

Abstract
Hepatoblastoma remains one of the most difficult childhood tumors to treat and is alarmingly understudied. We previously demonstrated that Proviral Insertion site in Maloney murine leukemia virus (PIM) kinases, specifically PIM3, are overexpressed in human hepatoblastoma cells and function to promote tumorigenesis. We aimed to use CRISPR/Cas9 gene editing with dual gRNAs to introduce large inactivating deletions in the PIM3 gene and achieve stable PIM3 knockout in the human hepatoblastoma cell line, HuH6. PIM3 knockout of hepatoblastoma cells led to significantly decreased proliferation, viability, and motility, inhibited cell-cycle progression, decreased tumor growth in a xenograft murine model, and increased animal survival. Analysis of RNA sequencing data revealed that PIM3 knockout downregulated expression of pro-migratory and pro-invasive genes and upregulated expression of genes involved in apoptosis and differentiation. Furthermore, PIM3 knockout decreased hepatoblastoma cancer cell stemness as evidenced by decreased tumorsphere formation, decreased mRNA abundance of stemness markers, and decreased cell surface expression of CD133, a marker of hepatoblastoma stem cell-like cancer cells. Reintroduction of PIM3 into PIM3 knockout cells rescued the malignant phenotype. Successful CRISPR/Cas9 knockout of PIM3 kinase in human hepatoblastoma cells confirmed the role of PIM3 in promoting hepatoblastoma tumorigenesis and cancer cell stemness.
Keywords: hepatoblastoma, PIM3 kinase, HuH6, CRISPR/Cas9 knockout, dual gRNA, cancer cell stemness
Introduction
Hepatoblastoma is the most common primary liver tumor in children (1). It remains alarmingly understudied with virtually no new therapies in the last 20 years, despite a recent doubling of its incidence (1). Regardless of significant advancements in tumor genetic profiling, there are currently no targeted molecular therapies for hepatoblastoma treatment.
Proviral Integration site for Maloney murine leukemia virus kinases, or PIM kinases (PIM1, PIM2, PIM3), are a family of serine/threonine kinases that are overexpressed in a number of human malignancies (2, 3), including hepatoblastoma (4), and function to promote tumorigenesis by affecting downstream proteins associated with cell survival (5), motility (6), and cell-cycle progression (7). We have previously demonstrated that PIM kinases are expressed in the long-term passage human hepatoblastoma cell line, HuH6, and in a human hepatoblastoma patient-derived xenograft line (4). We have shown that the small molecule pan-PIM inhibitor, AZD1208, led to decreased hepatoblastoma cell proliferation and survival, induced cell-cycle arrest in vitro, and decreased tumor growth in vivo (4). We have also shown that treatment of hepatoblastoma cells with AZD1208, led to decreased tumorsphere formation and CD133 cell surface expression, which are both characteristics of hepatoblastoma stem-cell like cancer cells (SCLCCs) (8).
PIM3, specifically, is thought to play a pivotal role in hepatoblastoma tumorigenesis. Hepatoblastoma cells with PIM3 overexpression yielded larger tumors in mice (9). Higher PIM3 expression in hepatoblastoma human tumors correlated with worse patient survival (9). Targeting PIM kinases has recently become attractive in multiple cancers, but there are no drugs that specifically target PIM3. RNA interference (RNAi), which reduces or knocks down gene translation by inducing rapid and reversible degradation of target transcripts, has also been employed to target PIM3. However, RNAi is short-lived, can inhibit undesired genes, and is limited by the fact that cells retain substantial residual target gene activity (10). In contrast, targeted genome editing techniques, such as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated nuclease 9 (Cas9), are powerful tools that allow for the permanent cleavage of specific DNA sequences rendering genes inoperative (11).
In the current study, we successfully used CRISPR/Cas9 gene editing with dual guide RNAs to establish a genetic knockout (KO) of PIM3 in hepatoblastoma cells. Using this novel cell line, we aimed to evaluate the role of PIM3 in hepatoblastoma tumorigenesis, cell-cycle progression, and maintenance of SCLCCs and the stem cell-like phenotype. The findings presented in this study provide evidence that loss of PIM3 decreased hepatoblastoma cell proliferation and motility, induced cell-cycle arrest, and decreased cancer cell stemness while upregulating genes involved in differentiation.
Materials and Methods
Cells and cell culture
Cells were maintained in culture under standard conditions at 37 °C and 5 % CO2. The human long-term passage hepatoblastoma cell line, HuH6, was acquired from Thomas Pietschmann (Hannover, Germany) (12) and maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10 % fetal bovine serum (HyClone, GE Healthcare Life Sciences, Logan, UT), 1 μg/mL penicillin/streptomycin (Gibco, Carlsbad, CA), and 2 mmol/L L-glutamine (Thermo Fisher Scientific, Waltham, MA). The HuH6 cell line was validated within the past 12 months using short tandem repeat analysis (Genomics Core, University of Alabama at Birmingham (UAB), Birmingham, AL) and determined to be free of mycoplasma.
Antibodies and reagents
Antibodies utilized included rabbit monoclonal anti-PIM1 (3247), anti-PIM2 (4730), anti-PIM3 (4165), and anti-cyclin D1 (92G2, 2978), and rabbit polyclonal anti-vinculin (4650) from Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH, clone 6C5, MAB374) was from EMD Millipore (Millipore Sigma, Burlington, MA). Mouse monoclonal anti-β-actin (A1978) was from Sigma Aldrich (St. Louis, MO).
Generation of stable CRISPR/Cas9-mediated PIM3 knockout cells
The CRISPR/Cas9 vector, pSpCas9(BB)-2A-GFP (pX458) was a gift from Dr. A. Joseph Tector and developed by Dr. Feng Zhang (Addgene plasmid # 48138) (13). We used Geneious software (Biomatters, Auckland, New Zealand) to design guide RNAs (gRNAs) to target sites crucial to protein function from the 5’ untranslated region through exon 3 of the PIM3 gene (Supplementary Figure S1 A–B). The selected gRNAs were evaluated with the MIT CRISPR Design Tool (http://crispr.mit.edu/) to assess for potential off-target sequences. The oligonucleotides were annealed at 37 °C for 30 minutes followed by 95 °C for 5 minutes and ramped down to 25 °C at a rate of 5 °C per minute. Annealed oligonucleotides were cloned into the CRISPR/Cas9 plasmid by digesting 1 μg of plasmid pX458 with BbsI (New England Biolabs, Ipswich, MA) in the presence of annealed oligonucleotides, T7 ligase, and ATP in a MyCycler™ thermal cycler (Bio-Rad, Hercules, CA) for 6 cycles of 37 °C for 5 minutes and 23 °C for 5 minutes. Ligation reaction was used to transform Invitrogen MAX Efficiency™ DH5α™ competent E. coli cells (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. The QIAprep® Miniprep (Qiagen, Germantown, MD) was used to isolate plasmid from one colony per treatment.
HuH6 cells were plated 24 hours prior to transfection with 2 × 106 cells per T150 flask. Transfection was carried out using FuGENE® HD Transfection Reagent (Promega, Madison, WI) per the manufacturer’s protocol with two plasmids per transfection. Briefly, the appropriate plasmid pair was incubated for 15 minutes at room temperature in Opti-MEM™ media (Thermo Fisher Scientific) with FuGENE® HD Transfection Reagent (Promega) in a 3:2 ratio of transfection reagent to DNA with 5 μg DNA from each plasmid (total of 10 μg DNA) per 1 × 106 cells and added to the cells while swirling the flask. Forty-eight hours after transfection, cells were sorted based on GFP expression using a FACSAria II cell sorter (BD Biosciences, San Jose, CA) into four 96-well plates per paired plasmid transfection with a single cell per well (Comprehensive Flow Cytometry Core, UAB, Birmingham, AL). To screen for CRISPR/Cas9-mediated deletions of PIM3 gene, genomic DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen) from HuH6 wild-type (WT) cells and those clones that grew to confluency and survived passaging into larger flasks. Pwo SuperYield DNA Polymerase, dNTPack (Sigma Aldrich) with GC-rich solution was utilized per manufacturer’s protocol to amplify the region of interest within the PIM3 gene using the following primers: (Forward: 5’-GGACCGACGCGACACG-3’, Reverse: 5’-TCCTTCACCACGTGCTTCAC-3’). The sizes of the PCR products were assessed using gel electrophoresis on a 2 % agarose gel. Individual bands were cut out and DNA was purified from the gel using the QIAquick Gel Extraction Kit (Qiagen). Nucleotide sequences of these DNA fragments were analyzed by Sanger sequencing (Genomics Core, UAB) and aligned to the human reference sequence using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). In addition, PIM3 expression in the HuH6 WT cells and selected PIM3 KO clones was assessed by Western blotting to confirm the absence of the PIM3 protein.
