

Abstract
Through catalyzing the ubiquitination of key regulatory proteins, cullin‐RING ubiquitin ligases (CRLs) play essential biological roles and their activities are controlled by multiple mechanisms including neddylation, the conjugation of NEDD8 to cullins. Upon neddylation, a CRL, such as the CUL1‐based CRL1, undergoes conformational changes that accelerate substrate ubiquitination. Given the structural diversity across subfamilies of CRLs and their substrates, to what extent neddylation modulates the activity of individual CRLs remains to be evaluated. Here, through reconstituting the CRL2 ubiquitination reaction in vitro, we showed that neddylation promotes CRL2VHL‐dependent degradation of both full‐length HIF1α and the degron peptide of HIF1α, resulting in more than 10‐fold increase in the rate of substrate ubiquitination. Consistently, pevonedistat (also known as MLN4924), an inhibitor of neddylation, inhibits the degradation of HIF1α in RCC4 cells stably expressing VHL in cycloheximide chase assays. However, such inhibitory effect of pevonedistat on HIF1α degradation was not observed in HEK293 cells, which was further found to be due to CRL2VHL‐independent degradation that was active in HEK293 but not RCC4 cells. After truncating HIF1α to its Carboxy‐terminal Oxygen‐Dependent Degradation (CODD) domain, we showed that pevonedistat inhibited the degradation of CODD and increased its half‐life by six‐fold in HEK293 cells. Our results demonstrate that neddylation plays a significant role in activating CRL2, and the cellular activity of CRL2VHL is better reflected by the degradation of CODD than that of HIF1α, especially under conditions where CRL2‐independent degradation of HIF1α is active.
Keywords: CODD, CRL2 ubiquitin ligase, neddylation, protein degradation, ubiquitination
1. INTRODUCTION
Ubiquitination, one of the most important posttranslational modifications, is catalyzed by sequential enzyme reactions involving E1 ubiquitin (Ub) activating enzymes, E2 conjugating enzymes, and E3 ligases. 1 , 2 As the largest family of E3 ligases, cullin‐RING ligases (CRLs) ubiquitinate ~20% of cellular proteins degraded by the ubiquitin‐proteasome system, 3 and aberrant regulations of CRLs have been found in various diseases, which provides potential therapeutic targets. 4 A CRL complex typically comprises four subunits, including a cullin, a RING protein, an adaptor protein, and an interchangeable substrate receptor. 5 Specifically, CUL2 forms a functional E3 ligase, referred to as CRL2, with the RING protein RBX1, the Elongin B/C adaptor complex, and a substrate receptor. 6 , 7 VHL is the most well‐known substrate receptor for CRL2, and its mutation has been implicated in cancers. 8 One of the canonical substrates of CRL2VHL is the hypoxia‐inducible factor 1α subunit (HIF1α), a key transcription factor responsible for cellular oxygen sensing. 9 Under normoxic conditions, HIF1α is hydroxylated by prolyl‐hydroxylase (PHD) and then recognized by CRL2VHL for ubiquitination and degradation. Under hypoxia, PHD is inhibited so that HIF1α accumulates and translocates to the nucleus.
To properly maintain protein homeostasis in cells, CRLs are tightly regulated by orchestrated pathways. 10 , 11 , 12 , 13 , 14 , 15 Posttranslational modification of cullin proteins by NEDD8, termed cullin neddylation, represents one of the most important mechanisms. Similar to the process of ubiquitin conjugation, NEDD8 is covalently conjugated to the C‐terminal WHB domain of cullin proteins through a cascade of enzymes comprising E1 NEDD8‐activating enzyme (NAE), E2 NEDD8‐conjugating enzyme, and E3 NEDD8 ligase. 16 Biochemical and structural studies have demonstrated that cullin neddylation promotes the activity of CRLs, leading to increased rates of substrate ubiquitination. 17 , 18 , 19 , 20 , 21 , 22 , 23 Interestingly, a recent CRISPR/Cas9 screen for mutations that reduce the small molecule‐induced degradation of CRL neo‐substrates identified COP9 signalosome, an enzyme that reverses NEDD8 modification, as being required for CRL4‐based but not CRL2‐based degraders. 24 This finding suggests that neddylation may exert a different role in CRL2 regulation. We thus decided to establish quantitative assays to evaluate the effect of cullin neddylation on CRL2 activities.
