

Abstract
Proteostasis, the balance of protein synthesis, folding and degradation, is essential to maintain cellular function and viability, and the many known intracellular chaperones are recognized as playing key roles in sustaining life. In contrast, the identity of constitutively secreted extracellular chaperones (ECs) and their physiological roles in extracellular proteostasis is less completely understood. We designed and implemented a novel strategy, based on the well‐known propensity of chaperones to bind to regions of hydrophobicity exposed on misfolding proteins, to discover new ECs present in human blood. We used a destabilized protein that misfolds at 37°C as “bait” to bind to potential ECs in human serum and captured the complexes formed on magnetic beads. Proteins eluted from the beads were identified by mass spectrometry and a group of seven abundant serum proteins was selected for in vitro analysis of chaperone activity. Five of these proteins were shown to specifically inhibit protein aggregation. Vitronectin and plasminogen activator‐3 inhibited both the in vitro aggregation of the Alzheimer's β peptide (Aβ1–42) to form fibrillar amyloid, and the aggregation of citrate synthase (CS) to form unstructured (amorphous) aggregates. In contrast, prothrombin, C1r, and C1s inhibited the aggregation of Aβ1–42 but did not inhibit CS aggregation. This study thus identified five novel and abundant putative ECs which may play important roles in the maintenance of extracellular proteostasis, and which apparently have differing abilities to inhibit the amorphous and amyloid‐forming protein aggregation pathways.
Keywords: C1r, C1s, extracellular chaperones, plasminogen activator inhibitor 3, proteostasis, prothrombin, vitronectin
Abbreviations
- A2AP
Alpha‐2‐antiplasmin
- Aβ1–42
Alzheimer's beta peptide 1–42
- AT3
antithrombin III
- bisANS
4, 4′‐dianilino‐1,1′‐binaphthyl‐5, 5′‐ disulfonic acid
- BSA
bovine serum albumin
- CLIC1
chloride intracellular channel protein 1
- CLU
clusterin
- C1r
complement component 1r
- C1s
complement component 1s
- PBS
phosphate‐buffered saline
- PAI‐3
plasminogen activator inhibitor‐3
- PT
prothrombin
- SOD
superoxide dismutatase 1
- ThioT
thioflavin T
- VTN
vitronectin
1. INTRODUCTION
Proteostasis refers to the ongoing regulation of protein synthesis, folding, localization, and clearance (degradation), both inside and outside cells. 1 This is crucial to maintain the right spatio‐temporal profile of protein molecules essential for organismal homeostasis. Many physical and chemical stresses in the body, and mutations, can lead to the misfolding of proteins, which then expose regions of hydrophobicity to the surrounding solvent and bind via hydrophobic interactions with other nearby misfolded protein molecules to form either soluble aggregates and/or insoluble deposits. The protein aggregates formed may either consist of β‐sheet‐rich fibrillar structures (amyloid) or be unstructured (amorphous). All of these protein structures have been found associated with disease states. For example, soluble oligomeric protein aggregates have been implicated as cytotoxic species in Alzheimer's disease (AD) and other neurodegenerative disorders and insoluble protein deposits play an important role in the pathologies of a variety of protein deposition diseases (e.g., diabetes type 2 and amyloidoses). 2 , 3 Chaperones are key players in proteostasis systems, acting to inhibit protein aggregation and toxicity, and assist in protein folding, translocation and turnover. 4
Decades of research has identified networks of intracellular chaperone and co‐chaperone proteins 5 that are upregulated in response to different forms of cellular stress including heat‐shock, pathogen attack, hypoxia, glucose deprivation, and ER stress. 6 However, relatively little is known about extracellular chaperones (ECs) that are normally secreted from cells and are present in mammalian body fluids, and previously only a small number of these had been identified. 7 The first mammalian EC identified was clusterin (CLU), 8 and in the 20 years since that discovery, only approximately a dozen other ECs have been reported. 7 , 9 , 10 Evidence from many different studies strongly suggests that constitutively secreted ECs patrol extracellular fluids for misfolded proteins, mask toxic hydrophobic regions on these species, and form stable soluble complexes with them to facilitate their cellular uptake via receptor‐mediated endocytosis and subsequent intracellular degradation. 2 , 7 It is, therefore, likely that ECs play a pivotal role in maintaining extracellular proteostasis and protecting us from protein aggregation related diseases.
As one example, the aggregation of Aβ to form toxic oligomers and subsequently deposit as insoluble amyloid plaques in the brains of Alzheimer's patients has been strongly implicated in the death of neurons that characterizes the disease. 11 Mutations in the human CLU gene are one of the highest known risk factors for AD. 12 , 13 , 14 CLU binds with high affinity to Aβ oligomers to potently inhibit their toxicity 15 , 16 and subsequent aggregation to form amyloid fibrils. 16 CLU is also strongly implicated in the clearance of neurotoxic Aβ forms from the brain. 17 CLU enhances the clearance of Aβ1–42 at the blood–brain barrier 18 via the receptor known as megalin/LRP2, 19 which is widely expressed in the brain, including in capillaries, astrocytes, and neurons. 20 CLU‐Aβ complexes bind to LRP2 at the cell surface and the complexes are subsequently internalized via receptor‐mediated endocytosis, transported to lysosomes, and degraded. 21 Consistent with an important clearance role in AD, the addition of purified CLU to cerebrospinal fluid from Alzheimer's patients increased the removal of Aβ1–42 from the supernatant by macrophage‐like U937 cells. 22 Furthermore, in mice, ablation of CLU expression shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. 17
Given that hundreds of intracellular chaperones and co‐chaperones have been identified, it stands to reason that we may not have yet identified even the major ECs that function in body fluids to protect us from undesirable protein aggregation and related serious disease pathologies. The currently very limited knowledge around even the identity of ECs, let alone their related biological functions, is acting as a roadblock to the future potential to harness these molecules in new therapies to treat debilitating proteinopathies such as AD, type 2 diabetes, and rheumatoid arthritis. There is, therefore, a critical need to identify new ECs and understand their chaperone functions.
Motivated by the above considerations, we designed a novel strategy to selectively retrieve from human serum a fraction enriched in proteins likely to be ECs, from which individual proteins could be further tested in vitro to identify genuine ECs. This strategy was based on the well‐known propensity of chaperones to bind to exposed regions of hydrophobicity on misfolding proteins. 23 Essentially, we identified a destabilized protein (cysteine‐free chloride intracellular channel protein 1; CLIC1), that when incubated for a period of 2 hr at 37°C, would misfold and expose hydrophobicity to solution. This “bait” protein was attached to magnetic beads which were then incubated with human serum at either 37°C (to induce bait protein misfolding) or at 4°C (as a control). The beads were subsequently washed before eluting bound proteins and analysing them by SDS PAGE. Separated proteins were digested in‐gel with trypsin and subsequently analyzed by reverse phase liquid chromatography and mass spectrometry to identify proteins present and their relative abundance (both within eluate fractions, and between the fractions eluted from beads incubated in serum at 37°C vs. 4°C).
We selected seven commercially available plasma proteins identified by this approach as more abundant in the eluate from beads incubated in serum at 37°C versus 4°C, and tested them in vitro for their abilities to inhibit (a) the formation of amyloid resulting from the aggregation of the Aβ1–42, and (b) the amorphous aggregation of CS. Of the seven proteins tested, five were confirmed to have in vitro chaperone activity. Two proteins, vitronectin (VTN), and plasminogen activator inhibitor 3 (PAI‐3), were shown to inhibit the amorphous aggregation of CS and the aggregation of Aβ1–42 to form amyloid. In contrast, the other three proteins, prothrombin (PT), C1r and C1s, specifically inhibited Aβ1–42 amyloid formation but not the amorphous aggregation of CS. Thus, herein, we report the first identification of five new putative ECs with differing chaperone activities that may play important roles in extracellular proteostasis and specific disease contexts.
2. RESULTS
2.1. When heated at 37°C, CLIC1 exposes increased hydrophobicity to solution and aggregates
When bound to magnetic beads and heated at 37°C, the level of exposed hydrophobicity on CLIC1 (measured as a change in bisANS fluorescence) increased progressively with the time of incubation, and began to plateau at ~ 60 min (Fig. 1a). Furthermore, when heated at 37°C in phosphate buffered saline (PBS), CLIC1 amorphously aggregated, measured as a time dependent increase in turbidity (absorbance at 360 nm) commencing at ~ 20 min and continuing to increase for the nearly 2 h time course (Fig. 1b). The similarity in the patterns of change seen in Figure 1a,b is consistent with the increased exposure of hydrophobic regions on CLIC1 playing a part in its aggregation when heated at 37°C in solution. These results established the suitability of CLIC1 as a “bait protein” that misfolds and aggregates at the mild physiological temperature of 37°C, and that will expose increased regions of hydrophobicity to solution under the same conditions, including when tethered to beads.