Immunoblotting
Cells or homogenized tumor specimens were lysed using radio-immunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (Sigma Aldrich), phosphatase inhibitors (Sigma Aldrich), and phenyl-methane-sulfonyl-fluoride (Sigma Aldrich). Immunoblotting, gel transfer, and immunodetection was performed as previously described (4). The Precision Plus Protein Kaleidoscope molecular weight marker (Bio-Rad) was used to confirm the expected size of target proteins. Antibodies were utilized according to the manufacturers’ recommendations. Equal protein loading was confirmed using vinculin, GAPDH or β-actin.
Proliferation, viability, and growth assays
Cell proliferation was measured using the CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). HuH6 WT or PIM3 KO cells (5 × 103 cells per well) were plated in 96-well plates. After 72 hours of incubation, 10 μL of CellTiter 96® reagent was added to each well and the absorbance was read at 490 nm using a microplate reader (BioTek Gen5, BioTek, Winooski, VT) to detect the formazan product. Cell viability was measured using the alamarBlue® Cell Viability Assay (Thermo Fisher Scientific). HuH6 WT or PIM3 KO cells (1.5 × 104 cells per well) were plated in 96-well plates and incubated for 72 hours prior to the addition of dye. At the time of assay completion, 10 μL of alamarBlue® reagent was added to each well and the absorbance was read at 562 nm (reduced reagent) and 595 nm (oxidized reagent) using a microplate reader (BioTek Gen5). For cell growth over time, HuH6 WT or PIM3 KO cells (5 × 104 cells per well) were plated in 12-well plates, incubated for 24, 48, 72, or 96 hours, stained with trypan blue (0.4%, Gibco), and counted with a hemocytometer. Doubling time was calculated as the duration in hours × log(2) / log(final cell count) - log(initial cell count) (14). Results of proliferation, viability, and cell growth from at least three biologic replicates were reported as mean fold change ± standard error of the mean (SEM).
Motility assays
For migration assays, 24-well culture plates (Corning Life Sciences) were utilized. The bottom of the 8 μm micropore Transwell® inserts were coated with collagen I (10 μg/mL, MP Biomedicals, Santa Ana, CA) overnight at 37 °C and then washed with phosphate-buffered saline (PBS). The inserts were placed in wells containing 350 μL media. HuH6 WT or PIM3 KO cells (3 × 104 cells per insert) were placed inside each insert and allowed to migrate for 24 hours. The inserts were fixed with 3 % paraformaldehyde and stained with 1 % crystal violet. Images of the inserts were obtained using a light microscope and the number of cells were counted using ImageJ (https://imagej.nih.gov/ij). Migration was reported as mean fold change in number of cells migrating through the membrane ± SEM.
Similar methods were used for invasion with the addition of coating the inside of the inserts with 50 μL of Matrigel™ (1 mg/mL, BD Biosciences) overnight at 37 °C. The inserts were placed in wells containing 350 μL media and 3 × 104 HuH6 WT or PIM3 KO cells were placed inside each insert and allowed to invade for 24 hours. Fixation, staining, image acquisition, and counting were performed as described above. Invasion was reported as mean fold change in number of cells invading through the Matrigel™ and membrane ± SEM.
Cell motility was also evaluated utilizing a monolayer wounding (scratch) assay as previously described (9). Briefly, HuH6 WT or PIM3 KO cells were plated and allowed to grow to near-confluency before a uniform scratch was made in the well with a sterile 200 μL pipette tip. Images of the scratch wound area were obtained at 0, 24, 48, and 72 hours. The area of the wound in pixels was quantified using ImageJ and data reported as mean fold change in scratch area compared to time zero ± SEM.
Analysis of the cell cycle
HuH6 WT or PIM3 KO cells (1 × 106) were plated, allowed to attach, and incubated for 24 hours. Cells were trypsinized, washed with PBS, and fixed in cold 100 % ethanol for at least 30 minutes. Following a second wash with PBS, cells were stained for 30 minutes at room temperature with a solution containing 20 μg/mL propidium iodide (Invitrogen) and RNAse A (0.2 mg/mL, Invitrogen) in 0.1 % Triton X (Active Motif, Carlsbad, CA). Data were obtained using the Attune™ NxT Flow Cytometer (Invitrogen™, Thermo Fisher, Eugene, OR) and analyzed with FlowJo software (FlowJo, LLC, Ashland, OR).
RNA extraction, library preparation, and sequencing
Total cellular RNA was extracted using the RNeasy kit (Qiagen) according to the manufacturer’s protocol. RNA was sent to the UAB Genomics Core for sample quality control (QC), library preparation, and sequencing. Briefly, the quality of the total RNA was assessed using the Agilent 2100 Bioanalyzer followed by 2 rounds of poly A+ selection and conversion to cDNA. The NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® library generation kit (New England Biolabs) was used per the manufacturer’s instructions. The libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for library quantitation (Kapa Biosystems, Woburn, MA). Subsequently, sequencing was performed on the Illumina NextSeq500 using the latest versions of the sequencing reagents and flow cells with single-end 75 bp reads. Raw and processed data were deposited in Gene Expression Omnibus (GEO, Accession # GSE164082).
RNA sequencing analysis
The raw RNA-Seq fastq reads were aligned to the human reference genome (GRCh38 p13 Release 32) from Gencode using STAR (version 2.7.3a) and the parameters --outReadsUnmapped Fastx --outSAMtype BAM SortedBy Coordinate --outSAMattributes All --outFilterIntronMotifs RemoveNoncanonicalUnannotated (15). Following alignment, Cufflinks (version 2.2.1) was used to assemble transcripts, estimate their abundances, and test for differential expression and regulation using parameters --library-type fr-firststrand -G –L (16, 17). Cuffmerge, which is part of Cufflinks, merged the Cufflinks transcripts across multiple samples using default parameters. Finally, Cuffdiff found significant changes in transcript expression, splicing and promoter usage using default parameters.
For generating network analysis of biological systems, a data set containing gene identifiers and corresponding expression values was uploaded into Ingenuity Pathway Analysis (IPA, https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/) (18). Each identifier was mapped to its corresponding object in Ingenuity’s Knowledge Base. A fold change cutoff of ± 2 was set to identify molecules whose expression was significantly differentially regulated. These molecules, called Network Eligible molecules, were overlaid onto a global molecular network developed from information contained in Ingenuity’s Knowledge Base. Networks of Network Eligible Molecules were then algorithmically generated based on their connectivity. The Functional Analysis identified the biological functions and/or diseases that were most significant to the entire data set. Molecules from the dataset that met the fold change cutoff of ± 2 and were associated with biological functions and/or diseases in Ingenuity’s Knowledge Base were considered for the analysis. Right-tailed Fisher’s exact test was used to calculate a p value determining the probability that each biological function and/or disease assigned to that data set is due to chance alone.
PIM3 rescue with transfection of PIM3 cDNA
The PIM3 expression vector, pcDNA3.1/V5-His-PIM3, was a generous gift from Dr. Jussi Taipale and was generated by PCR amplification and cloning into the pcDNA3.1/V5-HisC vector (19). The plasmid was sequenced for verification (Genomics Core, UAB). Empty vector (EV, pcDNA3.1/V5-HisC) was used as a control. Transfection was carried out using FuGENE® HD Transfection Reagent (Promega) per the manufacturer’s protocol. Briefly, HuH6 PIM3 KO cells were plated on the day prior to transfection. The appropriate plasmid was incubated for 15 minutes at room temperature in Opti-MEM™ media (Thermo Fisher Scientific) with FuGENE® HD Transfection Reagent (Promega) in a 3:2 ratio of transfection reagent to DNA with 7.5 μg DNA per 1 × 106 cells and added to the cells while swirling the flask. Cells were transfected with either pcDNA3.1/V5-HisC (EV) or pcDNA3.1/V5-HisC-PIM3 (PIM3 expression vector) for 48–72 hours prior to use in experiments. Lysates were made to perform immunoblotting, as described above, to assess for PIM3 expression.
Analysis of CD133 expression
HuH6 WT or PIM3 KO cells (1 × 106) were labeled with allophycocyanin (APC)-conjugated mouse immunoglobulin G1 (IgG1) anti-human CD133/1 (clone AC133, Miltenyi Biotec, Waltham, MA) according to the manufacturer’s instructions. Unlabeled cells were used as negative controls. The percent of cells positive for APC was determined via flow cytometry using the Attune™ NxT Flow Cytometer (Invitrogen™, Thermo Fisher).
