

Abstract

An implementation of the Interacting Quantum Atoms method for crystals is presented. It provides a real space energy decomposition of the energy of crystals in which all energy components are physically meaningful. The new package ChemInt enables one to compute intra-atomic and inter-atomic energies, as well as electron population measures used for quantitative description of chemical bonds in crystals. The implementation is tested and applied to characteristic molecular and crystalline systems with different types of bonding.
Introduction
Nowadays, many interesting materials, e.g., intermetallic compounds, belong to the families of chemical systems, where the usual valence approaches based on pure ionicity, covalency with 2-center 2-electron bonds, or “metallic bonding” are no longer valid. The interplay of all these features challenges our understanding with the emergence of new bonding scenarios beyond the well-accepted concepts.1−6 Since chemical bonding analysis represents a way to understand inter-atomic interactions, it is also a way to understand material properties and their relations to specific electron counts or partial structures. For this sake, chemical bonding parameters like atomic charges and bond orders must be consistently related to their energetic counterparts.
This is achieved in the Interacting Quantum Atoms (IQA) method.7−9 It represents an ideal framework to investigate covalent and ionic interactions on an equal footing. The sum of the individual atomic interaction energies forms a part of the recoverable total energy of the system. All routinely used quantum mechanical packages yield the total electronic energy. Usually this energy is given in terms of kinetic and potential energy contributions. Additionally, the potential energy is split into Coulomb and exchange-correlation parts. For deeper insight into the mutual interactions between the atoms, a suitable analysis of the total energy, respectively the wave function and its components, must be performed. A number of tools and methods are available for this task, but only the periodic energy decomposition analysis (pEDA)10 has been used to decompose the total energy of extended systems. The decomposition is done in Hilbert space. Also the Crystal Orbital Hamilton Populations (COHP)11 analysis does the decomposition in Hilbert space but decomposes the band structure energy instead. An alternative and appealing method to decompose the total energy components into contributions of and between position-space atomic regions is the approach of IQA. Unique features of IQA are that it is orbital invariant, methodologically independent of the type of basis set, applicable to correlated wave functions, and does not depend on an external reference state.
The IQA scheme is based on the idea that the one- and two-electron energy integrals computed over the total coordinate space can be evaluated separately over any set of nonoverlapping spatial domains. The connection to a sound chemical description is achieved by the utilization of domains defined by the Quantum Theory of Atoms in Molecules (QTAIM), i.e., by the atomic basins determined by the density gradient field.12 With this, by successive integration of the one-electron components over each atomic basin, the total one-electron energy is decomposed into atomic contributions. The contributions describe the kinetic energy of the atomic domain as well as the attractive energy between the electrons and the nucleus enclosed within the atomic domain. The integration of the two-electron energy components over the atomic basins yields the intra-atomic and inter-atomic contributions that are used in the bonding analysis to judge the ionic and covalent bonding character. The whole procedure was adopted for molecular wave functions based on the Gauss-type of basis sets in the program Promolden.13
Until now, the IQA method has not been implemented for solids. Nevertheless, on the basis of careful calibration studies using a zeroth order approximation of the full IQA interaction energies, the so-called point-charge approximation of the covalent interaction energy and the ionic interaction energy (Madelung energy), was employed to understand chemical bonding and site occupation in ternary half-Heusler (MgAgAs-type) phases.5 Finally, this scheme was employed to predict new phases with this kind of structure.6 This may show the strength and scientific potential of the solid-state implementation of the exact methodology to be presented below.
The developed ChemInt software is coupled with the DGrid package.14 This extends the capability to evaluate the molecular Gauss-type orbitals also to the utilization of Slater-type based molecular wave functions, as used in the ADF package,15 as well as to access the numerical atomic orbitals expanding the wave functions for molecular and crystalline solid state systems computed with the FHI-aims code.16 ChemInt is also capable to compute the delocalization indices for all those types of basis sets, including crystalline systems.17 In particular, the bielectronic exchange energy integrals between position-space atomic regions for solid state wave functions are extremely challenging due to the huge number of participating orbitals, which made parallelization of ChemInt a necessary feature.
Theory and implementation of IQA for crystalline solids are outlined, reliability of the code is tested with an extensive set of molecules, and the method is applied to prototype solid-state compounds exhibiting different bonding scenarios.
Methods
The total electronic energy of a molecule or solid with pairwise Coulomb interactions comprises the kinetic energy of the electrons T, electron–nucleus interactions Ene, electron–electron interactions Eee, and nucleus–nucleus interactions Enn
| 1 |
Given access to the first order density matrix ρ1(r; r′) and the pair density ρ2(r, r′), the energy can be written as
| 2 |
Suppose that the space is partitioned into a set of domains that exhaustively recover the total volume and are mutually exclusive. Then, the total energy can be decomposed as
| 3 |
The above energy terms can be written in compact form as the sum of domain self-energies and interdomain energies,
| 4 |
Domain self-energies are
| 5 |
| 6 |
where α ∈ A stands for all nuclei enclosed in domain A, and
| 7 |
Interdomain energies are
| 8 |
| 9 |
and
| 10 |
Notice that intradomain electron–electron terms are halved to represent total energies whereas interdomain terms represent nonequivalent interaction energies.
The approach to the energy decomposition given by eq 4 considers the crystal to be formed by interacting atoms, molecules, etc. One may, otherwise, take a reference unit formed by one or more domains, embedded in the crystal. We collect, in an additive manner, E = ∑GE(G), all energy contributions E(G) of reference unit G,
| 11 |
The interaction energy for reference unit G shall be organized by coordination spheres i,
| 12 |
where there are 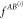 number of B neighbors
in the coordination sphere i of domain A of the reference unit. The last term condenses the sum over the
coordination spheres for each domain in the reference unit into a
single sum over coordination shells j for all domains
in the reference unit and, with
number of B neighbors
in the coordination sphere i of domain A of the reference unit. The last term condenses the sum over the
coordination spheres for each domain in the reference unit into a
single sum over coordination shells j for all domains
in the reference unit and, with 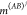 interaction
contributions
interaction
contributions  within that shell. The multiplicity factor
within that shell. The multiplicity factor  for
the shell of those interactions (i.e.,
Li–Cl(1) or Cl–Li(1)) takes the
form
for
the shell of those interactions (i.e.,
Li–Cl(1) or Cl–Li(1)) takes the
form
| 13 |
where
| 14 |
and nA, nB are the numbers of symmetry equivalent atoms A and B in the reference unit, respectively.
The pair density can be viewed as the product of two quasi-independent densities minus a term accounting for the difference with the true pair distribution ρ2(r, r′) = ρ(r)ρ(r′) – ρxc(r, r′).18 This leads in eq 8 to the Coulomb energy contribution
| 15 |
and the exchange-correlation energy contribution
| 16 |
With the exception of the exchange-correlation energy, all interdomain terms are classical. Thus, the interaction energy can be arranged as a sum of a classical electrostatic energy EclAB and a covalent energy Exc,
| 17 |
In the special case of the monodeterminantal wave function, the exchange-correlation density is directly given by the first order density matrix, ρxc = ρx = |ρ1(r; r′)|2.
Atomic Boundaries
Atomic domains defined by QTAIM theory are delimited by surfaces S that satisfy the zero-flux condition
| 18 |
where ρ(r) is the density at point r in the surface and n(r) is a normal vector to the surface. The manifold of all steepest ascent paths
| 19 |
terminating at given attractor (the common destination limt→∞r(t)) constitutes an atomic (QTAIM) basin. In the following, the QTAIM basins are used as the spatial domains over which the energetical decomposition is performed.
We note at this point that a representation of the surface compatible with atomic-centered grids is required to use the multi/bipolar expansions presented in the following sections. To alleviate the expense of the computation, we precomputed a trust sphere using the algorithm described by Rodríguez et al.19
Of fundamental relevance for solid state systems is achieving electroneutrality in the unit cell. In contrast to molecules, all atomic basins determined for a solid state system have finite volume. Thus, the atomic basins must recover the unit cell volume, a circumstance that seems to be problematic in case of atom-centered radial grids. We decided to use radial rays with symmetric spherical t-designs.20
Energy Integrals in the IQA Framework
The energy integrals for a system with perfect periodicity are performed over complex crystal orbitals ϕn,k for given band index n and reciprocal vector k
with R given by the unit cell vectors and the linear combination ψn,k(r – R) = ∑jcn,j(k) χj(r – aj) of atomic orbitals χj(r – aj), centered at aj = rj + R, and with coeficients cn,j(k) to be determined. In this paper, electronic structure calculations were done with FHI-aims, utilizing numerical atomic orbitals.
The band and translational symmetry labels
are condensed here to a single label i ≡ (n, k). With this, the crystal
orbitals ϕi determine the electron
density  as well as exchange
pair density
as well as exchange
pair density  , where
, where 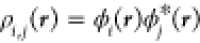 are overlap densities.
are overlap densities.
The density is replaced in eq 15, and the exchange pair density in eq 16,
| 20 |
with domains being now QTAIM atoms. Recall
from eq 7 that B = A integrals have a 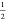 prefactor.
prefactor.
Intra-atomic Coulomb integrals (A = B) are computed with the Laplace expansion of 1/|r – r′| around the position of the atomic nucleus7
| 21 |
where 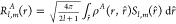 is
the angular (r̂ = (θ,ϕ)) integral
of the atomic density
is
the angular (r̂ = (θ,ϕ)) integral
of the atomic density
| 22 |
weighted by real spherical
harmonic Sl,m. The discriminant 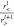 , with r< = min(r, r′) and r> = max(r, r′), ensures
the convergence of the expansion.
, with r< = min(r, r′) and r> = max(r, r′), ensures
the convergence of the expansion.
The expansion is valid also for the exchange integral7
| 23 |
where  . Atomic overlap densities ρi,jA are zero outside of the basin
like the atomic density above.
. Atomic overlap densities ρi,jA are zero outside of the basin
like the atomic density above.
The integral evaluation is more
efficiently performed in the basis
of overlap densities 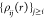 . The exchange term is diagonal in this
basis with coefficients dij = 2(2 – δij) for
monodeterminantal wave functions,9 and
no monadic diagonalization is required.