FIGURE 1.
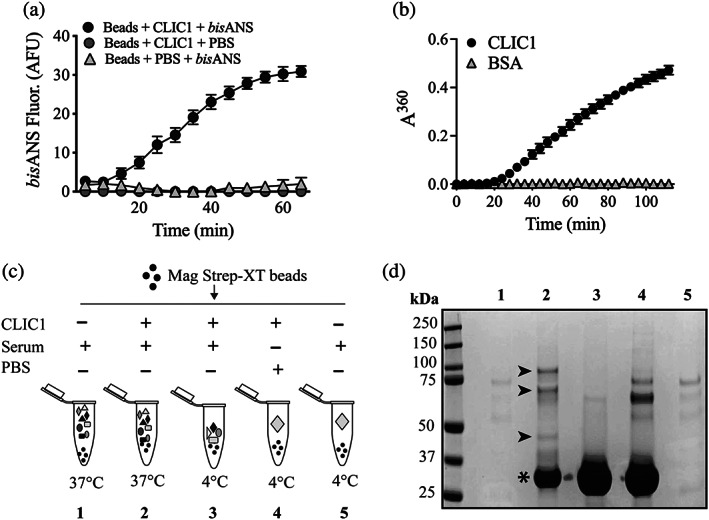
Fishing for new ECs in serum using CLIC1 as a bait protein. (a) Beads bearing bound CLIC1 were supplemented (or not) with 10 μM bisANS and then incubated at 37°C. Mean bisANS fluorescence (in arbitrary fluorescence units, AFU) ± range (n = 2) are plotted. In some cases, the error bars are too small to be visible. Result is representative of two independent experiments. (b) Amorphous aggregation of CLIC1 at 37°C measured as changes in absorbance (at 360 nm) over a period of 115 min. BSA was used as a non‐aggregating control protein. Mean absorbances (in arbitrary units, AU) at 360 nm ± SE (n = 3) are plotted. In many cases, the error bars are too small to be visible. (c) Schematic of experimental design using CLIC1 as bait to retrieve putative chaperones from human serum. Additives are indicated above each tube icon, and the temperature of incubation is indicated below the tubes. (d) Image of a completed SDS PAGE analysis of samples of bead eluates from beads treated as in (c). The numbering of lanes shown corresponds to the numbered treatments in (c). Molecular mass markers and their respective masses (kDa) are shown at the left of the image. Asterisk indicates the position of the major CLIC1 band; small arrowheads indicate the positions of bands enriched in the eluate from beads incubated with CLIC1 and serum at 37°C (compare Lane 2 with Lane 3)
2.2. Fishing putative chaperones from human serum
We reasoned it would be possible to retrieve (fish) potential chaperones from serum using CLIC1 as bait, with either of two slightly different approaches. In one approach, CLIC1 could be pre‐loaded onto beads, which are then added to serum and heated at 37°C to induce CLIC1 misfolding. In the second approach, the Streptactin beads and the Strep tag‐bearing CLIC1 protein could be added separately to the serum, which is then heated at 37°C to induce CLIC1 misfolding. In the second approach, the CLIC1 would be bound to the beads during the incubation, but would be free in solution prior to that binding (potentially being more sterically accessible to chaperones while in solution). Preliminary work suggested that either approach gave similar results (not shown). The experimental results presented here were obtained using the second approach, which is schematically outlined in Figure 1c. Controls included incubating mixtures of beads, CLIC1, and serum at 4°C, which was not expected to induce misfolding of CLIC1, and incubating mixtures of beads and serum, lacking CLIC1 (Figure 1c). Eluates from the beads treated as outlined in Figure 1c were subsequently analyzed by SDS PAGE. As expected, only limited nonspecific binding of serum proteins to the beads was detected in those mixtures in which the bait protein CLIC1 had not been added (Figure 1d, Lanes 1 and 5). For those mixtures supplemented with CLIC1, a major band at ~28 kDa representing CLIC1 was detected in the bead eluates (Figure 1d, Lanes 2–4, indicated by an asterisk). Some minor bands between ~60 and 75 kDa were detected in the bead eluate from the mixture containing only beads and CLIC1, which may represent minor contaminant proteins in the CLIC1 preparation (Figure 1d, Lane 4). These bands were much less evident in the bead eluate from the mixture that contained beads, CLIC1, and serum (Figure 1d, Lane 3), possibly because of competition from serum proteins for nonspecific binding to the beads. The most interesting comparison in Figure 1d is between Lanes 2 and 3, comparing bead eluates from mixtures of beads, CLIC1, and serum incubated at 37°C (Lane 2) versus 4°C (Lane 3). There are a series of protein bands detected in Lane 2 not visible in Lane 3 (indicated by black arrowheads in Lane 2), representing potential chaperones selectively interacting with CLIC1 at 37°C but not at 4°C.
Sections of gels like that shown in Figure 1d, above the position of the major CLIC1 band in Lanes 2 and 3, were excised from the gel and subjected to in‐gel tryptic digestion. Extracted peptides were then analyzed by nano‐LC–MS and the relative abundance of peptides derived from identified proteins compared between eluates from CLIC/beads incubated in serum at 37°C versus 4°C. The rationale was that during heating at 37°C, but to a much lesser extent at 4°C, CLIC1 would misfold and any chaperones present would bind to misfolded CLIC1. We, therefore, hypothesized that proteins enriched in the eluate from CLIC1/beads incubated at 37°C represent putative chaperones. We calculated the ratio of the normalized spectral abundance factor (NSAF) for peptides associated with specific proteins identified in the bead eluates from Treatments 2 (CLIC1 + serum at 37°C) and 3 (CLIC1 + serum at 4°C; Figure 1c), and detected 19 proteins with this NSAF ratio > 1.5 (Table 1). On the basis of their ready commercial availability as purified proteins, we selected seven candidates for further analysis (Table 1). The well‐known EC CLU, which was also detected by this method, was used as a positive control in subsequent protein aggregation assays.
TABLE 1.
Potential ECs identified using nano‐LC–MS, showing the ratio of the normalized spectral abundance factor (NSAF) of proteins bound to CLIC1/beads at 37 versus 4°C
| Protein | Uniprot identifier code | Ratio of NSAF (37°C:4°C) |
|---|---|---|
| C1r | P00736 | 50.20 |
| C4A | P0C0L4‐1 | 42 |
| Beta‐Ala‐his dipeptidase | Q96KN2 | 25.6 |
| Mannan‐binding lectin serine protease 1 | P48740‐1 | 23.6 |
| C1s | P09871 | 16.00 |
| Phosphatidyl‐glycan‐specific phospholipase D | P80108 | 15.4 |
| Coagulation factor V | P12259 | 13.3 |
| Cadherin 5 | P33151 | 11.3 |
| Lacritin | Q9GZZ8 | 11.3 |
| Alpha‐2‐antiplasmin (A2AP) | P08697‐1 | 9.23 |
| Plasminogen activator inhibitor III (PAI‐3) | P05154 | 7.18 |
| Proline‐rich protein 4 | P02810 | 7.18 |
| Prothrombin (PT) | P00734 | 6.36 |
| Statherin | P02808‐1 | 5.13 |
| Clusterin (CLU) | P10909 | 4.00 |
| Histidine‐rich glycoprotein | P04196 | 3.76 |
| Antithrombin III (AT3) | P01008 | 3.08 |
| Serum paraxonase | Q15166 | 2.29 |
| Vitronectin (VTN) | P04004 | 1.81 |
Note: Proteins selected for in vitro analysis of chaperone activity are shown in italics. The known EC clusterin, also detected by this method, is shown in bold.
2.3. Assessing the in vitro chaperone activities of putative extracellular chaperones
The seven selected proteins, together with CLU used as a known chaperone positive control, and SOD used as a non‐chaperone negative control protein, were next tested for their abilities to inhibit the in vitro aggregation of Aβ1–42 to form amyloid, and the aggregation of CS to form unstructured (amorphous) aggregates.
2.4. VTN and PAI‐3 inhibit both the aggregation of Aβ1–42 to form amyloid, and the amorphous aggregation of CS
Aβ1–42 aggregates to form amyloid, a fibrillar structure characterized by a highly ordered β‐sheet‐rich structure, which reacts strongly with the dye Thioflavin T (ThioT) to yield a bright fluorescence. The aggregation of Aβ1–42 to form amyloid was, therefore, monitored by measuring changes in ThioT fluorescence. When tested at a molar ratio of putative chaperone:Aβ1–42 = 1:10, VTN and PAI‐3 were even more effective than the known EC CLU in essentially completely inhibiting the formation of Aβ amyloid (Figure 2a,b). When tested at the same molar ratio, the non‐chaperone control protein superoxide dismutase (SOD) did not significantly affect Aβ1–42 aggregation. Furthermore, in assays measuring the amorphous aggregation of CS, when tested at a molar ratio of chaperone:CS = 1:5, VTN and PAI‐3 also inhibited CS aggregation to an extent comparable to CLU (Figure 2c,d).