2. RESULTS
We firstly developed a cycloheximide (CHX) chase assay to study the degradation of HIF1α, the well‐known natural substrate of CRL2VHL. We treated HEK293 cells with desferrioxamine (DFX), an inhibitor of prolyl hydroxylase, 25 to increase the basal level of HIF1α so it could be robustly detected by Western blot (WB). After washing off DFX, cells were treated with CHX for various periods of time and were harvested in SDS sample buffer. To access the effect of cullin neddylation on HIF1α degradation, we performed the CHX chase assay in the absence and presence of pevonedistat, a small molecule inhibitor of NAE. 26 As expected, the pevonedistat treatment eliminated neddylated CUL2, but surprisingly, the degradation of HIF1α was marginally affected in the absence of cullin neddylation (Figure 1a). We repeated the CHX chase assay using HEK293 Flp‐In T‐REX cells that express exogenous FLAGHIF1α, and similarly, the degradation of FLAGHIF1α was almost unaffected by the pevonedistat treatment (Figure 1b).
FIGURE 1.
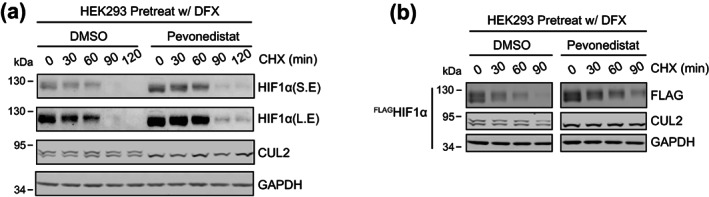
Eliminating cullin neddylation failed to inhibit the degradation of HIF1α in HEK293 cells. (a) HEK293 cells were pretreated with DFX for 5 h and pevonedistat or equivalent volume of DMSO for 1 h. After washout of inhibitors, cells were treated by CHX with or without pevonedistat for indicated time and were collected for WB analyses. (b) same as in (a) except that HEK293 cells containing tetracycline induced FLAGHIF1α were used
We rationalized two potential explanations for these unexpected results. First, the ubiquitination of HIF1α may not heavily rely on CUL2 neddylation. Because neddylation induces a conformational change in the cullin CTD and RBX1, 20 , 21 which potentially closes a gap between the CRL substrate binding site and the E2 ~ Ub bound to RBX1 5 and thus promotes substrate ubiquitination, 20 , 21 it is possible that a large substrate like HIF1α could undergo efficient ubiquitination by bridging the gap without the help of neddylation. Alternatively, it is possible that the degradation of HIF1α in HEK293 cells in the CHX chase assay was primarily cullin independent.
We tested the first possibility by comparing the rate of HIF1α ubiquitination with or without CUL2 neddylation in vitro. To obtain full‐length HIF1α protein, we expressed FLAGHIF1α in human RCC4 cells that naturally lack VHL and accumulate HIF1α in normoxia (Figure S1a,b), 9 extracted the FLAGHIF1α through immunoprecipitation and quantified its concentration by WB using recombinant FLAGIκBα as a standard (Figure S1c). The purified FLAGHIF1α was then mixed with recombinant CRL2VHL for in vitro ubiquitination. As a control, we also included a peptide substrate derived from the amino‐terminal oxygen‐dependent degradation (NODD) motif of HIF1α 27 that was used in our previous study. 28 As shown in Figure 2a,b, ubiquitination of full‐length FLAGHIF1α (~110 kDa) and the degron peptide (~4 kDa) was observed while reactions proceeded over time, only in the presence of CUL2. Importantly, the ubiquitination of both FLAGHIF1α and the degron peptide was accelerated by CUL2 neddylation, with an average 13‐ and 11‐fold increase in modification rate, respectively (Figure 2a–c). These results clearly show that, at least in vitro, neddylation promotes the CRL2VHL‐dependent substrate ubiquitination regardless of the substrate sizes. Following this experiment, we repeated the CHX chase assay for HIF1α degradation in RCC4 cells lacking VHL (VHL null) and RCC4 cells stably expressing VHL (+VHL) (Figure S1a). We firstly found that HIF1α disappeared only in RCC4 (+VHL) cells but not RCC4 (VHL null) cells (Figure 2d, upper left), suggesting that CRL2VHL‐dependent ubiquitination is the primary, if not only, mechanism for HIF1α degradation in these cells. We then asked if pevonedistat affects the degradation of HIF1α in RCC4 cells in the CHX chase assay. In contrast to results from HEK293 cells, pevonedistat inhibited the degradation of HIF1α in RCC4 (+VHL) cells and exhibited no effects on HIF1α levels in RCC4 (VHL null) cells (Figure 2d, right panel). Consistently, the degradation of exogenous FLAGHIF1α in RCC4 (+VHL) cells was also inhibited by pevonedistat (Figure 2e). These results, together with results from the in vitro assays, demonstrate that cullin neddylation is required for CRL2VHL‐dependent degradation of HIF1α.
