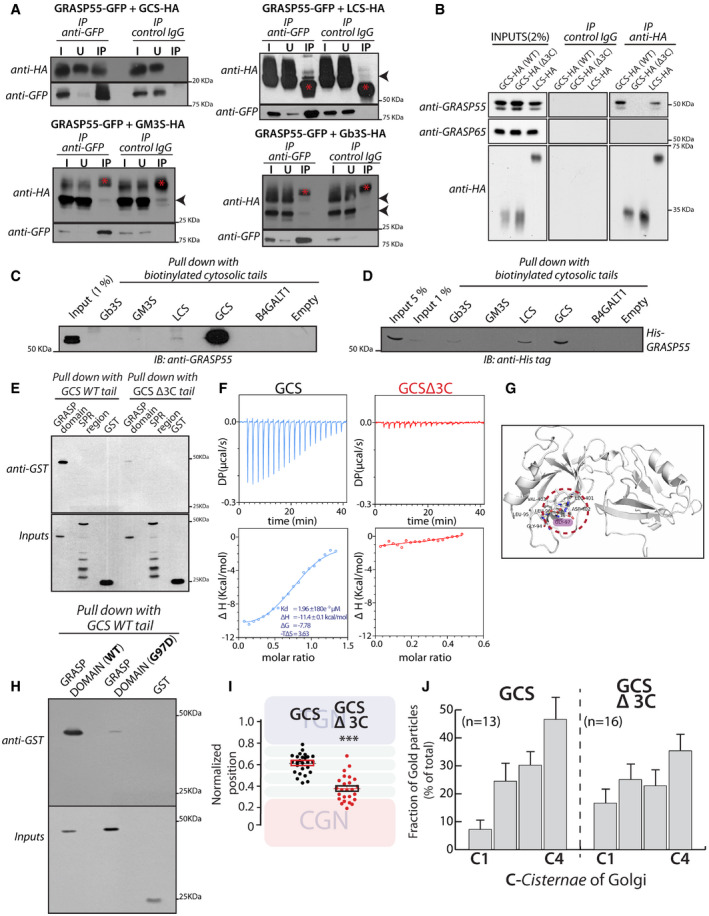
- HeLa cells co‐transfected with indicated HA‐tagged enzymes (GCS, LCS, GM3S, and Gb3S) and GRASP55‐GFP were lysed, immunoprecipitated with anti‐GFP antibody or control IgG, and were analyzed by Western blotting for interaction by immunoblotting (IB) with anti‐HA antibody. I represents 5% on the input lysate, U 5% of the unbound fraction, and IP the immunoprecipitate. Red asterisks indicate IgG bands, and arrow heads indicate the expected position of HA‐tagged enzymes
- WI‐26 fibroblasts transfected with the indicated HA‐tagged enzymes were lysed, immunoprecipitated with anti‐HA antibody or control IgG, and were analyzed by Western blotting for interaction by immunoblotting with the indicated antibodies.
- Chemically synthesized biotinylated peptides corresponding to cytosolic portions of glycosylation enzymes were bound to avidin beads and were used to pull down interactors from HeLa cell lysates and subjected to immunoblotting with anti‐GRASP55 antibody.
- The interaction of chemically synthesized biotinylated peptides, corresponding to cytosolic portions of glycosylation enzymes, with purified His‐tagged full‐length GRASP55 and their interaction was monitored by pulling down the biotinylated peptides bound to avidin beads followed by Western blotting with anti‐His tag antibody.
- Chemically synthesized biotinylated peptides corresponding to cytosolic portions of GCS (WT and Δ3C) and indicated purified GST‐tagged GRASP domain or SPR region of GRASP55 were incubated together, and their interaction was monitored by pulling down the biotinylated peptides with avidin beads followed by Western blotting with an anti‐GST tag antibody.
- ITC profile, representative of at least two independent experiments, for biotinylated GCS and GCS Δ3C cytosolic tails with recombinant GRASP55.
- The molecular basis of interaction between GRASP55 and ceramide glucosyltransferase C‐terminal peptide is studied by building a model of GRASP55:GCS peptide structure in the absence of the complex crystal structure. The carboxylate group of Leu of “LDV” motif retains conserved hydrogen bonds with the backbone of 95LLGV98motif of GRASP55. Gly97 residue which crucial to GRASP:GCS interaction is highlighted (pink).
- The interactions of chemically synthesized biotinylated peptides corresponding to cytosolic portions of GCS (WT) with the indicated purified GST‐tagged GRASP domain (WT) or GRASP domain (G97D) were monitored by pulling down the biotinylated peptides with avidin beads followed by Western blotting with an anti‐GST tag antibody.
- HeLa cells were transfected with either WT GCS or GCS Δ3C, treated with nocodazole (33 µM) for 3 h and labeled for enzymes, GM130, and TGN46 (to mark CGN and TGN respectively). Line scan analysis was performed as in Fig 3A and B, and the relative position of enzymes was quantitated and plotted. The data are mean ± SD (n = 30) representative of two experiments. **P < 0.01, ***P < 0.001(Student’s t‐test) and ns signifies not statistically significant.
- HeLa cells were transfected with either WT GCS or GCS Δ3C for 16 h and processed for cryoimmunolabeling. Distribution of indicated enzymes across the Golgi stack was quantified and represented as fraction of Gold particles in each cisterna (n indicated in the graph; data are Mean ± SEM).
Source data are available online for this figure.
GRASP55 is a peripheral membrane protein that is anchored to the Golgi membrane through its myristoylated N‐terminus. So, interaction with GRASP55 is likely mediated by cytosolically exposed portions of the enzymes. Many Golgi glycosylation enzymes are type II membrane proteins with a short N‐terminal cytosolic tail, a transmembrane domain, and the luminal enzymatic domain. Thus, the likely part to interact with GRASP55 is the short N‐terminal tail. GCS is an exception to this rule and is a multi‐transmembrane protein with both N‐ and C‐ terminal cytosolic tails and a large catalytic portion facing the cytosol. We noticed that the C‐terminal tail of GCS ends with a consensus class II PDZ domain interacting motif (Φ‐X‐Φ‐COOH, where Φ refers to hydrophobic amino acids and X to any amino acid) that can potentially bind to the N‐terminal tandem PDZ domains of GRASP55. We chemically synthesized the C‐terminal cytosolic tail of GCS and N‐terminal cytosolic tails of other GSL biosynthetic enzymes and studied their interaction with GRASP55 present in cell lysates as well as purified His‐tagged protein (list of cytosolic peptides used in this study for immunoprecipitation assays are listed in Appendix Table S5).
We found that GCS tail strongly bound to GRASP55 from both cell lysates and the purified protein unlike the tails of other GSL biosynthetic enzymes (Gb3S or GM3S) or the tail of B4GALT1 (see below for description of LCS binding) (Fig 4C and D). We used B4GALT1 a galactosyltransferase involved in protein glycosylation since it was unrelated to the GSL biosynthetic pathway. GRASP55 consists of two domains—N‐terminal tandem PDZ domains (GRASP domain) followed by a serine proline‐rich (SPR) region (30). The cytosolic tail of GCS interacted with the GST‐tagged GRASP domain of GRASP55 but not with GST‐tagged SPR region (Fig 4E). Deleting the Φ‐X‐Φ‐COOH motif in GCS cytosolic tail (GCS‐Δ3C tail) impaired its interaction with GST‐tagged GRASP domain of GRASP55 (Fig 4E). Further, deleting this motif also impaired the interaction of full‐length GCS (GCS‐Δ3C) with GRASP55 in cells as evidenced by co‐immunoprecipitation analysis (Fig 4B). Next to gain further insights into this interaction, we studied it by using isothermal titration calorimetry (ITC). By ITC, we found that the cytosolic tail of GCS interacts strongly with the GRASP domain of GRASP55 with a K d value of 2 µM (Fig 4F). Of note, the K d values of interaction between GRASP55 and the Golgin‐45 tail were approx. 0.27 µM (Zhao et al, 2017) and that of the LCS tail and GOLPH3 (another Golgi matrix protein that interacts with Golgi enzymes) were approx. 60 µM (preprint: Rizzo et al, 2019). There was no significant interaction of the GRASP domain of GRASP55 with the GCS tail deleted of Φ‐X‐Φ‐COOH motif or B4GALT1 tail (Fig 4F). These studies in sum suggest that the GCS C‐terminal tail directly and specifically interacts with the GRASP domain of GRASP55.
To identify the amino acid residues in GRASP55 that are critical for this interaction, we resorted to modeling. The crystal structure of the GRASP domain of GRASP55 bound to Golgin45 C‐terminal region (which contains a hydrophobic amino acid similar to GCS) showed that the peptide binds to a cleft between the PDZ1 and PDZ2 domains of the proteins (Zhao et al, 2017). A conserved pocket in the GRASP domain consisting of 95LLGV98 corresponding to the X‐Φ1‐G‐Φ2 motif (X: any amino acid, Φ: hydrophobic amino acid, G: glycine) acts as a binding site for the C‐terminal end of the interacting peptide forming hydrogen bonds with the last four residues of Golgin‐45 peptide. The X‐Φ1‐G‐Φ2 peptide has a strained left‐handed helix conformation, which is usually populated by glycine residues in the Ramachandran plot (Lee & Zheng, 2010). B‐factor analysis of the crystal structure of the GRASP55:Golgin‐45 complex revealed that the X‐Φ1‐G‐Φ2 motif forms a rigid loop (Appendix Fig S9A) where usually Gly is favored over Asp. Indeed, substitution of Gly to Asp abolished the GRASP55‐Golgin‐45 peptide interaction (Zhao et al, 2017). A model of the GCS peptide was first built using the backbone conformation of the Golgin‐45 peptide as a template and docked onto the cleft between PDZ1 and PDZ2 domains. The lowest energy model generated was analyzed to probe the protein:peptide interaction. The structural analysis also indicated the important contribution of the 95LLGV98 pocket to binding with the C‐terminal amino acids of the GCS peptide (Fig 4G). The predicted binding affinity (ΔG) between protein and peptide was –13.1 kcal/mol, indicating a favorable interaction, and when “LDV” was removed from the peptide, the binding affinity was reduced (ΔΔG = −1.5 kcal/mol) indicating the importance of the backbone‐mediated conserved hydrogen bonds in peptide binding. Given the overall similarity between the GRASP55‐Golgin‐45 peptide and GRASP55‐GCS peptide interactions, we tested whether the interactions show similar sensitivities to mutations. As mentioned before, the GRASP55‐Golgin‐45 peptide interaction is sensitive to the substitution of Gly97 to Asp (Zhao et al, 2017) and so we introduced the corresponding G97D mutation in the GRASP55 PDZ domain to examine its effect on the interaction with the GCS tail. The interaction of the mutant protein GCS C‐terminal tail was greatly diminished (Fig 4H). Thus, the interaction between GCS and GRASP55 is likely mediated by the C‐terminal LDV motif of GCS interacting with the 95LLGV98 pocket in the GRASP domain of GRASP55 with Gly97 playing a critical role in the process (Fig 4H). Next, we examined whether binding to GRASP55 is essential for intra‐Golgi localization of GCS. The last 3 C‐terminal amino acids of GCS were deleted (GCS‐Δ3C) and the localization of the mutant protein was studied. The intra‐Golgi localization of GCS‐Δ3C was altered, with it displaying a more cis‐Golgi localization compared with the WT enzyme (Fig 4I and J). Thus, direct interaction with GRASP55 is essential for the correct sub‐compartmentalization of GCS.