. The exchange term is diagonal in this
basis with coefficients dij = 2(2 – δij) for
monodeterminantal wave functions,9 and
no monadic diagonalization is required.
Two-center integrals (A ≠ B) are expanded around two poles, one at each atomic position, with a bipolar expansion21−23
| 24 |
where 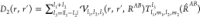 replaces
replaces 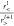 as the discriminant.7
as the discriminant.7
The exchange integral between two different basins is
| 25 |
Once the electronic structure problem
is solved by whatever method,
the self-energy of each atom (or fragment) in the unit cell is obtained.
The interaction energy of each of them, e.g., A,
with the rest of atoms of the lattice, appropriately classified by
increasing distance, B, is obtained (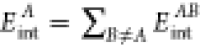 ). This infinite sum contains quickly
convergent
terms ExAB, which decay exponentially in insulators
and polynomially in metals, and a potentially divergent series made
up of Ecl electrostatic interactions.
By separating Ecl into a short-range,
penetration-like exponentially decaying term and a multipolar one
that is summed via the Ewald construction, one gets rid of divergences.
We have applied the Ewald construction with the QTAIM monopoles (net
atomic charges) via the Environ program24 and have summed directly the higher order multipolar corrections
from the IQA integrations up to near neighbors. Even if the QTAIM
monopoles are zero, those terms remain and constitute the well-known
electrostatic interaction between uncharged atoms or molecules.
). This infinite sum contains quickly
convergent
terms ExAB, which decay exponentially in insulators
and polynomially in metals, and a potentially divergent series made
up of Ecl electrostatic interactions.
By separating Ecl into a short-range,
penetration-like exponentially decaying term and a multipolar one
that is summed via the Ewald construction, one gets rid of divergences.
We have applied the Ewald construction with the QTAIM monopoles (net
atomic charges) via the Environ program24 and have summed directly the higher order multipolar corrections
from the IQA integrations up to near neighbors. Even if the QTAIM
monopoles are zero, those terms remain and constitute the well-known
electrostatic interaction between uncharged atoms or molecules.
Approximations Based on Population Measures
Inter-atomic bielectronic integrals are a particularly expensive step. As seen in eqs 24 and 25, they involve a bipolar expansion. For classic electrostatic energy integrals of distant basins it is equivalent to use a multipolar expansion25
| 26 |
where
| 27 |
is a real multipole moment
of the net charge
density ρt(r) = ∑AZAδ(r – RA) – ρ(r) in basin A. 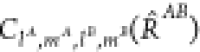 is an angular factor (cf. eq 25 in ref (7)). Technically, this approximation
is invalid if the circumsphere of the basin A (centered
at the nuclear position) intersects the circumsphere of basin B (with center at nucleus B). As a remark,
solid state systems normally have compact basins. This limits the
extent of the spheres, and it is safe to say that classic interactions
with neighbor atoms of the second and higher coordination spheres
are well approximated with a multipolar expansion in these cases.
Also, internuclear distances in the solid state systems are often
large, exceeding 2 Å.
is an angular factor (cf. eq 25 in ref (7)). Technically, this approximation
is invalid if the circumsphere of the basin A (centered
at the nuclear position) intersects the circumsphere of basin B (with center at nucleus B). As a remark,
solid state systems normally have compact basins. This limits the
extent of the spheres, and it is safe to say that classic interactions
with neighbor atoms of the second and higher coordination spheres
are well approximated with a multipolar expansion in these cases.
Also, internuclear distances in the solid state systems are often
large, exceeding 2 Å.
On the other hand, exchange integrals were proven numerically to lead to asymptotic convergent multipolar expansions, with low-order terms being a good approximation for second and further nearest neighbors.26 The appropriate expression has the same structure as for classic interactions
| 28 |
but in terms of the sum of overlap density multipoles
| 29 |
Further approximation of bielectronic integrals is to consider only the first term in the multipolar expansion. For the classic electrostatic term it is the same as assuming spherical density distribution in the atomic domain. By Gauss law, a point charge located at the nucleus position (for A and B) produces a physically equivalent classic force. The classic electrostatic energy,
| 30 |
is proportional
to the product of QTAIM net
charges  for both basins, where
for both basins, where 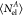 is the average electron population in basin A.
is the average electron population in basin A.
Inter-atomic exchange integrals also admit a similar expression26
| 31 |
that instead involves the delocalization index (DI) δAB between the atomic domains A and B
| 32 |
The delocalization index measures the number of electrons shared between domains (A and B) and is complemented by the localization index (LI)
| 33 |
Localization and delocalization indices must obey the following sum rule
| 34 |
The fluctuation of electron population σ2(A) in basin A is the result of electron pair sharing with atoms of the first coordination shell σ12(A), second coordination shell σ2(A), and so on.
Expressions 30 and 31 are the basis for our more generic scaled point-charge approximation (sPCA) for classic ionic and exchange (covalent) inter-atomic interactions. The scaling parameters scl and sx approximate the exact classic and exchange energy, respectively, with a scaled zeroth order multipolar approximation:
| 35 |
Computational Details
Density functional theory calculations (PBE functional27) for crystalline solids were performed with FHI-aims, an all-electron full-potential electronic-structure code. The numeric atom-centered basis sets used were of standard “tight” type. The computed unit cells and Brillouin zone samplings are listed in Table 1. An external output option of FHI-aims (version 200112) is used to generate DGrid compatible wave function information. ChemInt interfaces DGrid to evaluate wave function properties at discrete points in space, determines the atomic QTAIM basins, and performs the IQA analysis with this space partitioning.
Table 1. Crystallographic Information and Brillouin Zone Sampling for the Calculated Materialsa.
| system | space group type | lattice parameters [Å] | calculated cell | k-point mesh |
|---|---|---|---|---|
| β-N2b | P63/mmc | a = 4.050c | 1 × 1 × 1 | 5 × 5 × 3 |
| c = 6.604 | ||||
| CO2 | Pa3̅ | a = 5.494235 | 1 × 1 × 1 | 4 × 4 × 4 |
| diamond | Fd3̅m | a = 3.56660636 | 1 × 1 × 1 | 4 × 4 × 4 |
| BN (zincblende) | F4̅3m | a = 3.6137 | 1 × 1 × 1 | 4 × 4 × 4 |
| graphite | P63/mmc | a = 2.46438 | 2 × 2 × 1 | 4 × 4 × 3 |
| c = 6.711 | ||||
| BN-b | P63/mmc | a = 2.50432339 | 2 × 2 × 1 | 4 × 4 × 3 |
| c = 6.658852 | ||||
| MgB2 | P6/mmm | a = 3.084640 | 2 × 2 × 1 | 3 × 3 × 5 |
| c = 3.5199 | ||||
| LiCl | Fm3̅m | a = 5.1295241 | 1 × 1 × 1 | 4 × 4 × 4 |
| NaCl | Fm3̅m | a = 5.791542 | 1 × 1 × 1 | 4 × 4 × 4 |
| MgO | Fm3̅m | a = 4.21343 | 1 × 1 × 1 | 4 × 4 × 4 |
| Al | Fm3̅m | a = 4.049438 | 1 × 1 × 1 | 4 × 4 × 4 |
| Na | Im3®m | a = 4.23544 | 1 × 1 × 1 | 4 × 4 × 4 |
Lattice parameters are given for the crystallographic cell, and calculated cells are specified as multiples of the crystallographic cell.
The z coordinate for the Wyckoff site is calculated assuming a bond length of 1.108 Å and that the molecule center is located at the symmetry center of the space group P63/mmc, as in ref (45).
Lattice parameters taken from ref (46).
The set of molecules taken for the validation were computed with GAMESS,28 ADF,15 and FHI-aims,16 employing HF, LDA,29−31 PBE,27 BLYP,32,33 and B3LYP34 functionals. The basis set taken within each program is cc-pvtz (GAMESS), TZ2P (ADF), and standard “tight” (FHI-aims). For each molecule, the geometry was optimized before integrating the energy components. The geometry was fixed for molecules used for comparison against the solid phase (N2, CO2, neopentane, and phenalene). Only the four central atoms of phenalene were fixed while the positions of the other atoms were optimized.
Results and Discussion
IQA energy terms are examined for a number of molecular systems to evaluate the energy error. In subsequent exemplary application studies, some prototype compounds (diamond and zincblende structures, honeycomb networks, rocksalt structures, closest packings) covering covalent, ionic, and metallic situations were investigated.
Validation of the Implementation
Three approaches are explored to validate our implementation. First, similar results should be obtained with alternative implementations when comparable (molecular wave functions expanded with GTOs). In this document only the ethane system is shown (Table 2), but the equivalence was tested also with other simple systems reaching always similar accuracy. Second, properties like the volume, exchange-correlation energy, and total energy in the cell were checked against FHI-aims. The total energy requires an exhaustive computation of long- range interactions to be recovered. The electron count sum for the (crystallographic) unit cell provide a third way to check the validity of our implementation. Only the first is examined in this section and the other two in the next sections.
Table 2. Total Energy Components of Ethane Computed with ChemInt, Promolden, and GAMESSa.
| energy | ChemInt | Promolden | GAMESS |
|---|---|---|---|
| total energy, E | –79.731 | –79.732 | –79.730 |
| kinetic energy, T | 79.243 | 79.243 | –79.244 |
| total potential energy, Ene | –158.974 | –158.975 | –158.974 |
| electron–electron energy, Eee | 67.551 | 67.552 | 67.551 |
| Coulomb energy, ECoul | 80.528 | 80.529 | 80.528 |
| exchange-correlation energy, Exc | –12.977 | –12.977 | –12.977 |
The evaluated number of electrons per formula unit are 17.9992 (ChemInt) and 18.0001 (Promolden). Ex was re-scaled from the integration of the PBE functional.47 Energy in Ha units.
For the first check, we take the same molecular wave function and perform an energy decomposition analysis with two independent IQA implementations: our code and the Promolden code.13 We see that the integrated quantities have similar accuracy to those of Promolden and are consistent, beyond the chemical accuracy of approximately 1 mHa, with accurate full space integrations given by GAMESS (Table 2).