FIGURE 2.
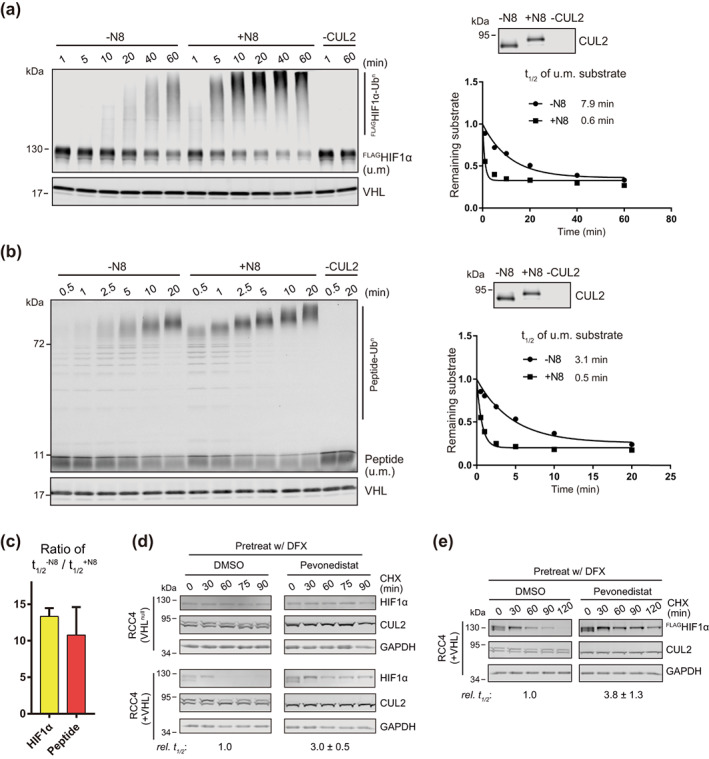
Neddylation is required for efficient CRL2VHL‐dependent degradation of HIF1α in vitro and in RCC4 (+VHL) cells. (a,b) Full‐length FLAGHIF1α purified from RCC4 (VHL null) cells stably expressing FLAGHIF1α (a) or HIF1α degron peptide (b) were ubiquitinated in vitro for indicated time with or without CUL2 neddylation. Samples without CUL2 served as negative controls. Intensities of unmodified (u.m.) FLAGHIF1α or degron peptide bands were normalized to intensities of VHL bands for regression analyses. WB of CUL2 from each group and regression curves of remaining unmodified substrates were shown on the right. (c) Average relative changes of substrate t 1/2 by neddylation from experiments in (a,b). Error bars: range of values, n = 2. (D) RCC4 (VHL null) and RCC4 (+VHL) cells were pretreated with DFX for 3 h and pevonedistat or equivalent volume of DMSO for 1 h. After washout of inhibitors, cells were treated by CHX with or without pevonedistat for indicated time and were collected for WB analyses. (e) Same assays as performed in (d) with FLAGHIF1α stably expressed in RCC4 (+VHL) cells. In (d,e), relative t 1/2 was shown as average ± range of values (n = 2)
The above findings also point out that the degradation of HIF1α in HEK293 cells observed in Figure 1 was likely due to cullin‐independent activities. To test this possibility, we treated HEK293 cells with VH‐298, a potent inhibitor for the VHL‐HIF1α interaction. 29 The VH‐298 treatment efficiently stabilized FLAGHIF1α, suggesting successful inhibition of the VHL‐HIF1α interaction (Figure 3a). However, in the CHX chase assay, FLAGHIF1α disappeared both in the absence and presence of VH‐298 (Figure 3b), indicating that HIF1α was primarily degraded through CRL2VHL‐independent pathways. As a result, the degradation of HIF1α in this assay did not reflect the activity of CRL2. We then hypothesized that, through truncating HIF1α, it is possible to remove motifs for cullin‐independent degradation and generate a substrate that depends on CRL2VHL for ubiquitination and degradation in HEK293 cells.
FIGURE 3.