Compared with GCS, the LCS tail showed a qualitatively weaker interaction with GRASP55 both for endogenous and recombinant protein (Fig 4C). Nevertheless, it was significantly above that of other GSL biosynthetic enzymes (GM3S or GB3S) and that of B4GALT1. This weak interaction was mediated by the GRASP domain of GRASP55 (Appendix Fig S9B). Nevertheless, ITC studies did not show a significant interaction between purified LCS cytosolic tail and the GRASP domain of GRASP55 (Appendix Fig S9C). Thus, in the case of LCS, while the full‐length enzyme co‐immunoprecipitated with GRASP55, the cytosolic tail of the enzyme itself shows only a weak or no interaction with the protein suggesting that the interaction between LCS and GRASP55 is likely to be indirect. The change in localization of LCS in the absence of GRASP55 correlates with the observed interaction with GRASP55 (Fig 4A and B). In summary, we conclude that GCS and LCS interact with GRASP55, and in the case of GCS, this interaction is direct and is essential for its intra‐Golgi localization.
GRASP55 compartmentalizes the enzymes by preventing their entry into retrograde carriers
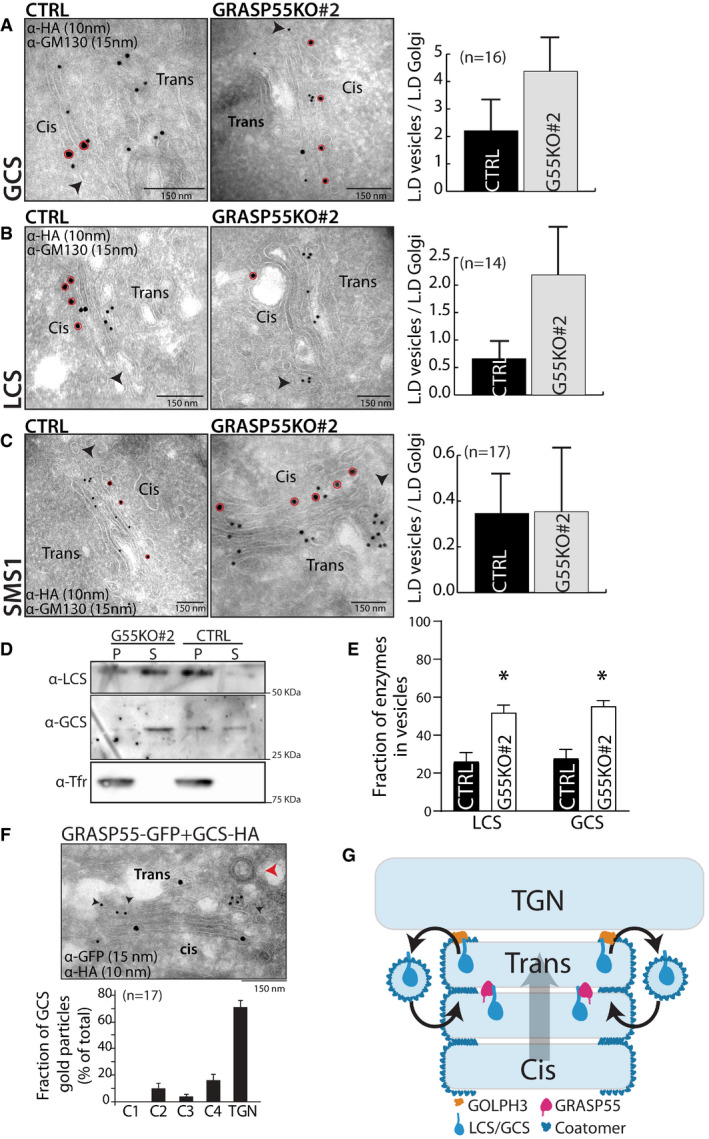
According to the cisternal maturation model, the retention of resident proteins in the Golgi is due to their continuous retrograde transport mediated by COPI in the face of anterograde flux of cargoes (Nakano & Luini, 2010; Glick & Luini, 2011). In the framework of this model, compartmentalization of enzymes to specific cisternae of the Golgi is achieved by a balance between the anterograde and retrograde flux of enzymes. Indeed, impairing the retrograde transport of enzymes promotes their forward transport leading to their localization to post‐Golgi compartments (Rizzo et al, 2013). So, to understand how this balance is affected in GRASP55‐depleted cells to change the steady‐state localization of GCS and LCS, we examined the distribution of GCS and LCS in peri‐Golgi vesicles/carriers. These vesicles depend on COPI for their formation, and entry into these vesicles is essential for the intra‐Golgi retrograde transport of proteins (Rizzo et al, 2013). In control HeLa cells, the distribution of enzymes in peri‐Golgi vesicles varied. For instance, the density of LCS in peri‐Golgi carriers was nearly the same as that of the cisterna while the density of GCS in vesicles was 1.8‐fold more than that of the cisterna. Further SMS1, an enzyme unaffected by GRASP55 depletion was depleted in peri‐Golgi vesicles compared with cisterna (density in vesicles was 0.3‐fold that of the cisterna) (Fig 5A–C). These differences in density of enzymes in vesicles correlate well with their observed distribution in the Golgi at steady state, i.e., an increased presence in peri‐Golgi vesicles correlates with increased cis/medial‐Golgi localization. For instance, GCS with higher relative density in peri‐Golgi vesicles also has a higher cis‐Golgi to TGN ratio than SMS1, which has a lower relative density in peri‐Golgi vesicles (Appendix Fig S1). This suggests that the presence of proteins in retrograde transport carriers could be a reliable indicator of intra‐Golgi distribution of proteins. Next, we studied the density of enzymes in peri‐Golgi vesicles in the absence of GRASP55. We found that the density of GCS in vesicles increased to 2.5‐fold compared with that present in the cisterna and that of LCS increased to twofold over that of the cisterna, while that of SMS1 was unaltered (Fig 5A–C). Thus, GCS and LCS that show an increased localization to cis/medial‐Golgi in the absence of GRASP55 also show an increased presence in vesicles in GRASP55 KO cells compared with control, while vesicle distribution of SMS1, whose intra‐Golgi localization is unaltered by the absence of GRASP55, remains unchanged. To further validate the increased entry of GCS and LCS into peri‐Golgi carriers in the absence of GRASP55, we resorted to in vitro budding assays to purify COPI‐coated vesicles from Golgi membranes using well‐established methods (Yang et al, 2002). The Golgi apparatus was purified from control and GRASP55 KO (#2) cells and was incubated with purified coatomer, myristoylated ARF1, and BARS (Brefeldin A ADP ribosylation substrate) to promote budding of COPI retrograde carriers from the Golgi. The budded vesicles were then separated from the Golgi apparatus by centrifugation. The Golgi apparatus was recovered in the pellet fraction while purified COPI vesicles remained in the supernatant fraction (Appendix Fig S10A). We then analyzed the presence of LCS and GCS in the supernatant fractions by Western blotting. We found that there were increased amounts of LCS and GCS in COPI vesicles that budded from the Golgi apparatus purified from GRASP55 KO cells as compared to control cells (Fig 5D and E), thus confirming our EM observations. The formation of these COPI vesicles depended on the presence of ARF1, ARFGAP, and BARS and did not contain transferrin receptor (a cargo that does not enter COPI vesicles) suggesting that the vesicles purified were indeed genuine COPI vesicles (Fig 5D, Appendix Fig S10A). This specific increase in amounts of GCS and LCS in the peri‐Golgi carriers in the absence of GRASP55 is consistent with the hypothesis that GRASP55 limits the retrograde transport of these enzymes.
Figure 5. GRASP55 compartmentalizes the enzymes by preventing their entry into retrograde carriers.
-
A–CControl and GRASP55 KO (#2) cells were transfected for 16 h with the indicated HA‐tagged enzymes and processed for cryoimmunolabeling with anti‐HA antibody (10‐nm gold particles) and anti‐GM130 antibody (15‐nm gold particles). Representative images of the distribution of HA‐tagged enzymes are shown. Red circles indicate GM130 labeling. Arrow heads represent the peri‐Golgi vesicles. Scale bar 150 nm. Quantification of the distribution of enzymes in vesicles represented as normalized linear density (n indicated in the graph; data are Mean ± SEM).
-
DCOPI vesicles were reconstituted using the two‐stage incubation system (detailed in the Methods section). After the second‐stage incubation, samples were centrifuged to obtain the pellet fraction that contains Golgi membranes and the supernatant fraction that contains reconstituted COPI vesicles. Both fractions were immunoblotted for LCS, GCS, and transferrin receptor (Tfr) to show their relative distributions on Golgi membranes and in COPI vesicles.
-
EThe COPI vesicle reconstitution system was performed as described above (in D), and then the fraction of LCS and GCS in COPI vesicles versus their total distribution (on Golgi membranes and in COPI vesicles) was calculated. The mean and standard error from three independent experiments are shown, *P < 0.05, Student’s t‐test.
-
FHeLa cells co‐transfected with HA‐tagged GCS and GRASP55‐GFP were incubated for 16 h and were processed for cryoimmunolabeling with anti‐HA antibody (10‐nm gold particles) and anti‐GFP antibody (15‐nm gold particles). Representative images of the distribution of GCS and GRASP55‐GFP are shown. Red arrowhead indicates the clathrin‐coated vesicle that marks the TGN area, and black arrowheads indicate the presence of GCS‐HA in TGN. Scale bar 150 nm. Distribution of GCS across the Golgi stack and TGN was quantified and represented as fraction of gold particles (n indicated in the graph; data are Mean ± SEM).
-
GModel represents GRASP55‐mediated compartmentalization of GCS and LCS. A cyclical and balanced activity of GRASP55 and GOLPH3 compartmentalizes LCS/GCS to the trans‐Golgi. The anterograde transport of enzymes (forward direction arrow; cis to trans direction) counterbalances their retrograde transport (reverse direction arrow; trans to cis direction) resulting in the compartmentalization of these enzymes.
Source data are available online for this figure.