The precision of IQA decomposition is mainly determined by errors in the atomic boundary, the quality of the grid, and the truncation of the Laplace/bipolar expansion. Another possible source of error can be differences in the evaluation of the energy in ChemInt with respect to the SCF program. The energy difference between reconstructing the total energy from IQA components against the SCF energy is a measure of the error of our method. For the evaluated molecular systems, this energy error per atom is always below chemical accuracy (see Figure 1) with a modest choice of integration parameters. The boundary is determined with a precision of more than 5 × 10–4 Å normal to the surface using an ODE integrator that preserves a local relative error of the same magnitude. The grid has 600 radial points and 5780 points distributed on a symmetric spherical t-design grid. A maximum expansion order of l = 4 (l = 6) for regions near (far from) the nucleus is chosen. Increasing the precision of the surface determination does not yield a better integration. As well, the length of the expansion chosen here is enough to approximate bielectronic integrals. From our observations, the quality of the grid is the most determining factor once the rest of the parameters have been fixed like here.
Figure 1.

Energy difference (per atom) between the reconstructed IQA energy and the SCF energy (energy error). The white band denotes the median. Boxes delimit the lower and upper quartiles Q3 and Q4, and fences delimit all energy errors from a sample that takes wave functions computed with GAMESS, ADF, or FHI-aims, employing HF, LDA, PBE, BLYP, and B3LYP functionals for every system. Ex was rescaled from the integration of the corresponding functional.47 All errors fall below 1 mHa.
Nevertheless, it is important to realize that periodic systems require a significantly larger number of orbitals than molecular systems. Computations with maximum resolution are not always feasible, and one must find a balance of precision and speed. Therefore, we are interested in the minimum required computation needed to achieve a reasonable accuracy level. For all the systems tested, we found that a surface represented with approximately 6000 points is enough to integrate the total volume in the crystallographic unit cell with a volume error ΔVcell = ∑AVA – Vcell below 0.200 Å3 if we combine it with a radial grid of at least 200 points (Table 3). The symmetrical t-design quadrature offers a more robust integration, devoid of occasional (large) volume integration errors present with Lebedev–Laikov grids. In particular, for half-Heusler LiMgN, the cell volume error would be 2.027 Å3 using a 5810-point Lebedev grid. Instead, it is 0.010 Å3 with a 5780-point t-design grid. The distribution of points offered by the new grids is more homogeneous, thus providing a better overall representation of basin surfaces. Besides this, volume errors take place at the boundary of the basins so the integration of the charge can, and this is the case from our observations, still be within chemically reasonable accuracy error (<0.01).
Table 3. Reconstructing the Crystallographic Unit Cell Volume from QTAIM Basin Volumes: Volume Error, ΔVcell, and Cell Charge, Qcell.
| compound | structure type | ΔVcell [Å3] | Qcell [e] |
|---|---|---|---|
| LiCl | NaCl | 0.104 | –0.005 |
| NaCl | NaCl | 0.168 | 0.009 |
| MgO | NaCl | 0.039 | 0.002 |
| CsCl | CsCl | 0.004 | –0.000 |
| C | diamond | –0.004 | 0.007 |
| BN | zincbende | 0.020 | 0.000 |
| C | graphite | 0.017 | 0.001 |
| BN | BN-b | 0.009 | 0.001 |
| Na | α-W (bcc) | –0.028 | 0.001 |
| Al | Cu (fcc) | 0.010 | 0.000 |
| MgB2 | AlB2 | 0.014 | 0.002 |
| LiMgN | MgAgAs | 0.010 | 0.006 |
| N2 | β-N2 | –0.007 | 0.008 |
| CO2 | CO2 | –0.073 | 0.000 |
Molecular Crystals
Molecular crystals are a suitable example to examine the type of strong bonds that one finds in molecules before shifting to the highly diverse bonds present in crystalline materials. For that matter, we analyze β-N2, and CO2 crystals. A priori, N–N and C–O bonds are only expected to differ slightly from a gas phase molecule. Indeed, covalent intramolecular energies in the solid phase bear a striking resemblance to bonds of equal internuclear distance between atoms in gas phase, as shown in Table 4. Intermolecular interactions in the solid barely debilitate the covalent stabilization provided by the N2 triple bonds. As well, the classic interaction EclNN between two nearest nitrogen atoms is almost unaltered. In this regard, the triple bond exhibited by a N2 molecule is transferable to a triple bond in β-N2.
Table 4. Comparison of IQA Two-Center Terms for Covalent Bonds in Molecular and Solid Phases β-N2 and CO2a.
| system | A–B | RAB | δAB | EnnAB | EneAB | EneBA | ECoulAB | EclAB | ExAB |
|---|---|---|---|---|---|---|---|---|---|
| N2 (mol.) | N–N | 1.108 | 3.041 | 23.399 | –21.944 | –21.944 | 20.716 | 0.227 | –0.906 |
| N2 (solid) | N–N(1) | 1.108 | 3.000 | 23.399 | –21.952 | –21.953 | 20.726 | 0.220 | –0.903 |
| CO2 (mol.) | C–O | 1.149 | 1.396 | 22.114 | –25.443 | –13.413 | 15.453 | –1.289 | –0.444 |
| CO2 (solid) | C–O(1) | 1.149 | 1.323 | 22.114 | –25.646 | –13.152 | 15.269 | –1.415 | –0.426 |
| CO2 (mol.) | O–O | 2.297 | 0.433 | 14.743 | –16.981 | –16.981 | 19.563 | 0.343 | –0.054 |
| CO2 (solid) | O–O(2) | 2.297 | 0.407 | 14.743 | –17.081 | –17.081 | 19.793 | 0.373 | –0.052 |
Nearest-neighbor interactions are denoted with A–B(1), second neighbors with A–B(2), and so on. Energies in Ha; distances in Å.
On the other hand, while the C–O bond is similar in both environments, the strong polarization of this bond permits ionic interactions with neighbor molecules, with O–O being the most intense interaction. The C–O bond is mainly altered by its classic ionic component, which varies by 0.126 Ha in comparison to 0.018 Ha for the covalent energy ExCO. In both scenarios, a strong contribution from dipolar and quadrupolar components to the classic energy is found. The same effect is observed for intramolecular O–O interactions but is more attenuated.
Diamond and Zincblende Structure
Convergence of Bielectronic Integrals
A steady convergence with increasing multipolar order is observed for intra-atomic bielectronic integrals in diamond and zincblende BN (Figures 2 and 3). The Coulomb energy ECoulAA shows a dominating monopolar term (l = 0) followed by zero l = 1, 2 terms as demanded by symmetry.48 The l = 3, 4 terms are nonzero due to nonspherical distribution of electrons in the basin. The l = 5 term vanishes again due to symmetry. These observations for diamond atoms are in close agreement with those of a carbon atom in methane or the quaternary atom in neopentane (cf. Supporting Information, Figure S2 and Figure S3).
Figure 2.

Diamond: Convergence
of bielectronic integrals inside the basin
of the carbon atom, with increasing multipolar order l,  and
and  . Only their magnitude is plotted. Dashed
lines indicate the trend of convergence for symmetry allowed terms.
Disconnected dots are not allowed by symmetry and are nonzero due
to numerical errors. Terms below 10–5 Ha are represented
as dots at the bottom.
. Only their magnitude is plotted. Dashed
lines indicate the trend of convergence for symmetry allowed terms.
Disconnected dots are not allowed by symmetry and are nonzero due
to numerical errors. Terms below 10–5 Ha are represented
as dots at the bottom.
Figure 3.

BN (zincblende): Convergence of bielectronic intrabasin integrals with increasing multipolar order l. For point and lines explanations, see Figure 2.
Convergence with l of nearest-neighbor inter-atomic electron–electron terms in diamond is slower than for intra-atomic terms (see Figures 2 and 4). The convergence of Coulomb and exchange components with the order of the multipolar expansion was examined to assess the possibility of truncating the series. Storage of higher-order terms contributes to a substantial increase of computation time, specially due to the increasing number of averaged overlap densities Rl,mi,j. Coulomb and exchange terms show the expected convergent behavior. However, terms of order l = 3, 4 are still important for the Coulomb integral. This forces us to take several orders in the expansion. This behavior could, as well, vary for other systems. Therefore, it is only a preliminary observation.
Figure 4.

Diamond: Convergence of bielectronic interbasin integrals with increasing bipolar order L = lA + lB. Labels in the top right indicate an interaction of atom A with an atom B of the ith coordination sphere as A–B(i).
The Coulomb integral ECoulAB between a boron atom and a nearest N atom is well approximated from the (lA, lB) = (0, 0) term (Figure 5). Higher order terms L = lA + lB > 0 clearly reflect the characteristics of the electron distribution in their respective basins. Terms with order L = 1, 2 result from the combinations {(0, 1), (1, 0), (1, 1), (2, 0), (0, 2)}. As we have seen above, since the symmetry in B and N sites is tetrahedral, l = 1, 2 terms are zero (Figure 3). The same argument justifies the absence of L = 5 terms. Thus, only with L = 0, 3, 4, and 6 are the corrresponding bicentric Coulomb terms nonzero. Only due to numerical accuracy are they nonzero. More distant interactions like the closest N–N and B–B exhibit the same pattern, but their contribution from higher L order terms is already small, especially for B–B interactions.
Figure 5.

BN (zincblende): Convergence of bielectronic interbasin integrals with increasing bipolar order L = lA + lB. For point and lines explanations, see Figure 2.
Self-Energies and Near Neighbor Interactions
Having a generic method to decompose atomic energies allows us to compare molecular and solid systems on the same footing. Diamond is a prototype of a covalent network with bonded quaternary carbon atoms. A carbon atom with the same coordination in a molecule is the quaternary carbon of neopentane.
Table 5 compares the population and intra-atomic energy components of this atom in the two environments. The average electron population of the quaternary C atom in neopentane is slightly lower than that in diamond carbon atoms. Both intra-atomic classic EclCC and exchange energies Ex are larger in diamond due to the higher population of the atomic basins.