FIGURE 2.
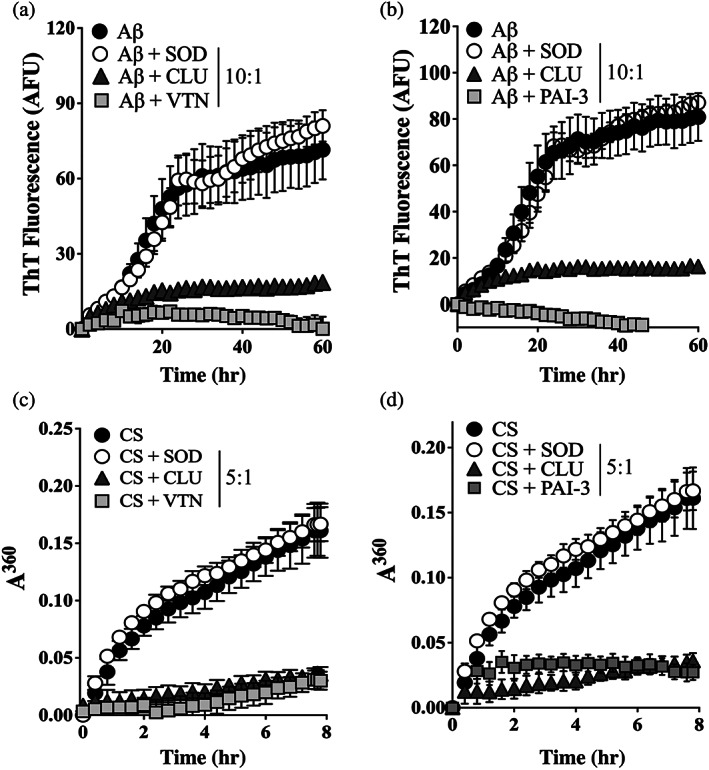
VTN and PAI‐3 inhibit the in vitro aggregation of Aβ1–42 and CS. (a & b) Aβ1–42 (10 μM), supplemented with ThioT (20 μM), was incubated for 60 hr in the presence or absence of 1 μM of (a) VTN or (b) PAI‐3, or superoxide dismutase‐1 (SOD, as non‐chaperone control protein), or clusterin (CLU, a chaperone control protein). Amyloid formation was monitored by measuring ThioT fluorescence. Mean ThioT fluorescence (in arbitrary fluorescence units, AFU) ± SEM (n = 3) are plotted. (c & d) CS (5 μM) was incubated at 43°C for 8 hr in the presence of absence of 1 μM SOD, CLU or (c) VTN, or (d) PAI‐3. Amorphous protein aggregation was monitored as increasing turbidity measured as absorbance at 360 nm (A360). Mean absorbance values ± SEM (n = 3) are plotted. In (a–d), the molar ratios of Aβ1–42 or CS to SOD/CLU/VTN/PAI‐3 are indicated in the respective keys, and in some cases, the error bars are too small to be visible. All results are representative of three independent experiments
2.5. PT, C1r, and C1s inhibit the aggregation of Aβ1–42 to form amyloid, but not the amorphous aggregation of CS
At a molar ratio of Aβ1–42:CLU of 10:1, CLU nearly abolished all ThioT fluorescence resulting from the aggregation of Aβ1–42 (Figure 3a–c). At this same molar ratio, PT, C1r, and C1s each partially inhibited Aβ1–42 amyloid formation. Under these conditions, relative to Aβ1–42 alone, the addition of PT had reduced the level of ThioT fluorescence at the 60 hr endpoint by ~35% (Figure 3a). The reduction in endpoint ThioT fluorescence was less for C1r and C1s, being ~23 and ~9%, respectively. However, both C1r and C1s significantly extended the lag phase preceding the increase in ThioT fluorescence (Figure 3b,c). When the amount of PT, C1r or C1s added to the aggregation reactions was increased to produce a ratio of Aβ:C1r/C1s = 5:1, the extent of the reduction in endpoint ThioT fluorescence was substantially greater, being ~84, ~67, and ~65%, respectively (Figure 3a–c). In contrast, even when present at a 1:1 molar ratio with the client protein, under the conditions tested, neither PT, C1r, or C1s significantly inhibited the amorphous aggregation of CS (Figure 3d,e). As expected, when also tested at a 1:1 ratio with CS, the non‐chaperone control protein SOD had no significant effect on CS aggregation, but CLU decreased the endpoint absorbance value measured by ~79% (Figure 3d,e).
FIGURE 3.
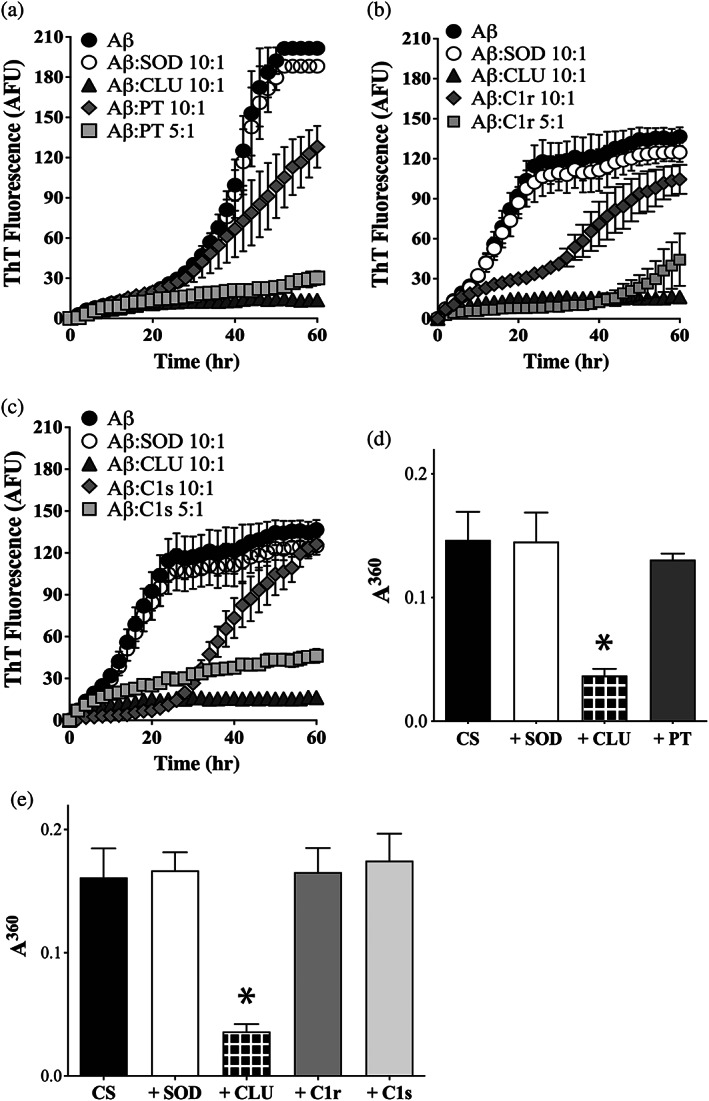
PT, C1r and C1s inhibit the aggregation of Aβ1–42 to form amyloid, but not the amorphous aggregation of CS. (a–c) Aβ1–42 (10 μM) was supplemented with ThioT (20 μM) and incubated for 60 hr in the absence or presence of 1 μM SOD (a non‐chaperone control protein) or CLU (a chaperone control protein). Aggregation reactions were also supplemented or not with either 1 μM or 2 μM of (a) PT, (b) C1r, or (c) C1s. In all cases, the molar ratio of Aβ1–42:test protein is indicated in the respective keys. Amyloid formation was monitored by measuring ThioT fluorescence. Mean ThioT fluorescence values (in arbitrary fluorescence units, AFU) ± SEM (n = 3) are plotted In (a–c), in some cases, the error bars are too small to be visible. (d, e) Endpoint absorbance values for experiments in which 5 μM CS was incubated for 8 hr in the presence or absence of 1 μM of SOD or CLU, or 5 μM of (d) PT or (e) C1r or C1s. Amorphous protein aggregation was monitored as turbidity measured as absorbance at 360 nm (A360). Mean endpoint absorbance values (i.e., at 8 hr) ± SEM (n = 3) are plotted. Each result is representative of two independent experiments. In (d) and (e), the asterisks indicate significant differences in A360 between the CS alone and +CLU treatments (Students t‐test, p < .01); other differences are not significant
2.6. Transmission electron microscopy confirmed that VTN, PAI‐3, PT, C1r, and C1s all specifically inhibited the formation of Aβ1–42 amyloid fibrils
The results of the ThioT fluorescence assays suggested that VTN, PAI‐3, PT, C1r, and C1s were all able to inhibit the formation of amyloid fibrils by Aβ1–42 (Figures 2 and 3). This was confirmed by transmission electron microscopy of samples taken from the 60 hr endpoint of aggregation reactions containing 10 μM Aβ1–42 supplemented or not with 1 μM SOD, CLU, VTN, PT, C1r, or C1s. When incubated alone, Aβ1–42 formed readily detected fibrillar structures (Figure 4a). Although the addition of the non‐chaperone control protein appeared to have some effects on the extent of self‐association of the Aβ1–42 amyloid fibrils formed, abundant fibrillar material was still detected (Figure 4b). As previously reported, 16 the addition of CLU to an Aβ1–42 aggregation reaction abolished the formation of fibrils (Figure 4c). Similarly, when supplemented with VTN, PAI‐3, PT, C1r, or C1s, most material detected was globular or unstructured in appearance, with very limited fibrillar material visible (Figure 4d–h).