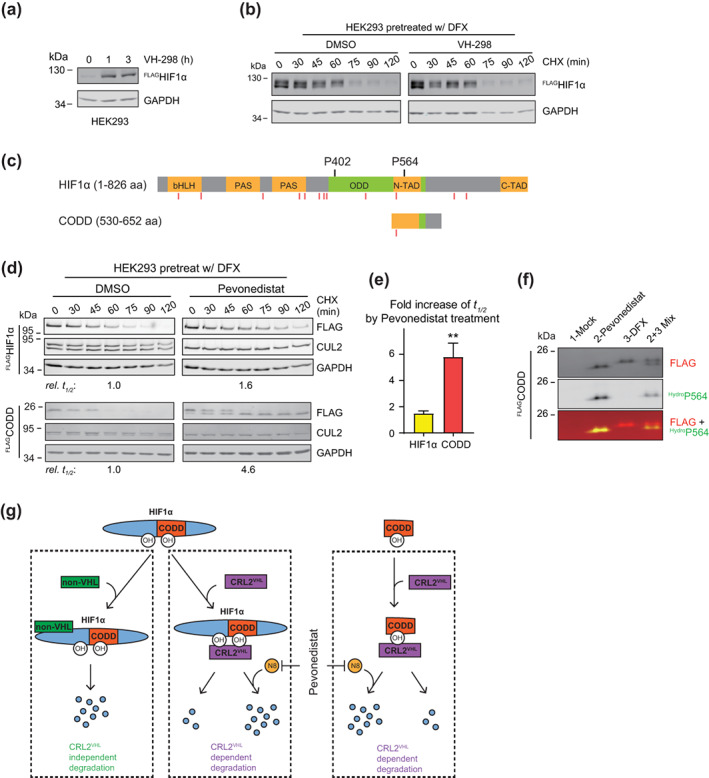
The degradation of CODD reports CRL2VHL activity in HEK293 cells in which VHL‐independent degradation of full‐length HIF1α is active. (a) HEK293 cells containing tetracycline induced FLAGHIF1α were treated by 100 μM VH‐298 for indicated time and were collected for WB analyses. (b) The same cells as in (a) were pretreated with DFX for 5 h and VH‐298 or equivalent volume of DMSO for 1 h. After washout of inhibitors, cells were treated by CHX with or without VH‐298 for indicated time and were collected for WB analyses. (c) Schematic illustration of the CODD truncation of HIF1α. Red vertical lines denote ubiquitination sites reported in GGBase. (d) Same assays as in Figure 1b with HEK293 cells containing tetracycline induced FLAGHIF1α or FLAGCODD. Relative t 1/2 of FLAGHIF1α or FLAGCODD elimination are shown below the corresponding blot. (e) Average changes in relative t 1/2 from experiments in (d). Error bars: SEM, n = 3, p < 0.01. (f) Unmodified (upper band) and hydroxylated (lower band) FLAGCODD separated on a 12.5% SDS‐PAGE gel. HEK293 cells with tetracycline induced expression of FLAGCODD were treated for 4 h with DMSO (Lane 1), or pevonedistat (Lane 2), or DFX (Lane 3), and were collected WB analyses. Equal volumes of samples in Lanes 2 and 3 were mixed and included in Lane 4. The bottom blot shows overlay of signals from FLAG and hydroxy‐HIF‐1α (Pro564) antibodies. (g) Summary of HIF1α degradation machineries. CRL2VHL binds the hydroxylated ODD domain of HIF1α or the hydroxylated CODD truncation to catalyze their ubiquitination and degradation. This CRL2VHL‐dependent degradation is accelerated by neddylation and can be inhibited by pevonedistat. CRL2VHL‐independent degradation acts through regions other than the HIF1α CODD domain and cannot be inhibited by pevonedistat
Essential to the binding of HIF1α to VHL is the hydroxylation of Pro402 and Pro564 in the ODD domain 30 (Figure 3c). We expressed the carboxy‐terminal ODD (CODD) domain (Figure 3c, S2) in HEK293 Flp‐In T‐REx cells and monitored its degradation in the CHX chase assay. Like FLAGHIF1α, FLAGCODD disappeared rapidly after the addition of CHX (Figure 3d, left panel). The pevonedistat treatment increased the half‐life (t 1/2) of FLAGHIF1α by ~50% (Figure 3d, e). In contrast and consistent with results in Figure 2, inhibition of neddylation led to much slower degradation of FLAGCODD (Figure 3d), with an average 5.8‐fold increase in t1/2 (Figure 3e). Notably, only a lower molecular weight band, representing the hydroxylated FLAGCODD 31 (Figure 3f), persisted in pevonedistat‐treated cells, while the upper band representing the nonhydroxylated FLAGCODD (Figure 3F) disappeared at a rate similar to that in cells with no pevonedistat (Figure 3d). These results suggest that the pevonedistat treatment does not change the rate at which CODD is hydroxylated, and once hydroxylated, CODD is quickly ubiquitinated and degraded in the presence of ongoing CUL2 neddylation. Taken together, we conclude that the degradation of CODD serves as a better reporter for CRL2VHL activity than that of full length HIF1α in HEK293 cells, one of the most heavily used cell lines in life sciences.