If absence of GRASP55 alters the distribution of the protein, does an increase in GRASP55 levels also influence their distribution? To test this, we overexpressed GRASP55‐GFP in cells along with GSL biosynthetic enzymes LCS and GCS. In the case of LCS, when GRASP55 was overexpressed, there was a change in localization of the enzyme, which was now present in endosome‐like structures along with the Golgi (Appendix Fig S10C). These structures were similar to those to which LCS localizes in the absence of GOLPH3, an adaptor that links LCS to COPI and thus promoting its retrograde transport through peri‐Golgi vesicles (46). This suggests that overexpression of GRASP55 likely inhibits retrograde transport in a way that is similar to the absence of GOLPH3. On the other hand, overexpression of GRASP55 did not shift GCS to a post‐Golgi localization (Appendix Fig S10B) but GCS was increasingly found in the TGN under these conditions (Fig 5F). This suggests that GCS behaves similar to LCS upon GRASP55 overexpression, i.e., its localization shifts to a forward position along the secretory pathway. This distribution of enzymes to TGN or post‐Golgi compartment caused by GRASP55 overexpression is consistent with an impairment of their retrograde transport. Thus, GRASP55 likely acts to inhibit retrograde transport of LCS and GCS, such that in its absence the enzymes shift to a cis/medial‐Golgi localization and when GRASP55 levels are increased, they shift to a TGN/post‐Golgi localization.
These observations suggest that GRASP55 prevents retrograde transport of GCS and LCS. In case of LCS, it is known that GOLPH3 promotes its retrograde transport. So we studied what happens to LCS when both GRASP55 and GOLPH3 are removed. GOLPH3KD led to the localization of LCS in endosomal structures (Appendix Fig S10C) as reported earlier (Rizzo et al, 2021). Double knockdown of GRASP55 and GOLPH3 also led to a similar phenotype (Appendix Fig S10C) suggesting that GOLPH3 likely acts downstream of GRASP55 such that the effect of GOLPH3 KD dominates over the effect of GRASP55 reduction.
We propose that GRASP55 inhibits retrograde transport of LCS and GCS by acting as a “retainer” that binds to and prevents their entry into retrograde transport carriers. While the action of adaptors, exemplified by GOLPH3, that promote entry into retrograde transport vesicles is well accepted, a retainer action in the Golgi apparatus has not been hypothesized before. A retainer molecule that prevents the entry of its interactors into peri‐Golgi vesicles can be expected to be absent from the peri‐Golgi vesicles unlike adaptor molecules (GOLPH3) that promote the sorting of their interactors into peri‐Golgi vesicles (Eckert et al, 2014). So, we analyzed the distribution of GRASP55 on the cisterna and on peri‐Golgi vesicles. We find that while nearly half of the Golgi‐localized GOLPH3 was found in peri‐Golgi carriers, only about 8% of the Golgi‐associated staining of GRASP55 is associated with peri‐Golgi vesicles, suggesting that GRASP55 is likely excluded from peri‐Golgi vesicles (Appendix Fig S10D).
Rationalizing these observations into a model, we propose that recycling adaptors and retainers act in an opposing manner to promote compartmentalization of enzymes in the Golgi apparatus. The binding to COPI either directly (Liu et al, 2018) or through recycling adaptors like GOLPH3 (Tu et al, 2008, 2012) promotes the retrograde transport of enzymes thus preventing their exit from the Golgi. On the other hand, retainers bind to their client molecules to prevent their entry into retrograde transport carriers and thus indirectly promote their anterograde transport by cisternal progression (Fig 5G). Thus, a cyclical and balanced transport of enzymes—in the retrograde direction by COPI machinery assisted by recycling adaptors and in the anterograde direction by cisternal flow assisted by retainers—compartmentalizes them to specific cisterna of the Golgi apparatus. Of note, this model explains how distinct molecular compositions of cisternae are achieved within the Golgi stack, which includes cis‐ to trans‐Golgi cisterna where COPI operates (Oprins et al, 1993) and not TGN where COPI coat is absent (Ladinsky et al, 1999) (Fig 5G).
Change in intra‐Golgi localization of GSL biosynthetic enzymes changes GSL output
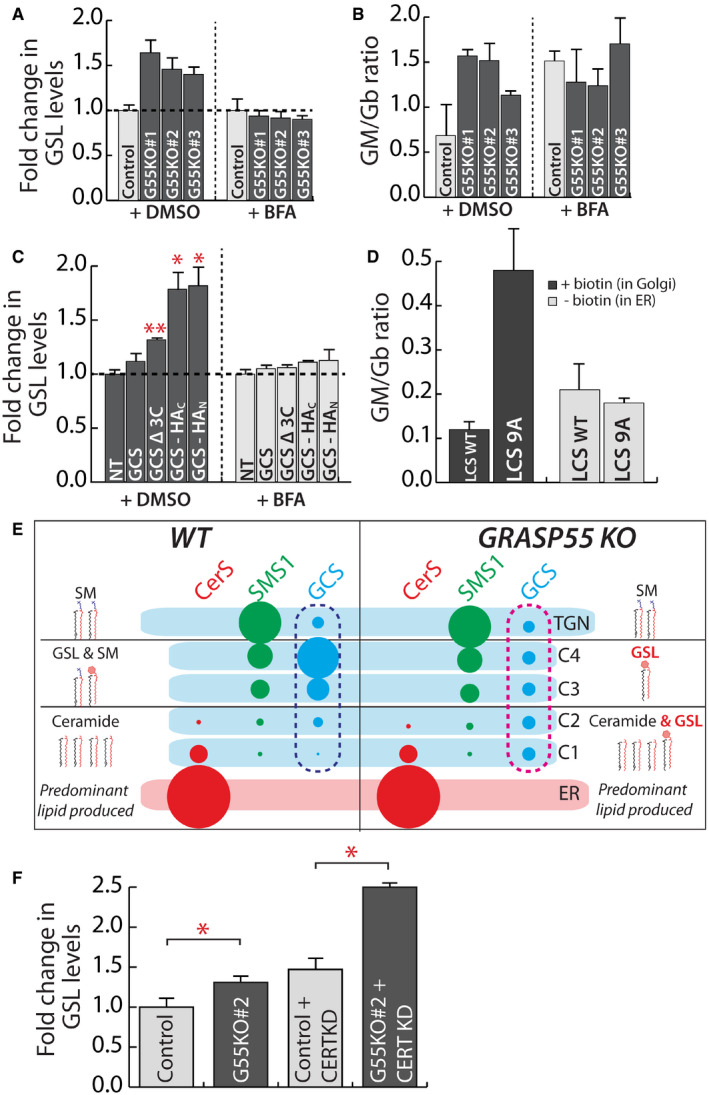
We then examined whether the change in localization of enzymes following reduction in GRASP55 levels contributes to the associated changes in GSL biosynthesis. When GSL biosynthesis was examined in Brefeldin‐A‐treated cells, the increased GSL production in GRASP55 KO cells and the increased GM/Gb ratio were not observed (Fig 6A and B), suggesting that compartmentalized localization of enzymes is essential to manifest GRASP55‐deletion‐induced alteration in GSL production. Of note, while data in Fig 2B suggested that the Gm/Gb ratio was strongly increased only in one clone (G55KO#1), here we observed an apparent increase of GM/Gb ratio in all three clones. This is probably due to the differences in time of chase. While data in Fig 2B were obtained after 24 h of chase, the data in Fig 6B were obtained after 8 h chase (toxicity of BFA prevented longer chase periods). Nevertheless, in both cases the GM/Gb ratio showed similar tendency with G55KO #1 > G55KO #2 > G55KO #3. Next, we examined GSL biosynthesis after expression of GCS and LCS mutants that have altered intra‐Golgi localization. We expressed three GCS mutants: a. GCS‐Δ3C that shows more cis‐Golgi presence than the WT enzyme (Fig 4J), b. GCS with a HA‐tag at the C‐terminus (GCS‐HAC) that is expected to block accessibility to the C‐terminal valine. GCS‐HAC distributes across the stack with significant presence in cis‐Golgi (30), and unlike GCS‐Δ3C, it was also partially localized to the ER (Appendix Fig S11A) and c. GCS with a HA‐tag in the N‐terminal tail (GCS‐HAN), which localizes to ER (Appendix Fig S11A). Thus, they have distinct but overlapping distributions along the secretory pathway. Expression of the wild‐type GCS construct in HeLa cells led to a 10% increase in the total GSLs produced compared with non‐transfected cells suggesting that expression of the construct does not overwhelm the biosynthetic system. The expression of GCS‐Δ3C, which localizes to the cis‐Golgi unlike wild‐type GCS, led to a 30% increase in GSLs produced, a difference that is quantitatively similar to what is observed between control and GRASP55 KO cells. This suggests that the increased production of GSLs under GRASP55 KO conditions can be explained by the shift in localization of GCS. Interestingly, an increasing ER localization of GCS (GCS‐HAC and GCS‐HAN) led to a larger increase in GSL production (2.3‐ and 2.6‐fold increase, respectively) (Fig 6C). The observed differences in GSL production are not due to differences in expression since addition of Brefeldin A neutralized these differences (Fig 6C), implying that changes in localization were indeed the cause for the observed increase in GSL production.
Figure 6. Change in intra‐Golgi localization of GSL biosynthetic enzymes changes GSL output.
-
A, BControl and GRASP55 KO clones were pre‐treated with DMSO or BFA (5 μg/ml) for 30 min and SL output monitored by [3H]‐sphingosine pulse‐chase assay (8 h chase). Total GSL levels were quantified and expressed as fold changes with respect to control (A). The GM/Gb ratio was calculated and represented (B). Data are mean ± SD of two independent experiments.
-
CHeLa cells were transfected with indicated GCS constructs for 16 h and SL output monitored by [3H]‐sphingosine pulse‐chase assay (8 h chase) in the presence or absence of BFA (5 μg/ml). Total GSL levels were quantified and expressed as fold changes with respect to control. NT refers to non‐transfected cells. Data are mean ± SD of two independent experiments. *P < 0.05, **P < 0.01, (Student’s t‐test).
-
DHeLa cells KO for LCS were transfected with indicated LCS RUSH constructs for 16 h, and expressed enzymes were retained in the ER (‐biotin) or placed in the Golgi by the addition of biotin (40 μM) (+biotin) and SL output monitored by [3H]‐sphingosine pulse‐chase assay. The ratio of GM/Gb is represented. Data are mean ± SD of two independent experiments.
-
ESchematic representation of how the change in localization of GCS in GRASP55 KO with respect to control (pink dashed box versus blue dashed box) from trans‐ to cis‐Golgi results in preferential access to ceramide and thus an increased production of GSLs.
-
FCERT was silenced using siRNAs for 72 h in control and GRASP55 KO (#2) clone before subjecting to [3H]‐sphingosine pulse‐chase assay. GSLs were quantified and represented as fold change with respect to control. Values are mean ± SD (n = 3). *P < 0.05, (Student’s t‐test).