FIGURE 4.
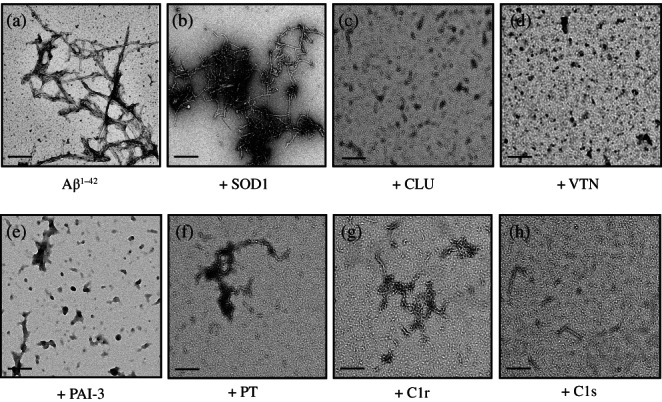
Transmission electron microscopy images of samples collected at the 60 hr endpoint from 10 μM Aβ1–42 aggregation reactions. In (a), Aβ1–42 was aggregated alone without any additions. Other samples were supplemented with 1 μM of (b) SOD, (c) CLU, (d) VTN, or (e) PAI‐3, or 2 μM of (f) C1r, (g) C1s, or (h) PT. In all cases, the black scale bars (bottom left of panels) represent 200 nm
2.7. α‐2‐Antiplasmin and antithrombin III have very limited abilities to inhibit the aggregation of Aβ1–42 to form amyloid, or the amorphous aggregation of CS
Of the seven putative chaperone candidates tested in the in vitro protein aggregation assays, α‐2‐antiplasmin (A2AP) and antithrombin III (AT3) were the only two that demonstrated very limited ability to inhibit the aggregation of either Aβ1–42 or CS (Figure 5). In Aβ1–42 (20 μM) aggregation assays, 2 μM SOD (Aβ1–42:SOD = 10:1) did not reduce the endpoint level of ThioT fluorescence, but in some assays slightly increased the ThioT fluorescence at earlier time points (Figure 5a). In contrast, relative to Aβ1–42 alone, 2 μM CLU (Aβ1–42:CLU = 10:1) reduced the level of endpoint ThioT fluorescence developed by between 79 and 93% (Figure 5a,b). Even when 4 μM A2AP was used (to give Aβ1–42:A2AP = 5:1), the level of ThioT fluorescence actually increased at early time points, and at the 60 min endpoint was ~20% lower than Aβ1–42 alone. Similarly, when Aβ1–42 aggregation reactions were supplemented with an even higher concentration of AT3 (10 μM; Aβ1–42:AT3 = 2:1), there was some reduction in ThioT fluorescence evident especially between 0 and 30 min, but at the 60 min endpoint there was no significant difference between the levels of ThioT fluorescence obtained for Aβ1–42 alone versus wells containing both Aβ1–42 and AT3 (Figure 5b).
FIGURE 5.
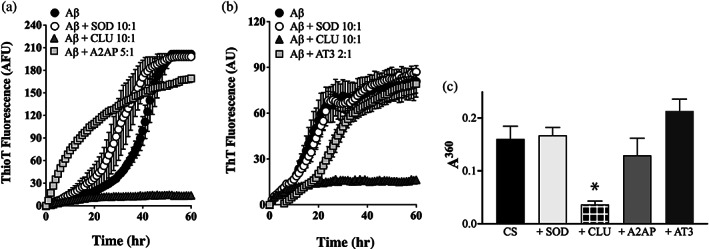
A2AP and AT3 have very limited abilities to inhibit the aggregation of Aβ1–42 or CS. (a, b) Aβ1–42 (10 μM) was supplemented with ThioT (20 μM) and incubated for 60 hr in the absence or presence of 1 μM SOD (a non‐chaperone control protein) or CLU (a chaperone control protein). Aggregation reactions were also supplemented or not with (a) 2 μM A2AP or (b) 5 μM AT3; the molar ratio of Aβ1–42:test protein are indicated in the respective keys. Amyloid formation was monitored by measuring ThioT fluorescence. Mean ThioT fluorescence values (in arbitrary fluorescence units, AFU) ± SEM (n = 3) are plotted. In (a) and (b), in some cases, the error bars are too small to be visible. Results shown are representative of three independent experiments. (c) Endpoint absorbance values following an 8 hr incubation of 5 μM CS in the presence or absence of 1 μM of SOD or CLU, or 5 μM of A2AP or AT3. Amorphous protein aggregation was monitored as turbidity measured as absorbance at 360 nm (A360). Mean endpoint absorbance values ± SEM (n = 3) are plotted. The asterisk indicates a significant difference in A360 between the CS alone and +CLU treatments (Student's t‐test, p < .01); other differences are not significant. Each result is representative of two independent experiments
Furthermore, under the conditions tested, even when present at a 1:1 molar ratio with CS, the aggregation endpoint absorbance values for CS samples supplemented with either A2AP or AT3 were not significantly different from that of wells containing CS alone (Figure 5c). As expected, when also tested at a 1:1 ratio with CS, the non‐chaperone control protein SOD had no significant effect on CS aggregation, but CLU decreased the endpoint absorbance value measured by ~79% (Figure 5c). Collectively, these results indicate that unlike CLU, VTN, PAI‐3, PT, C1r, and C1s (see Figures 2 and 3), both A2AP and AT3 have very limited abilities to inhibit either amyloid formation by Aβ1–42 or the amorphous aggregation of CS, indicating that they lack chaperone activity and were presumably retrieved by virtue of them binding non‐specifically to exposed regions of hydrophobicity on the misfolding “bait” protein.
3. DISCUSSION
This study was based on two premises, (a) that there are likely to be multiple proteins present in blood and other body fluids that have a previously unknown chaperone activity, and (b) the method we devised, involving retrieving proteins that selectively bind to a misfolding protein, would allow us to quickly retrieve and identify putative chaperone proteins from human serum. The results presented strongly support the validity of both these premises. SDS PAGE analyses indicated that, relative to the control incubated at 4°C (where little protein misfolding is expected), there was an enrichment of specific proteins in the eluate retrieved from the magnetic beads incubated in human serum for 2 hr at 37°C with the temperature‐labile bait protein, CLIC1 (Figure 1d). The identity and relative abundance of individual proteins in the eluates from beads incubated with CLIC1 and serum was established using nano‐LC–MS. On the basis of the ratio of the NSAF of proteins in the eluates from beads incubated with CLIC1 and serum at 37 versus 4°C seven commercially available proteins were selected as potential ECs and tested for chaperone activity in protein aggregation assays. The known EC CLU was also identified by this approach (Table 1). We recently reported that two ECs, neuroserpin, and transthyretin, have a chaperone activity that, unexpectedly, preferentially inhibited amyloid formation, but was far less effective at inhibiting the amorphous aggregation of proteins. 24 Therefore, as an initial screen for chaperone activity in the current study, we tested the ability of the selected seven proteins (and CLU as a known chaperone positive control) to inhibit both the formation of fibrillar amyloid by Aβ1–42, and the amorphous aggregation of CS.