3. DISCUSSION
Biochemical and structural studies have provided important insights into the impact of neddylation on the activity and conformation of CRLs such as CRL1 and CRL5. 17 , 18 , 19 , 20 , 32 , 33 , 34 Given that various substrate receptor modules and substrates are engaged by CRLs, 3 , 35 , 36 considerable structural diversity across CRLs exists, and therefore, to what extent neddylation modulates the activity of individual CRLs remains an open question. Here, we developed quantitative assays to study the effect of neddylation on CRL2VHL‐mediated ubiquitination and degradation. Using RCC4 (+VHL) cells as the experimental model, we found that cullin neddylation promoted the CRL2VHL‐dependent degradation of HIF1α. Using in vitro assays, we showed that regardless of significant difference in substrate sizes, neddylation resulted in >10‐fold increase in rates of CRL2VHL substrate ubiquitination, an effect consistent with previous findings using CRL1. 33 , 34
Unexpectedly, our CHX chase assay revealed little effect of neddylation inhibition on the degradation rate of HIF1α in HEK293 cells. We found this was because the CRL‐independent pathway predominated in HIF1α degradation in HEK293 when HIF1α accumulated to a higher level (Figure 3). Consistent with this explanation, VHL‐independent degradation has been reported previously, 30 , 37 , 38 , 39 , 40 , 41 and pevonedistat failed to increase HIF1α levels in several gastric cancer cell lines that contain higher basal levels of HIF1α. 42 The activity of this CRL‐independent HIF1α degradation varies in different cell lines, as it was not detected in RCC4 cells (Figure 2d). In light of these findings, we suggest that when quantifying rates of HIF1α degradation using a CHX chase assay, pevonedistat treatment should be included as a control to access if HIF1α is primarily degraded by the CRL‐dependent pathway in the cell line and the experimental condition used. If pevonedistat fails to inhibit HIF1α degradation, indicating CRL‐independent degradation predominates, then we suggest using the CODD truncation of HIF1α as an alternative reporter for CRL2VHL activity (Figure 3g). The t 1/2 of CODD was significantly increased upon neddylation inhibition (Figure 3e), and furthermore, hydroxylated CODD migrated faster than unmodified CODD on a 12.5% SDS‐PAGE gel (Figure 3f). These features not only make CODD a relevant model for studying CRL2‐dependent protein degradation, but also provide the advantage of monitoring the CODD hydroxylation rate in the same assay.
4. MATERIALS AND METHODS
4.1. Generation of stable cell lines
HEK293 Flp‐In T‐REx cells (Thermo Fisher Scientific) stably integrating FLAGHIF1α and FLAGCODD were generated as described previously. 15 The expression of integrated genes were induced by 2 μg/ml tetracycline for 24 h. RCC4 (VHL null) and RCC4 (+VHL) cells (Sigma‐Aldrich) overexpressing FLAGHIF1α were generated via lentivirus‐mediated integration as described previously. 15
4.2. Antibodies
Antibodies for HIF1 α (Novus, NB100‐449), CUL2 (Thermo Fisher, 51–1800), GAPDH (Santa Cruz, sc‐47724), FLAG (Sigma‐Aldrich, F1804), VHL (Cell Signaling Technology, 68547S), and hydroxy‐HIF‐1α (Pro564) (Cell Signaling Technology, 3434S) were used in this study. Primary antibodies used were shown next to each blot. All FLAGHIF1α and FLAGCODD were blot with FLAG antibody, except that hydroxy‐HIF‐1α (Pro564) antibody was also used to detect hydroxylated FLAGCODD (Figure 3f).
4.3. HIF1α and CODD degradation assay
Cells were grown in 6‐well plates and treated with 200 μM DFX for indicated time to stabilize HIF1α, FLAGHIF1α, or FLAGCODD. Pevonedistat (10 μM, MedChemExpress), or VH‐298 (100 μM, Tocris Bioscience), or DMSO was added to the medium for 1 h and was then washed out. To monitor protein degradation, 60 μg/ml CHX along with Pevonedistat (1 μM) or VH‐298 (100 μM) or DMSO was added to the medium. At different time points post the start of the CHX treatment, cells were washed with PBS and lysed in 2× SDS sample buffer. Cell lysates were collected, sonicated, and fractionated by SDS‐PAGE for WB analyses. FLAG or HIF1α signals were measured using the Image Studio software (LI‐COR Biosciences), normalized to GAPDH signals from the same sample, and fit to a single exponential in Prism to calculate half‐lives.
