

Abstract
Red fluorescent proteins (RFPs) are powerful tools used in molecular biology research. Although RFP can be easily monitored in vivo, manipulation of RFP by suitable nanobodies binding to different epitopes of RFP is still desired. Thus, it is crucial to obtain structural information on how the different nanobodies interact with RFP. Here, we determined the crystal structures of the LaM2‐mCherry and LaM4‐mCherry complexes at 1.4 and 1.9 Å resolution. Our results showed that LaM2 binds to the side of the mCherry β‐barrel, while LaM4 binds to the bottom of the β‐barrel. The distinct binding sites of LaM2 and LaM4 were further verified by isothermal titration calorimetry, fluorescence‐based size exclusion chromatography, and dynamic light scattering assays. Mutation of the residues at the LaM2 or LaM4 binding interface to mCherry significantly decreased the binding affinity of the nanobody to mCherry. Our results also showed that LaM2 and LaM4 can bind to mCherry simultaneously, which is crucial for recruiting multiple operation elements to the RFP. The binding of LaM2 or LaM4 did not significantly change the chromophore environment of mCherry, which is important for fluorescence quantification assays, while several GFP nanobodies significantly altered the fluorescence. Our results provide atomic resolution interaction information on the binding of nanobodies LaM2 and LaM4 with mCherry, which is important for developing detection and manipulation methods for RFP‐based biotechnology.
Keywords: mCherry, nanobody, red fluorescent protein, structure
Short abstract
Abbreviations
- CDR
complementarity determining region
- DLS
dynamic light scattering
- FP
fluorescent protein
- GFP
green fluorescent protein
- ITC
isothermal titration calorimetry
- MST
microscale thermophoresis
- RFP
red fluorescent protein
1. INTRODUCTION
Fluorescent proteins (FPs) are the most extensively studied and widely used genetic tools in molecular biology research. FPs can be easily expressed in almost all kinds of cells, and the fusion of FPs generally does not affect the function of other proteins. 1 , 2 , 3 Compared to jellyfish‐derived green FPs (GFPs), red FPs (RFPs) have several advantages when applied in imaging due to their long‐wavelength excitation, lower light scattering, and decreased autofluorescence. 4 , 5 , 6 , 7 , 8 , 9 Although many genetically encoded RFP animal strains have been established to facilitate live observation, the manipulation of RFPs is still desired, which may be improved by the development of RFP‐specific nanobodies. 10
Nanobodies, first discovered by Hamers‐Casterman in 1993, 11 are single domain antibodies derived from the heavy chain variable regions (VHH) of Camelidae atypical immunoglobulins. Nanobodies are the smallest functional fragments derived from a naturally occurring immunoglobulin. Unlike monoclonal antibodies, nanobodies can be easily produced in prokaryotic expression systems. Because of their small size (12–15 kDa) and high stability and solubility, nanobodies are widely used for industrial 12 in vitro diagnostic and clinical applications. 13 The small size also allows nanobodies to be genetically encoded as chimera proteins and delivered to cells by fusion plasmids. Typically, the long CDR3 region enables nanobodies to bind to antigens with high specificity and affinity similar to those of traditional antibodies. 14 , 15 The small size of nanobodies also goes beyond traditional IgG antibodies in several specific applications, including binding with the smooth PD‐L1 protein surface, 16 inserting into canyons on the HIV envelope that are not accessible to IgG 17 to neutralize a broad range of HIV‐1 strains, and effectively blocking the entry of SARS‐CoV‐2 spike protein. 18 , 19 , 20
Kirchhofer et al. first developed a series of GFP nanobodies that can induce subtle opposing changes in the chromophore environment. 21 The GFP‐specific nanobodies GBP1 (GFP enhancer) and GBP4 (GFP minimizer) were suitable for monitoring protein expression, subcellular localization, and translocation. Our previous work also showed that the chimeric GFP nanobody GFP‐enhancer‐(GGGGS)4‐LaG16 increased the binding affinity of GFP and was suitable for GFP‐tagged target protein purification. 22 Tang et al. developed a GFP nanobody‐based system for the selective manipulation of diverse GFP‐labeled cells across transgenic lines. 23 Later, Tang et al. achieved direct optogenetic control of GFP expression in neurons by Cre/loxP recombination through the binding of the GFP‐specific nanobody Cre chimera protein to GFP. 24 Herce et al. designed a cell‐permeable nanobody system to label and manipulate intracellular antigens in living cells. 25 Simpson performed PROTAC degradation of a GPF fusion protein with an anti‐GFP nanobody conjugated to the Halo‐tag. 26 Prole and Taylor developed methods to visualize and manipulate intracellular signaling through GFP and GFP nanobodies. 27 In the existing solved GFP nanobody structures, most of the nanobody binding epitopes of GFP are different. GFP‐enhancer, 21 GBP‐minimizer, 21 and Sb44 28 bind to the different epitopes surrounding GFP's β‐barrel. While Nb2 29 and LaG16 22 bind to the same epitope of GFP, they shared only 29.7% identical CDR sequences. These complex structures provided important structural information for the further development of GFP manipulation tools.
Although many GFP nanobody‐related protein visualization and manipulation applications have been introduced, few RFP nanobodies have been reported. Fridy et al. generated a series of nanobodies (named LaMs) that bind specifically to mCherry through a high‐throughput screening method. 10 To develop an in vivo RFP manipulation system, the design of two or more nanobodies fused with other manipulating components that can interact with different epitopes of the RFP surface at the same time is required. However, the lack of structural information on the detailed interaction interfaces between RFP and specific nanobodies hinders the design and application of manipulation of RFP or RFP fusion proteins by high‐affinity antibodies. Here, we determined the crystal structure of the LaM2‐mCherry and LaM4‐mCherry complexes and clarified the details of the binding of these two nanobodies to mCherry. We also verified the simultaneous binding of LaM2 and LaM4 to RFP by a series of orthogonal molecular biology assays. Our results provide crucial atomic resolution interaction information for the further development of methods to manipulate RFP or RFP fusion proteins in vivo.