Table 5. IQA Monocentric Integrals Inside Basins with Td Point Symmetrya.
| system | A | ⟨NeA⟩ | λA | σ2(A) | TA | EneAA | ECoulAA | EclAA | ExAA |
|---|---|---|---|---|---|---|---|---|---|
| neopentane | C | 5.929 | 3.775 | 2.154 | 37.781 | –89.694 | 19.154 | –70.540 | –4.566 |
| diamond | C | 5.999 | 3.820 | 2.180 | 37.917 | –90.128 | 19.522 | –70.606 | –4.602 |
| BN | B | 2.839 | 2.075 | 0.765 | 23.700 | –51.753 | 7.923 | –43.830 | –3.008 |
| N | 9.161 | 7.517 | 1.643 | 55.650 | –138.162 | 36.638 | –101.524 | –7.019 |
Energies in Ha units.
Carbon atoms have a similar inter-atomic distance in diamond and neopentane, approximately 1.545 Å. To facilitate the discussion, the C – C distance in neopentane is fixed to be exactly the same as in diamond. This way, the nucleus–nucleus repulsion is identical. Full relaxation of the geometry does not change our conclusions. Inter-atomic interaction energies are presented in Table 6. As for intra-atomic terms ECoulCC, electron–electron Coulomb energies are larger in the solid phase. This is counterbalanced by Ene to finally yield a smaller classic energy. In diamond, electron–nucleus interactions are symmetrical, EneAB = Ene. The sum of all classic electrostatic terms nearly cancels to yield EclCC = 0.019 Ha for neopentane and Ecl = 0.014 Ha for diamond because the electroneutrality of the atom makes the L = 0 term vanish. The classical L > 0 energy term Ecl,L≠0AB has to be positive for neutral nonoverlapping charge distributions.8
Table 6. IQA Two-Center Terms for Relevant Interactions in Neopentane, Diamond, and Zincblende BNa.
| system | A–B | m | RAB | δAB | EnnAB | EneAB | EneBA | ECoulAB | EclAB | ExAB |
|---|---|---|---|---|---|---|---|---|---|---|
| neopentane | C–C(1) | 4 | 1.544 | 0.957 | 12.335 | –12.058 | –12.094 | 11.836 | 0.019 | –0.288 |
| diamond | C–C(1) | 4 | 1.544 | 0.914 | 12.335 | –12.217 | –12.217 | 12.113 | 0.014 | –0.284 |
| C–C(2) | 12 | 2.522 | 0.042 | 7.554 | –7.551 | –7.551 | 7.549 | 0.001 | –0.006 | |
| BN | B–N(1) | 4 | 1.563 | 0.357 | 11.848 | –15.502 | –6.697 | 8.765 | –1.586 | –0.106 |
| N–N(2) | 6 | 2.553 | 0.152 | 10.158 | –13.290 | –13.291 | 17.390 | 0.967 | –0.030 | |
| B–B(2) | 6 | 2.553 | 0.002 | 5.183 | –2.943 | –2.943 | 1.671 | 0.968 | –0.000 |

Exchange energies ExCC are almost identical in neopentane and diamond. Conversely, the nearest-neighbor DI δCC in diamond is smaller due to higher delocalization of the wave function (more distant neighbors) in the solid.
Diamond presents a DI of δCC = 0.914 between nearest-neighbor atoms. The delocalization goes down to δCC = 0.044 for second nearest-neighbors. The variation of DIs for the third coordination shell obtained for equivalent interactions is too large to prevent a detailed numerical examination. Up to first and second nearest neighbors the sum rule (34) is shown in Table 7.
Table 7. Cumulative
Electron Populations  of Atomic Basins in Diamond and Boron Nitride
(Zincblende) from Successive Inclusion of Delocalization Shells α
with DI δαa.
of Atomic Basins in Diamond and Boron Nitride
(Zincblende) from Successive Inclusion of Delocalization Shells α
with DI δαa.
| α | coordination sphere | δα | ⟨NelA⟩cum |
|---|---|---|---|
| 0 | C | – | 3.820 |
| 1 | C–C(1) | 0.914 | 5.648 |
| 2 | C–C(2) | 0.044 | 5.912 |
| 0 | B | – | 2.075 |
| 1 | B–N(1) | 0.357 | 2.789 |
| 2 | B–B(2) | 0.002 | 2.801 |
| 0 | N | – | 7.517 |
| 1 | N–B(1) | 0.357 | 8.231 |
| 2 | N–N(2) | 0.155 | 9.161 |
Boron nitride represents an example of a diamond-type structure with polar bonds. While the nearest-neighbor electrostatic interaction EclCC=0.014 Ha in diamond represents a destabilization, in B–N it is a stabilizing classic interaction between opposite charged atoms with an energy contribution of Ecl=–1.586 Ha (Table 6).
The nearest-neighbor exchange interaction ExBN is only one-third of the analogous nearest neighbors interaction Ex in diamond. Note that this decrease can be estimated (31) from the strong decrease of the delocalization index from δCC = 0.957 to δBN = 0.357.
For BN, in overall, classic electrostatic energies between nearest neighbors are much larger than the exchange ones. This fact reveals the high importance of classic interactions in the short-range regime.
The closest B–N interactions involve 0.36 electron pair sharing (Table 7) that parallels the value of 0.37 obtained before.49 Nitrogen atoms share 0.15 electron pairs with any other nearest nitrogen atom (to be compared with 0.35 electron pairs). The sum-rule for N is approached including interactions up to the second coordination shell (see Table 7). On the other hand, boron atoms have no significant electron sharing with other boron atoms. There are, however, 0.038 electrons still missing to recover the average number of electrons in the boron basin.
The localization indices are in agreement with results obtained before by some of us (Table 6).
Additive Interaction Energies
The
additive interaction
energy (cf. equation 11) for a 2-atom reference unit G = {C1, C2} ≡
C1C2 in diamond can be approximately obtained as follows. The covalent
interaction energies  between nearest neighbors (−0.284
× 4 = −1.136 Ha) and
between nearest neighbors (−0.284
× 4 = −1.136 Ha) and 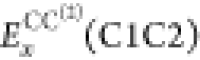 between second nearest neighbors (−
0.006 × 12 = −0.072 Ha) sum to a total covalent interaction
energy of Ex(C1C2) =
−1.208 Ha. Higher neighbors are omitted due to a very small
value of remaining bond fluctuations
between second nearest neighbors (−
0.006 × 12 = −0.072 Ha) sum to a total covalent interaction
energy of Ex(C1C2) =
−1.208 Ha. Higher neighbors are omitted due to a very small
value of remaining bond fluctuations 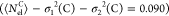 . A kind
of lower bound of the neglected
covalent interaction can be estimated with the zeroth order approximation
if we assign all remaining electrons to the third coordination shell,
. A kind
of lower bound of the neglected
covalent interaction can be estimated with the zeroth order approximation
if we assign all remaining electrons to the third coordination shell, 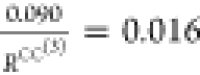 Ha. The stabilizing
covalent interactions
are counterbalanced by electrostatic interactions Ecl(C1C2) = 0.014 × 4 + 0.001 × 12 = 0.068 Ha
(in diamond) from contributions between first and second nearest neighbors,
respectively. The total interactions considered amount to Eint(C1C2) = −1.140 Ha.
Ha. The stabilizing
covalent interactions
are counterbalanced by electrostatic interactions Ecl(C1C2) = 0.014 × 4 + 0.001 × 12 = 0.068 Ha
(in diamond) from contributions between first and second nearest neighbors,
respectively. The total interactions considered amount to Eint(C1C2) = −1.140 Ha.
For zincblende,
again, a diatomic reference unit G = {B1, N1} ≡
B1N1 is chosen. The approximate covalent interaction energy up to
second nearest neighbor terms sums nearest neighbor B–N(1) interactions (− 0.106 × 4 = −0.424 Ha),
second nearest neighbors N–N(2) interactions (−
0.030 × 6 = −0.180 Ha), and second nearest neighbor B–B(2) interactions (0.000 × 6 = 0 Ha) giving Ex(B1N1) = −0.604 Ha. While the
electron sum rule is already well satisfied for N, some 0.038 electrons
are missing on B. If we use this number to approximate the maximum
additional covalent energy, the third nearest neighbor B–N(3) interaction with a distance 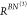 may be estimated to contribute
maximally
may be estimated to contribute
maximally  . It is interesting that
a non-negligible
covalent N–N(2) interaction has been found, which
makes up 30% of the covalent bond energy. It can be considered as
the residual bonding of the incompletely occupied N2.161– atoms, which is a necessary consequence of the polar bonding B–N(1) leading to the small DI δBN = 0.357.
. It is interesting that
a non-negligible
covalent N–N(2) interaction has been found, which
makes up 30% of the covalent bond energy. It can be considered as
the residual bonding of the incompletely occupied N2.161– atoms, which is a necessary consequence of the polar bonding B–N(1) leading to the small DI δBN = 0.357.
The procedure applied for the covalent part
is not feasible for
the electrostatic part, because of the long-range of the electrostatic
interactions and the well-known resulting nonconvergence of the simple
coordination series energy in real space. The electrostatic part of
the interaction per reference unit can only be approximated by the
point charge approximation using the Madelung constant of zincblende fM,R = 1.63806.
The validity of the PCA for this procedure can be estimated by determining
the PCA scale factor scl for the computed
first and second nearest neighbor Ecl(B1N1)
energies using Q atomic net charges and the corresponding
distances 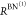 , and
, and 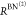 . It turns out that the PCA energy values
are rather close to the exact IQA ones (
. It turns out that the PCA energy values
are rather close to the exact IQA ones (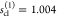 ,
,  ), such that the PCA is well justified.
The Madelung energy per unit cell G = B1N1 is calculated
according to
), such that the PCA is well justified.
The Madelung energy per unit cell G = B1N1 is calculated
according to  Ha, where R is
the first-neighbor
B–N distance. Although l = 1, 2 (dipoles,
quadrupoles) are forbidden by symmetry, there are contributions from
classic electrostatic terms with L > 0 due to
the
large atomic net charges present in the system. Owing to their absence
in the Madelung formula, they must be accounted at least for nearest
neighbors. We compute them with Ecl(G) – Ecl,L=0(G) = −0.026 Ha, where the last term
is just the point-charge contribution (eq 30). The total electrostatic energy is then Ecl = −2.617 Ha. From these values, the
total interaction energy amounts to Eint(B1N1) = −3.221 Ha. The fraction of covalent bond energy per G = B1N1 with respect to the total interaction energy
Ha, where R is
the first-neighbor
B–N distance. Although l = 1, 2 (dipoles,
quadrupoles) are forbidden by symmetry, there are contributions from
classic electrostatic terms with L > 0 due to
the
large atomic net charges present in the system. Owing to their absence
in the Madelung formula, they must be accounted at least for nearest
neighbors. We compute them with Ecl(G) – Ecl,L=0(G) = −0.026 Ha, where the last term
is just the point-charge contribution (eq 30). The total electrostatic energy is then Ecl = −2.617 Ha. From these values, the
total interaction energy amounts to Eint(B1N1) = −3.221 Ha. The fraction of covalent bond energy per G = B1N1 with respect to the total interaction energy  yields a 19% contribution of covalent interaction.