Next, we examined whether the next branch point in the SL metabolic pathway viz GB3‐GM3 branch was also sensitive to enzyme localization. The key enzyme regulating this branch point is LCS, whose localization is again controlled by GRASP55. While we find that GRASP55 and LCS interact, there is no convincing evidence for their direct interaction so mutating key residues to decompartmentalize the protein similar to what was achieved for GCS is not possible. While studying the molecular basis of LCS localization, it was found that the 14 amino acid cytosolic tail of LCS contains the information needed to localize the protein in the Golgi (preprint: Rizzo et al, 2019). So, we performed alanine mutagenesis of most of the cytosolic tail of LCS (LCS9A) except for the membrane proximal 5 amino acid region essential for interaction with GOLPH3 and the subsequent retention of LCS in Golgi. We found that while LCS9A was still retained in the Golgi apparatus as expected, it localized in a de‐compartmentalized manner with no clear trans‐Golgi localization as observed with LCS‐WT (Appendix Fig S11B). We do not know the reason why these mutations lead to a de‐compartmentalized localization of LCS, but they provide an opportunity to test whether compartmentalization of LCS regulates the flux across Gb3‐GM3 branches similar to what was observed with GCS. To this end, we analyzed the GSL output following the expression of LCS‐WT and LCS9A constructs in LCS‐KO HeLa cells (Yamaji & Hanada, 2014). We found that the expression of LCS9A in Golgi favored the production of gangliosides over globosides as compared to LCS‐WT. Thus, the GM/Gb ratio of 0.12 that is observed in case of LCS‐WT‐expressing cells changed to 0.47 in case of LCS9A‐expressing cells (Fig 6D). The differences were nullified when the enzymes were retained in the ER (Fig 6D) suggesting that the observed differences were likely due to an altered intra‐Golgi localization of these enzymes. Thus, a change in LCS localization observed in GRASP55 KO conditions may contribute to the observed increase in GM/Gb ratio in these cells. From these data, we conclude that altered localization of key enzymes of the GSL pathway can reproduce the effects on SL biosynthesis following GRASP55 deletion and that localization of enzymes in the Golgi can control cargo flux across competing biosynthetic pathways.
To explain the effects of GRASP55 KO on GSL biosynthesis based on these results, we thus considered the localization of these respective enzymes. As discussed earlier, most of the GSL biosynthesis likely happens in the medial/trans‐Golgi where along with the GSL enzymes, a substantial portion of SMS1 is also localized (Fig 1B). This likely leads to a competition between SMS1 and GCS for Cer that is transported by vesicular transport to the medial/trans‐Golgi (Fig 6E). By moving GCS to the cis‐Golgi, as happening in GRASP55 KO cells, the enzyme now gets preferential access to Cer and resulting in an increased production of GSLs (Fig 6E). Indeed, in CERT KD cells where the non‐vesicular transport of Cer is blocked and Cer reaches the Golgi mostly by vesicular transport, the effect on GSL biosynthesis resulting from the absence of GRASP55 is further increased (Fig 6F) suggesting that GCS and SMS1 compete for Cer that is transported by vesicular transport to the Golgi. The effect of GRASP55 KO on GSL output can also be explained by a similar logic. While both GM3S and Gb3S are localized mainly to the medial/trans‐Golgi, significant amounts of GM3S can also be seen in cis/medial‐Golgi unlike Gb3S (Appendix Fig S1). Thus, when LCS is moved toward the cis/medial‐Golgi, ganglioside biosynthesis by GM3S is favored over globoside biosynthesis likely due to preferential access of GM3S to the substrate LacCer.
Thus, these data provide a model of how compartmentalization of enzymes in the Golgi contributes to faithful glycan output. When two enzymes compete for a common substrate and are localized in the same compartment, the product of a glycoenzyme with more affinity for substrate and/or increased expression dominates the glycan output. On the other hand, if one of the competing enzymes is present in an earlier part of the secretory pathway relative to the other, it gets preferential access to the substrate before its competitor and thus promotes flux across the pathway catalyzed by it resulting in a glycan output that has a bias toward the product of this enzyme. Thus, positioning of enzymes in the Golgi regulates the pattern of glycan distribution in the output.
Discussion
Here, we describe how GRASP55 compartmentalizes two key GSL biosynthetic enzymes—GCS and LCS by binding to these enzymes and preventing their entry into retrograde transport carriers. This action of GRASP55 opposes COPI‐mediated retrograde transport of residents, and a balance between these two actions compartmentalizes the enzymes to trans‐Golgi. Translocation of the enzymes to cis‐Golgi in the absence of GRASP55 provides them a privileged access to cargoes thus favoring the reactions catalyzed by them. This is the first time a molecule that specifically regulates intra‐Golgi localization of glycosylation enzymes is described and that acts by preventing the entry of cargoes into COPI carriers. This study has important implications for the cell biology of Golgi organization as well as glycobiology which we discuss below.
Mechanisms regulating Golgi compartmentalization
The retention of glycosylation enzymes in the Golgi apparatus is due to their continuous retrograde transport by the COPI machinery. How the same retrograde transport machinery achieves the compartmentalization of enzymes to different cisterna was not clear. The discovery of an enzyme adaptor—GOLPH3 acting at the trans‐Golgi—suggested that trans‐ and cis‐Golgi‐localized enzymes may have differential modes of interacting with the COPI machinery. While the cis‐Golgi‐localized proteins interacted with COPI directly (Liu et al, 2018), the trans‐Golgi residents may require an adaptor recruited specifically to the trans‐Golgi, thus allowing the same COPI machinery to compartmentalize enzymes to two different sub‐Golgi compartments. Our discovery of GRASP55 actively prevents entry of enzymes into COPI vesicles and shows this process is more elaborate and with both retaining and recycling adaptors required for appropriate localization to the Golgi. Indeed, in the absence of GRASP55 and with only GOLPH3 present, the compartmentalization of LCS is altered. Thus, compartmentalization of Golgi residents is a dynamic process resulting from a balance between their anterograde and retrograde flux mediated by retaining and recycling adaptors, respectively (Fig 5G). Recently, GRASP proteins have been suggested to regulate COPI vesicle dynamics (Xiang et al, 2013; Grond et al, 2020). This activity of GRASP55 may further contribute to oppose the retrograde transport of residents. Of note, GRASP proteins have been linked to the Golgi localization of p24 (Barr et al, 2001) and the transport of cargoes through the Golgi (D'Angelo et al, 2009). To prove, whether these effects are also linked to the role of GRASP55‐mediated sorting into COPI vesicles needs to be investigated. Given the well‐known regulation of GRASP55 and GOLPH3 activities by phosphorylation in response to various stimuli, we expect that regulation of glycosylation by altering Golgi compartmentalization may turn out to be a regulated physiological phenomenon.
We also note that there are several other Golgi‐resident proteins with canonical PDZ domain‐binding motifs, which includes other enzymes and channels. Thus, GRASP55 (and possibly GRASP65) may play a wider role in the localization of several proteins in the Golgi.
Regulation of glycosylation by GRASP55
GRASP55 was originally discovered as Golgi stacking protein (Shorter et al, 1999; Xiang & Wang, 2010), but recent studies have unequivocally shown that absence of GRASP proteins does not affect Golgi stacking (Grond et al, 2020; Zhang & Seemann, 2021) leaving the role of this protein obscure. In this light, the contribution of GRASP55 to conventional as well as unconventional secretion (D'Angelo et al, 2009; Kim et al, 2016; Chiritoiu et al, 2019) and glycosylation (Xiang et al, 2013; Jarvela & Linstedt, 2014) become prominent areas to investigate. Its effect on glycosylation was proposed to be indirect through regulation of the kinetics of intra‐Golgi transport of cargoes (Xiang et al, 2013) and/or ribbon formation (Jarvela & Linstedt, 2014). For GRASP55‐mediated regulation of GSL biosynthesis, neither is a likely explanation since kinetics of GSL biosynthesis (Appendix Fig S5A) is very different from that of protein glycosylation and downregulation of several matrix proteins known to fragment Golgi ribbon does not affect GSL biosynthesis (Fig 2A). Instead, we show that absence of GRASP55 leads to a change in the localization of LCS and GCS (Fig 3). This alters SL production by providing a privileged early access of substrates to these enzymes and/or providing them a more permissive environment (Hayashi et al, 2018; Ishibashi et al, 2018) and thus increases GSL production (Fig 6E). Thus, our study here provides a mechanistic view of how GRASP55 regulates glycosylation and identifies an important role for this protein in the Golgi function.
It is well known that presence of two Grasp proteins is a feature of vertebrates while other organisms have only one Grasp protein and plants have none at all (Vinke et al 2011). Since this single Grasp protein is mostly localized in the cis‐Golgi similar to GRASP65 (Vinke et al 2011), the medial‐Golgi‐localized GRASP55 is probably an evolutionary innovation of vertebrates. Analysis of GCS sequences showed that the C‐terminal 3 amino acid PDZ domain‐binding motif is conserved only in vertebrates (Appendix Fig S9D) thus paralleling the evolution of GRASP55. This co‐evolution of a protein and the interaction motif on its client suggests the evolution of a regulatory mechanism for GSL biosynthesis. The absence of classical PDZ binding motifs in other organisms may likely reflect the absence of such a regulation in these organisms.
Given the close link between GSL metabolism and cancers, we analyzed whether GRASP55 expression is altered in cancers. There were no significant alterations at the genomic level (mutations or copy number alterations) at the GRASP55 loci, but expression levels of GRASP55 were moderately low in acute myeloid leukemia, kidney cancers, and thymoma compared with other cancer types (www.cbioportal.org). Acute myeloid leukemic cells are known to have increased levels of GSLs compared with non‐transformed lymphocytes (Wang et al, 2012). Whether GRASP55 contributes to this phenomenon is worthy of exploration. Further, mouse insertional mutagenesis studies have found that inactivation of GRASP55 promotes tumorigenesis in liver and colorectal cancer models (Ref: Candidate Cancer Gene Database). Thus, studying the contribution of GRASP55 to cancer through its role in the regulation of GSL biosynthesis could be an interesting area to explore.
SL biosynthesis
SL biosynthesis depends on CERT‐mediated transfer of ceramide from ER to TGN for SM biosynthesis (Hanada et al, 2003) and FAPP2‐mediated transfer of GlcCer from cis‐Golgi to TGN for Gb3 biosynthesis (D'Angelo et al, 2013). Earlier reports had shown that GCS and SMS1 are present in cis‐ and trans‐Golgi respectively (Halter et al, 2007). Surprisingly, we observe that GCS is present in the medial/trans‐Golgi. We ascribe the observed difference in localization of GCS to the use of constructs with a tag that blocks the C‐terminus (important for interaction with PDZ domain of GRASP55) in the previous studies (Halter et al, 2007). This revised localization of GCS has implications for the model of SL biosynthesis: If GCS is localized to trans‐side why is the activity of GCS not sensitive to CERT KD? While GCS is indeed present in the trans‐Golgi, a large fraction of SMS1 is present in a distinct compartment, the TGN. CERT‐mediated transfer of ceramide likely happens at the TGN, and thus, SMS1 depends on ceramide delivered by CERT unlike GCS.