2. RESULTS
2.1. The overall structure of the LaM2‐mCherry and LaM4‐mCherry complexes
To gain insight into the binding sites of nanobodies to RFPs, we purified recombinant LaM2, LaM4 and RFP mCherry and then determined the crystal structures of the LaM2‐mCherry and LaM4‐mCherry complexes. The crystal of the complex contains mCherry and LaM2 or LaM4 at a 1:1 stoichiometry. The overall structure of LaM2‐mCherry was refined to 1.39 Å resolution and that of LaM4‐mCherry was refined to 1.92 Å resolution. The crystallographic data are shown in Table 1. The binding interface of CDRs 1–3 of LaM2/LaM4 and mCherry was well defined. LaM2‐mCherry crystallized in the space group P212121, and the asymmetric unit contained one LaM2 nanobody and one mCherry molecule. The Matthews coefficient was approximately 2.11 Å3/Da, and the solvent content was 41.58%. LaM4‐mCherry crystallized in space group C121, and the asymmetric unit contained one LaM4 nanobody and one mCherry molecule. The Matthews coefficient was approximately 2.14 Å3/Da, and the solvent content was 42.42%.
TABLE 1.
Data collection and refinement statistics. The values in parentheses are for the high‐resolution shells
| LaM2‐mCherry | LaM4‐mCherry | |
|---|---|---|
| PDB entry | 6IR2 | 6IR1 |
| Space group | P 21 21 21 | C 1 2 1 |
| a, b, c (Å) | 49.817, 61.046, 109.21 | 156.489, 41.665, 53.182 |
| α, β, γ (°) | 90, 90, 90 | 90, 90.628, 90 |
| Resolution (Å) | 29.4–1.393 (1.443–1.393) | 32.55–1.919 (1.987–1.919) |
| I/δ (I) | 7.5 (2.3) | 8.0 (3.0) |
| Completeness (%) | 97.51 (93.96) | 92.93 (90.98) |
| Number of unique reflections | 65,713 (6,230) | 24,709 (2,411) |
| Rwork | 0.1950 (0.2682) | 0.1799 (0.2581) |
| Rfree | 0.2157 (0.2935) | 0.2319 (0.3110) |
| RMSD | ||
| Bond length (Å) | 0.006 | 0.013 |
| Bond angles (°) | 1.34 | 1.51 |
| Average B, all atoms (Å2) | 20.23 | 31.98 |
Figure 1a shows the overall structure of the LaM2‐mCherry complex, and Figure 1b shows the overall structure of the LaM4‐mCherry complex. The binding sites of LaM2 and LaM4 on mCherry were different. Figure 1c shows the superposed structures of LaM2‐mCherry and LaM4‐mCherry. LaM2 binds to the side of the β‐barrel (the fourth and fifth β‐sheets of the 11 total β‐sheets), while LaM4 binds to the bottom of the β‐barrel (both the amino and carboxyl termini of RFP are at the bottom). The binding modes of the nanobodies are also very different. Figure 1d compares the binding of LaM2 and LaM4. Although the constant domains of the nanobodies are similar, the CDRs are totally different. The CDR3 of nanobodies is longer than that in IgG, and therefore, while only a loop in the IgG secondary structure typically interacts with the antigen, an α‐helix in the nanobody may also emerge and provide an additional interaction mode with the antigen. CDR3 and CDR1 of LaM2 contain two α‐helices: residues 123–126 (Ser‐Glu‐Asn‐Asp) and residues 42–45 (Thr‐Phe‐Ser‐Asp). CDR3 of LaM4 contains an α‐helix consisting of residues 109–111 (Gln‐Arg‐Leu). Additionally, the surface potentials of LaM2 and LaM4 are quite different; LaM2 has a large negative patch in CDR1 that contributes to ionic interactions with mCherry, while the binding of LaM4 to mCherry does not involve similar ionic interactions (Figure 1e).
FIGURE 1.
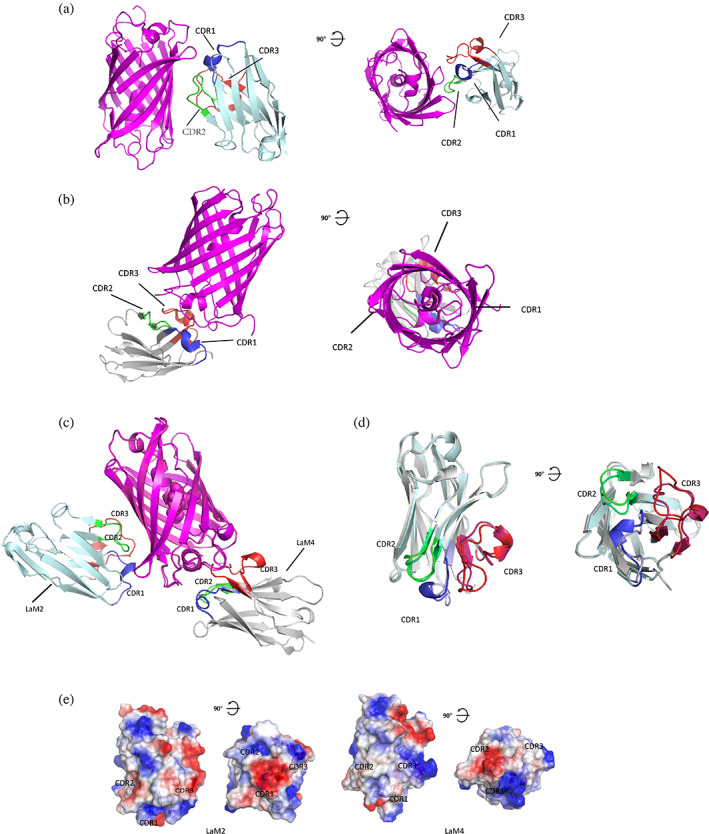
Structure of mCherry: mCherry‐specific nanobody complex determined by X‐ray crystallography. (a) Overall structure of LaM2‐mCherry complex. mCherry is shown in purple. LaM2 is shown in light blue, and CDR1–3s are shown in blue, green, and red (PDB ID: 6IR2). (b) Overall structure of the LaM4‐mCherry complex. mCherry is shown in purple. LaM4 is shown in gray, and the complementarity determining regions (CDRs) are shown in blue (CDR1), green (CDR2), and red (CDR3) (PDB ID: 6IR1). (c) Alignment of LaM2 (light blue)‐mCherry and the LaM4 (gray)‐mCherry complex. (d) Alignment of nanobodies LaM2 and LaM4. (e) Comparison of the surface potential of nanobodies LaM2 and LaM4 (blue: positive, red: negative)
2.2. Details of the binding sites of LaM2/LaM4 to mCherry
Since the resolution of both nanobody‐mCherry complex crystals was high enough, the binding sites between LaM2/LaM4 and mCherry were clearly defined. The detailed interaction interfaces of LaM2 and LaM4 with mCherry are shown in Figure 2. In the LaM2‐mCherry complex, all the CDRs 1–3 of LaM2 contributed to the binding to mCherry.