Based on this fraction, the interaction in BN (zincblende) is characterized
by dominating 81% ionic interactions. Still, the covalency is non-negligible,
and it represents the local type of bonding, while the electrostatic
Coulomb interaction is a collective type of bonding as discussed previously.50 While the covalent bonding A–B is a local
property of A and B, the electrostatic interaction A–B has
long-range consequences, alternating stabilizing and destabilizing
terms. These two different types of bonding cannot be discussed on
the same footing, i.e., in a local picture. For this reason, the energies
per reference unit have been calculated for characterization.
yields a 19% contribution of covalent interaction.
Based on this fraction, the interaction in BN (zincblende) is characterized
by dominating 81% ionic interactions. Still, the covalency is non-negligible,
and it represents the local type of bonding, while the electrostatic
Coulomb interaction is a collective type of bonding as discussed previously.50 While the covalent bonding A–B is a local
property of A and B, the electrostatic interaction A–B has
long-range consequences, alternating stabilizing and destabilizing
terms. These two different types of bonding cannot be discussed on
the same footing, i.e., in a local picture. For this reason, the energies
per reference unit have been calculated for characterization.
Honeycomb Networks
Honeycomb networks can be found in graphite, hexagonal boron nitride, and the intermetallic superconductor MgB2. Table 8 shows nearest-neighbor interactions in the three hexagonal systems. In addition to the σ bonds discussed in the previous section, they might exhibit a certain degree of π double bond character. In graphite, it has been shown,51 based on delocalization indices, that the shortest C–C bonds with a DI δCC = 1.20 have partial double bond character, whereas in MgB2 the number of shared electron pairs is the same as for a single bond δBB = 1.00. For comparison, similar values of δCC = 1.202 in graphite and δBB = 0.983 in MgB2 are obtained.
Table 8. IQA Two-Center Terms for Relevant Interactions in Hexagonal Graphite, BN, and MgB2a.
| A–B | m | RAB | δAB | EnnAB | EneAB | EneBA | ECoulAB | EclAB | ExAB |
|---|---|---|---|---|---|---|---|---|---|
| C–C(1)ab | 3 | 1.423 | 1.202 | 13.391 | –13.098 | –13.102 | 12.846 | 0.037 | –0.372 |
| C–C(2)ab | 6 | 2.464 | 0.054 | 7.731 | –7.692 | –7.692 | 7.655 | 0.002 | –0.007 |
| C–C(3)ab | 3 | 2.845 | 0.037 | 6.696 | –6.679 | –6.681 | 6.665 | 0.001 | –0.004 |
| C–C(4)c | 1 | 3.356 | 0.019 | 5.677 | –5.709 | –5.709 | 5.746 | 0.004 | –0.003 |
| C–C(5)c | 6 + 3 | 3.645 | 0.006 | 5.227 | –5.245 | –5.247 | 5.266 | 0.001 | –0.001 |
| B–N(1)ab | 3 | 1.446 | 0.452 | 12.810 | –16.751 | –7.065 | 9.244 | –1.762 | –0.135 |
| B–B(2)ab | 3 | 2.504 | 0.004 | 5.283 | –2.936 | –2.935 | 1.632 | 1.043 | –0.000 |
| N–N(2)ab | 3 | 2.504 | 0.212 | 10.354 | –13.604 | –13.605 | 17.877 | 1.021 | –0.040 |
| B–N(3)ab | 3 | 2.892 | 0.008 | 6.405 | –8.419 | –3.564 | 4.685 | –0.893 | –0.001 |
| B–N(4)c | 2 | 3.329 | 0.006 | 5.563 | –7.329 | –3.110 | 4.101 | –0.776 | –0.001 |
| N–N(5)c | 3 | 3.630 | 0.024 | 7.144 | –9.411 | –9.410 | 12.403 | 0.726 | –0.003 |
| B–B(1)ab | 3 | 1.781 | 0.983 | 7.429 | –8.392 | –8.392 | 9.522 | 0.166 | –0.249 |
| B–B(3)ab | 6 | 3.085 | 0.052 | 4.289 | –4.953 | –4.953 | 5.722 | 0.106 | –0.006 |
| B–B(5)ab | 3 | 3.562 | 0.017 | 3.714 | –4.303 | –4.303 | 4.987 | 0.095 | –0.002 |
| Mg–B(2)c | 12 | 2.504 | 0.061 | 12.681 | –14.830 | –10.964 | 12.824 | –0.290 | –0.013 |
| Mg–Mg(3)ab | 3 | 3.085 | 0.003 | 24.704 | –21.364 | –21.364 | 18.475 | 0.450 | –0.000 |
The reference units corresponding
to 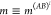 are G = C1C2, G = B1N1, and G = MgB1B2. Energies in Ha;
distances in Å units. Key: ab, intralayer interaction; c, interlayer interaction
are G = C1C2, G = B1N1, and G = MgB1B2. Energies in Ha;
distances in Å units. Key: ab, intralayer interaction; c, interlayer interaction
Graphite nearest-neighbor C–C bonds display
higher ExAB energies (covalent
bond
energies) than in the other systems with honeycomb networks. The nearest-neighbor
B–N covalent bond is particularly weak. In the honeycomb layer,
the Ex proportion of
the B–N covalent bond compared to the C–C covalent bond,  , parallels the proportion
seen in diamond
and zincblende BN,
, parallels the proportion
seen in diamond
and zincblende BN, 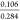 . Conversely, classic
electrostatic interactions
are different for each system studied here. BN is largely stabilized
from collective classical interactions between ions. This finding
provides conclusive support for interpreting B–N as a very
polar bond. However, destabilizing second-neighbor ionic interactions
overcome the stabilization achieved by nearest neighbors. In the long-distance
limit, those classic ionic interactions would tend to partially cancel.50 Graphite only has high order multipole contributions
to the classic electrostatic energy. Thus, already for first neighbors,
a low value of Ecl = 0.037 Ha is obtained.
In MgB2, nearest B–B interactions display a considerable
classic (interionic) destabilization that is compensated by Mg–B
classic electrostatic stabilization.
. Conversely, classic
electrostatic interactions
are different for each system studied here. BN is largely stabilized
from collective classical interactions between ions. This finding
provides conclusive support for interpreting B–N as a very
polar bond. However, destabilizing second-neighbor ionic interactions
overcome the stabilization achieved by nearest neighbors. In the long-distance
limit, those classic ionic interactions would tend to partially cancel.50 Graphite only has high order multipole contributions
to the classic electrostatic energy. Thus, already for first neighbors,
a low value of Ecl = 0.037 Ha is obtained.
In MgB2, nearest B–B interactions display a considerable
classic (interionic) destabilization that is compensated by Mg–B
classic electrostatic stabilization.
As in zincblende BN (ExNN = −0.030 Ha), among second-neighbor interactions only N–N still has a notable exchange contribution Ex = −0.040 Ha in the hexagonal phase.
Phase Stability: Cubic versus Hexagonal Structures
Diamond and graphite are the main allotropes of carbon. The experimental difference in enthalpies is known to be quite small, ΔH298 K = +1.895 kJ/mol,52 requiring an exquisite degree of accuracy that challenges DFT functionals. Albeit the relative stability problem is already solved,53 the internal forces leading to a greater stability of graphite are not completely known. The additional stability might result from the delocalized π bonding framework. However, it is not clear if the higher coordinated carbon atoms (via σ bonds) in diamond should be less stable. The origin of graphite stability could as well be attributed to a particular local property like a different hybridization. In the spirit of a recent publication that includes those contributions in an analytical model,54 we perform a numerical exploration to elucidate why nature chooses graphite over diamond.
Similar to the case
of diamond discussed in the Diamond and Zincblende
Structure section, the case of graphite involves a combination
of stabilizing covalent bond contributions 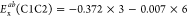 – 0.004 × 3 = −1.170
Ha for ortho, meta, and para neighbors for the chosen C1C2 reference
unit. The covalent part of the C–C interaction in the c direction amounts to
– 0.004 × 3 = −1.170
Ha for ortho, meta, and para neighbors for the chosen C1C2 reference
unit. The covalent part of the C–C interaction in the c direction amounts to  =
=  Ha to be added to yield final Ex(C1C2) = −1.182 Ha. The destabilizing
electrostatic interactions amount to
Ha to be added to yield final Ex(C1C2) = −1.182 Ha. The destabilizing
electrostatic interactions amount to  =
= 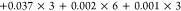 = +0.126 Ha (intralayer)
and
= +0.126 Ha (intralayer)
and  Ha (interlayer) summing up to Ecl(C1C2) = +0.139 Ha. A total interaction energy of Eint(C1C2) = −1.043 Ha is obtained. Comparison
of the total interaction energy to that of the diamond modification Eint(C1C2) = −1.140 Ha (Ex(C1C2) = −1.208 Ha, Ecl(C1C2) = +0.068 Ha) reveals that the covalent interactions
up to third nearest neighbors are energetically smaller, and the destabilizing
electrostatic ones are larger. This yields an interaction energy preference
for the diamond structure of ΔEint(C1C2) = 0.097 Ha.
Ha (interlayer) summing up to Ecl(C1C2) = +0.139 Ha. A total interaction energy of Eint(C1C2) = −1.043 Ha is obtained. Comparison
of the total interaction energy to that of the diamond modification Eint(C1C2) = −1.140 Ha (Ex(C1C2) = −1.208 Ha, Ecl(C1C2) = +0.068 Ha) reveals that the covalent interactions
up to third nearest neighbors are energetically smaller, and the destabilizing
electrostatic ones are larger. This yields an interaction energy preference
for the diamond structure of ΔEint(C1C2) = 0.097 Ha.