Of the seven selected proteins tested, five were demonstrated to have chaperone activity in vitro. Two of these proteins, VTN and PAI‐3, appear to have a chaperone activity similar to CLU in that they inhibited both the formation of amyloid by Aβ1–42 and the amorphous aggregation of CS (Figures 2 and 4). In contrast, three of the other selected proteins, PT, C1r, and C1s, all showed a preferential ability to inhibit Aβ1–42 amyloid formation, but had very limited ability to inhibit the amorphous aggregation of CS (Figures 3 and 4). This analysis therefore suggests that the chaperone activities of PT3, C1r, and C1s are distinct from that of CLU, VTN, and PAI‐3, and are likely to be more similar to that of neuroserpin and transthyretin. 24 Clearly further work involving expanding the range of aggregating client protein models tested is needed to more fully characterize the individual chaperone activities of VTN, PAI‐3, PT3, C1r, and C1s. However, the results reported here clearly establish that all five of these proteins potently inhibit the formation of amyloid by Aβ1–42, and VTN and PAI‐3 also inhibit the amorphous aggregation of CS (Figures 2, 3, 4). Only two of the seven proteins selected for analysis, A2AP and AT3, were shown to lack any substantive ability to inhibit the aggregation of either Aβ1–42 or CS (Figure 5). This means that of seven proteins selected by our “fishing for chaperones” method, five were later confirmed as having the ability to interact with misfolded Aβ1–42 and potently inhibit its formation of amyloid, and two of these proteins were also shown to inhibit the amorphous aggregation of CS. This suggests that the approach used in this study could be applied to successfully identify previously unknown chaperones present in other body fluids (e.g., cerebrospinal fluid, tears, semen, and milk).
Previously identified mammalian ECs that are relatively abundant in body fluids (present at concentrations of μg‐mg/ml) include CLU, α2‐macroglobulin, haptoglobin, transthyretin, SAP, caseins (in milk), and possibly proSAAS, although estimates of the concentration of this protein in plasma and CSF vary widely. 25 α2‐Macroglobulin and haptoglobin were both identified in the bead eluates but were not a focus of the current study. The concentrations in human body fluids of the five putative new ECs identified in this study are given in Table 2.
TABLE 2.
Reported concentrations of CLU and putative new ECs in human body fluids
| Protein | Concentration in body fluids (μg/ml) | ||||
|---|---|---|---|---|---|
| Serum | Cerebrospinal fluid | Saliva | Urine | Semen | |
| Clusterin (CLU) | 125 26 | 2 27 | 0.0008 28 | 7.06 29 | 400 30 |
| Vitronectin (VTN) | 610 31 | 0.025–0.072 32 | 0.0003 28 | 0.002–0.003 33 | 1,350 34 |
| Plasminogen activator inhibitor 3 (PAI‐3) | 6.3 35 | 0.05 35 | 0.03 35 | 0.13 35 | 220 35 |
| Prothrombin (PT) | 154 36 | 210–960 37 | _ | 99–117 38 | _ |
| C1r | 5.5 31 | _ | _ | _ | _ |
| C1s | 93 31 | _ | _ | _ | _ |
Note: (−) Indicates not measured.
Other ECs present at much lower concentrations in body fluids (e.g., at ng/ml levels) include neuroserpin, 39 progranulin, 40 and 7B2. 41 It is probable that any ECs present at these very low levels would be difficult to identify using the method adopted in the current study because they would have to compete with much more abundant ECs when binding to the misfolding bait protein, and would thus likely be present in the bead eluate in only trace amounts. It could be argued that the more abundant ECs are likely to play more substantive roles in stabilizing and clearing potentially dangerous misfolding proteins from body fluids than those ECs present in trace amounts. This does not exclude, however, that some ECs may play critical roles in local environments where they are, for example, released from cells in response to signaling (e.g., the release of 7B2 from secretory granules in neuroendocrine cells 41 ). It is interesting to note in this context that CLU is found in mast cell granules that are released locally at sites of inflammation. 42
Previous reports had identified individual ECs amounting to a cumulative total of around one dozen in number. The results of the current single study suggest the addition of a further five abundant ECs to this growing family. Amongst these five new likely ECs, three of them are previously best known for their roles in the complement system. The first of these, VTN, also shares many structural and functional similarities with CLU. Secreted VTN is a 459‐residue acidic glycoprotein, which is an extensively glcosylated ~52 kDa polypeptide that migrates in SDS PAGE with an apparent mass of ~75 kDa. 43 Secreted CLU is a 427‐residue acidic glycoprotein, which is an extensively glycosylated ~50 kDa polypeptide and also migrates at ~75 kDa in SDS PAGE. VTN (also known as “S protein”) binds to complement membrane attack complex (MAC) that has failed to insert into membranes, to form the soluble SC5b‐9 complex, which is cleared by the kidneys. 44 The first report of the CLU protein in humans identified it as co‐localizing with the SC5b‐9 in deposits within the kidneys of patients suffering from glomerulonephritis, 26 and CLU has been shown to inhibit the assembly of MAC in vitro. 45 At sites of complement activation, where MAC are assembled and may be actively killing cell targets, pro‐inflammatory damaged proteins, and debris are likely to be abundant. Therefore, perhaps it is not surprising that in addition to its ability to terminate the cell lytic activity of the MAC and assist in the disposal of SC5b‐9 complexes, VTN's newly identified chaperone activity may also allow it to bind to damaged and potentially toxic proteins and debris to facilitate their safe disposal.
C1r and C1s are both well‐characterized initiator proteases of the antibody‐dependent classical complement pathway, that are activated when they complex with C1q molecules bound multivalently to the Fc regions of antibodies on opsonized surfaces. 46 Results from the current study clearly show that both these proteins have the ability to inhibit amyloid formation by Aβ1–42 (Figures 3, 4 and 4), but they were unable to efficiently inhibit the amorphous aggregation of CS (Figure 3). Such behavior is consistent with our recent report of neuroserpin and TTR as two ECs that preferentially interact with amyloid‐forming client proteins. 24 For each of these proteins, further testing of their abilities to inhibit the aggregation of different client proteins is required to better characterize their chaperone activities. It is worth noting, however, that in a mouse model of AD, the expression of both C1r and C1s shows a significant age‐dependent increase at the time that Aβ plaques start to accumulate, 46 consistent with the two proteins playing a protective role to inhibit Aβ aggregation.
A fourth identified putative EC is PAI‐3 (SERPINA5), which is a widely expressed protease inhibitor of broad specificity suggested to play roles in hemostasis, host defense, and male reproduction. 47 In an interesting parallel, CLU is an inhibitor of matrix metalloproteases (MMPs), including MMP‐9 48 and possibly MT6‐MMP/MMP‐25. 49 Proteases are active at sites of injury and inflammation, where damage to extracellular proteins is expected. Therefore, perhaps evolution has selected molecules such as CLU and PAI‐3 with complementary protease inhibitor and chaperone activities to aid in the clearance of damaged proteins and protect against both proteotoxicity and excessive proteolysis at such locations.
Lastly, we identified PT as a putative EC that preferentially intacts with amyloid‐forming Aβ. PT is a plasma glycoprotein produced in the liver that cleaves fibrinogen to fibrin, which combines with platelets to form blood clots. It is interesting to note that platelets contain Aβ that is released upon platelet activation, and that after thrombosis in normal mice, Aβ was detected inside blood vessels in the visual cortex and around capillaries in the entorhinal cortex. 50 This may be relevant in AD where small thrombosis events might contribute to disease pathology. Thus, the ability of PT to inhibit Aβ aggregation may be relevant in the context of AD. Another pertinent observation is that select mutations in the fibrinogen Aα gene result in the aggregation of mutant fibrinogen to form amyloid, leading to the most prevalent form of hereditary renal amyloidosis. 51 In the future, the ability of PT to inhibit amyloid formation by fibrinogen should be examined, as this could be relevant to the development of hereditary renal amyloidosis.
In summary, we developed a novel approach to preferentially retrieve chaperones from human serum, and using this technique selected seven human plasma proteins showing enhanced binding to a misfolding protein “bait.” Further analysis confirmed that five of these seven proteins specifically inhibited protein aggregation in vitro. Two putative new ECs, VTN and PAI‐3, could inhibit both the amorphous aggregation of CS and the formation of amyloid by Aβ. The remaining three putative ECs, C1r, C1s, and PT, inhibited Aβ amyloid formation but not the amorphous aggregation of CS, suggesting that they may preferentially interact with amyloid‐forming species. The chaperone activities of these newly identified putative ECs may prove to be important at both the level of organismal proteostasis and in a variety of serious disease contexts. We also suggest that the method we developed to quickly identify these putative new ECs may be widely applicable to a variety of other body fluids, and its widespread use could accelerate our discoveries of new members of the growing EC family. In this regard, our results suggest that future studies of those proteins listed in Table 1, but not tested further here, are likely to identify additional currently unknown plasma ECs.
4. MATERIALS AND METHODS
4.1. Chemicals
All chemicals and reagents were purchased from Sigma Aldrich (Australia), were of analytical grade, and prepared in MilliQ water (Millipore; Australia) unless otherwise stated. MagStrep “type3” XT beads were purchased from IBA Life Sciences, Germany. Roche cOmplete protease inhibitor tablets were purchased from Sigma Aldrich (Australia).