FIGURE 2.
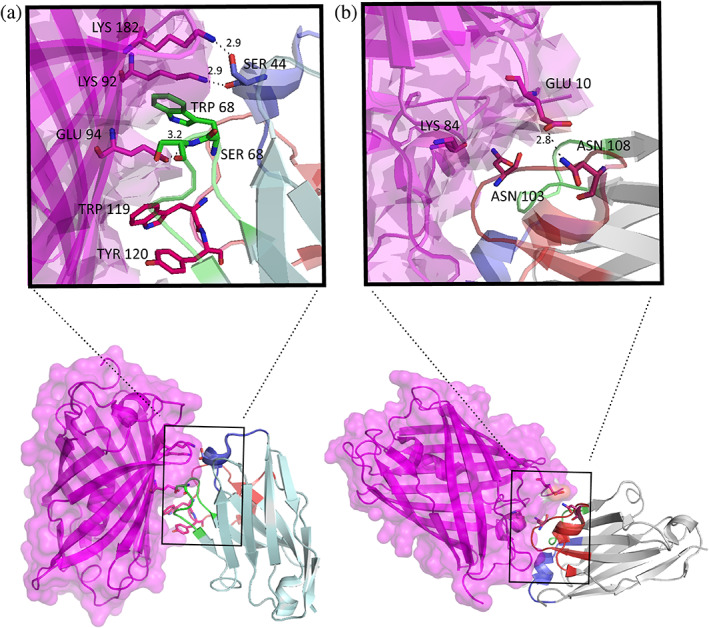
The detailed binding interfaces of nanobodies to mCherry. (a) The detailed interactions of LaM2 complementarity determining regions (CDRs) with mCherry. (b) The detailed interactions of LaM4 CDRs with mCherry
In LaM2 CDR1, two hydrogen bonds were formed between Ser44 and mCherry's Lys92 (2.9 Å) and Lys182 (2.9 Å). In CDR2, there was a strong hydrophobic interaction between Ser66, Trp67, and Ser68 and mCherry, there were also two hydrogen bonds between Ser66 (2.7 Å), Ser68 (2.6 Å), and mCherry's Glu94, and a hydrogen bond between His70 and mCherry's Thr180 (3.0 Å). In CDR3, the Trp119 and Tyr120 side chains were inserted in the pocket of mCherry, and a hydrogen bond formed between Tyr120 and mCherry's Thr106 (2.8 Å).
In the LaM4‐mCherry complex, the main interactions were between CDR3 of LaM4 and mCherry. There were strong hydrogen bonds between Asn103 and mCherry's Lys84 (2.9 Å), Asn108 and mCherry's Glu10 (2.8 Å), Gly102 and mCherry's Tyr38 (2.8 Å), and Leu101 and mCherry's Asp81 (3.0 Å). There was also a hydrogen bond between CDR1 Arg28 and mCherry's Gln188 (3.0 Å).
2.3. Validation of the thermodynamics and binding affinity of the nanobody to mCherry by site‐directed mutagenesis
To further clarify the detailed driving forces of the binding between the nanobodies and mCherry, we performed structurally guided site‐directed mutagenesis and studied the binding affinity of the mutated nanobodies to mCherry. We first used isothermal titration calorimetry (ITC) to measure the binding affinity and thermodynamic parameters because it is a label‐free and in‐solution method and is regarded as the gold standard for protein–protein interactions (Figure 3, Table 2). Both LaM2 and LaM4 showed high binding affinity to mCherry; the K D of LaM2‐mCherry was 3.02 nM and that of LaM4‐mCherry was 22.5 nM (Figure 3a,b). Then, we mutated some residues that contributed to the binding of mCherry.
FIGURE 3.
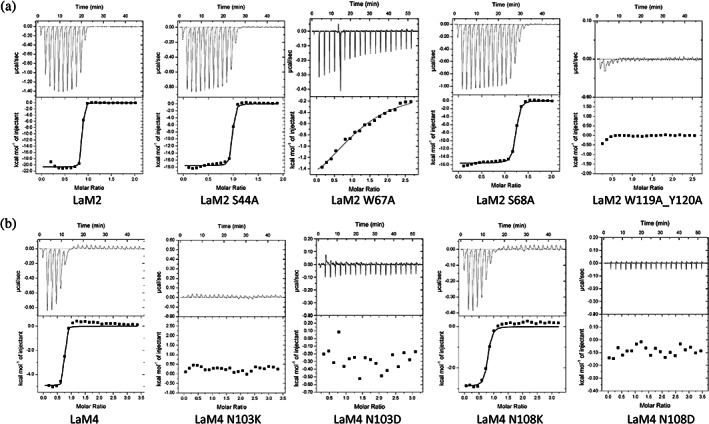
Validation of the interaction between nanobodies (including site‐directed mutants) and mCherry by isothermal titration calorimetry (ITC) at 25°C. (a) LaM2 series nanobodies (wild type, S44A, W67A, S68A, and W119A/Y120A) and (b) LaM4 series nanobodies (wild type, N103K, N103D, N108K, and N108D)
TABLE 2.