To sum up, we identify a novel molecular pathway regulated by GRASP55 that compartmentalizes specific glycosylation enzymes and by this action modulates the competition between reactions to achieve a specific cellular glycan profile.
Materials and Methods
Reagents
All reagents and chemicals were molecular biology grade. Methanol (Cat # 9093) and chloroform (Cat # 9180) were purchased from JT Baker, USA. Silica‐gel high‐performance TLC (HPTLC) plates (Cat #1055830001) were purchased from Merck, Germany. Fatty acid‐free Bovine Serum Albumin (Cat #A8806), HA Peptide (Cat #I2149), and Brefeldin A (Cat #B7651) were purchased from Sigma‐Aldrich, Germany. Protein A Sepharose CL‐4B (Cat #17‐0780‐01) was purchased from GE Healthcare Life Sciences, USA. Anti‐HA magnetic beads (Cat #88836) were purchased from ThermoFisher Scientific, USA. Sphingosine, [3‐3H]‐, D‐erythro > 97% (Cat #NET1072050UC) was purchased from PerkinElmer, USA. Lipofectamine 2000 (Cat #11668027) and Lipofectamine LTX with PLUS (Cat #15338100) were purchased from ThermoFisher Scientific, USA. TransIT‐LT1 (Cat #MIR 2305) was purchased from Mirus, USA. RPMI 1640 (Cat #21875), DMEM (Cat #41965), DMEM/F‐12 (Cat #11320033), and FBS (Cat #10437036) were purchased from Gibco/ThermoFisher Scientific, USA. BODIPY™ FL C5‐Ceramide (Cat #D3521) was purchased from ThermoFisher Scientific, USA. Protein A gold 15 nm and Protein A gold 10 nm were acquired from Cell Microscopy Core, UMC Utrecht. Biotin (Cat #29129) was purchased from Pierce, USA. Bacterial strains E. coli (DH5α) (Cat #18265017) and E. coli (BL‐21‐DE3) (Cat #C600003) were purchased from Thermo Fisher Scientific, USA. All the siRNAs and qPCR primers indicated in Appendix Tables S3 and S6 respectively were purchased from Sigma‐Aldrich, Germany.
Cell lines
HeLa‐M (human cervical cancer cells, female origin) was a kind gift from Prof. Paul Lehner, University of Cambridge. Wild‐type and GRASP55 knockout Human Fibroblasts (WI‐26) were a kind gift from Markus Plomann, Institute for Biochemistry, University of Cologne. TALEN LCS‐KO HeLa cell line (human cervical cancer cells, female origin) was kind gift from Kentaro Hanada, National Institute of Infectious Diseases, Japan. HeLa‐M and GRASP55 KO HeLa cell lines (see below) were cultured in RPMI‐1640 supplemented with 10% FCS. TALEN LCS‐KO cell lines were cultured in DMEM supplemented with 10% FCS. Wild‐type and GRASP55 knockout Human Fibroblasts (WI‐26) were cultured in DMEM: Nutrient Mixture F‐12 (DMEM/F‐12) supplemented with 10% FCS. All media were supplemented with 100 U/ml penicillin/streptomycin and 2 mM l‐glutamine. All cells were grown in controlled atmosphere (5% CO2 and 95% air) at 37°C. Mycoplasma contamination was not observed in cell cultures as observed by DAPI staining. Cell cultures between 3 and 15 passages were used for the experiments, and the cells were cultured to 80% confluence for the experiments unless indicated otherwise.
Generation of GRASP55 Knockout cell lines by CRISPR‐CAS9
To generate HeLa‐M cell lines in which GRASP55 expression was abolished, we performed genome editing using CRISPR/Cas9 system. We obtained a pool of three plasmids each encoding guide RNA (gRNA) sequence designed to target GRASP55 coding sequence and pSpCas9 ribonuclease (Cat # SC‐401106; Santa Cruz Biotechnology, Inc.). The list of gRNA sequences are reported in Appendix Table S1. These plasmids also encoded EGFP allowing positive selection of transfected cells. Hela‐M cells were transfected with pooled plasmid for 48 h, EGFP‐positive cells were isolated by FACS, and single cells were sorted into each well of 96 well plates. The single cells were maintained in optimal culture conditions for 10 days, by replenishing fresh media every 48 h. After 10 days, colonies formed from single cells were trypsinized and moved to 48 well plates and expanded to 6 well plates. Clones were collected, and protein lysates were subjected to SDS–PAGE analysis and Western blotting analysis using GRASP55‐specific antibody to assess the presence of GRASP55 protein.
Generation of expression constructs
GCS‐HA was generated by inserting 9aa HA‐ tag into the C‐terminal cytoplasmic tail of GCS between the two indicated Gly residues DPTISWRTGRYRLRCGGTAEEILDV. The following oligonucleotide primers used:
F:5′GCTGGAGAACTGGTCGCTACAGATTACGCTGTGGGTACCCATACGATGTTCCAGATTACGCTGGTACAGCAGAGGAAATCCTAGATGTATGATAACTCG‐3′R:5′CGAGTTATCATACATCTAGGATTTCCTCTGCTGTACCAGCGTAATCTGGAACATCGTATGGGTACCCACAGCGTAATCTGTAGCGACCAGTTCTCCAGC‐3′
GCS Δ3C (del of three C‐terminal amino acids) was obtained by PCR‐amplifying GCS with the following primers to introduce a stop codon after the isoleucine residue at position −4 (DPTISWRTGRYRLRCGGTAEEILDV):
F: 5′‐CATCGCGGATCCATGGCGCTGCTGGACCTG‐3′ and
R: 5′‐GATCCGCTCGAGTTATCAGATTTCCTCTGCTGTACCAGCGTAATC‐3′
The PCR‐generated fragment for both constructs was digested with EcoRI and XhoI (New England Biolabs, USA) and cloned into pcDNA4b‐3xHA expression vector. LCS9A Mutant (mutation of 9 amino acids to alanine in cytosolic tail of LCS) was generated by PCR amplification of LCS‐WT‐RUSH with following primers to introduce alanine into 9 amino acids (MAAAAAAAAAPRRSLLA):
F: 5′‐CACAACCCGGGAGGCGCGCCATGGCAGCAGCAGCAGCAGCAGCAGCAGCA‐3′
R: 5′‐ GCGAGCAGCGAGCGGCGCGGTGCTGCTGCTGCTGCTGCTGCTGCTGC −3′. The PCR‐generated megaprimer was then annealed into LCS‐WT‐RUSH at 66°C and extension at 72°C for 25 cycles.
Plasmids and siRNA transfection
HeLa‐M cells, Wi‐26 cells, LCS‐KO cells, and GRASP55 KO cells were transfected with plasmid vectors using TransIT‐LT1 or Lipofectamine LTX reagents. A list of plasmids used in this study can be found in Appendix Table S2. Knockdown experiments for Hela‐M cells were carried out using a pool of 4 siRNAs or 2 siRNAs using Lipofectamine 2000 according to manufacturer’s instructions (used at concentration of 100nM for the target gene). Expression or knockdown efficiencies (> 85%) were checked after every experiment either by indirect immunofluorescence or immunoblotting or by qPCR analysis. A list of siRNA sequences used in this study can be found in Appendix Table S3.
Cell lysis, Western blotting, and analysis
Cells were washed three times with ice‐cold PBS and lysed immediately at 4°C in RIPA lysis buffer (0.1% Triton X‐100, 20 mM Tris‐HCl, pH 8.0, 0.1% SDS, 0.05% sodium deoxycholate, 150 mM NaCl, 10 mM Na3VO4, 40 mM β‐glycerophosphate, and 10 mM NaF) and complete protease/phosphates inhibitors (Roche). Cell lysates were clarified at 20,000 g for 10 min at 4°C to eliminate detergent‐insoluble pellet. To visualize GCS on SDS–PAGE, the protein lysates prepared as described above were treated with 250 mM of DTT in sample buffer (62.5 mM Tris‐HCL, pH 6.8, 2% SDS, 10% glycerol, 0.001% bromophenol, and 125 mM dithiothreitol) at 37°C for 30 min. The lysate was immediately processed for SDS–PAGE and immunoblotting with antibodies. A complete list of primary and secondary antibodies are given in Appendix Table S4. The Western blots were then exposed to X‐ray films, and exposure time was varied to obtain optimal signal.
Immunoprecipitation and peptide pull down assay
Total lysates were prepared using IP lysis buffer (150 mM NaCl, 25 mM Tris‐HCl pH 7.5, 1% Triton X, 10 mM Na3VO4, 40 mM β‐glycerophosphate, 10 mM NaF, and protease cocktail inhibitor from Roche). The protein concentrations were quantified using BCA Protein Assay kit (Pierce). 1mg of protein was used for precipitation with antibodies conjugated to either Protein A sepharose or magnetic dynabeads or to monomeric avidin beads (Pierce) in case of cytosolic peptides, and incubated at 4°C overnight. The beads were then washed 5 times in IP lysis buffer, and bound proteins were subjected to SDS–PAGE and immunoblotted. The list of cytosolic peptides used in this study are reported in Appendix Table S5.
Reconstitution of COPI vesicle formation
A two‐stage incubation system was performed essentially as previously described (Yang et al, 2005). In brief, the first stage involved incubating prewashed (3 M KCl) Golgi membrane (0.4 mg/ml) with coatomer (6 μg/ml), ARF1 (6 μg/ml), and 2 mM GTP in 500 μl of assay buffer (25 mM Hepes‐KOH, pH 7.2, 50 mM KCl, 2.5 mM Mg(OAc)2, 1 mg/ml soybean trypsin inhibitor, and 200 mM sucrose) for 15 min at 37°C. Afterward, the Golgi membrane was pelleted (12,000× g at 4°C for 10 min) and then resuspended in 100 μl of assay buffer for the second‐stage incubation, which had ARFGAP1 (6 μg/ml) and BARS (6 μg/ml) added for 10 min at 37°C. Samples were then centrifuged for 10 min at 12,000 g and 4°C, with the pellet fraction containing the Golgi membrane and the supernatant fraction containing COPI vesicles.
Protein purification
Recombinant proteins were induced to express with 0.3 mM isopropyl β‐D‐thiogalactoside in BL21 (DE3) competent bacterial cells for 16 h at 22°C. Cells were harvested at 7,519 g for 30 min at 4°C, lysed in lysis buffer (cold PBS with 1 mM DTT, 1% triton X‐100, phosphatase, and protease inhibitors), and purified with Glutathione Sepharose 4B beads (GE Healthcare, USA). Recombinant GST‐tagged proteins were eluted in elution buffer (50 mM Tris, 100 mM NaCl, 50 mM reduced glutathione, and pH 8.0); purified recombinant proteins have been concentrated and the buffer has been exchanged by using Vivaspin TURBO 4 filters (Sartorius, UK) at 4°C and 4,000× g. The recombinant purified proteins were quantified by Bradford assay and were assessed for contaminants by SDS–PAGE.