In vitro extreme limiting dilution analysis
HuH6 WT or PIM3 KO cells were plated into 96-well ultra-low attachment plates using serial dilutions from 5000 to 1 cell per well with at least 12 replicates per dilution. Cells were plated into Dulbecco’s Modified Eagle’s Medium/Ham’s F12 supplemented with 2 mmol/L L-glutamine (Thermo Fisher Scientific), 1 μg/mL penicillin/streptomycin (Gibco), 20 ng/mL epidermal growth factor (EMD Millipore), 20 ng/mL beta-fibroblast growth factor (EMD Millipore), 2 % B27 supplement (Gibco), and 2.5 μg/mL amphotericin B (HyClone). Once spheres were present in the wells containing the most cells, all wells were counted. The presence or absence of spheres in each well was quantified by a single researcher blinded to the treatment groups. Extreme limiting dilution analysis software was utilized to analyze the data (http://bioinf.wehi.edu.au/software/elda/) (20).
Quantitative real-time PCR (qPCR)
For synthesis of cDNA, 1 μg of RNA was used in a 20 μL reaction mixture utilizing an iScript cDNA Synthesis kit (Bio-Rad) according to the supplier’s instructions. Resulting reverse transcription products were stored at −20 °C until further use. For quantitative real-time PCR, SsoAdvanced™ SYBR® Green Supermix (Bio-Rad) was utilized according to manufacturer’s protocol. Probes specific for the transcription factors octamer-binding transcription factor 4 (Oct4), homeobox protein Nanog, and sex determining region Y-box 2 (Sox2), as well as for β-actin were obtained (Applied Biosystems, Foster City, CA). Nestin primers (Forward: 5’-TCCAGGAACGGAAAATCAAG-3’, Reverse: 5’-GCCTCCTCATCCCCTACTTC-3’) were designed using Primer3 web version 4.1.0 (21) and checked for non-specific binding using the basic local alignment search tool (BLAST, NCBI). qPCR was performed with 10 ng cDNA in 20 μL reaction volume. Amplification was done using an Applied Biosystems 7900HT cycler (Applied Biosystems) and cycling conditions were 95 °C for 2 min, followed by 39-cycle amplification at 95 °C for 5 s and 60 °C for 30 s. Samples were analyzed in triplicate with β-actin utilized as an internal control. Gene expression was calculated using the ΔΔCt method (22) and reported as mean fold change ± SEM.
In vivo tumor growth and animal survival
Animal studies were approved by the UAB Institutional Animal Care and Use Committee (IACUC-021420) and conducted within institutional, national, and NIH guidelines. Six-week old athymic nude mice (Envigo) were maintained in the specific pathogen-free facility with standard 12-hour light/dark cycles and access to chow and water ad libitum. Both male and female mice were used to account for sex as a biological variable. Two studies were performed. In the first study, HuH6 WT cells or PIM3 KO cells (2.5 × 106 in 25 % Matrigel™, BD Biosciences) were injected subcutaneously into the right flank of 14 male animals (randomized to either group). In the second study, HuH6 WT cells and PIM3 KO cells (2.5 × 106 in 25 % Matrigel™, BD Biosciences) were injected subcutaneously into the right flank and left flank, respectively, of 7 female animals. Tumor volumes were measured three times weekly with calipers and calculated with the standard formula (width2 x length) / 2, where the length is the largest measurement. Tumor growth and animal survival were monitored over the period of 6 weeks (for the first study) and 7 weeks (for the second study) or until IACUC parameters for euthanasia were met. The investigators were not blinded to the group assignment. Animals were humanely euthanized with CO2 and cervical dislocation, and the tumors were harvested and prepared for further study.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded xenograft tumor specimens were cut into 5 μm sections and baked at 70 °C for one hour on positive slides. Immunohistochemistry for Ki-67 staining was performed as previously described (4). Slides were counterstained with hematoxylin. A negative control (rabbit IgG, 1 μg/mL, EMD Millipore) was included with each run. Stained slides were evaluated, and Ki-67 staining quantified by a board-certified pathologist (E.M.M.) who was blinded to the two groups. The number of Ki-67 positive cells per 500 cells in a representative section of each tumor was counted and the mean ± SEM calculated and reported (23).
Statistical analysis
All experiments were performed with a minimum of three biologic replicates. Data were reported as mean ± SEM of separate experiments. Student’s t-test (two-sided) or analysis of variance (ANOVA) was used to compare means between groups as appropriate, with p≤0.05 determined to be statistically significant. Survival curves were generated using SPSS (version 25, IBM®) and Kaplan-Meier analysis was performed with log-rank test to determine survival significance.
Results
Establishment of stable CRISPR/Cas9-mediated PIM3 knockout hepatoblastoma cell lines
We designed guide RNAs (gRNAs) to successfully target sites from the 5’ untranslated region through exon 3 of the PIM3 gene (Supplementary Material, Figure S1 A) with minimal potential off-target effects. The sequences of the gRNAs used in this study (from 5’ to 3’) as well as the sequences of the oligonucleotides that were annealed together to clone in these gRNAs are listed in Supplementary Material, Table S1. HuH6 human hepatoblastoma cells were transfected with two plasmids per transfection (1A and 1B, 2A and 2B, 3A and 3B, Supplementary Material, Figure S1 A) to achieve a double gRNA knockout resulting in large inactivating deletions of the PIM3 gene. Transfected cells were sorted based on GFP expression into four 96-well plates per paired plasmid transfection with a single cell per well. Of 1152 individual cells plated, 24 clones (9 clones transfected with plasmids containing gRNAs 1A and 1B, 8 clones transfected with 2A and 2B, and 17 clones transfected with 3A and 3B) grew to confluence and survived propagation and passage into larger flasks. We used both PCR, to screen these clones and confirm deletion of the PIM3 gene, as well as immunoblotting, to verify absence of PIM3 protein expression.
Of 24 total clones probed for PIM3 expression, three clones showed stable PIM3 KO. PCR products using PIM3 primers designed as described above were ran on a 2 % agarose gel (Supplementary Material, Figure S1 C). The band for the unedited HuH6 WT DNA of interest had an expected size of 828 bp and appeared between 750 and 1000 bp. PIM3 KO resulted in the generation of lower molecular weight DNA fragments, which migrated faster. Clone B11 (transfected with plasmids containing gRNAs 3A and 3B) had a smaller band, which appeared between 500 and 750 bp. Clones D2 (transfected with plasmids containing gRNAs 1A and 1B) and E8 (transfected with plasmids containing gRNAs 2A and 2B) also had smaller bands, indicating that the CRISPR/Cas9 machinery with the double gRNAs introduced large deletions into the genomic sequence. The bands were excised, and sequencing results aligned to the human reference sequence using BLAST. A representative analysis is illustrated in the Supplementary Material, Figure S1 D. The primer pair positions are shown along with sequencing results of the HuH6 WT and B11 PIM3 KO clone bands. The gRNAs 3A and 3B were also aligned and their positions shown.
PIM3 protein expression of the unedited HuH6 WT cells and the three KO clones, B11, D2, and E8, was assessed by immunoblotting. The resulting absence of PIM3 protein expression confirms the CRISPR/Cas9-mediated KO of PIM3 gene (Figure 1 A). Taken together, these findings confirmed the generation of stable HuH6 PIM3 KO cell line. We sought to determine how the deletion of PIM3 protein affected the abundance of the two other members of the PIM kinase family, PIM1 and PIM2. Western blotting of HuH6 PIM3 KO clones revealed no change in PIM1 and PIM2 expression (Figure 1 A).
Figure 1. PIM3 knockout decreased proliferation, viability, and motility in hepatoblastoma cells.