4.2. Proteins
BSA and CS were purchased from Sigma Aldrich. Vitronectin was purchased from ProSci (Poway, CA). Complement components C1r and C1s were purchased from Immuno Pty Ltd. (Wentworth Falls, NSW, Australia). Prothrombin, alpha‐2‐antiplasmin and antithrombin III were purchased from BioVision (Milpitas, CA). Plasminogen activator inhibitor‐3 was purchased from Sapphire Bioscience (Rosehill, NSW, Australia). Purified SOD was a kind gift from Dr. Luke McAlary (Illawarra Health & Medical Research Institute, University of Wollongong, NSW, Australia). Alzheimer's β peptide 1–42 was purchased from Anaspec (Fremont, CA). Human plasma clusterin was purified as described before 52 by immunoaffinity chromatography of plasma prepared from human blood kindly donated by Wollongong Public Hospital (Wollongong, Australia).
4.3. Recombinant CLIC1 expression and purification
The bait protein we used in this study was a mutant cysteine‐free chloride intracellular channel protein 1 (CLIC1) in which four cysteine residues have been replaced with alanines (described in Goodchild et al. 53 ). A sequence encoding this protein fused at the C‐terminus with the Strep‐tag II sequence (WSHPQFEK; see Supporting Information) was cloned into the pET‐28c (+) plasmid (Novagen, Australia). The plasmid was transformed into Escherichia coli BL21 cells for protein expression, that were cultured in Luria Broth at 37°C and, once the optical density reached 0.65 at 600 nm, expression was induced with the addition of 250 μM isopropyl β‐d‐1‐thiogalactopyranoside (IPTG). The culture was grown for 4 hr before centrifugation and collection of the bacterial pellet. The bacterial pellet was lysed by resuspending in Buffer W (100 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, pH 8.0) containing 0.1% vol/vol Triton X100, lysozyme (0.5 mg/mL), and cOmplete protease inhibitor (Roche, Sydney), and incubating on ice for 30 min. Cellular disruption was completed by sonication on ice using a Branson 250 sonicator set to 2 min at 50% amplitude (2s on, 2s off). The bacterial lysate was centrifuged 24,000g for 30 min at 4°C. Supernatant was removed and supplemented with 2.5 μg/ml avidin (to block any free biotin that could interfere with subsequent purification) and then passed over a 5 ml Streptactin XT Superflow column (IBA Lifesciences, Germany). Recombinant CLIC1 was purified as per the manufacturer's instructions. Briefly, the Streptactin XT column was equilibrated with five column volumes of Buffer W. The filtered cell lysate was loaded onto the column at 1 ml/min, before the column was washed with five column volumes of Buffer W. Protein was eluted with five column volumes of Buffer BXT (100 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, 50 mM biotin, pH 8.0) and collected fractions were analyzed for purity using SDS‐PAGE and stained with Imperial Protein Stain (Thermo Fisher Scientific, Australia). Fractions were pooled and concentrated, dialyzed into PBS (137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8 mM Na2HPO4, pH 7.4) containing 0.01% (wt/vol) sodium azide, and stored at 4°C.
4.4. Confirmation that CLIC1 misfolds and aggregates at 37°C
The susceptibility of CLIC1 to misfolding and aggregation at 37°C was confirmed in two different assays. A fluorescent probe (4, 4′‐dianilino‐1,1′‐binaphthyl‐5, 5′‐ disulfonic acid; bisANS) was used to assess changes in the extent of surface‐exposed hydrophobicity on CLIC1 bound to MagStrep “type3” XT beads. Briefly, 2 μg of CLIC1 (bearing the C‐terminal Strep‐tag II) was incubated with 2 μl MagStrep “type3” XT beads for 1 hr at 4°C. These beads were then incubated in duplicate with 100 μl of 10 μM bisANS in PBS in the wells of a 96‐well clear, flat‐bottom 96 microplate (Greiner; Kremsmunster, Austria) and changes in fluorescence measured at (ex) 405/(em) 520+/10 nm using a POLARstar Omega plate reader (BMG Labtech, Australia). Readings from all samples were corrected for the background fluorescence of 10 μM bisANS in PBS. In separate experiments, the aggregation of CLIC1 in solution was monitored as a time‐dependent increase in turbidity. CLIC1 or BSA at 75 μM in PBS was incubated at 37°C for ~115 min and turbidity was measured as absorbance at 360 nm using a POLARstar Omega plate reader (BMG Labtech, Australia). All sample readings were corrected for the absorbance of PBS alone.
4.5. Retrieving potential extracellular chaperones from human serum
Human blood, collected according to the University of Wollongong human ethics approval HE02/080, was allowed to clot at room temperature, and then centrifuged at 500 g for 20 min to pellet cells and clotted material. The supernatant (serum) was then removed by pipette and stored frozen at ‐20°C until use. MagStrep “type 3” XT beads (5 μl) were washed three times with Buffer W (100 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, pH 8.0), using a magnetic separator. Beads were then suspended in 400 μl of human serum (containing Roche protease inhibitor and ~20–30 μg of avidin) to which had been added either 200 μl of a stock solution of CLIC1 in PBS at 1.5 mg/ml, or 200 μl PBS, and then incubated for 2 hr at either 4 or 37°C on a rotary shaker (Bioline, Australia). Subsequently, beads were washed with Buffer W + 0.1% (vol/vol) Triton X100 for a total of 16 hr on a rotary shaker, with four buffer changes, before eluting CLIC1 and bound serum proteins using 17.5 μl of Buffer BXT (100 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, 50 mM biotin pH 8.0). The elution was repeated twice more, using 25 μl on each occasion of a 1:1 mixture of Buffer W and Buffer BXT, and all eluates were combined.
4.6. Sample preparation for proteomics analysis
4.6.1. SDS PAGE and preparation of excised gel pieces
Eluted samples (described above) of 25 μl were supplemented with 8 μl of 4X SDS loading buffer (200 mM Tris–HCl, 400 mM DTT, 8% SDS (wt/vol), 6 mM bromophenol blue, 4.3 M glycerol), reduced with the addition of 1 μl β‐mercaptoethanol, boiled for 5 min, and then separated by 10% SDS‐PAGE and stained with Imperial protein stain (Thermo Fisher Scientific, Australia). The gel was then fixed in 5% (vol/vol) acetic acid, before excising horizontal sections of lanes cut into 1–2 mm lengths, and incubating these in 100 μl of 100 mM NH4HCO3, pH 8.0, for 10 min. Subsequently, gel pieces were incubated for 20 min in 100 μl of 50 mM NH4HCO3/50% (vol/vol) acetonitrile with occasional vortexing to remove the protein stain, and finally placed in 100 μl of 100% (vol/vol) acetonitrile and dried by vacuum centrifugation. The gel pieces were then rehydrated and chemically reduced by incubating them for 30 min at 50°C in 50 μl of 10 mM DTT in 100 mM NH4HCO3. Subsequently, excess DTT buffer was removed, and then to alkylate proteins, 50 μl of 20 mM iodoacetamide in 100 mM NH4HCO3 was added to the gel pieces, which were then incubated in the dark at room temperature for 40 min before transferring the gel pieces to 100% acetonitrile and vacuum centrifugation as described above.
4.6.2. In‐gel tryptic digestion
The gel pieces prepared as described above were then digested overnight at 37°C with trypsin (12.5 ng/μl; V5111, Promega, WI) in a sufficient volume of 100 mM NH4HCO3 to cover the gel pieces. The digestion was terminated with the addition of 2 μl of formic acid. Tryptic peptides were extracted twice with 50% (vol/vol) acetonitrile/2% (vol/vol) formic acid and dried under vacuum centrifugation. The peptides were resuspended in 0.1% formic acid and desalted on a pre‐equilibrated C18 Omix Tip (Agilent, CA), eluted in 50% (vol/vol) acetonitrile, 0.1% (vol/vol) formic acid, and dried under vacuum centrifugation.