Isothermal titration calorimetry (ITC)
ITC experiments were performed in a buffer containing 300 mM NaCl, 10 mM Bicine pH 8.5, and 1 mM DTT. Biotinylated peptides were synthesized and delivered as lyophilized powder with a biotin moiety located at the N or C‐terminus (Charite Universitaetsmedizin Berlin, Germany). The peptides were dissolved in buffer, centrifuged at 14,000× g for 10 min, and only the supernatant was used. The dissolved peptide concentrations were calculated based upon their absorbance at 280 nm and their corresponding molar extinction coefficient. Experiments consisted of titrations of 20 injections of 2 μl of titrant (peptides) into the cell containing GRASP domain protein at a 25‐fold lower concentration. Typical concentrations for the titrant were around 2.5 mM for experiments depending on the affinity. Experiments were performed at 25°C and a stirring speed of 1,000 rpm on a MicroCal PEAQ‐ITC (Malvern Panalytic). All data were processed using MicroCal PEAQ‐ITC Analysis Software and fit to a one‐site binding model after background buffer subtraction.
GRASP55‐GCS peptide modeling
Model of GCS peptide was first built using backbone conformation of Golgin45 peptide as a template and fit into the same cleft of GRASP55, where Golgin45 peptide is located using COOT (Emsley & Cowtan, 2004). This initial complex structure was further refined by the Docking2 module of Rosetta protocols through local docking search (Lyskov & Gray, 2008; Chaudhury et al, 2011; Lyskov et al, 2013). The lowest energy model generated by this protocol was analyzed to probe the protein:peptide interaction. PRODIGY software was used to calculate the protein–protein binding energy (Vangone & Bonvin, 2015; Xue et al, 2016).
Immunofluorescence and confocal microscopy
Indirect Immunofluorescence was performed as follows: cells were grown on 24 mm coverslips, washed with PBS, and fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, USA) for 30 min at room temperature (RT). The cells were then permeabilized and blocked in blocking buffer (0.05% saponin and 0.5% BSA in PBS) for 30 min at RT followed by incubation with specific primary antibodies (see Appendix Table S4) for 1 h at RT and washed with PBS. Cells were subsequently labeled with appropriate Alexa Fluor‐conjugated secondary antibodies (Appendix Table S4). The coverslips were mounted using mowiol, and images are acquired using confocal microscope Zeiss LSM700.
Line scan analysis
The images were acquired with a pinhole set to 1 airy unit and under non‐saturation conditions using a 63× objective (1.4 NA). Images were 8 bit with dimensions of 512 × 512 pixels, and each pixel corresponded to an area of 132 × 132 nm2. The line scan analysis was performed as described previously (Rizzo et al, 2013) using the Zen software system (Carl Zeiss). In brief, images of stacks stained for GM130, TGN46, and enzyme of interest tagged with HA were acquired as described earlier. Only cells with a moderate level of expression were considered for the analysis. Golgi stacks with clearly separated GM130‐stained and TGN46‐stained zones were identified and used for the analysis. A line was drawn in the middle of the stacks along the cis‐trans direction, and the fluorescence intensity of each stained marker along this line was plotted. At least 30 stacks were examined per treatment, and a representative data are shown for analysis. The normalization of the distances was carried out by considering the start of the GM130 peak as 0, and the end of the TGN peak as 1. The images of Golgi stacks were processed using the “Image with Zoom” function of Metamorph 7.7.3.0 (Universal Imaging), for presentation.
Cell profiler analysis
Control and GRASP55 KO cells were stained ShTxB‐Cy3 and ChTxB‐AlexaFluor 488 and images were acquired as described above using a 20× objective (NA 0.5). Images were 8 bit with dimensions of 512 × 512 pixels, and each pixel corresponded to an area of 623 × 623 nm2. For quantification experiments, 10–15 random fields were imaged with the same microscope settings (i.e., laser power and detector gain). The integrated intensity of fluorescence of each cell was calculated for each channel after the cells were segmented by Cell Profiler (Carpenter et al, 2006). Since only a fraction of the cells were positive for staining, the normalized intensity analysis was performed by taking top 10% of total cells in all the conditions and normalized on the background intensity.
Flow cytometry analysis
Control and GRASP55 KO cells were subjected to trypsin digestion, fixed with 4% paraformaldehyde, washed, and resuspended with PBS. Cells were incubated with bacterial toxins for 1 h at 4°C. Then, cells were extensively washed with PBS and incubated with fluorescence‐labeled secondary antibodies when required or directly analyzed by BD FACS ARIAIII cell sorter (BD Biosciences). Cells incubated with secondary antibody alone, or unlabeled cells, were used as a negative control. The cell surface expression of GSLs of selected cells was further analyzed in the gated region by BD FACS ARIAIII cell sorter (BD Biosciences). Bacterial toxins used are described in (Table S4).
Electron microscopy
Ultrastructural analysis of Golgi morphology
Briefly, control and GRASP55 knockout cells were grown in 35 mm plastic dishes to 80% confluence. Cells were then fixed with 1% Glutaraldehyde (Electron Microscopy Sciences) in 0.2 M HEPES pH 7.3 at RT for 1 h. The fixative was replaced with 1% BSA in PBS, and cells were carefully detached using a plastic cell scraper, collected into microfuge tubes, and centrifuged to obtain the pellet. All samples were then washed three times in 0.2 M HEPES pH 7.3 and post‐fixed 30 min in 1% Osmium Tetroxide in the dark at 4°C in the same buffer. They were then washed three times in distilled water and post‐fixed 25 min in 1% Osmium Tetroxide and 1.5% Potassium Ferrocyanide in the dark at room temperature in HEPES 0.2 M pH 7.3. After washing three times in distilled water, they were stained with 0.5% uranyl acetate over night at 4°C. The pellets were dehydrated in graded steps of ethanol (50, 70, 90, and 100%), 2 times with 100% of acetone, and embedded into Epon. Thin sections (60 nm) were cut on a Leica UC7 ultramicrotome and examined with 120 kV Philips Tecnai 12 Biotwin electron microscope (FEI, Eindhoven, The Netherlands) using a VELETA digital camera.
Cryo‐immuno EM
Control or GRASP55 Knockout cells were transfected using indicated HA‐tagged construct for 16 h. The cells were then fixed with 2% formaldehyde and 0.2% glutaraldehyde in PHEM buffer (0.1 M) pelleted by centrifugation, embedded in 12% gelatin, cooled on ice, and cut into 1‐mm3 cubes at 4°C. The cubes were immersed in 2.3 M sucrose at 4°C overnight and then frozen in liquid nitrogen. Fifty‐nanometer sections were cut with a diamond knife on a UC7 Leica cryo‐ultramicrotome. The sections were picked up in a mix of 2% methyl cellulose and 2.3 M sucrose (1:1), as previously described (Pulvirenti et al, 2008), and collected on grids covered with Formvar carbon supporting film (Electron Microscopy Sciences, PA, USA). The grids were first incubated with the rabbit anti‐GM130 and/or mouse anti‐HA polyclonal antibodies and then incubated with different sizes of Protein A gold (10 and 15 nm) to reveal antigen staining. After labeling, the sections were treated with 1% glutaraldehyde and embedded in methyl cellulose uranyl acetate for 10 min on ice. The excess of methyl cellulose uranyl acetate was removed, and the sections were dried at room temperature before their analysis at 120 kV in a Philips Tecnai 12 Biotwin electron microscope (FEI, Eindhoven, The Netherlands) using a VELETA digital camera. The polarity of the Golgi stacks was defined by compositional (cis‐Golgi marker GM130) parameter. For quantitation (performed with iTEM image acquisition software), cis, medial, or trans‐Golgi was defined as previously described (Rizzo et al, 2013). TGN was defined as the area in front of the trans cisterna up to a distance that equals the thickness of the Golgi stack. Vesicles were round profiles of 50–80 nm in diameter, present within 200 nm from the rims of the stack. The distribution of enzyme in the Golgi stack was expressed as the fraction of gold particles in each cisterna of the Golgi stack and TGN.
Lipid analysis
HPLC‐Mass spectrometry
Sphingolipids of control and GRASP55 KO cells were analyzed by liquid chromatography–tandem mass spectrometry (LC‐MS/MS) as described earlier (Bielawski et al, 2006). Briefly, cells were washed twice with ice‐cold PBS, and lipids were extracted with 2 ml ethyl acetate/isopropyl alcohol/water (60:30:10%, v/v/v) solvent mixture (Bielawski et al, 2006). The lipid extracts were analyzed with a Quantum Ultra triple quadrupole mass spectrometer connected to an Accela HPLC and Accela autosampler using a solvent gradient at the Lipidomics Core at Stony Brook University. Ceramides were identified through MRM analysis with soft fragmentation. Calibration curves were generated for each lipid and used for quantitative analysis of lipids in the samples. Inorganic phosphate (Pi) released from total phospholipids was measured by using a colorimetric method. Briefly, following a lipid extraction by using the Bligh and Dyer method, samples were dried under a stream of nitrogen gas. Then, a mixture of sulfuric and hydrochloric acid was added to the sample to ash the organic content. Samples were heated overnight at 160°C. Then, water, ammonium molybdate, and ascorbic acid were subsequently added to samples and incubated for 30 min at 45°C. The absorbance was measured at 600 nm by using a Spectramax M5 plate reader. A calibration curve was created and utilized to quantify the inorganic lipid content for each sample and used as normalization for relative quantification of sphingolipids.
Radioactive pulse‐chase assay to monitor SL biosynthesis
Radioactive pulse‐chase assay was performed as described earlier (D'Angelo et al, 2013). Briefly, HeLa‐M cells or human fibroblasts wi‐26 or GRASP55 KO cells were pulse labeled for 2 h with [3H]‐sphingosine (final concentration of 0.1 μCi/ml; Perkin Elmer) in serum‐free DMEM containing 1% fatty acid‐free BSA followed by a chase for indicated times in complete media. Lipids were then extracted from the cells, resuspended in chloroform, spotted onto silica‐gel high‐performance TLC (HPTLC) plates (Merck, Germany), resolved using a mixture of chloroform, methanol, and water (65:25:4 v/v/v) and quantified using GINA® (Raytest, Germany) software analysis.