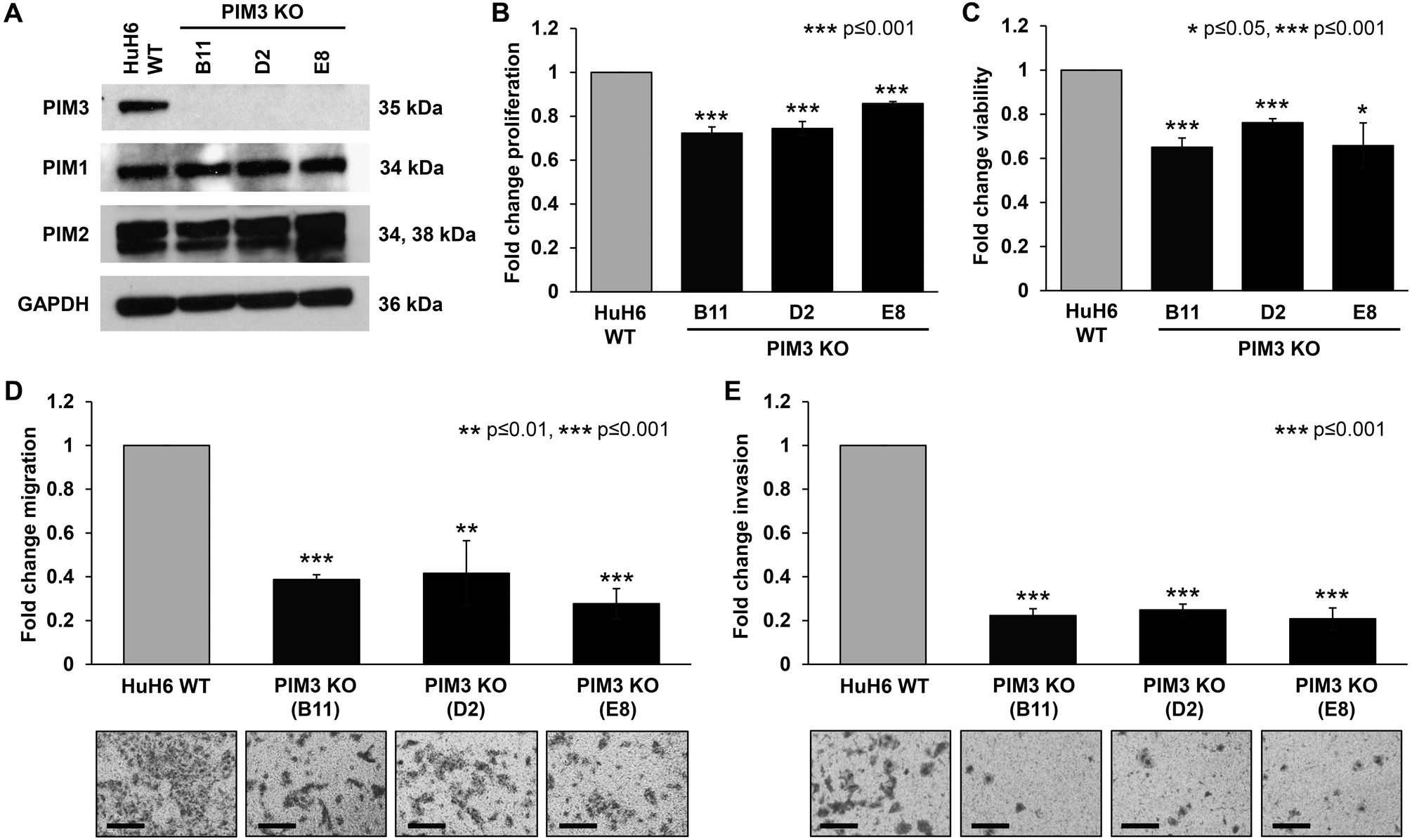
(A) Immunoblotting for PIM kinases in HuH6 wild-type (WT) and PIM3 knockout (KO) cells. PIM3 protein expression was absent as expected in PIM3 KO clones. There was no change in PIM1 and PIM2 protein expression in PIM3 KO cells compared to HuH6 WT cells. GAPDH was used as a loading control. (B) Proliferation and (C) viability were measured using CellTiter 96® and alamarBlue® assays, respectively. All three clones of PIM3 KO cells exhibited significantly decreased (B) proliferation and (C) viability compared to HuH6 WT cells. Data represent at least three biologic replicates and are reported as mean ± standard error of the mean. (D) Migration and (E) invasion were also assessed. HuH6 WT or PIM3 KO cells were seeded into modified Boyden chambers. Inserts were coated on the bottom with collagen which acted as a chemoattractant and a layer of Matrigel™ was added to the top of the insert for invasion. After 24 hours, photographs were taken with representatives shown (panels to the bottom of the graphs) and migration and invasion from at least three biologic replicates reported as mean fold change in number of cells migrating or invading, respectively ± standard error of the mean. Scale bars represent 100 μm. PIM3 KO cells exhibited a significant decrease in (D) migration and (E) invasion compared to HuH6 WT cells.
CRISPR/Cas9-mediated PIM3 kinase knockout decreased proliferation, viability, and motility of hepatoblastoma cells
We aimed to assess the role of PIM3 kinase in hepatoblastoma by exploring the phenotypic effects of the genetic KO of PIM3. PIM3 KO cells did not differ in cell morphology from that of the HuH6 WT cells (Supplementary Material, Figure S2). PIM3 KO cells exhibited significantly decreased proliferation (28 ± 3 % for clone B11, 26 ± 3 % for clone D2, and 14 ± 1 % for clone E8, p=0.001, Figure 1 B) and viability (35 ± 4 % for clone B11, 24 ± 2 % for clone D2, and 34 ± 10 % for clone E8, p≤0.001 for clones B11 and D2 and p≤0.05 for clone E8, Figure 1 C) compared to HuH6 WT cells.
We next sought to assess migration and invasion as early steps in the metastatic cascade, since tumor invasion and metastasis are hallmarks of aggressive hepatoblastoma. Migration was significantly decreased with PIM3 KO (61 ± 2 % for clone B11, 58 ± 15 % for clone D2, and 72 ± 7 % for clone E8, p≤0.01, Figure 2 D). Finally, PIM3 KO resulted in significantly decreased invasion through Matrigel™ (78 ± 3 % for clone B11, 75 ± 3 % for clone D2, and 79 ± 5 % for clone E8, p≤0.001, Figure 2 E). Representative images of migration and invasion inserts are presented below the graphs (Figure 2 D, E). Given these findings, we chose to focus on clone B11 for further experimentation which was referred to as PIM3 KO for the remainder of this study.
Figure 2. PIM3 knockout decreased growth over time in hepatoblastoma cells and inhibited their progression through the cell cycle.

(A) PIM3 knockout (KO) cells exhibited a slower growth rate compared to HuH6 wild-type (WT) cells over the course of 96 hours. (B) Doubling time was significantly longer for PIM3 KO cells compared to HuH6 WT cells. Data represent at least three biologic replicates and are reported as mean ± standard error of the mean. (C) HuH6 WT or PIM3 KO cells were stained with propidium iodide and were analyzed using flow cytometry to evaluate progression through the cell cycle. FlowJo software was used for analysis. Quantification of average percent cells in each phase of the cell cycle across all replicates revealed a significant increase in G1 phase and a decrease in S phase in PIM3 KO cells compared to HuH6 WT cells. (D) Representative histograms showing percentages of one experiment. (E) Tabular results (mean percent cells in phase ± SEM) of the cell cycle analysis from three biologic replicates are provided.
Motility was also assessed using a cell monolayer wounding (scratch) assay. PIM3 KO cells (clone B11) resulted in significantly larger wound (scratch) area over time compared to HuH6 WT cells (0.03 ± 0.003 fold change in wound healing at 72 hours vs. 0.4 ± 0.04, PIM3 KO vs. WT cells, p≤0.001, Supplementary Material, Figure S3). These data corroborate a decrease in cell motility with PIM3 KO.
In addition to changes in proliferation, viability, and motility, PIM3 KO cells (clone B11) exhibited decreased growth over time and a slower growth rate compared to HuH6 WT cells over the course of 96 hours (5.7 ± 0.01 fold change in growth on average vs. 11.3 ± 0.03, PIM3 KO vs. WT cells, p≤0.001, Figure 2 A). Doubling time was 38 ± 0.13 hours for PIM3 KO cells compared to 27 ± 0.01 hours for HuH6 WT cells (p≤0.001, Figure 2 B).
PIM3 kinase knockout inhibited cell-cycle progression in hepatoblastoma cells
To further examine the phenotypic changes observed with PIM3 KO and understand how cell growth is inhibited, cell-cycle progression was analyzed using flow cytometry. PIM3 KO resulted in an arrest in the G0/G1 phase and inhibition of progression through the cell cycle. There was an increase in the percentage of cells in the G1 phase (p≤0.05, Figure 2 C, E) accompanied by a decrease in the percentage of cells in the S phase in PIM3 KO compared to HuH6 WT cells (p≤0.01, Figure 2 C, E). Representative histograms of a single experiment are presented in Figure 2 D. Cell cycle data from three biologic replicates are presented in tabular form with mean ± SEM reported (Figure 2 E).
Differential gene expression analysis of hepatoblastoma cells upon PIM3 knockout
The effect of PIM3 KO was further investigated at the genomic level. We examined the differences in gene expression in PIM3 KO compared to HuH6 WT cells by RNA sequencing and observed that loss of PIM3 altered the expression of 746 genes overall (Figure 3 A). At an adjusted p value of less than 0.05 and fold change cut off of greater or less than 2, there were 378 differentially upregulated and 368 differentially downregulated genes (Figure 3 B).