4.7. Reverse phase C18 liquid chromatography mass spectrometry
Lyophilized peptides were resuspended in 0.1% (vol/vol) formic acid and bath sonicated for 20 min. The resuspended peptides were then centrifuged at 14,000g for 15 min to remove any insoluble debris, and the clarified peptides were analyzed by LC–MS/MS. The peptides were separated on an Ultimate 3,000 nanoLC (Thermo Fisher Scientific, Australia) fitted with the Acclaim PepMap RSLC column (Thermo Fisher Scientific, Australia), making use of a 60 min gradient (2–75% (vol/vol) acetonitrile, 0.1% (vol/vol) formic acid) running at a flow rate of 300 nl/min. Peptides eluted from the nano LC column were subsequently ionized into the Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Australia). The electrospray source was fitted with a 10 μm emitter tip (New Objective, Woburn, MA) and maintained at a 1.6 kV electrospray voltage. The temperature of the capillary was set to 250°C. Precursor ions were selected for MS/MS fragmentation using a data‐dependent “Top 10” method operating in Fourier transformation acquisition mode with higher energy collision fragmentation. FT‐MS analysis on the Q Exactive Plus was carried out at 70,000 resolution, and an AGC target of 1 × 106 ions in full MS. MS/MS scans were carried out at 17,500 resolution with an AGC target of 2 × 104 ions. Maximum injection times were set to 30 and 50 ms, respectively. The ion selection threshold for triggering MS/MS fragmentation was set to 25,000 counts and an isolation width of 2.0 Da was used to perform HCD fragmentation with normalised collision energy of 27.
Raw spectra files were processed using the Proteome Discoverer software 2.4 (Thermo) incorporating the Sequest search algorithm. Peptide identifications were determined using a 20‐ppm precursor ion tolerance and a 0.1 Da MS/MS fragment ion tolerance for FT‐MS and HCD fragmentation. Carbamidomethylation modification of cysteines was considered a static modification while oxidation of methionine, deamidation of asparagine and glutamine, and acetyl modification on N‐terminal residues were set as variable modifications allowing for a maximum of two missed cleavages. The data were processed through Percolator for estimation of false discovery rates. Protein identifications were validated employing a q‐value of .01. The relative abundance of proteins within each sample was calculated by label‐free quantitation using NSAFs as described in Zybailov et al. 54
4.8. Protein aggregation assays
All protein aggregation assays were performed in clear, flat‐bottom microplates. Aβ1–42 aggregation assays were performed in 384‐well microplates (Greiner Bio‐One, Interpath, Australia), and CS aggregation assays used Corning 1,536‐well microplates (Sigma Aldrich, Australia), using a total volume of 50 or 10 μl/well, respectively. Amyloid formation by Aβ1–42 was monitored by measuring changes in ThioT fluorescence. Briefly, Aβ1–42 was diluted from a 200 μM stock solution in 100 mM Na2HPO4, pH 7.8, into PBS to give a final concentration of 10 μM. This was then supplemented or not with (a) 1 μM of VTN, PAI‐3, SOD (a non‐chaperone control protein), or CLU (a known chaperone protein), (b) 1 μM or 2 μM of C1r, C1s, or A2AP, or (c) 5 μM AT3, and incubated at 37°C with double orbital shaking (200 rpm). All samples were supplemented with 20 μM ThioT. Changes in the ThioT fluorescence of triplicate wells were measured using a POLARstar Omega plate reader (BMG Labtech, Australia) with band pass filters (Ex 440 ± 10 nm, Em 480 ± 10 nm). All readings were background corrected by subtracting the fluorescence of 20 μM ThioT in PBS.
4.8.1. Amorphous aggregation of CS
CS (5 μM) in CS aggregation assay buffer (50 mM Tris–HCl, 5 mM HEPES, pH 8) was heated at 43°C for 3–4 hr to induce amorphous protein aggregation, monitored as a time‐dependent increase in turbidity (measured as absorbance at 360 nm). Wells containing CS were supplemented or not with 1 μM of SOD (a non‐chaperone control protein) or CLU (a known chaperone protein), or up to 5 μM of putative chaperones (VTN, PAI‐3, C1r, C1s, AT3 or A2AP) before incubation at 43°C. The changes in absorbance of triplicate wells was measured using a Spectrostar microplate reader (BMG Labtech, Australia) and samples were shaken (double orbital, 100 rpm) for 20 s prior to each measurement. All absorbance values were background corrected by subtracting the absorbance of a matching volume of CS aggregation assay buffer alone.
4.9. Transmission electron microscopy (TEM)
To prepare samples for TEM analysis, 10 μM Aβ1–42 in PBS was aggregated for 60 hr as described above, supplemented or not with test proteins. CLU, SOD, VTN, and PAI‐3 were added to a final concentration of 1 μM, while the final concentration added for PT, C1r, and C1s was 2 μM. At the 60 hr endpoint of aggregation, 10 μl samples were taken from wells and applied to Formvar‐coated copper grids. The samples were next stained with 2% (wt/vol) uranyl acetate, and imaged using a FEI Tecnai T12 transmission electron microscope (Cryogenic Electron Microscopy Facility, Molecular Horizons, University of Wollongong). Images were analyzed using the SIS Megaview II Image Capture system (Olympus, Australia).
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare that are relevant to the content of this article.
AUTHOR CONTRIBUTIONS
Nicholas Geraghty: Investigation (equal); writing – review and editing (equal). Sandeep Satapathy: Investigation (equal); writing – review and editing (supporting). Megan Kelly: Investigation (supporting); writing – review and editing (supporting). Flora Cheng: Investigation (supporting). Albert Lee: Investigation (supporting); supervision (supporting); writing – review and editing (supporting). Mark Wilson: Conceptualization (lead); investigation (supporting); project administration (lead); supervision (lead); writing – review and editing (lead).
ETHICS STATEMENT
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the University of Wollongong (2002; HE02/080).
CONSENT TO PARTICIPATE
Written consent was obtained in advance from blood donors, as required by HE02/080.
CONSENT FOR PUBLICATION
All authors have consented to publication of this article.
Supporting information
Appendix S1: Supporting information is provided separately and contains the nucleotide sequence encoding CLIC1‐Strep tag II and an image of an SDS‐PAGE analysis of purified CLIC1
ACKNOWLEDGEMENTS
The authors are grateful to The Illawarra Health and Medical Research Institute (Wollongong, Australia), and Molecular Horizons (University of Wollongong, Wollongong, NSW, Australia), for providing us with access to laboratory infrastructure and administrative support needed to carry out this work. SS is thankful to the Graduate Research School, University of Wollongong, for their generous support in the form of a PhD Scholarship.
Geraghty NJ, Satapathy S, Kelly M, Cheng F, Lee A, Wilson MR. Expanding the family of extracellular chaperones: Identification of human plasma proteins with chaperone activity. Protein Science. 2021;30:2272–2286. 10.1002/pro.4189
DATA AVAILABILITY STATEMENT
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 55 partner repository with the dataset identifier PXD027042 and 10.6019/PXD027042. Username: reviewer_pxd027042@ebi.ac.uk; Password: f9SrPQAh.