MALDI‐MS
Total lipid extracts were prepared using a standard MTBE protocol followed by a methylamine treatment for sphingo‐ and glycosphingolipids analysis by mass spectrometry. Briefly, cell pellet was resuspended in 100 μl H2O. 360 μl methanol and 1.2 ml of MTBE were added and samples were placed for 10 min on a vortex at 4°C followed by incubation for 1 h at room temperature on a shaker. Phase separation was induced by the addition of 200 μl of H2O. After 10 min at room temperature, samples were centrifuged at 1,000 g for 10 min. The upper (organic) phase was transferred into a glass tube, and the lower phase was re‐extracted with 400 μl artificial upper phase [MTBE/methanol/H2O (10:3:1.5, v/v/v)]. The combined organic phases were dried in a vacuum concentrator. Lipids were then resuspended in 500 μl of CHCl3 and divided into two aliquots for a further methylamine treatment. 500 µl of freshly prepared monomethylamine reagent [methylamine/H2O/n‐butanol/methanol (5:3:1:4, (v/v/v/v)] was added to the dried lipid extract and then incubated at 53°C for 1 h in a water bath. Lipids were cooled to room temperature and then dried. The dried lipid extract was then extracted by n‐butanol extraction using 300 μl water‐saturated n‐butanol and 150 μl H2O. The organic phase was collected, and the aqueous phase was re‐extracted twice with 300 μl water‐saturated n‐butanol. The organic phases were pooled and dried in a vacuum concentrator. Lipids were then resuspended in 500 µl of CHCl3 and analyzed by MALDI‐MS. 30 mg/ml 2,5‐DHB was freshly prepared in acetonitrile/water solution (50:50 v/v) with 0.1% TFA. An equivalent volume of sample solution (50 µl) was then mixed with matrix before deposition on the MALDI target. All mass spectrometry analysis for the identification of lipids (m/z 600–1,800) was obtained using an Applied Biosystems 4800 MALDI‐TOF/TOF mass spectrometer equipped with a 200 Hz tripled frequency Nd:YAG pulsed laser with 355 nm wavelength. Measurements were performed in positive ion reflection mode at an accelerating potential of 20 kV. Each mass spectrum was obtained by applying a laser energy of 4,600 watts/cm2, averaging 4,000 single laser shots/spectrum.
FRAP assay
Control and GRASP55 (#2) knockout cells were plated on 35‐mm glass bottom microwell dishes (MatTech, USA) and transfected with CERT‐YFP (1 μg) for 16 h. FRAP was performed using Zeiss LSM 710 confocal microscope equipped with an environmental control system set to 37°C and 5% CO2. FRAP experiments were performed by bleaching CERT‐YFP in the Golgi area (50 bleaching iterations) followed by imaging every 7.5 s for 4 min (2% laser power). Recovery of fluorescence was calculated as ratio of Golgi intensity and that of total cell after background correction.
Ceramide transport assay
Control and GRASP55 knockout cells were grown on coverslips to 80% confluence. Cells were washed with DMEM‐HEPES containing 10% FCS for three times and incubated with BODIPY ceramide (5 µM) in fatty acid‐free BSA for 30 min on ice. After incubation, cells were washed with DMEM‐HEPES for three times and followed by imaging. The laser conditions used were 488 nm (excitation) and 620 nm (emission), and fluorescence was recorded every 2 min for a period of 20 min. The fluorescence of bodipy Ceramide in the Golgi was determined followed by normalization to the maximum.
RNA extraction and Real‐Time PCR
Total RNA was extracted from HeLa‐M cells (Control and GRASP55 Knockout) using the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. The yield and the integrity of RNA were determined by spectrophotometer NanoDrop 2000c (Thermo Scientific) and by TAE agarose gel electrophoresis, respectively. RNA (1 μg) was reverse transcribed using QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions and subjected to qPCR with gene‐specific primers (Appendix Table S6) in the presence of LightCycler®480 SYBR Green I Master Mix (Roche, Switzerland) on a LightCycler®480 II detection system (Roche, Switzerland). Analyses were carried out on biological triplicate samples for each experiment, and they were processed separately. The thermal profile consisted of 10 min at 95°C pre‐incubation, and 40 cycles at 10 s at 95°C, 10 s at 60°C, and 10 s at 72°C. The qPCR data were normalized to the average of the reference gene human hypoxanthine–guanine phosphoribosyltransferase 1 (HPRT1). The fold changes in the relative quantifications were calculated according to the ΔΔCt method. All the commercial kits used are reported in (Appendix Table S7).
Experimental conditions for RUSH experiments
LCS‐KO cells were transiently transfected with LCS or LCS9A constructs at 37°C in absence of biotin for 16 h, to localize the proteins to endoplasmic reticulum (ER). Their ER exit was promoted by the addition of biotin (40 µM) for 6 h. For pulse‐chase assay experiments (Fig 4C), biotin was replaced every 3 h and maintained throughout chase period for a total of 24 h.
Statistics
Error bars correspond to either standard deviation (SD) or standard error of mean (SEM) as indicated in figure legends. All the statistical evaluations were done using GraphPad Prism built‐in tests (unpaired two‐tailed Student’s t‐tests), and as indicated, significance values are all marked as follows *P < 0.05, **P < 0.01, and ***P < 0.001 (ns, not significant). All the measurements reported are from distinct samples.
Author contributions
PP contributed to development of the idea, conducted most of the experiments, analysis and interpretation of data, and contributed to writing the manuscript; IA conducted the experiments on peptide binding using purified proteins; MP was involved in labeling and acquisition of EM and confocal microscopy images; RR assisted in acquisition and analyses of electron micrographs; DR conducted mRNA analysis; GT was involved in EM sample preparation and acquisition of images; LC conducted MALDI‐MS and lipidomics measurements; CG performed the ITC experiments; GV assisted PP in morphological analysis of Golgi; ND was involved in cloning and construct preparation; J‐SY performed vesicle budding assay and was guided in this by VWH; PH was involved in synthesis of biotinylated peptides; JN provided the WT and GRASP55 KO fibroblasts, performed the protein–protein interaction studies, and was guided by MP; TN, J‐II, MJH‐C, LMO, and YAH contributed to MS analysis of lipids; AB performed in silico modeling of GRASP55‐GCS peptide interaction and was guided by MT; AL contributed to development of the idea of how Golgi organization may play role in regulating glycosylation and the mode of action of GRASP55 in regulating enzyme compartmentalization, critical analysis of the data throughout the entire project and also provided inputs in manuscript preparation. GDA contributed to setting up the system to monitor SL biosynthesis, provided important inputs on the data to understand and interpret changes in lipid biosynthesis, and also provided critical inputs in manuscript preparation. SP developed the idea, designed and supervised the entire project, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Antonella De Matteis for valuable discussions, Nina Dathan for help with cloning and construct preparation, and Francesco Russo for assistance with statistical analysis. We thank the Bioimaging Facility of the Institute of Biochemistry and Cell Biology and the joint FACS Facility of the Institute of Genetics and Biophysics (IGB‐CNR, Naples) and IBBC‐CNR for access to technologies and Pasquale Barba for assistance with FACS. PP thanks Samba Siva Rao. We thank Dr. Bruno Correia, EPFL, Switzerland, for help with the analysis of protein–protein interaction studies. We thank Drs. Ahamarshan, Corda, Colanzi, Grimaldi, Jung, Krishnamoorthy, Polishchuk, Qadri, and Roy for critical comments on the manuscript. We thank MEDINTECH, the Italian Node of Euro‐Bioimaging (Preparatory Phase II –INFRADEV), Italian Cystic Fibrosis Research Foundation (FFC #6‐2019), POR Campania project 2014‐2020 (C.I.R.O.), and CTC project for financial support. R.R. acknowledges financial support from Fondazione Italiana per la Ricerca sul Cancro (FIRC Fellowship 15111). A.L. acknowledges financial support from the, AIRC (Projects IG 15767 and IG 20786), TERABIO, PON‐IMPARA, and S.A.T.I.N. G.D.A. acknowledges financial support from EPFL institutional fund, Kristian Gerhard Jebsen Foundation, and from the Swiss National Science Foundation (SNSF) (grant number, 310030_184926).
The EMBO Journal (2021) 40: e107766
Data availability
Source data are provided with the manuscript, and if not, they are available from the corresponding author upon reasonable request. Targeted lipidomics data from this study are available at Metabolomics Workbench database (https://www.metabolomicsworkbench.org/) with the following ID=ST001877.