Thus, the interaction energies do not explain the obtained total energy difference ΔEFHI-aims(C1C2) = 0.009 Ha between both modifications giving a preference to the graphite modification. This finding suggests that, at least for the current calculation, the preference of the graphite modification is caused by intra-atomic electron reorganization terms. Indeed, inclusion of the intra-atomic energies using ΔE(C1C2) = E(C1C2, diamond) – E(C1C2, graphite) = ΔEself(C1C2) + ΔEint(C1C2) resolves this seeming discrepancy and yields the energetical preference of the hexagonal phases (Table 9). In the framework of Valence Bond Theory, one would argue, the sp3 hybridization of carbon in diamond requires more energy than the sp2 hybridization in graphite. Just as in the solid phase, the 4-connected central carbon atom in neopentane and the 3-connected one in phenalene display a consistent self-energy difference of ΔEself(2 × C1) = 0.068 Ha, thereby demonstrating that the hybridization of the carbon atom is related to the stability of graphite over diamond. This indicates that the relative stability of carbon allotropes is not ruled by nonlocal contributions (π delocalization) but by intra-atomic electron redistribution energies often coined as “hybridization energies”.
Table 9. Relative Phase Stability of Cubic and Hexagonal Phases for C and BN Compoundsa.
| phase | Ex(G) | Ecl(G) | ΔEint(G) | ΔEself(G)b | ΔE(G) | ΔEFHI-aims(G) |
|---|---|---|---|---|---|---|
| C (cubic) | –1.208 | 0.068 | 0 | 0.146 | 0.049 | 0.009 |
| C (hex.) | –1.182 | 0.139 | 0.097 | 0 | 0 | 0 |
| BN (cubic) | –0.604 | –2.617 | 0 | B: 0 | 0.012 | 0.005 |
| N: 0.120 | ||||||
| BN (hex.) | –0.539 | –2.645 | 0.037 | B: 0.071 | 0 | 0 |
| N: 0 |
Values for a reference unit: G = C1C2 and G = B1N1. ΔE(G) values are referred to the most stable phase. Energies in Ha units.
Due to numerical difficulties integrating the total kinetic energy with mHa accuracy, atomic kinetic energies TA were scaled to recover the total kinetic energy from FHI-aims.
The covalent interaction part per diatomic unit G = B1N1 is calculated according to  =
=  = −0.528
Ha for ortho (3 B–N(1)), meta (3 B–B(2) + 3 N–N(2)), and para neighbors (3 B–N(3)) respectively.
The B–N(4) and N–N(5) interactions
in the c direction amount to
= −0.528
Ha for ortho (3 B–N(1)), meta (3 B–B(2) + 3 N–N(2)), and para neighbors (3 B–N(3)) respectively.
The B–N(4) and N–N(5) interactions
in the c direction amount to  Ha to yield a total covalent bond
energy Ex(B1N1) = −0.539
Ha.
Since the Madelung constant for this structure is not fixed by symmetry,
it cannot be found tabulated in the literature. Therefore, the ionic
electrostatic interaction energy
Ha to yield a total covalent bond
energy Ex(B1N1) = −0.539
Ha.
Since the Madelung constant for this structure is not fixed by symmetry,
it cannot be found tabulated in the literature. Therefore, the ionic
electrostatic interaction energy  Ha was computed
with the Environ program
using the QTAIM net charges. This value is slightly larger than
Ha was computed
with the Environ program
using the QTAIM net charges. This value is slightly larger than 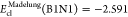 Ha obtained
for cubic phase. The low symmetry
of hexagonal BN allows classic contributions not present in the cubic
phase Ecl,L>0(B1N1)
=
0.123 Ha that in sum counteract the point-charge Madelung energy.
Thus, classic energy components account for Ecl(B1N1) = −2.645 Ha. The portion of covalent interactions
Ha obtained
for cubic phase. The low symmetry
of hexagonal BN allows classic contributions not present in the cubic
phase Ecl,L>0(B1N1)
=
0.123 Ha that in sum counteract the point-charge Madelung energy.
Thus, classic energy components account for Ecl(B1N1) = −2.645 Ha. The portion of covalent interactions  of 17% is smaller than 19% obtained for
the cubic variant. The total interaction energy obtained amounts to Eint(B1N1) = −3.184 Ha, which is of smaller
size than Eint(B1N1) = −3.221 Ha
obtained for cubic BN by 37 mHa. In contrast, the total energy difference
per BN unit computed with FHI-aims is ΔEFHI-aims(B1N1) = 0.005 Ha energy preference to hexagonal
BN. So, the total energies obtained from FHI-aims yield a preference
for the hexagonal variants for both cases: carbon, and BN. In contrast,
for both cases, the summed interaction energies yield a preference
of the cubic structures. This seeming discrepancy is resolved again
taking the intra-atomic energies into account as well. According to
this finding it is the intra-atomic energies and not the interaction
ones that are responsible for the preferred stability of the hexagonal
phases (Table 9). For
BN, an interesting competition between B and N species is found. Intra-atomic
energies indicate a preference of B species for the zincblende environment,
while N species prefer the hexagonal environment. In the end the higher
stabilization of the N species in the hexagonal environment dominates,
and from ΔE(B1N1) = E(B1N1,
cubic) – E(B1N1, hex.), the preference for
the hexagonal phase is obtained.
of 17% is smaller than 19% obtained for
the cubic variant. The total interaction energy obtained amounts to Eint(B1N1) = −3.184 Ha, which is of smaller
size than Eint(B1N1) = −3.221 Ha
obtained for cubic BN by 37 mHa. In contrast, the total energy difference
per BN unit computed with FHI-aims is ΔEFHI-aims(B1N1) = 0.005 Ha energy preference to hexagonal
BN. So, the total energies obtained from FHI-aims yield a preference
for the hexagonal variants for both cases: carbon, and BN. In contrast,
for both cases, the summed interaction energies yield a preference
of the cubic structures. This seeming discrepancy is resolved again
taking the intra-atomic energies into account as well. According to
this finding it is the intra-atomic energies and not the interaction
ones that are responsible for the preferred stability of the hexagonal
phases (Table 9). For
BN, an interesting competition between B and N species is found. Intra-atomic
energies indicate a preference of B species for the zincblende environment,
while N species prefer the hexagonal environment. In the end the higher
stabilization of the N species in the hexagonal environment dominates,
and from ΔE(B1N1) = E(B1N1,
cubic) – E(B1N1, hex.), the preference for
the hexagonal phase is obtained.
Rocksalt-Type Structures
Ionic crystals should present situations with extremely polar bonds. As a case in point we consider LiCl, NaCl, and MgO.
Table 10 shows short-range interactions in the rocksalt-type structure for various compounds. Even nearest-neighbor inter-atomic interactions have a small energy contribution from electron sharing. One can foresee from the delocalization indices that this is the case. Classical interactions take over the exchange component for those systems. As expected from their large QTAIM charges, the classic energy only converges when long-range interactions are also included.
Table 10. IQA Two-Center Terms for Relevant Interactions in LiCl, NaCl, and MgOa.
| A–B | m | RAB | δAB | EnnAB | EneAB | EneBA | ECoulAB | EclAB | ExAB |
|---|---|---|---|---|---|---|---|---|---|
| Li–Cl(1) | 6 | 2.565 | 0.044 | 10.523 | –11.076 | –7.374 | 7.762 | –0.165 | –0.007 |
| Li–Li(2) | 6 | 3.627 | 0.000 | 1.313 | –0.920 | –0.920 | 0.645 | 0.118 | –0.000 |
| Cl–Cl(2) | 6 | 3.627 | 0.084 | 42.164 | –44.393 | –44.393 | 46.741 | 0.118 | –0.012 |
| Na–Cl(1) | 6 | 2.896 | 0.063 | 34.173 | –35.919 | –31.452 | 33.061 | –0.138 | –0.010 |
| Na–Na(2) | 6 | 4.095 | 0.000 | 15.635 | –14.391 | –14.391 | 13.246 | 0.099 | –0.000 |
| Cl–Cl(2) | 6 | 4.095 | 0.044 | 37.344 | –39.274 | –39.272 | 41.301 | 0.100 | –0.005 |
| Mg–O(1) | 6 | 2.106 | 0.125 | 24.116 | –29.218 | –20.674 | 25.050 | –0.725 | –0.029 |
| Mg–Mg(2) | 6 | 2.979 | 0.001 | 25.579 | –21.931 | –21.931 | 18.803 | 0.520 | –0.000 |
| O–O(2) | 6 | 2.979 | 0.090 | 11.368 | –13.803 | –13.803 | 16.759 | 0.522 | –0.014 |
The net charges of QTAIM atoms are ±0.90e, ± 0.88e, and ±1.71e in LiCl, NaCl, and MgO, respectively. Energies in Ha; distances in Å units.
The additive energy for a reference unit G = LiCl
is computed as follows. The covalent interaction energy from interactions
with the first and second coordination sphere is Ex(LiCl) = −0.007 × 6 + 0.0
× 6–0.012 × 6 = −0.114 Ha where six Li–Cl(1), six Li–Li(2), and six Cl–Cl(2) interactions are considered. The lattice electrostatic
energy assuming point charges is  Ha based on a Madelung summation. The effects
of nonsphericity of the charge distribution around ions has an effect
that is appreciated only for nearest neighbor interactions, as seen
from their scaling factors of scl(1)=0.98 and scl = 1.00.
To correct the classic energy for first-neighbors, Li–Cl(1), higher order multipolar contributions, taken from the
IQA inter-atomic energy as
Ha based on a Madelung summation. The effects
of nonsphericity of the charge distribution around ions has an effect
that is appreciated only for nearest neighbor interactions, as seen
from their scaling factors of scl(1)=0.98 and scl = 1.00.
To correct the classic energy for first-neighbors, Li–Cl(1), higher order multipolar contributions, taken from the
IQA inter-atomic energy as 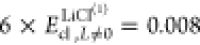 Ha,
are included. The total electrostatic
energy is therefore Ecl(LiCl) = −0.2848
Ha. Thus, the covalent part contributes
Ha,
are included. The total electrostatic
energy is therefore Ecl(LiCl) = −0.2848
Ha. Thus, the covalent part contributes  to the
interaction energy Eint(LiCl) = −0.398
Ha.
to the
interaction energy Eint(LiCl) = −0.398
Ha.