Figure 3. RNA sequencing of PIM3 knockout and HuH6 wild-type cells.

(A) Volcano plot of genes expressed in PIM3 knockout (KO) compared to HuH6 wild-type (WT) cells. Log2 fold changes were plotted on the x-axis and −Log10 p values were plotted on the y-axis. The dotted horizontal line represents a p value of 0.05, and the vertical dotted lines represent a Log2 fold change of ± 1. Downregulated genes (fold change<−2 and p≤0.05) are shown in blue and upregulated genes (fold change>+2 and p≤0.05) are shown in orange. Non-significant (NS) genes (p value > 0.05) are shown in black. (B) PIM3 KO of hepatoblastoma cells followed by RNA sequencing analysis revealed changes of expression of 746 genes. Genes associated with PIM3 KO were analyzed with Ingenuity Pathway Analysis (IPA) and top (C) biological functions and (D) canonical pathways presented. A positive z-score (orange bars) indicates activation or a positive correlation and a negative z-score (blue bars) indicates inactivation or a negative correlation. The dotted line in (D) represents a predicted p value of 0.05. The ratio indicates the number of proteins from the dataset that map to the pathway divided by the total number of proteins that map to the same pathway. An asterisk marks CCR5 signaling as one of the top inactivated pathways. FXR: Farnesoid X receptor; RXR: Retinoid X receptor; LXR: Liver X receptor; LPS: Lipopolysaccharide; IL-1: Interleukin-1; Inhibit.: Inhibition; VDR: Vitamin D receptor; GPCR: G protein-coupled receptor; CDK5; Cyclin dependent kinase 5; BMP: Bone morphogenetic protein; eNOS: Endothelial nitric oxide synthase; NF-κB: Nuclear factor kappa B; CRH: Corticotropin releasing hormone; TGF-β: Transforming growth factor beta.
The differentially regulated genes (p<0.05 and fold change cutoff ± 2) associated with PIM3 KO were analyzed using the Ingenuity Pathway Analysis (IPA) tool and a list of relevant biological functions and canonical pathways generated (Supplementary Material, Tables S2 and S3). IPA analysis of PIM3 KO cells compared to their WT counterparts showed upregulation of genes linked to biologic functions including cell death and differentiation (Figure 3 C, orange bars). Analysis of PIM3 KO compared to WT cells also showed downregulation of genes associated with cell movement, proliferation, and growth (Figure 3 C, blue bars). The downregulated canonical pathways associated with PIM3 KO, according to IPA, unveiled CCR5 signaling, which is known to facilitate tumor progression through inducing cancer cell homing to metastatic sites (24), as one of the top inactivated pathways (Figure 3 D, asterisk). Notably, IPA also highlighted the downregulation of the cell cycle G1/S checkpoint regulation and multiple cancer-associated signaling pathways including PI3K/AKT, TGF-β, and mTOR signaling in the PIM3 KO cells (Figure 3 D, blue bars). On the contrary, canonical pathways that were activated in the PIM3 KO cells comprised of those regulating cellular differentiation, such as FXR/RXR and LXR/RXR activation (Figure 3 D, orange bars). These findings underline the oncogenic activity of PIM3 kinase and highlight its role in de-differentiation of hepatoblastoma cells.
Addition of PIM3 rescued the PIM3 kinase knockout phenotype
To further demonstrate that PIM3 KO was responsible for the anti-tumor phenotype observed and address the potential off-target effects that may occur with the CRISPR/Cas9 system, a rescue experiment was performed. HuH6 PIM3 KO cells were transfected with empty vector (EV) or PIM3 cDNA expressing plasmid. Both PIM3 KO and rescue of expression were observed by immunoblotting (Figure 4 A). Introduction of PIM3 cDNA in KO cells returned proliferation to 88 ± 1 % (p≤0.001, PIM3 rescue vs. PIM3 KO, Figure 4 B) and migration to 94 ± 3 % (p≤0.001, PIM3 rescue vs. PIM3 KO, Figure 4 C) of baseline. Representative images of migration inserts are presented to the bottom of the graph (Figure 4 C). EV transfected controls did not affect proliferation or migration of PIM3 KO cells (Figure 4 B, C).
Figure 4. PIM3 re-introduction rescued the anti-proliferative and anti-migratory phenotype observed with PIM3 knockout.
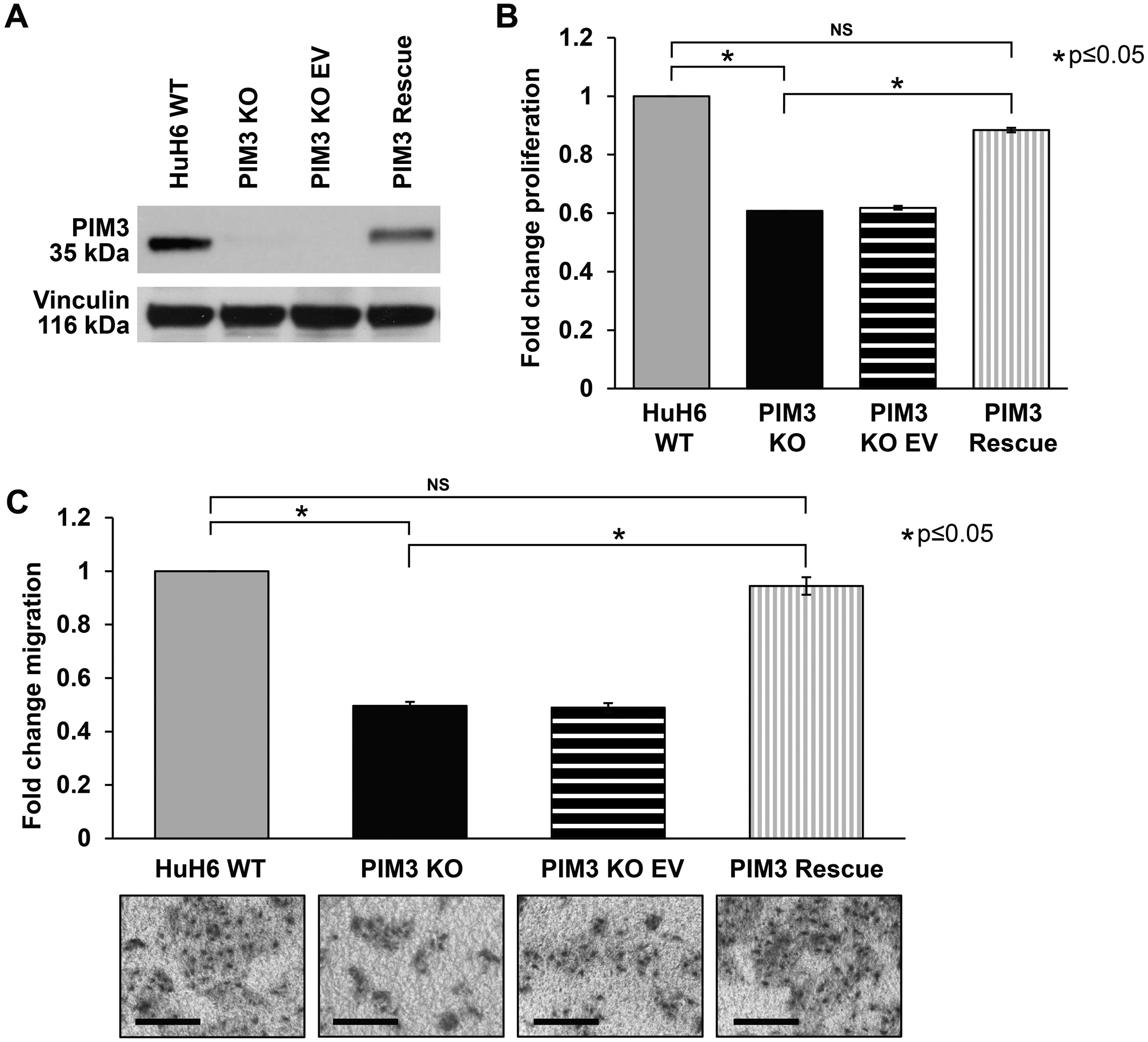
(A) PIM3 knockout (KO) cells were transfected with an empty vector (EV) control or PIM3 cDNA expressing plasmid. Immunoblotting confirmed both absence of PIM3 protein expression in PIM3 KO cells and rescue of expression in PIM3 KO-PIM3 cDNA (PIM3 rescue). (B) Introduction of PIM3 cDNA in PIM3 KO cells returned proliferation to almost baseline levels. (C) Migration was also evaluated. Introduction of PIM3 cDNA in PIM3 KO cells returned migration to baseline levels. Representative images of migration inserts are presented to the bottom of the graph. Scale bars represent 100 μm. EV transfected controls did not affect (B) proliferation or (C) migration of PIM3 KO cells. Data represent at least three biologic replicates and are reported as mean ± standard error of the mean. NS: non-significant.