4.4. Preparation of FLAGHIF1α protein for in vitro ubiquitination
RCC4 (VHL null) cells stably expressing FLAGHIF1α were lysed by sonication in Pierce IP lysis buffer (ThermoFisher Scientific) containing 1 mM DTT and 1× protease inhibitor cocktail (PIC, Roche). The cell lysates were cleared by centrifugation at 15,000 g for 15 min, and the resulting supernatant was incubated with anti‐FLAG beads (Sigma‐Aldrich) for 2 h at 4°C. After washing the beads three times, bound FLAGHIF1α was eluted with 3× FLAG peptide (Sigma‐Aldrich) and was concentrated using an Amicon Ultra‐4 Centrifugal Filter (Millipore). To determine the concentration of the purified FLAGHIF1α, serial dilutions of recombinant FLAGIκBα with a known concentration, along with diluted aliquots of the purified FLAGHIF1α, were analyzed using WB with the FLAG antibody. The concentration of FLAGHIF1α was then calculated by fitting the WB signal to the standard curve of FLAGIκBα. Recombinant FLAGIκBα was generated by expressing FLAGIκBα on a pGEX vector in E. coli cells, followed by purification with glutathione agarose beads, thrombin cleavage, and gel filtration chromatography. 28 , 43 , 44
4.5. In vitro neddylation and ubiquitination
Neddylation reactions were performed for 1 h at room temperature in 1× reaction buffer [30 mM Tris (pH 7.5), 5 mM MgCl2, 2 mM DTT, 2 mM ATP and 1× PIC (Roche)] containing 0.25 μM NAE, 3 μM UBC12, 0.2 μM CUL2•RBX1 (R&D Systems) and 0.2 μM NEDD8 (R&D Systems). NEDD8 was opted out for the no neddylation group. CUL2•RBX1 was opted out for the negative control. In vitro ubiquitination was performed at room temperature in 1× reaction buffer containing 0.5 μM Ub‐E1, 0.2 μM Cdc34, 30 μM ubiquitin, 0.1 μM CUL2•RBX1, 0.1 μM VHL•ElonginB/C, and 0.1 μM purified FLAGHIF1α or 0.1 μM TAMRA labeled HIF1α degron peptide [Ac‐KLRREPDALTLLA‐(hydroxylated‐P)‐AAGDTIISLDFGSNGRRASYK(TAMRA)‐amide]. 28 At indicated time points, aliquots of the reaction mixtures were withdrawn and mixed with 4× SDS sample buffer. Samples were fractionated with SDS‐PAGE and analyzed by WB on an Odyssey Imager (LI‐COR Biosciences), or by fluorescence scan (Typhoon scanner). Levels of unmodified FLAGHIF1α or peptide were normalized to levels of VHL and then fit to a single exponential in Prism to calculate half‐lives.
FUNDING
This study was supported by National Institutes of Health grant R35 GM138016 (to Xing Liu) and American Heart Association Career Development Award (to Xing Liu).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Kankan Wang: Data curation (equal); formal analysis (lead); investigation (lead); methodology (lead); validation (lead); writing – original draft (lead); writing – review and editing (supporting). Kurt M. Reichermeier: Conceptualization (supporting); data curation (supporting); investigation (supporting); methodology (supporting); writing – original draft (supporting). Xing Liu: Conceptualization (lead); data curation (equal); formal analysis (supporting); funding acquisition (lead); methodology (supporting); project administration (lead); supervision (lead); writing – review and editing (lead).
Supporting information
FIGURE S1 Expression and purification of FLAGHIF1α in RCC4 cells. (a) RCC4 (VHL null) and RCC4 (+VHL) cells were treated with DMSO or pevonedistat for 2 h and analyzed by WB with VHL and GAPDH antibodies. (b) The indicated RCC4 cells were infected with lentiviruses expressing FLAGHIF1α, treated with DFX for the indicated time periods, and lysed in 2× SDS sample buffer for WB analyses with FLAG and GAPDH antibodies. (c) Concentration of FLAGHIF1α protein stock purified from RCC4 cells was quantified for the in vitro ubiquitination assay. FLAGHIF1α was immunoprecipitated from RCC4 (VHL null) cells as shown in (b), eluted with 3× FLAG peptide, concentrated and aliquoted. Recombinant FLAGIκBα with a known concentration was serially diluted and analyzed by WB together with purified FLAGHIF1α. The concentration of diluted FLAGHIF1α stock were determined by fitting the intensity of the FLAGHIF1α band to the standard curve of FLAGIκBα. Number in brackets is the calculated value.
FIGURE S2. Amino acid sequence of CODD. Related to Figure 3c‐f.