Similarly, for a G = NaCl reference unit,
covalent
interactions contribute with Ex(NaCl) = −0.09 Ha. The classic energy, assuming point
charges, is  Ha, that is corrected by nonspherical terms
Ha, that is corrected by nonspherical terms  Ha.
The corrected classic energy Ecl(NaCl)
= −0.230 Ha entails a 72% percent
of the interaction energy Eint(NaCl) =
−0.320 Ha.
Ha.
The corrected classic energy Ecl(NaCl)
= −0.230 Ha entails a 72% percent
of the interaction energy Eint(NaCl) =
−0.320 Ha.
MgO exhibits a stronger covalent stabilization
than either LiCl
or NaCl, Ex(MgO) = −0.258
Ha. However, this increase is paralleled by stronger classical interactions  Ha generated by interacting point charges
of ±1.71 e. The correction from nonspherical
terms is also enhanced,
Ha generated by interacting point charges
of ±1.71 e. The correction from nonspherical
terms is also enhanced,  Ha.
In overall, the total classic energy
is Ecl(MgO) = −1.215 Ha. Thus,
the covalent part contributes less,
Ha.
In overall, the total classic energy
is Ecl(MgO) = −1.215 Ha. Thus,
the covalent part contributes less, 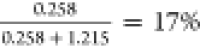 , to the
interaction energy Eint(MgO) = −1.473
Ha than the previous systems
with rocksalt-type structure.
, to the
interaction energy Eint(MgO) = −1.473
Ha than the previous systems
with rocksalt-type structure.
With respect to interaction energies, the sequence of increasing covalent character is MgO (17%) < NaCl (28%) < LiCl (29%).
Scaled Point-Charge Approximation (sPCA)
The kind of approximation discussed here has been used previously to predict the stability of phases with highly symmetric MgAgAs structure6 and to discuss Fe–Fe bonding in FeGa3.4
The validity of those approximations is tested against formally exact integrals from IQA method. Table 11 compares the zero order (point charge) approximation for interactions where largest deviations are expected, that is, between nearest neighbors.
Table 11. Scaled Point-Charge Approximation (sPCA) (See Eq 35) of Inter-Atomic Bielectronic Integrals for Nearest-Neighbor Interactionsa.
| System | RAB | QA | QB | δAB | EclAB | scl | ExAB | sx |
|---|---|---|---|---|---|---|---|---|
| β-N2 | 1.108 | 0.003 | 0.003 | 2.999 | 0.220 | b | –0.903 | 0.630 |
| CO2 | 1.149 | 2.280 | –1.140 | 1.323 | –1.415 | 1.182 | –0.426 | 0.699 |
| Graphite | 1.422 | 0.000 | –0.001 | 1.202 | 0.037 | b | –0.372 | 0.832 |
| BN (hex.) | 1.446 | 2.214 | –2.213 | 0.452 | –1.762 | 0.982 | –0.135 | 0.816 |
| Diamond | 1.545 | 0.001 | 0.001 | 0.914 | 0.014 | b | –0.284 | 0.907 |
| BN (cubic) | 1.561 | 2.160 | –2.160 | 0.357 | –1.587 | 1.004 | –0.106 | 0.881 |
| MgB2 | 1.782 | –0.813 | –0.813 | 0.983 | 0.166 | 0.845 | –0.249 | 0.852 |
| MgO | 2.106 | 1.711 | –1.712 | 0.129 | –0.725 | 0.984 | –0.029 | 0.884 |
| LiCl | 2.566 | 0.897 | –0.899 | 0.044 | –0.165 | 0.991 | –0.007 | 0.819 |
| Al | 2.863 | 0.000 | 0.000 | 0.273 | 0.0013 | b | –0.054 | 1.071 |
| NaCl | 2.896 | 0.875 | –0.878 | 0.064 | –0.1376 | 0.980 | –0.010 | 0.866 |
| CsCl | 3.540 | 0.826 | –0.827 | 0.126 | –0.1019 | 0.996 | –0.017 | 0.915 |
| Na | 3.667 | 0.000 | 0.000 | 0.108 | 0.0008 | b | –0.013 | 0.836 |
Energies in Ha, distances in Å, and charges in e units.
For interactions between (nearly)
noncharged atomic species, the scaling parameters scl become quite large, because the electrostatic interaction
is then no longer dominated by a monopolar  term. Nevertheless, the absolute
error
of this assumption is typically small, because the interactions are
weak.
term. Nevertheless, the absolute
error
of this assumption is typically small, because the interactions are
weak.
Molecular crystals like β-N2 or CO2 present rather short interactions that can not be modeled with the point-charge approximation. Rather like molecules, both solids show very strong covalent bonds as evidenced by their DIs and inter-atomic ExAB. This is in agreement with chemical wisdom. Covalent bonds break the spherical symmetry of the density leading to larger contributions from higher order multipoles to the classic energy. Even more, in N2 the atomic charge is zero but there is still a 0.220 Ha classic destabilization.
Graphite and diamond feature fractional double bonds and covalent single bonds, respectively, which display a corresponding increase of inter-atomic distances that leads to a corresponding decrease of EclCC.
An inflection point is seen for zincblende BN. Despite having also covalent single bonds like diamond, the classic energy closely follows the point-charge approximation scl ≈ 1, indicating that the electron density is close to the spherical one.
Ionic and metallic crystals typically display longer inter-atomic distances and conceptual bond orders <1. Therefore, their covalent and ionic interactions are expected to be accurately approximated with the simple scaled point-charge equations applying an intermediate scaling factor sx ≈ 0.75–0.85.
Conclusions
The new software package ChemInt was developed to decompose the energy of crystalline solids according to the Interacting Quantum Atoms approach. The QTAIM basins are chosen as domains for the energy decomposition. Other possible spatial partitions (i.e., Becke fuzzy atoms, Hirshfeld atoms, ...) deserve future research and will assess the robustness of our conclusions. A number of key issues were addressed which limited the accuracy of the integration. The package ChemInt was used for the examination of some prototype crystalline solids. For the ionic solids studied, covalent interactions contribute 15–30% to the interaction energy. The scale parameters of our scaled point-charge approximation were obtained from the formally exact integration of covalent and classic inter-atomic interaction energies. While certain trends for these parameters are already visible, further investigations are needed to faithfully predict them for all kinds of bonding situations. The phase stability of graphite over diamond and hexagonal BN over zincblende BN was obtained in terms of chemically meaningful energy components. Future explorations along this line are a promising area of research. The obvious limitations of the application to very large systems can be significantly reduced in the future with the exploitation of crystal symmetries and treating only valence electrons. The new program may contribute toward a better understanding of interactions in crystalline materials, in particular in those which do not follow traditional valence rules. Extending the IQA method to planewave bases is perfectly possible, and it can be implemented on any electron structure package because it does not depend on the representation of the wave function in terms of atomic orbitals.
Acknowledgments
D.M.C. thanks J. L. Casals-Sainz for suggesting spherical t-design grids as an alternative to Lebedev–Laikov grids. A.M.P. and E.F. thank the Spanish MICINN, Project PGC2018-095953-B-I00, for funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.1c06574.
Computational details of ChemInt calculations and supporting IQA results (PDF)
Open access funded by Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- Hume-Rothery W.Materials Science and Engineering; McGraw Hill: 1967; pp 3–23. [Google Scholar]
- Fredrickson D. C.; Lee S.; Hoffmann R. The Nowotny Chimney Ladder Phases: Whence the 14 Electron Rule?. Inorg. Chem. 2004, 43, 6159–6167. 10.1021/ic049897h. [DOI] [PubMed] [Google Scholar]
- Yannello V. J.; Fredrickson D. C. Generality of the 18- n Rule: Intermetallic Structural Chemistry Explained through Isolobal Analogies to Transition Metal Complexes. Inorg. Chem. 2015, 54, 11385–11398. 10.1021/acs.inorgchem.5b02016. [DOI] [PubMed] [Google Scholar]
- Wagner F. R.; Cardoso-Gil R.; Boucher B.; Wagner-Reetz M.; Sichelschmidt J.; Gille P.; Baenitz M.; Grin Y. On Fe–Fe Dumbbells in the Ideal and Real Structures of FeGa3. Inorg. Chem. 2018, 57, 12908–12919. 10.1021/acs.inorgchem.8b02094. [DOI] [PubMed] [Google Scholar]
- Bende D.; Grin Y.; Wagner F. R. Covalence and Ionicity in MgAgAs-type Compounds. Chem. - Eur. J. 2014, 20, 9702–9708. 10.1002/chem.201400299. [DOI] [PubMed] [Google Scholar]
- Bende D.; Wagner F. R.; Sichevych O.; Grin Y. Chemical Bonding Analysis as a Guide for the Preparation of New Compounds: The Case of VIrGe and HfPtGe. Angew. Chem., Int. Ed. 2017, 56, 1313–1318. 10.1002/anie.201610029. [DOI] [PubMed] [Google Scholar]
- Martín Pendás Á.; Blanco M. A.; Francisco E. Two-electron Integrations in the Quantum Theory of Atoms in Molecules. J. Chem. Phys. 2004, 120, 4581–4592. 10.1063/1.1645788. [DOI] [PubMed] [Google Scholar]
- Blanco M. A.; Martín Pendás Á.; Francisco E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. 10.1021/ct0501093. [DOI] [PubMed] [Google Scholar]
- Martín Pendás Á.; Francisco E.; Blanco M. A. Two-electron Integrations in the Quantum Theory of Atoms in Molecules with Correlated Wave Functions. J. Comput. Chem. 2005, 26, 344–351. 10.1002/jcc.20173. [DOI] [PubMed] [Google Scholar]
- Raupach M.; Tonner R. A Periodic Energy Decomposition Analysis Method for the Investigation of Chemical Bonding in Extended Systems. J. Chem. Phys. 2015, 142, 194105. 10.1063/1.4919943. [DOI] [PubMed] [Google Scholar]
- Dronskowski R.Computational Chemistry of Solid State Materials; John Wiley & Sons, Ltd.: 2005. [Google Scholar]
- Bader R. F. W.Atoms in Molecules: A Quantum Theory (International Series of Monographs on Chemistry); Clarendon Press: 1990. [Google Scholar]
- Martín Pendás Á.; Francisco E.. Promolden: A QTAIM/IQA code. Available from the authors upon request by writing to ampendas@uniovi.es.