PIM3 knockout decreased hepatoblastoma cancer cell stemness
To evaluate the effect of PIM3 KO on hepatoblastoma cancer cell stemness, we examined i) the expression of cell surface marker CD133, a glycoprotein previously identified as a marker for hepatoblastoma SCLCCs (8), ii) the ability of cells to form tumorspheres in serum- and attachment-free conditions, and iii) the abundance of mRNA of common markers of cell stemness. Flow cytometry demonstrated that PIM3 KO led to a decrease in percent CD133 positive cells compared to HuH6 WT (62 ± 1.3 % vs. 76 ± 0.5 %, PIM3 KO vs. WT, p≤0.05, Figure 5 A). Representative contour plots with negative staining controls for each cell line are shown in Figure 5 B. In addition, PIM3 KO resulted in reduced ability of hepatoblastoma cells to form spheres in non-adherent serum-free conditions. Using an extreme limiting dilution analysis, we found that PIM3 KO cells formed spheres less frequently and at higher cell concentrations than HuH6 WT cells (p≤0.001, Figure 5 C), indicating a decrease in cancer cell stemness.
Figure 5. PIM3 knockout decreased hepatoblastoma cancer cell stemness.
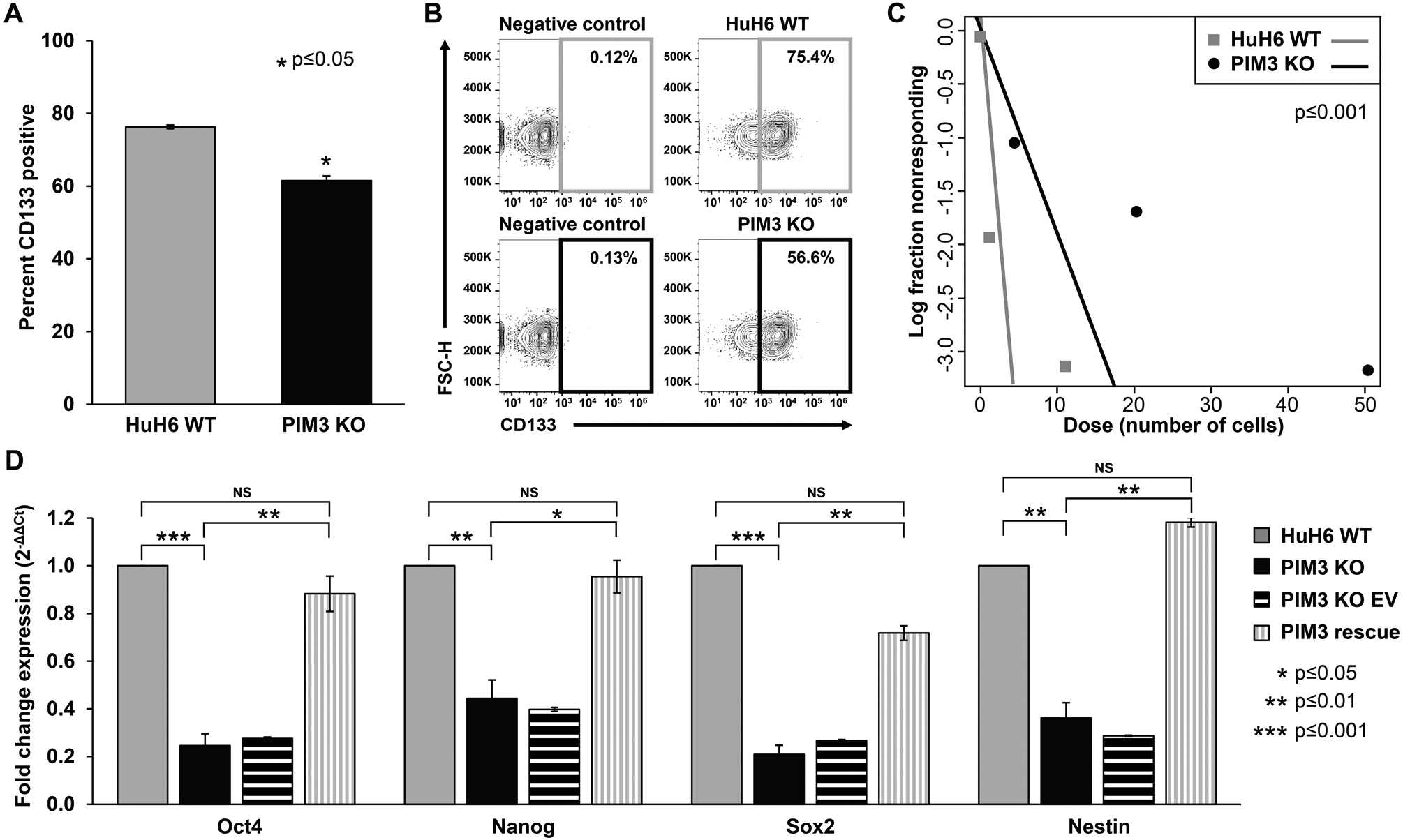
(A) CD133 cell surface expression was determined using flow cytometry. PIM3 knockout (KO) cells had significantly decreased CD133 expression compared to HuH6 wild-type (WT) cells. (B) Representative contour plots with negative staining controls for each cell line are shown. (C) Cells were plated for an extreme limiting dilution assay with decreasing numbers of cells per well. PIM3 KO cells formed spheres less frequently and at higher cell concentrations than HuH6 WT cells, indicating decreased tumorsphere formation ability. (D) Real-time PCR was used to examine the mRNA abundance for Oct4, Nanog, Sox2, and nestin. Gene expression was normalized to β-actin and calculated as fold change to HuH6 WT using the ΔΔCt method. PIM3 KO led to decreased mRNA abundance of these markers compared to HuH6 WT cells, while re-introduction of PIM3 cDNA in PIM3 KO cells led to rescue of mRNA abundance and a return to levels comparable to those seen in HuH6 WT cells. Data represent at least three biologic replicates and are reported as mean ± standard error of the mean. NS: non-significant.
Finally, we evaluated the mRNA abundance of four markers of cancer cell stemness, Oct4, Nanog, Sox2, and nestin. Quantitative real-time PCR (qPCR) comparing HuH6 WT to PIM3 KO cells demonstrated significantly lower mRNA abundance of these markers with PIM3 KO (p≤0.05, Figure 5 D), further indicating that PIM3 KO is associated with a reduced stem-like phenotype.
We again performed a rescue experiment to further demonstrate that PIM3 KO was responsible for the loss of stemness phenotype observed and address the potential off-target effects that may occur with the CRISPR/Cas9 system. When PIM3 cDNA was re-introduced into PIM3 KO cells, gene expression of the four stemness markers returned to levels comparable to those seen in HuH6 WT cells and there was no significant difference between PIM3 rescue and HuH6 WT cells (Figure 5 D). EV transfected controls did not affect mRNA abundance of the four markers (Figure 5 D). Similarly, when PIM3 cDNA was re-introduced into PIM3 KO cells and CD133 cell surface expression evaluated using flow cytometry, we observed an increase in the percentage of CD133 positive cells returning to the baseline seen in HuH6 WT cells. (Supplementary Material, Figure S4).
PIM3 knockout decreased in vivo tumor growth and increased animal survival
We next examined the effect of PIM3 KO on primary tumor growth in vivo. Male mice injected subcutaneously with PIM3 KO cells exhibited a significantly decreased percent tumor volume compared to mice injected with HuH6 WT cells (701 ± 166 % vs. 1401 ± 212 %, PIM3 KO vs. WT, p≤0.05, Figure 6 A). Representative tumor photos are presented in Figure 6 B. Relative tumor growth at the end of the 6-week study period was significantly decreased in mice bearing PIM3 KO tumors (2.7 ± 0.3 fold change vs 3.7 ± 0.2, p=0.02, Supplementary Figure S5). All mice bearing PIM3 KO tumors survived to the end of the experiment, compared to those with WT tumors who had a median survival of 31 days (range 27–42 days, p=0.002, Figure 6 C).
Figure 6. PIM3 knockout decreased in vivo tumor growth and increased animal survival in a xenograft model of hepatoblastoma.