REFERENCES
- 1. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. [DOI] [PubMed] [Google Scholar]
- 2. Yerbury JJ, Stewart EM, Wyatt AR, Wilson MR. Quality control of protein folding in extracellular space. EMBO Rep. 2005;6:1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fink AL. Protein aggregation ‐ folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–R23. [DOI] [PubMed] [Google Scholar]
- 4. Yerbury JJ, Ooi L, Dillin A, et al. Walking the tightrope: Proteostasis and neurodegenerative disease. J Neurochem. 2016;137:489–505. [DOI] [PubMed] [Google Scholar]
- 5. Kim YE, Hipp MS, Bracher A, Hayer‐Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. [DOI] [PubMed] [Google Scholar]
- 6. Zuo D, Subjeck J, Wang XY. Unfolding the role of large heat shock proteins: New insights and therapeutic implications. Front Immunol. 2016;7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wyatt A, Yerbury J, Ecroyd H, Wilson M. Extracellular chaperones and proteostasis. Annu Rev Biochem. 2013;82:295–322. [DOI] [PubMed] [Google Scholar]
- 8. Humphreys DT, Carver JA, Easterbrook‐Smith SB, Wilson MR. Clusterin has chaperone‐like activity similar to that of small heat‐shock proteins. J Biol Chem. 1999;274:6875–6881. [DOI] [PubMed] [Google Scholar]
- 9. Cristóvão JS, Figueira AJ, Carapeto AP, Rodrigues MS, Cardoso I, Gomes CM. The S100B alarmin is a dual‐function chaperone suppressing amyloid‐β oligomerization through combined zinc chelation and inhibition of protein aggregation. ACS Chem Nerosci. 2020;11:2753–2760. [DOI] [PubMed] [Google Scholar]
- 10. Coker AR, Purvis A, Baker D, Pepys MB, Wood SP. Molecular chaperone properties of serum amyloid P component. FEBS Lett. 2000;473:199–202. [DOI] [PubMed] [Google Scholar]
- 11. Duran‐Aniotz C, Moreno‐Gonzalez I, Gamez N, et al. Amyloid pathology arrangements in Alzheimer's disease brains modulate in vivo seeding capability. Acta Neuropath Commun. 2020;9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bettens K, Brouwers N, Engelborghs S, et al. Both common variations and rare non‐synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener. 2012;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 14. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Narayan P, Orte A, Clarke RW, et al. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid‐beta1‐40 peptide. Nat Struct Mol Biol. 2012;19:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yerbury JJ, Poon S, Meehan S, et al. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007;21:2312–2322. [DOI] [PubMed] [Google Scholar]
- 17. Wojtas AM, Kang SS, Olley BM, et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc Natl Acad Sci U S A. 2017;114:E6962–E6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer's amyloid β‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zlokovic BV, Martel CL, Matsubara E, et al. Glycoprotein 330/megalin: Probable role in receptor‐mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid b at the blood‐brain and blood‐cerebrospinal fluid barriers. Proc Natl Acad Sci U S A. 1996;93:4229–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alvira‐Botero X, Carro EM. Clearance of amyloid‐β peptide across the choroid plexus in alzheimer's disease. Curr Aging Sci. 2010;3:219–229. [DOI] [PubMed] [Google Scholar]
- 21. Hammad SM, Ranganathan S, Loukinova E, Twal WO, Argraves WS. Interaction of apolipoprotein j‐amyloid beta‐peptide complex with low density lipoprotein receptor‐related protein‐2 megalin ‐ a mechanism to prevent pathological accumulation of amyloid beta‐peptide. J Biol Chem. 1007;272:18644–18649. [DOI] [PubMed] [Google Scholar]
- 22. Yerbury JJ, Wilson MR. Extracellular chaperones modulate the effects of Alzheimer's patient cerebrospinal fluid on AŒ≤ 1‐42 toxicity and uptake. Cell Stress Chaperones. 2010;15:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poon S, Rybchyn MS, Easterbrook‐Smith SB, Carver JA, Pankhurst GJ, Wilson MR. Mildly acidic pH activates the extracellular molecular chaperone clusterin. J Biol Chem. 2002;277:39532–39540. [DOI] [PubMed] [Google Scholar]
- 24. West J et al. Neuroserpin and transthyretin are extracellular chaperones that preferentially inhibit amyloid formation. Sci Adv. 2021; (revised manuscript under review). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chaplot K, Jarvela TS, Lindberg I. Secreted chaperones in neurodegeneration. Front Aging Neurosci. 2020;12:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murphy B, Kirszbaum L, Walker ID, d'Apice JF. SP‐40,40, a newly identified normal human serum protein found in the SC5b‐9 complex of complement and in the immune deposits in glomerulonephritis. J Clin Invest. 1988;81:1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Polihronis M, Paizis K, Carter G, Sedal L, Murphy B. Elevation of human cerebrospinal fluid clusterin concentration is associated with acute neuropathology. J Neurol Sci. 1993;115:230–233. [DOI] [PubMed] [Google Scholar]
- 28. Hirtz C, Vialaret J, Nowak N, Gabelle A, Deville de Périère D, Lehmann S. Absolute quantification of 35 plasma biomarkers in human saliva using targeted MS. Bioanalysis. 2016;8:43–53. [DOI] [PubMed] [Google Scholar]
- 29. Ghiggeri GM, Bruschi M, Candiano G, et al. Depletion of clusterin in renal diseases causing nephrotic syndrome. Kidney Int. 2002;62:2184–2194. [DOI] [PubMed] [Google Scholar]
- 30. Choi N‐H, Tobe T, Hara K, Yoshida H, Tomita M. Sandwich ELISA for quantitative measurement of SP40,40 in seminal plasma and serum. J Immunol Meth. 1990;131:159–163. [DOI] [PubMed] [Google Scholar]
- 31. Berglund L, Björling E, Oksvold P, et al. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–2027. [DOI] [PubMed] [Google Scholar]
- 32. Fukushima Y, Tamura M, Nakagawa H, Itoh K. Induction of glioma cell migration by vitronectin in human serum and cerebrospinal fluid. J Neurosurg. 2007;107:578–585. [DOI] [PubMed] [Google Scholar]
- 33. Carreras‐Planella L, Cucchiari D, Cañas L, et al. Urinary vitronectin identifies patients with high levels of fibrosis in kidney grafts. J Nephrol. 2021;34:861–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bronson RA, Preissner KT. Measurement of vitronectin content of human spermatozoa and vitronectin concentration within seminal fluid. Fertil Steril. 1997;68:709–713. [DOI] [PubMed] [Google Scholar]
- 35. Laurell M, Christensson A, Abrahamsson PA, Stenflo J, Lilja H. Protein C inhibitor in human body fluids: Seminal plasma is rich in inhibitor antigen deriving from cells throughout the male reproductive system. J Clin Invest. 1992;89:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shapiro SS, Martinez J. Human prothrombin metabolism in normal man and in hypocoagulable subjects. J Clin Invest. 1969;48:1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lewczuk P, Wiltfang J, Lange M, Jahn H, Reiber H, Ehrenreich H. Prothrombin concentration in the cerebrospinal fluid is not altered in Alzheimer's disease. Neurochem Res. 1999;24:1531–1534. [DOI] [PubMed] [Google Scholar]
- 38. Furie B, Diuguid CF, Jacobs M, Diuguid DL, Furie BC. Randomized prospective trial comparing the native prothrombin antigen with the prothrombin time for monitoring oral anticoagulant therapy. Blood. 1990;75:344–349. [PubMed] [Google Scholar]
- 39. Nielsen HM, Minthon L, Londos E, et al. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology. 2007;69:1569–1579. [DOI] [PubMed] [Google Scholar]
- 40. Meeter LHH, Patzke H, Loewen G, et al. Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement Geriatr Cogn Dis Extra. 2016;6:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peinado JR, Sami F, Rajpurohit N, Lindberg I. Blockade of islet amyloid polypeptide fibrillation and cytotoxicity by the secretory chaperones 7B2 and proSAAS. FEBS Lett. 2013;587:3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laslop A, Steiner HJ, Egger C, et al. Glycoprotein‐III (clusterin, sulfated glycoprotein‐2) in endocrine, nervous, and other tissues ‐ immunochemical characterization, subcellular localization, and regulation of biosynthesis. J Neurochem. 1993;61:1498–1505. [DOI] [PubMed] [Google Scholar]
- 43. Preissner KT. Structure and biological role of vitronectin. Annu Rev Cell Biol. 1991;7:275–310. [DOI] [PubMed] [Google Scholar]
- 44. Ogawa A, Yorioka N, Yamakido M. Immunohistochemical studies of vitronectin, C5b‐9, and vitronectin receptor in membranous nephropathy. Nephron. 1994;68:87–96. [DOI] [PubMed] [Google Scholar]
- 45. Jenne DE, Tschopp J. Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: Identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc Natl Acad Sci U S A. 1989;86:7123–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mortensen SA, Sander B, Jensen RK, et al. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc Natl Acad Sci U S A. 2017;114:986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang H, Geiger M. Cell penetrating SERPINA5 (protein C inhibitor, PCI): More questions than answers. Semin Cell Dev Biol. 2017;62:187–193. [DOI] [PubMed] [Google Scholar]
- 48. Jeong S, Ledee DR, Gordon GM, et al. Interaction of clusterin and matrix metalloproteinase‐9 and its implication for epithelial homeostasis and inflammation. Am J Pathol. 2012;180:2028–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsuda A, Itoh Y, Koshikawa N, Akizawa T, Yana I, Seiki M. Clusterin, an abundant serum factor, is a possible negative regulator of MT6‐MMP/MMP‐25 produced by neutrophils. J Biol Chem. 2003;278:36350–36357. [DOI] [PubMed] [Google Scholar]
- 50. Kucheryavykh LY, Dávila‐Rodríguez J, Rivera‐Aponte DE, et al. Platelets are responsible for the accumulation of β‐amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res Bull. 2017;128:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chapman J, Dogan A. Fibrinogen alpha amyloidosis: Insights from proteomics. Exp Rev Proteomics. 2019;16:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson MR, Easterbrook‐Smith SB. Clusterin binds by a multivalent mechanism to the fc and fab regions of IgG. Biochim Biophys Acta. 1992;1159:319–326. [DOI] [PubMed] [Google Scholar]
- 53. Goodchild SC, Angstmann CN, Breit SN, Curmi PMG, Brown LJ. Transmembrane extension and oligomerization of the CLIC1 chloride intracellular channel protein upon membrane interaction. Biochemistry. 2011;50:10887–10897. [DOI] [PubMed] [Google Scholar]
- 54. Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae . J Proteome Res. 2006;5:2339–2347. [DOI] [PubMed] [Google Scholar]
- 55. Perez‐Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information is provided separately and contains the nucleotide sequence encoding CLIC1‐Strep tag II and an image of an SDS‐PAGE analysis of purified CLIC1
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 55 partner repository with the dataset identifier PXD027042 and 10.6019/PXD027042. Username: reviewer_pxd027042@ebi.ac.uk; Password: f9SrPQAh.