- Kohout M.DGrid, ver. 5.2; Dresden: 2021.
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Blum V.; Gehrke R.; Hanke F.; Havu P.; Havu V.; Ren X.; Reuter K.; Scheffler M. Ab Initio Molecular Simulations with Numeric Atom-Centered Orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. 10.1016/j.cpc.2009.06.022. [DOI] [Google Scholar]
- Baranov A. I.; Kohout M. Electron Localization and Delocalization Indices for Solids. J. Comput. Chem. 2011, 32, 2064–2076. 10.1002/jcc.21784. [DOI] [PubMed] [Google Scholar]
- Chen H.; Friesecke G. Pair Densities in Density Functional Theory. Multiscale Model. Simul. 2015, 13, 1259–1289. 10.1137/15M1014024. [DOI] [Google Scholar]
- Rodríguez J. I.; Köster A. M.; Ayers P. W.; Santos-Valle A.; Vela A.; Merino G. An Efficient Grid-Based Scheme to Compute QTAIM Atomic Properties without Explicit Calculation of Zero-Flux Surfaces. J. Comput. Chem. 2009, 30, 1082–1092. 10.1002/jcc.21134. [DOI] [PubMed] [Google Scholar]
- Womersley R. S.Contemporary Computational Mathematics - A Celebration of the 80th Birthday of Ian Sloan; Springer International Publishing: Cham, Switzerland, 2018; pp 1243–1285. [Google Scholar]
- Blanco M. A.Métodos Cuánticos Locales para la Simulación de Materiales Iónicos. Fundamentos, algoritmos y aplicaciones. Ph.D. thesis, University of Oviedo: 1997. [Google Scholar]
- Kay K. G.; Todd H. D.; Silverstone H. J. Bipolar Expansion for r12nYlm(θ12,ϕ12). J. Chem. Phys. 1969, 51, 2363–2367. 10.1063/1.1672353. [DOI] [Google Scholar]
- Buehler R. J.; Hirschfelder J. O. Bipolar Expansion of Coulombic Potentials. Phys. Rev. 1951, 83, 628–633. 10.1103/PhysRev.83.628. [DOI] [Google Scholar]
- Luaña Víctor.Environ: local environment determination of arbitrary positions in a crystal; Oviedo: 1992. [Google Scholar]
- Kosov D. S.; Popelier P. L. A. Atomic Partitioning of Molecular Electrostatic Potentials. J. Phys. Chem. A 2000, 104, 7339–7345. 10.1021/jp0003407. [DOI] [Google Scholar]
- Francisco E.; Menéndez Crespo D.; Costales A.; Martín Pendás Á. A Multipolar Approach to the Interatomic Covalent Interaction Energy. J. Comput. Chem. 2017, 38, 816–829. 10.1002/jcc.24758. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Barca G. M. J.; Bertoni C.; Carrington L.; Datta D.; De Silva N.; Deustua J. E.; Fedorov D. G.; Gour J. R.; Gunina A. O.; Guidez E.; et al. Recent Developments in the General Atomic and Molecular Electronic Structure System. J. Chem. Phys. 2020, 152, 154102. 10.1063/5.0005188. [DOI] [PubMed] [Google Scholar]
- Dirac P. A. M. Note on Exchange Phenomena in the Thomas Atom. Math. Proc. Cambridge Philos. Soc. 1930, 26, 376–385. 10.1017/S0305004100016108. [DOI] [Google Scholar]
- Bloch F. Bemerkung zur Elektronentheorie des Ferromagnetismus und der elektrischen Leitfähigkeit. Eur. Phys. J. A 1929, 57, 545–555. 10.1007/BF01340281. [DOI] [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: a Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. 10.1139/p80-159. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Correlation Energy of an Inhomogeneous Electron Gas: A Coordinate-Space Model. J. Chem. Phys. 1988, 88, 1053–1062. 10.1063/1.454274. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Downs R. T.; Somayazulu M. S. Carbon Dioxide at 1.0 GPa. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1998, 54, 897–898. 10.1107/S0108270198001140. [DOI] [Google Scholar]
- Bindzus N.; Straasø T.; Wahlberg N.; Becker J.; Bjerg L.; Lock N.; Dippel A.-C.; Iversen B. B. Experimental Determination of Core Electron Deformation in Diamond. Acta Crystallogr., Sect. A: Found. Adv. 2014, 70, 39–48. 10.1107/S2053273313026600. [DOI] [PubMed] [Google Scholar]
- Dobrzhinetskaya L. F.; Wirth R.; Yang J.; Green H. W.; Hutcheon I. D.; Weber P. K.; Grew E. S. Qingsongite, Natural Cubic Boron Nitride: The First Boron Mineral from the Earth’s Mantle. Am. Mineral. 2014, 99, 764–772. 10.2138/am.2014.4714. [DOI] [Google Scholar]
- Didier C.; Pang W. K.; Guo Z.; Schmid S.; Peterson V. K. Phase Evolution and Intermittent Disorder in Electrochemically Lithiated Graphite Determined Using in Operando Neutron Diffraction. Chem. Mater. 2020, 32, 2518–2531. 10.1021/acs.chemmater.9b05145. [DOI] [Google Scholar]
- Li M.-R.; Deng Z.; Lapidus S. H.; Stephens P. W.; Segre C. U.; Croft M.; Paria Sena R.; Hadermann J.; Walker D.; Greenblatt M. Ba3(Cr0.97(1)Te0.03(1))2TeO9: in Search of Jahn-Teller Distorted Cr(II) Oxide. Inorg. Chem. 2016, 55, 10135–10142. 10.1021/acs.inorgchem.6b01047. [DOI] [PubMed] [Google Scholar]
- Tsirelson V.; Stash A.; Kohout M.; Rosner H.; Mori H.; Sato S.; Lee S.; Yamamoto A.; Tajima S.; Grin Y. Features of the Electron Density in Magnesium Diboride: Reconstruction from X-ray Diffraction Data and Comparison with TB-LMTO and FPLO Calculations. Acta Crystallogr., Sect. B: Struct. Sci. 2003, 59, 575–583. 10.1107/S0108768103012072. [DOI] [PubMed] [Google Scholar]
- Ievinǎ A.; Straumanis M.; Karlsons K. Präzisionsbestimmung von Gitterkonstanten hygroskopischer Verbindungen (LiCl, NaBr). Z. Phys. Chem. 1938, 40B, 146–150. 10.1515/zpch-1938-4009. [DOI] [Google Scholar]
- Walker D.; Verma P. K.; Cranswick L. M.; Jones R. L.; Clark S. M.; Buhre S. Halite-sylvite Thermoelasticity. Am. Mineral. 2004, 89, 204–210. 10.2138/am-2004-0124. [DOI] [Google Scholar]
- Ewais E. M.; El-Amir A. A.; Besisa D. H.; Esmat M.; El-Anadouli B. E. Synthesis of Nanocrystalline MgO/MgAl2O4 Spinel Powders from Industrial Wastes. J. Alloys Compd. 2017, 691, 822–833. 10.1016/j.jallcom.2016.08.279. [DOI] [Google Scholar]
- Barrett C. S. X-ray Study of the Alkali Metals at Low Temperatures. Acta Crystallogr. 1956, 9, 671–677. 10.1107/S0365110X56001790. [DOI] [Google Scholar]
- Streib W. E.; Jordan T. H.; Lipscomb W. N. Single-crystal X-Ray Diffraction Study of β Nitrogen. J. Chem. Phys. 1962, 37, 2962–2965. 10.1063/1.1733125. [DOI] [Google Scholar]
- Schuch A. F.; Mills R. L. Crystal Structures of the Three Modifications of Nitrogen 14 and Nitrogen 15 at High Pressure. J. Chem. Phys. 1970, 52, 6000–6008. 10.1063/1.1672899. [DOI] [Google Scholar]
- Francisco E.; Casals-Sainz J. L.; Rocha-Rinza T.; Martín Pendás Á. Partitioning the DFT Exchange-Correlation Energy in Line with the Interacting Quantum Atoms Approach. Theor. Chem. Acc. 2016, 135, 170. 10.1007/s00214-016-1921-x. [DOI] [Google Scholar]
- Gelessus A.; Thiel W.; Weber W. Multipoles and Symmetry. J. Chem. Educ. 1995, 72, 505–508. 10.1021/ed072p505. [DOI] [Google Scholar]
- Bende D.Chemical Bonding Models and Their Implications for Bonding-Property Relations in MgAgAs-Type and Related Compounds.; Ph.D. Dissertation; p 97. [Google Scholar]
- Martín Pendás Á.; Casals-Sainz J. L.; Francisco E. On Electrostatics, Covalency, and Chemical Dashes: Physical Interactions versus Chemical Bonds. Chem. - Eur. J. 2019, 25, 309–314. 10.1002/chem.201804160. [DOI] [PubMed] [Google Scholar]
- Wagner F. R.; Baranov A. I.; Grin Y.; Kohout M. A Position-Space View on Chemical Bonding in Metal Diborides with AlB2 Type of Crystal Structure. Z. Anorg. Allg. Chem. 2013, 639, 2025–2035. 10.1002/zaac.201200523. [DOI] [Google Scholar]
- Barin I.Thermochemical Data of Pure Substances; Wiley: 1995; Vol. I. [Google Scholar]
- Grochala W. Diamond: Electronic Ground State of Carbon at Temperatures Approaching 0 K. Angew. Chem., Int. Ed. 2014, 53, 3680–3683. 10.1002/anie.201400131. [DOI] [PubMed] [Google Scholar]
- Popov I. V.; Görne A. L.; Tchougréeff A. L.; Dronskowski R. Relative Stability of Diamond and Graphite as Seen Through Bonds and Hybridizations. Phys. Chem. Chem. Phys. 2019, 21, 10961–10969. 10.1039/C8CP07592A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.