(A) HuH6 wild-type (WT) or PIM3 knockout (KO) cells were injected into the right flank of male athymic nude mice (n=7 per group) and tumor volume monitored over time. Mice injected subcutaneously with PIM3 KO cells exhibited a significantly decreased percent tumor volume compared to mice injected with HuH6 WT cells. (B) Representative photos of tumors from both groups are shown. (C) Survival was significantly increased in the mice bearing PIM3 KO tumors compared to those bearing HuH6 WT tumors. (D) Immunoblotting confirmed PIM3 protein expression and absence thereof in HuH6 WT and PIM3 KO tumor xenografts, respectively. PIM3KO tumors exhibited lower expression of cyclin D1 protein compared to WT tumors, indicating cell-cycle arrest. β-actin was used as a loading control. (E) Formalin-fixed, paraffin-embedded tumor xenografts were stained for Ki-67, a marker of cell proliferation. The number of Ki-67 positive cells per 500 cells in a representative section of each tumor was counted and the mean was calculated and reported for each group ± standard error of the mean. Tumors from mice injected with PIM3 KO had significantly less Ki-67 staining than those injected with HuH6 WT. (F) Representative photomicrographs of HuH6 WT (top panel) and PIM3 KO (bottom panel) tumors are shown with an IgG negative control in the bottom left corner (bottom panel). Scale bars represent 100 μm.
Using immunoblotting, PIM3 protein expression and KO was confirmed in tumors harvested from both WT and PIM3 KO groups, respectively (Figure 6 D). To determine the effect on cell-cycle progression in PIM3 KO tumors, we evaluated protein expression levels of cyclin D1, a key cell-cycle regulator and a marker of G1/S progression (25). PIM3 KO tumors exhibited lower expression of cyclin D1 protein compared to WT tumors (Figure 6 D).
IHC for Ki-67 was performed to determine the effect of PIM3 KO on proliferation. Tumors from mice injected with PIM3 KO had significantly less Ki-67 staining (35 ± 7 vs. 51 ± 3 Ki-67 positive cells/500 cells, PIM3 KO vs. WT tumors, p=0.039) indicating decreased proliferation (Figure 6 E). Representative photomicrographs of Ki-67 IHC slides are presented in Figure 6 F along with the appropriate negative staining control (insert, bottom panel).
To account for potential bias based upon animal sex, we performed an additional experiment with female mice. To comply with the three R’s (Replacement, Reduction and Refinement), we utilized a bilateral flank model. PIM3 KO cells resulted in smaller tumors than those of HuH6 WT cells (464 ± 112 mm3 vs. 1515 ± 184 mm3, PIM3 KO vs. WT, p≤0.05, Supplementary Figure S6 A). Relative tumor growth at the end of the 43-day study period was decreased to 3.3 ± 0.5 fold change in PIM3 KO tumors compared to 5.1 ± 0.4 in WT tumors (p=0.016, Supplementary Material, Figure S6 B).
Discussion
PIM proteins are a highly conserved family of kinases that differ in their tissue distribution (26). PIM1 has been shown to be overexpressed in numerous solid tumors as well as leukemia and lymphoma (27–29), while increased PIM2 levels have been predominantly detected in hematologic malignancies (30). PIM3 has been shown to be highly expressed in malignant tumors of endodermal origin such as the pancreas and liver (31, 32). Finally, in some cancers, such as germ-cell tumors, all three PIM members have been found to be overexpressed, which suggests that there is only a partial redundancy and overlap in function among them. A potential functional redundancy was also suggested by the high degree of sequence homology between the 3 PIM kinases at the amino acid level (71 % between PIM1 and PIM3 and 44 % between PIM2 and PIM3) (33), which could explain the minimal phenotype alterations in PIM3−/− mice (34) and the compensatory activation of PIM2 in transgenic mice lacking PIM1 gene (35). In the current study, stable CRISPR/Cas9 KO of PIM3 did not lead to a change in protein expression levels of PIM1 and PIM2, suggesting that these two family members were not compensating for the loss of PIM3.
A growing body of evidence suggests that PIM3 is the specific family member to play a role in hepatoblastoma tumorigenesis and tumor maintenance. PIM3 has been reported to be aberrantly expressed in various malignant lesions, but not normal tissues, of the pancreas, prostate, ovary, and liver (31, 32, 36, 37). PIM3 was shown to accelerate pancreatic cancer development and promote tumor neovascularization and subsequent tumor growth (38). Qu et al. reported PIM3 expression as a critical risk factor in the development and prognosis of prostate cancer with high expression of PIM3 being associated with significantly decreased patient survival (36). PIM3 was also reported to be a prognostic marker in ovarian cancer and PIM3 overexpression was associated with increased proliferation and migration of ovarian cancer cells (37). Finally, in human hepatocellular carcinoma (HCC) cell lines, knockdown of PIM3 by siRNA reduced cell growth and induced apoptosis (32). Consistent with these previous reports, in this study, we showed that PIM3 KO inhibited hepatoblastoma cell proliferation, viability, and motility, decreased tumor growth in vivo, and decreased cancer cell stemness. Overall, these findings confirm and extend previous studies showing that knockdown of PIM3 by siRNA and pan-PIM inhibition, with AZD1208, decreased hepatoblastoma tumorigenicity and the stem cell-like phenotype (4, 8).
The novelty of this study lies in the application of CRISPR/Cas9 technology to achieve stable genetic KO of PIM3 in human hepatoblastoma cells. Different approaches for PIM3 targeting have been described including small molecule inhibitors. Most of the PIM inhibitors developed thus far are ATP competitive inhibitors targeting the ATP-binding pocket to inhibit protein function. These compounds inhibit all three family members (PIM1, PIM2, and PIM3), mainly because of the similarity in protein structure, and no PIM3-specific inhibitor has been described. There are disadvantages to relying solely upon pharmacologic inhibition for experimental studies. Proteins are still present with the use of pharmacological inhibition, and although their function is impaired, it is still possible for the drug-inhibited proteins to interact with binding partners (39). In addition, potential off-target kinase inhibition exists. For example, Nair et al. performed a kinase selectivity screening of the PIM2 inhibitor, JP11646, and found that, in addition to inhibiting PIM1 and PIM3 at a comparable half maximal inhibitory concentration (IC50), at least 5 other kinases were inhibited including GSK3β and CDK9 (40). GSK3β, which has been shown to play a role in tumorigenesis independent of PIM (41), was also identified as the most potent off-target kinase with the pan-PIM inhibitor LGB321, prior to the authors’ efforts of compound optimization (42). These limitations highlight the importance of implementing genetic tools to complement investigations employing pharmacological interventions.
In the past few years, CRISPR/Cas9 has emerged as a powerful genome engineering tool for introducing stable loss or gain-of-function alterations in a targeted gene in mammalian cells. Although the use of one gRNA is frequently sufficient to knock out the target gene, exon skipping and alternative splicing may occur, resulting in the production of fully or partially functional proteins, and allowing the cells to bypass single Cas9‐induced double-strand breaks (43). To maximize the likelihood of knocking out a target gene, a two-guide strategy has been described (44). In this study, we used dual gRNAs targeting two distinct sites to introduce double cuts which resulted in the elimination of the DNA between the two targeted sites as confirmed by PCR and Sanger sequencing. Another limitation to the CRISPR/Cas9 system is off-target mutagenesis which may confound the interpretation of results (45). We first addressed this potential issue by using different combinations of gRNAs and by analyzing multiple independent single cell clones by Western blotting, PCR, and sequencing. We also employed rescue experiments which have been widely described (46). These experiments were achieved by genetic complementation using a plasmid expressing PIM3 cDNA to rule out clonal artifacts and ensure that the phenotype observed was specific to the on-target activity.
PIM kinases act via multiple mechanisms to prevent apoptosis and promote viability, motility, and cell growth, ultimately resulting in tumorigenesis and tumor maintenance (2, 5, 6). In the current study, knockout of PIM3 affected both the malignant phenotype and the genome. The doubling time was significantly increased by 29% in PIM3 KO cells, and the level of expression of 746 genes was significantly altered. Among significantly downregulated genes following PIM3 KO were those involved in signaling pathways that promote cancer cell survival, proliferation, and movement according to IPA. The integration of genome editing, RNA sequencing, and the IPA bioinformatics tool revealed findings that implicate the role of PIM3 at the whole transcriptome level. In addition to its effects on hepatoblastoma cells in vitro, PIM3 KO was shown to have significant anti-tumor effects on hepatoblastoma cell line xenografts in vivo. In these experiments, PIM3 KO resulted in a significant growth inhibition and smaller tumors compared to WT tumors. In addition, mice bearing PIM3 KO tumors survived a median of 12 days longer than those with WT tumors. Decreased nuclear staining of Ki-67, a marker of proliferation, was also observed with PIM3 KO, further substantiating the role of PIM3 in promoting proliferation. Although we did not intend for a side-by-side comparison, both male and female mice were included in these in vivo studies to confirm that these tumors will propagate in both sexes and that the PIM3 KO effect seen is not sex-dependent. In both humans and mice, males are shown to be slightly more likely to develop hepatoblastoma (47, 48), although sex has not been shown to impact outcome (49). Further, both unilateral and bilateral flank injections were employed to exclude abscopal effects such as tumor-tumor distant interactions or tumor-derived factors capable of governing tumor progression at another site, which have been reported but are poorly understood (50).