References
- Akintayo A, Stanley P (2019) Roles for Golgi glycans in oogenesis and spermatogenesis. Front Cell Dev Biol 7: 98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Bannykh SI, Rowe T, Balch WE (1995) Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol 131: 875–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Preisinger C, Kopajtich R, Korner R (2001) Golgi matrix proteins interact with p24 cargo receptors and aid their efficient retention in the Golgi apparatus. J Cell Biol 155: 885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beznoussenko GV, Parashuraman S, Rizzo R, Polishchuk R, Martella O, Di Giandomenico D, Fusella A, Spaar A, Sallese M, Capestrano MG et al (2014) Transport of soluble proteins through the Golgi occurs by diffusion via continuities across cisternae. Elife 3: e2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielawski J, Szulc ZM, Hannun YA, Bielawska A (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high‐performance liquid chromatography‐tandem mass spectrometry. Methods 39: 82–91 [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD (2007) Heparan sulphate proteoglycans fine‐tune mammalian physiology. Nature 446: 1030–1037 [DOI] [PubMed] [Google Scholar]
- Biswas A, Thattai M (2020) Promiscuity and specificity of eukaryotic glycosyltransferases. Biochem Soc Trans 48: 891–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn JB, D'Souza Z, Lupashin VV (2019) Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett 593: 2466–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J et al (2006) Cell profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang IJ, He M, Lam CT (2018) Congenital disorders of glycosylation. Ann Transl Med 6: 477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, Gray JJ (2011) Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 6: e22477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiritoiu M, Brouwers N, Turacchio G, Pirozzi M, Malhotra V (2019) GRASP55 and UPR control interleukin‐1beta aggregation and secretion. Dev Cell 49: 145–155 e4 [DOI] [PubMed] [Google Scholar]
- D’Angelo G, Uemura T, Chuang C‐C, Polishchuk E, Santoro M, Ohvo‐Rekilä H, Sato T, Di Tullio G, Varriale A, D’Auria S et al (2013) Vesicular and non‐vesicular transport feed distinct glycosylation pathways in the Golgi. Nature 501: 116–120 [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Prencipe L, Iodice L, Beznoussenko G, Savarese M, Marra P, Di Tullio G, Martire G, De Matteis MA, Bonatti S (2009) GRASP65 and GRASP55 sequentially promote the transport of C‐terminal valine‐bearing cargos to and through the Golgi complex. J Biol Chem 284: 34849–34860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy WG, Rothman JE (1985) Compartmental organization of the Golgi stack. Cell 42: 13–21 [DOI] [PubMed] [Google Scholar]
- Eckert ES, Reckmann I, Hellwig A, Rohling S, El‐Battari A, Wieland FT, Popoff V (2014) Golgi phosphoprotein 3 triggers signal‐mediated incorporation of glycosyltransferases into coatomer‐coated (COPI) vesicles. J Biol Chem 289: 31319–31329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fisher P, Spencer H, Thomas‐Oates J, Wood AJ, Ungar D (2019) Modeling Glycan processing reveals golgi‐enzyme homeostasis upon trafficking defects and cellular differentiation. Cell Rep 27: 1231–1243.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, Luini A (2011) Models for Golgi traffic: a critical assessment. Cold Spring Harb Perspect Biol 3: a005215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grond R, Veenendaal T, Duran JM, Raote I, van Es JH, Corstjens S, Delfgou L, El Haddouti B, Malhotra V, Rabouille C (2020) The function of GORASPs in Golgi apparatus organization in vivo. J Cell Biol 219: e202004191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halter D, Neumann S, van Dijk SM, Wolthoorn J, de Maziere AM, Vieira OV, Mattjus P, Klumperman J, van Meer G, Sprong H (2007) Pre‐ and post‐Golgi translocation of glucosylceramide in glycosphingolipid synthesis. J Cell Biol 179: 101–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M (2003) Molecular machinery for non‐vesicular trafficking of ceramide. Nature 426: 803–809 [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM (2018) Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol 19: 175–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Nemoto‐Sasaki Y, Matsumoto N, Hama K, Tanikawa T, Oka S, Saeki T, Kumasaka T, Koizumi T, Arai S et al (2018) Complex formation of sphingomyelin synthase 1 with glucosylceramide synthase increases sphingomyelin and decreases glucosylceramide levels. J Biol Chem 293: 17505–17522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi Y, Ito M, Hirabayashi Y (2018) Regulation of glucosylceramide synthesis by Golgi‐localized phosphoinositide. Biochem Biophys Res Commun 499: 1011–1018 [DOI] [PubMed] [Google Scholar]
- Ishii M, Suda Y, Kurokawa K, Nakano A (2016) COPI is essential for Golgi cisternal maturation and dynamics. J Cell Sci 129: 3251–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiman A, Thattai, M (2020) Golgi compartments enable controlled biomolecular assembly using promiscuous enzymes. Elife 9: e49573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvela T, Linstedt AD (2014) Isoform‐specific tethering links the Golgi ribbon to maintain compartmentalization. Mol Biol Cell 25: 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Noh SH, Piao H, Kim DH, Kim K, Cha JS, Chung WY, Cho HS, Kim JY, Lee MG (2016) Monomerization and ER relocalization of GRASP is a requisite for unconventional secretion of CFTR. Traffic 17: 733–753 [DOI] [PubMed] [Google Scholar]
- Klumperman J (2011) Architecture of the mammalian Golgi. Cold Spring Harb Perspect Biol 3: a005181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama‐Koganeya A, Nabetani T, Miura M, Hirabayashi Y (2011) Glucosylceramide synthase in the fat body controls energy metabolism in Drosophila . J Lipid Res 52: 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Mastronarde DN, McIntosh JR, Howell KE, Staehelin LA (1999) Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J Cell Biol 144: 1135–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Zheng JJ (2010) PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signal 8: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Doray B, Kornfeld S (2018) Recycling of Golgi glycosyltransferases requires direct binding to coatomer. Proc Natl Acad Sci USA 115: 8984–8989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, Chou FC, Conchuir SO, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P et al (2013) Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8: e63906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, Gray JJ (2008) The RosettaDock server for local protein‐protein docking. Nucleic Acids Res 36: W233–W238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni HJ, Giraudo CG, Daniotti JL (2002) Understanding the stepwise synthesis of glycolipids. Neurochem Res 27: 629–636 [DOI] [PubMed] [Google Scholar]
- Nairn AV, Aoki K, dela Rosa M, Porterfield M, Lim J‐M, Kulik M, Pierce JMichael, Wells L, Dalton S, Tiemeyer M et al (2012) Regulation of glycan structures in murine embryonic stem cells: combined transcript profiling of glycan‐related genes and glycan structural analysis. J Biol Chem 287: 37835–37856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn AV, York WS, Harris K, Hall EM, Pierce JM, Moremen KW (2008) Regulation of glycan structures in animal tissues: transcript profiling of glycan‐related genes. J Biol Chem 283: 17298–17313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Luini A (2010) Passage through the Golgi. Curr Opin Cell Biol 22: 471–478 [DOI] [PubMed] [Google Scholar]
- Oprins A, Duden R, Kreis TE, Geuze HJ, Slot JW (1993) Beta‐COP localizes mainly to the cis‐Golgi side in exocrine pancreas. J Cell Biol 121: 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanikou E, Day KJ, Austin J, Glick BS (2015) COPI selectively drives maturation of the early Golgi. Elife 4: e13232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parashuraman S, D'Angelo G (2019) Visualizing sphingolipid biosynthesis in cells. Chem Phys Lipids 218: 103–111 [DOI] [PubMed] [Google Scholar]
- Popoff V, Adolf F, Brugger B, Wieland F (2011) COPI budding within the Golgi stack. Cold Spring Harb Perspect Biol 3: a005231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothukuchi P, Agliarulo I, Russo D, Rizzo R, Russo F, Parashuraman S (2019) Translation of genome to glycome: role of the Golgi apparatus. FEBS Lett 593: 2390–2411 [DOI] [PubMed] [Google Scholar]
- Pulvirenti T, Giannotta M, Capestrano M, Capitani M, Pisanu A, Polishchuk RS, San Pietro E, Beznoussenko GV, Mironov AA, Turacchio G et al (2008) A traffic‐activated Golgi‐based signalling circuit coordinates the secretory pathway. Nat Cell Biol 10: 912–922 [DOI] [PubMed] [Google Scholar]
- Rabouille C, Klumperman J (2005) Opinion: The maturing role of COPI vesicles in intra‐Golgi transport. Nat Rev Mol Cell Biol 6: 812–817 [DOI] [PubMed] [Google Scholar]
- Rizzo R, Parashuraman S, Mirabelli P, Puri C, Lucocq J, Luini A (2013) The dynamics of engineered resident proteins in the mammalian Golgi complex relies on cisternal maturation. J Cell Biol 201: 1027–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo R, Russo D, Kurokawa K, Sahu P, Lombardi B, Supino D, Zhukovsky M, Vocat A, Pothukuchi P, Kunnathully V et al (2019) The Glyco‐enzyme adaptor GOLPH3 links intra‐Golgi transport dynamics to glycosylation patterns and cell proliferation. bioarxiv 10.1101/870477 [PREPRINT] [DOI] [Google Scholar]
- Rizzo R, Russo D, Kurokawa K, Sahu P, Lombardi B, Supino D, Zhukovsky MA, Vocat A, Pothukuchi P, Kunnathully V et al (2021) Golgi maturation‐dependent glycoenzyme recycling controls glycosphingolipid biosynthesis and cell growth via GOLPH3. EMBO J 40: e107238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd P, Karlsson NG, Khoo KH, Packer NH (2015) Glycomics and glycoproteomics. In Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH (eds), Essentials of glycobiology, pp 653–666. New York: Cold Spring Harbor; [Google Scholar]
- Russo D, Capolupo L, Loomba JS, Sticco L, D'Angelo G (2018a) Glycosphingolipid metabolism in cell fate specification. J Cell Sci 131: jcs219204 [DOI] [PubMed] [Google Scholar]
- Russo D, Della Ragione F, Rizzo R, Sugiyama E, Scalabri F, Hori K, Capasso S, Fioriniello Sticco LS, De Gregorio R, Granata I et al (2018b) Glycosphingolipid metabolic reprogramming drives neural differentiation. EMBO J 37: e97674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryczko MC, Pawling J, Chen R, Abdel Rahman AM, Yau K, Copeland JK, Zhang C, Surendra A, Guttman DS, Figeys D et al (2016) Metabolic reprogramming by hexosamine biosynthetic and Golgi N‐Glycan branching pathways. Sci Rep 6: 23043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Watson R, Giannakou ME, Clarke M, Warren G, Barr FA (1999) GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell‐free system. EMBO J 18: 4949–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P (2011) Golgi glycosylation. Cold Spring Harb Perspect Biol 3: a005199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L, Chen L, Banfield DK (2012) A conserved N‐terminal arginine‐motif in GOLPH3‐family proteins mediates binding to coatomer. Traffic 13: 1496–1507 [DOI] [PubMed] [Google Scholar]
- Tu L, Tai WC, Chen L, Banfield DK (2008) Signal‐mediated dynamic retention of glycosyltransferases in the Golgi. Science 321: 404–407 [DOI] [PubMed] [Google Scholar]
- Vangone A, Bonvin AM (2015) Contacts‐based prediction of binding affinity in protein‐protein complexes. Elife 4: e07454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A (1998) Factors controlling the glycosylation potential of the Golgi apparatus. Trends Cell Biol 8: 34–40 [DOI] [PubMed] [Google Scholar]
- Varki A (2017) Biological roles of glycans. Glycobiology 27: 3–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Kornfeld S (2015) Historical background and overview. In Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH (eds), Essentials of glycobiology, pp 1–18. New York: Cold Spring Harbor; [PubMed] [Google Scholar]
- Vinke FP, Grieve AG, Rabouille C (2011) The multiple facets of the Golgi reassembly stacking proteins. Biochem J 433: 423–433 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wen L, Ma X, Chen Z, Yu Y, Zhu J, Wang Y, Liu Z, Liu H, Wu D et al (2012) High expression of lactotriaosylceramide, a differentiation‐associated glycosphingolipid, in the bone marrow of acute myeloid leukemia patients. Glycobiology 22: 930–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Munro S (2014) Membrane trafficking. The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled‐coil proteins. Science 346: 1256898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Wang Y (2010) GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J Cell Biol 188: 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Zhang X, Nix DB, Katoh T, Aoki K, Tiemeyer M, Wang Y (2013) Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat Commun 4: 1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A (2016) PRODIGY: a web server for predicting the binding affinity of protein‐protein complexes. Bioinformatics 32: 3676–3678 [DOI] [PubMed] [Google Scholar]
- Yamaji T, Hanada K (2014) Establishment of HeLa cell mutants deficient in sphingolipid‐related genes using TALENs. PLoS One 9: e88124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lee SY, Gao M, Bourgoin S, Randazzo PA, Premont RT, Hsu VW (2002) ARFGAP1 promotes the formation of COPI vesicles, suggesting function as a component of the coat. J Cell Biol 159: 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lee SY, Spano S, Gad H, Zhang L, Nie Z, Bonazzi M, Corda D, Luini A, Hsu VW (2005) A role for BARS at the fission step of COPI vesicle formation from Golgi membrane. EMBO J 24: 4133–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CJ, Rosenwald AG, Willingham MC, Skuntz S, Clark J, Kahn RA (1994) Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J Cell Biol 124: 289–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Seemann J (2021) Rapid degradation of GRASP55 and GRASP65 reveals their immediate impact on the Golgi structure. J Cell Biol 220: e202007052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Li B, Huang X, Morelli X, Shi N (2017) Structural basis for the interaction between Golgi reassembly‐stacking protein GRASP55 and Golgin45. J Biol Chem 292: 2956–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]