The binding specificity of LaM2 and LaM4 series nanobodies to mCherry by ITC
| n | △H (kcal/mol) | △S (cal/mol/°) | −T△S (kcal/mol) | △G (kcal/mol) | K D (nM) | |
|---|---|---|---|---|---|---|
| LaM2 | 0.83 ± 0.01 | −20.62 ± 0.17 | −30.1 | 8.97 | −11.65 ± 0.17 | 3.02 ± 1.88 |
| LaM2‐S44A | 0.94 ± 0.01 | −17.60 ± 0.15 | −21.8 | 6.50 | −11.10 ± 0.15 | 7.19 ± 2.45 |
| LaM2‐W67A | 1.23 ± 0.07 | −2.12 ± 0.19 | 13.1 | −3.91 | −6.03 ± 0.19 | 3.86 × 104 ± 6.60 × 104 |
| LaM2‐S68A | 1.19 ± 0.01 | −15.60 ± 0.09 | −17.4 | 5.19 | −10.41 ± 0.09 | 23.64 ± 3.94 |
| LaM2‐W119AY120A | — | — | — | — | — | — |
| LaM4 | 0.69 ± 0.01 | −4.93 ± 0.15 | 18.5 | −5.51 | −10.44 ± 0.15 | 22.50 ± 34.60 |
| LaM4‐N103K | — | — | — | — | — | — |
| LaM4‐N103D | — | — | — | — | — | — |
| LaM4‐N108K | 0.74 ± 0.02 | −2.89 ± 0.09 | 21.8 | −6.50 | −9.39 ± 0.09 | 132.27 ± 63.59 |
| LaM4‐N108D | — | — | — | — | — | — |
Abbreviation: ITC, isothermal titration calorimetry.
When the two residues of LaM2 CDR1 (Ser44) and CDR2 (Ser68) that form hydrogen bonds with mCherry were individually replaced by Ala, the binding affinity with mCherry was only slightly reduced (Figure 3a). The side chain of LaM2 Trp67 was inserted into a hydrophobic hole in mCherry, and the W67A mutation abolished the hydrophobic interaction and significantly reduced the binding affinity to mCherry. When Trp119 and Tyr120 in CDR3 were replaced by Ala simultaneously, the binding with mCherry was totally abolished (Figure 3a), indicating that this region was crucial for mCherry binding.
For LaM4, the surface of N103 seemed to be complementary to the surface near mCherry Lys84, and a hydrogen bond seemed to form between N108 and mCherry's Glu10. To confirm which interaction was dominant, we constructed LaM4 N103D, N103K, N108D, and N108K point mutation nanobodies and tested their binding affinity to mCherry by ITC. Both the N103D, N108D, and N103K mutations abolished the interaction with mCherry, while N108K still had high binding affinity (Figure 3b). These results suggest that Asn103 and mCherry binding occurs mainly through Van der Waals forces complementary to the protein surface because if Asn103 interacts with mCherry mainly through hydrogen bonds or salt bridges, the binding affinity should still be strong when mutated to Asp; however, the LaM4 N103D mutation totally abolished the interaction, similar to the N103K mutation. When Asn108 mutated to Lys, the interaction was only slightly weakened, while the Asp mutation totally abolished the interaction with mCherry, indicating that the interaction between N108 and mCherry's Glu10 was mainly through the specific hydrogen bond.
2.4. Validation of the simultaneous binding of LaM2 and LaM4 to mCherry
The crystal structure of the LaM2‐mCherry and LaM4‐mCherry complexes showed that the binding regions of LaM2 and LaM4 to mCherry did not overlap, so we assumed that LaM2 and LaM4 could bind to mCherry simultaneously. We confirmed this assumption by ternary ITC and F‐SEC experiments. The K D of LaM2 titrated into the LaM4‐mCherry complex obtained by gel filtration was 8.33 nM, similar to that obtained for LaM2 directly titrated into mCherry (Figure 4a, Table 3), indicating that the binding of LaM4 did not significantly affect LaM2. The K D of LaM4 titrated into the LaM2‐mCherry complex was 261 nM, a 10‐fold decrease compared to titration into mCherry alone (Figure 4b, Table 3), indicating that the binding of LaM2 induces an allosteric change in mCherry's binding interface with LaM4. We proved this assumption by the analysis of crystal structure data, and the binding of LaM2 slightly shifted the loop position of mCherry β‐barrel's two large bottom loops, which are crucial for the binding to LaM4’s CDR1 (around Arg28) and CDR3 (around Leu101).
FIGURE 4.