ACKNOWLEDGMENTS
We thank Dr. Jörg Klug and Dr. Andreas Meinhardt (Justus‐Liebig Universität, Giessen, Germany) for their mentorship to Kurt M. Reichermeier. We thank Dr. Raymond Deshaies and Dr. Rati Verma (Amgen Inc., formerly California Institute of Technology) for critical reading of the manuscript.
Wang K, Reichermeier KM, Liu X. Quantitative analyses for effects of neddylation on CRL2VHL substrate ubiquitination and degradation. Protein Science. 2021;30:2338–2345. 10.1002/pro.4176
Funding information National Institutes of Health, Grant/Award Number: R35GM138016; American Heart Association, Grant/Award Number: 20CDA35270030
REFERENCES
- 1. Hershko A. The ubiquitin system for protein degradation. Annu Rev Biochem. 1992;61:761–807. [DOI] [PubMed] [Google Scholar]
- 2. Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–387. [DOI] [PubMed] [Google Scholar]
- 3. Petroski MD, Deshaies RJ. Function and regulation of cullin‐RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. [DOI] [PubMed] [Google Scholar]
- 4. Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase‐targeted therapies. Nat Rev Drug Discov. 2014;13:889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng N, Schulman BA, Song L, et al. Structure of the Cul1‐Rbx1‐Skp1‐F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. [DOI] [PubMed] [Google Scholar]
- 6. Nguyen HC, Yang H, Fribourgh JL, Wolfe LS, Xiong Y. Insights into Cullin‐RING E3 ubiquitin ligase recruitment: Structure of the VHL‐EloBC‐Cul2 complex. Structure. 2015;23:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cardote TAF, Gadd MS, Ciulli A. Crystal structure of the Cul2‐Rbx1‐EloBC‐VHL ubiquitin ligase complex. Structure. 2017;25:901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 2015;15:55–64. [DOI] [PubMed] [Google Scholar]
- 9. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature. 1999;399:271–275. [DOI] [PubMed] [Google Scholar]
- 10. Wang K, Deshaies RJ, Liu X. Assembly and regulation of CRL ubiquitin ligases. Adv Exp Med Biol. 2020;1217:33–46. [DOI] [PubMed] [Google Scholar]
- 11. Schmidt MW, McQuary PR, Wee S, Hofmann K, Wolf DA. F‐box‐directed CRL complex assembly and regulation by the CSN and CAND1. Mol Cell. 2009;35:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pierce NW, Lee JE, Liu X, et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell. 2013;153:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu S, Zhu W, Nhan T, Toth JI, Petroski MD, Wolf DA. CAND1 controls in vivo dynamics of the cullin 1‐RING ubiquitin ligase repertoire. Nat Commun. 2013;4:1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zemla A, Thomas Y, Kedziora S, et al. CSN‐ and CAND1‐dependent remodelling of the budding yeast SCF complex. Nat Commun. 2013;4:1641. [DOI] [PubMed] [Google Scholar]
- 15. Liu X, Reitsma JM, Mamrosh JL, Zhang Y, Straube R, Deshaies RJ. Cand1‐mediated adaptive exchange mechanism enables variation in F‐Box protein expression. Mol Cell. 2018;69:773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rabut G, Peter M. Function and regulation of protein neddylation. 'Protein modifications: Beyond the usual suspects' review series. EMBO Rep. 2008;9:969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu K, Chen A, Pan ZQ. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1‐CUL1 complex to promote ubiquitin polymerization. J Biol Chem. 2000;275:32317–32324. [DOI] [PubMed] [Google Scholar]
- 18. Read MA, Brownell JE, Gladysheva TB, et al. Nedd8 modification of Cul‐1 activates SCFbeta TrCP‐dependent ubiquitination of Ikappa Balpha. Mol Cell Biol. 2000;20:2326–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morimoto M, Nishida T, Honda R, Yasuda H. Modification of cullin‐1 by ubiquitin‐like protein Nedd8 enhances the activity of SCF(skp2) toward p27(kip1). Biochem Biophys Res Commun. 2000;270:1093–1096. [DOI] [PubMed] [Google Scholar]
- 20. Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin‐RING ligases: Conformational control of conjugation. Cell. 2008;134:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saifee NH, Zheng N. A ubiquitin‐like protein unleashes ubiquitin ligases. Cell. 2008;135:209–211. [DOI] [PubMed] [Google Scholar]
- 22. Ohh M, Kim WY, Moslehi JJ, et al. An intact NEDD8 pathway is required for Cullin‐dependent ubiquitylation in mammalian cells. EMBO Rep. 2002;3:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boh BK, Smith PG, Hagen T. Neddylation‐induced conformational control regulates cullin RING ligase activity in vivo. J Mol Biol. 2011;409:136–145. [DOI] [PubMed] [Google Scholar]