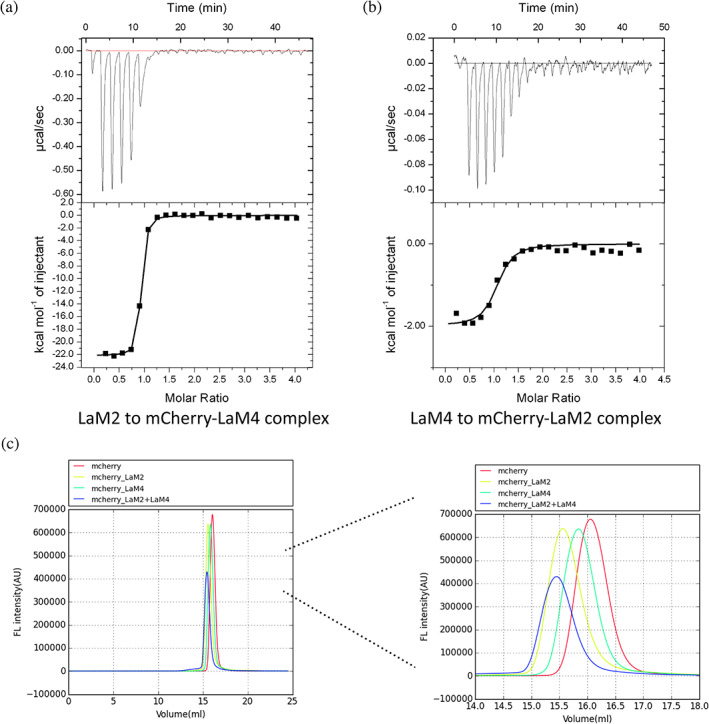
Isothermal titration calorimetry (ITC) and fluorescence‐based size exclusion chromatography (F‐SEC) assays confirmed that LaM2 and LaM4 can bind simultaneously to mCherry. (a) The ITC result of LaM2 binding to the LaM4‐mCherry preformed complex. (b) The ITC result of LaM4 binding to the LaM2‐mCherry preformed complex. (c) The F‐SEC results of mCherry (red), LaM2‐mCherry (light green), LaM4‐mCherry (dark green), and the premixed LaM2‐mCherry complex mixed with LaM4 (blue). The right panel shows an enlargement of 14–17 ml of F‐SEC to clearly show the change in protein complex size
TABLE 3.
The binding specificity of LaM2 to the LaM4‐mCherry complex and LaM4 to the LaM2‐mCherry complex by ITC
| n | △H (kcal/mol) | △S (cal/mol/°) | −T△S (kcal/mol) | △G (kcal/mol) | K D (nM) | |
|---|---|---|---|---|---|---|
| LaM2 to LaM4‐mCherry | 0.866 ± 0.003 | −22.14 ± 0.15 | −37.3 | 11.12 | −11.02 ± 0.15 | 8.33 ± 1.27 |
| LaM4 to LaM2‐mCherry | 1.010 ± 0.036 | −1.99 ± 0.10 | 23.8 | −7.09 | −9.08 ± 0.10 | 216.40 ± 92.28 |
Abbreviation: ITC, isothermal titration calorimetry.
We also observed the formation of a ternary complex of LaM2‐mCherry‐LaM4 by fluorescence‐based size exclusion chromatography (F‐SEC), 30 which can directly show the size of the biological macromolecule complex under physiological conditions. The F‐SEC results (Figure 4c) also confirmed that a stable complex of 1:1:1 LaM2‐mCherry‐LaM4, 1:1 LaM2‐mCherry, and 1:1 LaM4‐mCherry formed if these proteins were mixed in a proper ratio. It is worth noting that although LaM2 and LaM4 are similar in size, there was a certain difference in the position of the peak after binding with mCherry, which may be due to the different 3D shapes of the LaM2‐mCherry and LaM4‐mCherry complexes.
We determined the size distributions of mCherry alone, LaM2‐mCherry, LaM4‐mCherry, and LaM2‐mCherry‐LaM4 by dynamic light scattering (DLS) experiments. The results showed that mCherry alone was very high uniformity, centered at approximately 6 nm, and when complexes were formed with the respective nanobodies, the size increased to approximately 10–11 nm (Figure 5a).
FIGURE 5.
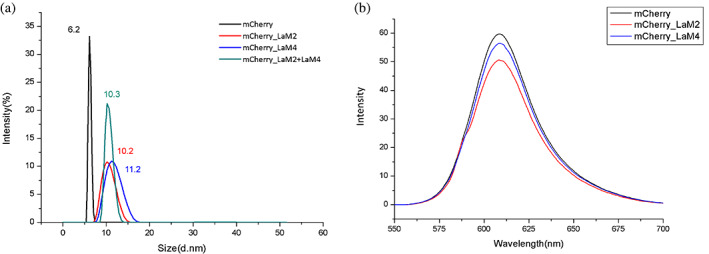
The top of Figure 5b's rectangular box was cut. The size and fluorescence intensity of mCherry binding with nanobodies. (a) The size distributions of mCherry (black), LaM2‐mCherry (red), LaM4‐mCherry (blue), and LaM2‐mCherry‐LaM4 (green) by dynamic light scattering. (b) The emission spectra of mCherry (black), LaM2‐mCherry (red), and LaM4‐mCherry (blue)
In contrast to some GFP nanobodies (GFP enhancer and minimizer), 21 the binding of LaM2 and LaM4 did not significantly affect the chromophore environment of mCherry, resulting in few changes in mCherry fluorescence properties (Figures 5b and S1). This feature ensured that the optical activity of mCherry would not change significantly with the binding of LaM2/LaM4. Thus, quantification by RFP fluorescence remains accurate when manipulated through the binding of LaM2/LaM4 chimeric operators.
3. DISCUSSION
While the molecular weight of a nanobody is only approximately one tenth that of IgG, nanobodies still provide a relatively large binding interaction interface. We calculated and compared the buried surface areas of LaM2 and LaM4 to mCherry and five nanobodies of GFP (GBP1 enhancer PDB ID: 3K1K, GBP4 minimizer PDB ID: 3G9A, LaG16, PDB ID: 6LR7, Nb2, PDB ID: 7E53, and Sb44, PDB ID: 6LZ2) to GFP, in addition to a representative PD‐L1 nanobody KN035 (PDB ID: 5JDS) entering clinical trials by PISA 16 (Table 4). All of these complexes have similar buried surface areas of approximately 600–850 Å2, which is comparable to that of IgG and provides high affinity and specificity. We also compared the buried surface areas of two hapten nanobodies (CorNb‐Cortisone, PDB ID: 6ITQ 31 and MTX Nb‐MTX, PDB ID: 3QXV 32 ). Since these hapten antigens are relatively small and cannot provide a large surface for binding, the buried surface areas are relatively small, between 300 and 400 Å2; however, in contrast to the small buried surface areas of protein antigens, over 50% of the hapten total surface is buried, showing the effectiveness of their interactions with specific antigens.
TABLE 4.
Comparison of typical buried surface areas after specific nanobody binding to their antigens
In addition to the delivery of plasmids encoding nanobodies, unlike IgG, nanobodies can easily enter the cell membrane through a nonendocytic delivery system using a poly‐Arg tag 25 and thus may have additional advantages over IgG‐based chimeric manipulation systems.
Simulations based on the crystal structures show that LaM4 can bind to the DsRed tetramer, and the binding sites are not on the DsRed self‐multimerization interface; thus, the binding of LaM4 does not affect tetramerization. Therefore, it is possible to design chimeric proteins linking functional operation components with LaM4 and develop a self‐assembling macromolecular machine based on the RFP tetramer.
4. CONCLUSION
In summary, we have obtained the details of how nanobodies LaM2 and LaM4 bind to mCherry's different epitopes at atomic resolution via structural biology techniques. Additionally, our thermodynamic and molecular biology assays verified the crucial residues for the nanobody‐RFP interaction. The binding of LaM2 or LaM4 did not significantly change the fluorescence of mCherry, which is important for fluorescence quantification assays. LaM2 and LaM4 can bind simultaneously to mCherry, which is crucial for recruiting multiple operation elements to the RFP. These results provide important basic information for the development of a LaM2/LaM4‐based RFP manipulation system and provide strategies to further optimize the binding affinity of nanobodies to RFP.
5. MATERIALS AND METHODS
5.1. Protein expression, purification, and characterization
The coding sequences of LaM2 and LaM4 were optimized based on favored codon usage in Escherichia coli and were synthesized by Genewiz (Suzhou, China). For crystallization and binding assays, DNA encoding LaM2 and LaM4 was subcloned into the pET28a‐SUMO vector with an N‐terminal 6xHis tag followed by a SUMO tag or a pET21a‐derived vector with an N‐terminal 10xHis tag, respectively. The plasmids were transformed into E. coli strain BL21 (DE3) for expression. The bacteria were cultured in LB medium at 37°C until the OD600 reached 0.8. Recombinant protein expression was induced by the addition of 0.2 mM isopropyl‐d‐1‐thiogalactopyranoside and incubation for an additional 18 hr at 18°C. The cells were harvested and resuspended in NiA buffer containing 20 mM imidazole, 5% glycerol, 150 mM NaCl, and 100 mM Tris–HCl, pH 7.5. The His10‐tagged recombinant LaM2/LaM4 and their respective mutants were initially purified by Ni‐NTA affinity purification using a HisTrap HP column (Qiagen) and eluted with NiB buffer containing 300 mM imidazole, 5% glycerol, 150 mM NaCl, and 100 mM Tris pH 7.5. For crystallization, the His‐SUMO tag was removed by incubation with recombinant ULP1 overnight at 4°C. The cleaved tag fragment and ULP1 were removed by passing through a HisTrap HP column. LaM2/LaM4 was further purified by SEC on a Superdex75 Increase column (Cytiva), and the buffer was exchanged to gel filtration buffer: 10 mM HEPES, pH 7.4 and 100 mM NaCl. The purity and molecular weight of the target proteins were verified by SDS–PAGE.
5.2. Site‐directed mutagenesis
Site‐directed mutagenesis was carried out by employing a PCR‐based mutagenesis site‐directed method (2x Phanta Master Mix, Vazyme Biotech Co., Ltd.) using His10‐LaM2 and His10‐LaM4 as the template. The sequences of the primers used to generate these mutants are displayed in Table S1. All site‐directed mutagenesis constructs were confirmed by DNA sequencing (RuiDi, Shanghai, China).
5.3. Crystallization and data collection
Concentrated LaM2 or LaM4 was mixed with mCherry at a molar ratio of 2:1 and incubated for 1 hr at 4°C. The LaM2‐mCherry and LaM4‐mCherry complexes were separated from the excess LaM2/LaM4 by SEC using a Superdex75 Increase column (Cytiva). The nanobody‐mCherrry complex was then concentrated to 15 mg/ml in gel filtration buffer containing 10 mM HEPES pH 7.4 and 100 mM NaCl. Crystals of the LaM2‐mCherry complex were obtained by the sitting‐drop vapor diffusion method at 293 K in drops containing a mixture of 1 μl of protein solution and 1 μl of reservoir solution, which consisted of 0.2 M magnesium chloride hexahydrate, 0.1 M Tris–HCl, pH 8.5, and 25% wt/vol polyethylene glycol 350. Crystals of the LaM4‐mCherry complex were obtained in 0.2 M lithium sulfate monohydrate, 0.1 M Bis‐Tris, pH 5.5, and 25% wt/vol polyethylene glycol 3,350.
Cryoprotection was performed by adding glycerol to the reservoir buffer at a 20% concentration. X‐ray diffraction data were collected at 100 K in beamlines BL17U1 33 and BL19U1, 34 Shanghai Synchrotron Radiation Facility, Chinese Academy of Sciences.
5.4. Determination and refinement of protein structure
Diffraction images were indexed and processed by HKL2000. 35 The structures of LaM2‐mCherry and LaM4‐mCherry were obtained by molecular replacement using the Phaser program from the CCP4 crystallography package 36 with mCherry (PDB ID: 2H5Q 5 ) and a GFP nanobody (PDB ID: 3K1K 21 ) as the search model. Structure refinement was performed by Refmac 37 and Phenix. 38 The model was refined by COOT. 39 The crystallographic parameters of LaM2‐mCherry (PDB ID: 6IR2; 1.39 Å) and LaM4‐mCherry (PDB ID: 6IR1; 1.92 Å) are listed in Table 1. The related figures were drawn by PyMOL. 40
5.5. Isothermal titration calorimetry
The thermodynamic parameters of the binding of LaM2/LaM4 and their respective mutants to mCherry were determined by ITC using VP‐ITC or ITC200 calorimetry (MicroCal VP‐ITC, Malvern). In a typical experiment, each titration was performed by injecting a 12 μl aliquot of protein sample into the cell containing another reactant (detailed concentration information is listed in Table S2) at a time interval of 120 s to ensure that the titration peak returned to baseline. Altogether, 23 aliquots were titrated in each individual experiment. The stoichiometry of binding (n), the association constant Ka, and the binding enthalpy ΔH were evaluated using MicroCal Origin 7.0 software with a one‐site binding model.