- 24. Mayor‐Ruiz C, Jaeger MG, Bauer S, et al. Plasticity of the Cullin‐RING ligase repertoire shapes sensitivity to ligand‐induced protein degradation. Mol Cell. 2019;75:849–858. [DOI] [PubMed] [Google Scholar]
- 25. Kageyama Y, Koshiji M, To KK , Tian YM, Ratcliffe PJ, Huang LE. Leu‐574 of human HIF‐1alpha is a molecular determinant of prolyl hydroxylation. FASEB J. 2004;18:1028–1030. [DOI] [PubMed] [Google Scholar]
- 26. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8‐activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. [DOI] [PubMed] [Google Scholar]
- 27. Hon WC, Wilson MI, Harlos K, et al. Structural basis for the recognition of hydroxyproline in HIF‐1 alpha by pVHL. Nature. 2002;417:975–978. [DOI] [PubMed] [Google Scholar]
- 28. Diaz S, Li L, Wang K, Liu X. Expression and purification of functional recombinant CUL2*RBX1 from E. coli . Sci Rep. 2021;11:11224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frost J, Galdeano C, Soares P, et al. Potent and selective chemical probe of hypoxic signalling downstream of HIF‐alpha hydroxylation via VHL inhibition. Nat Commun. 2016;7:13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yee Koh M, Spivak‐Kroizman TR, Powis G. HIF‐1 regulation: Not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. [DOI] [PubMed] [Google Scholar]
- 31. MacKenzie ED, Selak MA, Tennant DA, et al. Cell‐permeating alpha‐ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase‐deficient cells. Mol Cell Biol. 2007;27:3282–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Podust VN, Brownell JE, Gladysheva TB, et al. A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proc Natl Acad Sci U S A. 2000;97:4579–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baek K, Krist DT, Prabu JR, et al. NEDD8 nucleates a multivalent cullin‐RING‐UBE2D ubiquitin ligation assembly. Nature. 2020;578:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin‐RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rusnac DV, Zheng N. Structural biology of CRL ubiquitin ligases. Adv Exp Med Biol. 2020;1217:9–31. [DOI] [PubMed] [Google Scholar]
- 37. Minet E, Mottet D, Michel G, et al. Hypoxia‐induced activation of HIF‐1: role of HIF‐1α‐Hsp90 interaction. FEBS Lett. 1999;460:251–256. [DOI] [PubMed] [Google Scholar]
- 38. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau‐independent hypoxia‐inducible factor‐1 alpha‐degradative pathway. J Biol Chem. 2002;277:29936–29944. [DOI] [PubMed] [Google Scholar]
- 39. Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF‐1alpha and is required for O(2)‐independent and HSP90 inhibitor‐induced degradation of HIF‐1alpha. Mol Cell. 2007;25:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia‐inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species‐dependent. J Biol Chem. 2011;286:6963–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flugel D, Gorlach A, Kietzmann T. GSK‐3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28‐dependent degradation of HIF‐1alpha. Blood. 2012;119:1292–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lan H, Tang Z, Jin H, Sun Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci Rep. 2016;6:24218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reitsma JM, Liu X, Reichermeier KM, et al. Composition and regulation of the cellular repertoire of SCF ubiquitin ligases. Cell. 2017;171:1326–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garsamo M, Zhou Y, Liu X. Using in vitro fluorescence resonance energy transfer to study the dynamics of protein complexes at a millisecond time scale. J Vis Exp. 2019;14:145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Expression and purification of FLAGHIF1α in RCC4 cells. (a) RCC4 (VHL null) and RCC4 (+VHL) cells were treated with DMSO or pevonedistat for 2 h and analyzed by WB with VHL and GAPDH antibodies. (b) The indicated RCC4 cells were infected with lentiviruses expressing FLAGHIF1α, treated with DFX for the indicated time periods, and lysed in 2× SDS sample buffer for WB analyses with FLAG and GAPDH antibodies. (c) Concentration of FLAGHIF1α protein stock purified from RCC4 cells was quantified for the in vitro ubiquitination assay. FLAGHIF1α was immunoprecipitated from RCC4 (VHL null) cells as shown in (b), eluted with 3× FLAG peptide, concentrated and aliquoted. Recombinant FLAGIκBα with a known concentration was serially diluted and analyzed by WB together with purified FLAGHIF1α. The concentration of diluted FLAGHIF1α stock were determined by fitting the intensity of the FLAGHIF1α band to the standard curve of FLAGIκBα. Number in brackets is the calculated value.
FIGURE S2. Amino acid sequence of CODD. Related to Figure 3c‐f.