5.6. Fluorescence‐based SEC
The oligomeric state of the tested samples in buffers was recorded by F‐SEC. We used 100 μl 0.1 mg/ml mCherry as control. For the LaM2‐mCherry and LaM4‐mCherry complexes, 50 μl of 0.2 mg/ml mCherry (approximately 7 μM) and 0.2 mg/ml (approximately 14 μM) LaM2 or LaM4 were mixed in equal volumes (the final concentration of mCherry was 3.5 μM, and the final concentration of LaM2/LaM4 was 7 μM) and incubated on ice for 1 hr. For the LaM2‐LaM4‐mCherry complex, 50 μl 0.2 mg/ml mCherry (approximately 7 μM) was mixed with 25 μl 0.4 mg/ml LaM2 and 25 μl 0.4 mg/ml LaM4 and incubated on ice for 1 hr. After high‐speed centrifugation, the supernatants were loaded onto a Superdex 200 Increase size‐exclusion column (Cytiva) equilibrated with SEC buffer (20 mM HEPES pH 7.0, 150 mM NaCl). The fluorescence of each sample was recorded by a fluorometer (excitation, 587 nm; emission, 610 nm for mCherry fluorescence). The data were processed and normalized by FSEC plotter software.
5.7. Emission spectrum measurements
The emission spectra of mCherry (0.1 mg/ml) and LaM2/LaM4‐mCherrry (mCherry 0.1 mg/ml with excess nanobodies) were recorded using a fluorescence spectrophotometer (Varian Cary Eclipse). The excitation wavelength was 587 nm. The emission spectrum was recorded between 550 and 700 nm. The spectra data were analyzed with Origin.
5.8. DLS assay
The particle size distributions of mCherry, the LaM2‐mCherry complex, the LaM4‐mCherry complex, and the LaM2‐LaM4‐mCherry complex were measured by a Nano‐size‐Zeta potential analyzer (Malvern Instruments, ZS90‐2026). The test temperature was 25°C, and the test angle was 90°.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Yu Ding: Conceived and supervised the study. Ziying Wang, Long Li, Rongting Hu, and Rui Liu: Performed protein purification, verification, crystallization, and crystal data analysis; Ziying Wang, Peiyu Zhong, Yiran Zhang, Shihao Cheng and He Jiang: Performed the protein–protein interaction assay.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGMENTS
The authors thank Prof M. Hattori of Fudan University for help with the F‐SEC experiment. The authors would also like to thank the staff members of the Large‐scale Protein Preparation System at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, Shanghai Advanced Research Institute, Chinese Academy of Science for providing technical support and assistance in ITC data collection and analysis. This work was supported by grants 32070939, 91527305, and 82030106 from the National Natural Science Foundation of China; grants 19ZR1405200 and 20JC1410900 from Shanghai Science and Technology Commission; an innovative research team of high‐level local universities in Shanghai and a key laboratory program of the Shanghai Municipal Education Commission (ZDSYS14005).
Wang Z, Li L, Hu R, Zhong P, Zhang Y, Cheng S, et al. Structural insights into the binding of nanobodies LaM2 and LaM4 to the red fluorescent protein mCherry . Protein Science. 2021;30:2298–2309. 10.1002/pro.4194
Funding information National Natural Science Foundation of China, Grant/Award Numbers: 32070939, 82030106, 91527305; Science and Technology Commission of Shanghai Municipality, Grant/Award Numbers: 19ZR1405200, 20JC1410900; Shanghai Municipal Education Commission, Grant/Award Number: ZDSYS14005
DATA AVAILABILITY STATEMENT
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. [DOI] [PubMed] [Google Scholar]
- 2. Zimmer M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem Rev. 2002;102:759–782. [DOI] [PubMed] [Google Scholar]
- 3. Mishin AS, Belousov VV, Solntsev KM, Lukyanov KA. Novel uses of fluorescent proteins. Curr Opin Chem Biol. 2015;27,1–9. 10.1016/j.cbpa.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 4. Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. [DOI] [PubMed] [Google Scholar]
- 5. Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Novel chromophores and buried charges control color in mFruits. Biochemistry. 2006;45:9639–9647. [DOI] [PubMed] [Google Scholar]
- 6. Shcherbakova DM, Subach OM, Verkhusha VV. Red fluorescent proteins: Advanced imaging applications and future design. Angew Chem Int Ed Engl. 2012;51:10724–10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eason MG, Damry AM, Chica RA. Structure‐guided rational design of red fluorescent proteins: Towards designer genetically‐encoded fluorophores. Curr Opin Struct Biol. 2017;45:91–99. [DOI] [PubMed] [Google Scholar]
- 8. Hoffman RM. Strategies for in vivo imaging using fluorescent proteins. J Cell Biochem. 2017;118:2571–2580. [DOI] [PubMed] [Google Scholar]
- 9. Miyawaki A, Shcherbakova DM, Verkhusha VV. Red fluorescent proteins: Chromophore formation and cellular applications. Curr Opin Struct Biol. 2012;22:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fridy PC, Li YY, Keegan S, et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods. 2014;11(12):1253–1260. 10.1038/nmeth.3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hamers‐Casterman C, Atarhouch T, Muyldermans S, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446–448. 10.1038/363446a0 [DOI] [PubMed] [Google Scholar]
- 12. Leow C, Fischer K, Leow C, Cheng Q, Chuah C, McCarthy J. Single Domain Antibodies as New Biomarker Detectors. Diagnostics. 2017;7(4):52. 10.3390/diagnostics7040052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scully M, Cataland S, Peyvandi F, et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. New England Journal of Medicine, 2019;380(4):335–346. 10.1056/nejmoa1806311 [DOI] [PubMed] [Google Scholar]
- 14. Zimmermann I, Egloff P, Hutter CA, et al.Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife. 2018;7. 10.7554/elife.34317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zimmermann I, Egloff P, Hutter CA, et al. Generation of synthetic nanobodies against delicate proteins. Nature Protocols. 2020;15:1707–1741. 10.1038/s41596-020-0304-x [DOI] [PubMed] [Google Scholar]
- 16. Zhang F, Wei H, Wang XX, et al. Structural basis of a novel PD‐L1 nanobody for immune checkpoint blockade. Cell Discovery. 2017;3. 10.1038/celldisc.2017.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weiss RA, Verrips CT. Nanobodies that neutralize HIV. Vaccines. 2019;7:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huo JD, Le Bas A, Ruza RR, et al. Neutralizing nanobodies bind SARS‐CoV‐2 spike RBD and block interaction with ACE2. Nature Structural & Molecular Biology. 2020;27;846–854. 10.1038/s41594-020-0469-6 [DOI] [PubMed] [Google Scholar]
- 19. Schoof M, Faust B, Saunders RA Reuben A, et al. An ultrapotent synthetic nanobody neutralizes SARS‐CoV‐2 by stabilizing inactive Spike. Science. 2020;370(6523):1473–1479. 10.1126/science.abe3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiang YF, Nambulli S, Xiao ZY, et al. Versatile and multivalent nanobodies efficiently neutralize SARS‐CoV‐2. Science. 2020;370:1479–1484. 10.1126/science.abe4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirchhofer A, Helma J, Schmidthals K, et al. Modulation of protein properties in living cells using nanobodies. Nature Structural & Molecular Biology. 2010;17:133–138. 10.1038/nsmb.1727 [DOI] [PubMed] [Google Scholar]
- 22. Zhang ZY, Wang Y, Ding Y, Hattori M. Structure‐based engineering of anti‐GFP nanobody tandems as ultra‐high‐affinity reagents for purification. Scientific Reports. 2020;10: 10.1038/s41598-020-62606-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang JC, Szikra T, Kozorovitskiy Y, et al. A Nanobody‐Based System Using Fluorescent Proteins as Scaffolds for Cell‐Specific Gene Manipulation. Cell. 2013;154:928–939. 10.1016/j.cell.2013.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang JC, Rudolph S, Dhande O, et al. Cell type–specific manipulation with GFP‐dependent Cre recombinase. Nature Neuroscience. 2015;18:1334–1341. 10.1038/nn.4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herce HD, Schumacher D, Schneider AF, et al. Cell‐permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nature Chemistry. 2017;9:762–771. 10.1038/nchem.2811 [DOI] [PubMed] [Google Scholar]
- 26. Simpson L, Macartney T, Nardin A, et al. Inducible Degradation of Target Proteins Through a Tractable Affinity‐Directed PROtein Missile (AdPROM) System. SSRN Electronic Journal. 2020. 10.2139/ssrn.3552150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prole DL, Taylor CW. A genetically encoded toolkit of functionalized nanobodies against fluorescent proteins for visualizing and manipulating intracellular signalling. BMC Biol. 2019;17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cai HM, Yao HB, Li TT, et al. An improved fluorescent tag and its nanobodies for membrane protein expression, stability assay, and purification. Communications Biology. 2020;3(1). 10.1038/s42003-020-01478-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhong PY, Wang ZY, Cheng SH, et al. Structural insights into two distinct nanobodies recognizing the same epitope of green fluorescent protein. Biochem. Biophys. Res. Commun. 2021;565:57–63. 10.1016/j.bbrc.2021.05.089 [DOI] [PubMed] [Google Scholar]
- 30. Hattori M, Hibbs RE, Gouaux E. A fluorescence‐detection size‐exclusion chromatography‐based thermostability assay for membrane protein precrystallization screening. Structure. 2012;20:1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ding LL, Wang ZY, Zhong PY, et al. Structural insights into the mechanism of single domain VHH antibody binding to cortisol. FEBS Letters. 2019;593:1248–1256. 10.1002/1873-3468.13398 [DOI] [PubMed] [Google Scholar]
- 32. Fanning SW, Horn JR. An anti‐hapten camelid antibody reveals a cryptic binding site with significant energetic contributions from a nonhypervariable loop. Protein Sci. 2011;20:1196–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang QS, Zhang KH, Cui Y, et al.Upgrade of macromolecular crystallography beamline BL17U1 at SSRF. Nuclear Science and Techniques. 2018;29. 10.1007/s41365-018-0398-9 [DOI] [Google Scholar]
- 34. Zhang WZ, Tang JC, Wang SS, Wang ZJ, Qin WM, He JH. The protein complex crystallography beamline (BL19U1) at the Shanghai Synchrotron Radiation Facility. Nuclear Science and Techniques. 2019;30. 10.1007/s41365-019-0683-2 [DOI] [Google Scholar]
- 35. Otwinowski Z, Minor W. Processing of X‐ray diffraction data. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- 36. Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 37. Vagin AA, Steiner RA, Lebedev AA, et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallographica Section D Biological Crystallography. 2004;60:2184–2195. 10.1107/s0907444904023510 [DOI] [PubMed] [Google Scholar]
- 38. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography. 2010;66(2):213–221. 10.1107/s0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development ofCoot. Acta Crystallographica Section D Biological Crystallography. 2010;66:486–501. 10.1107/s0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeLano WL, Lam JW. PyMOL: A communications tool for computational models. Abstr Pap Am Chem Soc. 2005;230:U1371–U1372. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.