PIM kinases also promote cell-cycle progression, which contributes to increased proliferation and accelerated tumor development. Cyclin-dependent kinase inhibitor 1 (p21cip1/waf1 or p21) binds to and prevents the activation of D-type cyclins/CDK complexes, which are required for progression of the cell cycle at G1. PIM kinases have been shown to stabilize the cell-cycle inhibitor p21 in the cytoplasm by directly phosphorylating it (4), leading to accumulation of p21 in the cytoplasm and subsequent cell-cycle progression. Transgenic mice expressing human PIM3 selectively in the liver had accelerated cell-cycle progression and, when exposed to the hepatocarcinogen, diethylnitrosamine, developed HCC at a two-fold rate and with greater tumor burden than wild type mice (51). In hepatoblastoma, AZD1208 treatment of HuH6 cells led to a decrease in phosphorylation of p21 at Thr145 and resulted in cell-cycle arrest (4). Consistent with these findings, in the current study, we show that PIM3 KO induced cell-cycle arrest in G1 phase in vitro, and that PIM3 KO tumors exhibited decreased expression of cell cycle regulatory protein cyclin D1. These findings are supported by the downregulation of cell-cycle progression genes following PIM3 KO based on the analysis of RNA sequencing data and further confirm that PIM3 plays an important role in hepatoblastoma cell proliferation and tumor development.
SCLCCs are thought to be responsible for tumor maintenance, chemoresistance, and disease recurrence (52), but the molecular mechanism of SCLCC induction and maintenance in hepatoblastoma is not fully understood. Hepatoblastoma SCLCCs may be identified by their ability to form tumorspheres (53) and by their expression of the cell surface marker CD133 (8). We have previously shown that overexpression of PIM3 increased tumorsphere formation in both a human hepatoblastoma cell line and a hepatoblastoma patient-derived xenograft, suggesting that PIM3 may play a role in promoting SCLCCs. In the current study, we show evidence that PIM3 KO resulted in decreased cancer cell stemness, as seen by decreased tumorsphere formation and mRNA abundance of Oct4, Nanog, Sox2, and nestin, all known markers of stemness in many cancer types (54–57). PIM3 KO also decreased expression of the cell surface marker, CD133, a marker of SCLCCs in hepatoblastoma (8). High expression of CD133 in human hepatoblastoma samples has been shown to correlate with advanced stage disease and poor response to therapy (58). The observation that PIM3 KO led to an upregulation of genes involved in cellular differentiation that were identified by IPA analysis further supports the role of PIM3 in maintaining the SCLCC population and promoting cancer cell stemness. Previous studies showed that pharmacologic PIM inhibition in hepatoblastoma cells reduced CD133 expression and arrested the growth of CD133-enriched hepatoblastoma tumor xenografts (8). Our current and previous findings suggest a promising clinical application for PIM inhibition since SCLCCs represent a subpopulation of cells within the tumor mass that must be effectively targeted to prevent hepatoblastoma recurrence and metastasis.
A limitation of the current study that must be addressed was the use of only one cell line. One of the major challenges in studying hepatoblastoma is the lack of cell lines and model systems. Although 15 hepatoblastoma cell lines have been reported in the literature (59), the majority lack validation. For example, HepG2 cells have been described in the literature by multiple researchers to be hepatoblastoma cells, but they are derived from a 15-year-old with HCC and the gene expression signature is more consistent with HCC (60). The HuH6 cell line is well-established and commonly used as it is the only commercially available long-term passage hepatoblastoma cell line. Our laboratory has previously developed and utilized human hepatoblastoma PDXs (4). However, PDX cells do not propagate well in culture, presenting numerous technical challenges for the CRISPR/Cas9 knockout and rendering the growth of single clones difficult.
In conclusion, KO of PIM3 demonstrated inhibitory effects on the tumorigenicity of hepatoblastoma cells, including decreased cell proliferation and motility in vitro and decreased tumor growth in vivo, induced cell-cycle arrest, and decreased cancer cell stemness. This novel cell line provided a means to further delineate the role of PIM3 in promoting hepatoblastoma tumorigenesis and the stem cell-like phenotype.
Supplementary Material
Acknowledgements
The authors wish to thank Vidya Sagar Hanumanthu and the UAB Comprehensive Flow Cytometry Core (supported by NIH P30 AR048311 and NIH P30 AI27667), Dr. Michael Crowley and the UAB Genomics Core (supported by NIH CA013148), and Dr. Anita Hjemeland’s laboratory for their assistance with the qPCR.
Funding
This project was made possible by funding from the National Cancer Institute of the National Institutes of Health under award numbers T32 CA229102 (RM and LVB), T32 CA091078 (LLS), T32 CA183926 (APW), 5T32GM008361 (CHQ), P30 AR048311 and P30 AI027767 to the Flow Cytometry Core, and CA013148 to the UAB Genomics Core. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Other funding sources include Cannonball Kids cancer, Sid Strong Foundation, Elaine Roberts Foundation, Open Hands Overflowing Hearts, and Starr Fund-Vince Lombardi Cancer Foundation (EAB), and the Society of University Surgeons (RM).
Footnotes
Conflict of Interest
The authors declare no competing financial interests in relation to the work described.
References
- 1.Feng J, Polychronidis G, Heger U, Frongia G, Mehrabi A, Hoffmann K. Incidence trends and survival prediction of hepatoblastoma in children: a population-based study. Cancer Commun (Lond). 2019;39(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang X, Song M, Kundu JK, Lee MH, Liu ZZ. PIM Kinase as an Executional Target in Cancer. J Cancer Prev. 2018;23(3):109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34(1):136–59. [DOI] [PubMed] [Google Scholar]
- 4.Stafman LL, Mruthyunjayappa S, Waters AM, Garner EF, Aye JM, Stewart JE, et al. Targeting PIM kinase as a therapeutic strategy in human hepatoblastoma. Oncotarget. 2018;9(32):22665–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Popivanova BK, Li YY, Zheng H, Omura K, Fujii C, Tsuneyama K, et al. Proto-oncogene, Pim-3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad-mediated apoptosis. Cancer Sci. 2007;98(3):321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santio NM, Eerola SK, Paatero I, Yli-Kauhaluoma J, Anizon F, Moreau P, et al. Pim Kinases Promote Migration and Metastatic Growth of Prostate Cancer Xenografts. PLoS One. 2015;10(6):e0130340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008;68(13):5076–85. [DOI] [PubMed] [Google Scholar]
- 8.Stafman LL, Williams AP, Garner EF, Aye JM, Stewart JE, Yoon KJ, et al. Targeting PIM Kinases Affects Maintenance of CD133 Tumor Cell Population in Hepatoblastoma. Transl Oncol. 2019;12(2):200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stafman LL, Waldrop MG, Williams AP, Aye JM, Stewart JE, Mroczek-Musulman E, et al. The presence of PIM3 increases hepatoblastoma tumorigenesis and tumor initiating cell phenotype and is associated with decreased patient survival. J Pediatr Surg. 2019;54(6):1206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mocellin S, Provenzano M. RNA interference: learning gene knock-down from cell physiology. J Transl Med. 2004;2(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillory LA, Stewart JE, Megison ML, Nabers HC, Mroczek-Musulman E, Beierle EA. FAK Inhibition Decreases Hepatoblastoma Survival Both In Vitro and In Vivo. Transl Oncol. 2013;6(2):206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephenson FH, ScienceDirect. Calculations for molecular biology and biotechnology : a guide to mathematics in the laboratory, Chapter 3 - Cell Growth: Academic Press; 2010. [Google Scholar]
- 15.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer A, Green J, Pollard J, Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, et al. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009;5(3):e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–8. [DOI] [PubMed] [Google Scholar]
- 21.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. [DOI] [PubMed] [Google Scholar]
- 22.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270(1):41–9. [DOI] [PubMed] [Google Scholar]
- 23.Graham D, Magee H, Kierce B, Ball R, Dervan P, O’Meara A. Evaluation of Ki-67 reactivity in neuroblastoma using paraffin embedded tissue. Pathol Res Pract. 1995;191(2):87–91. [DOI] [PubMed] [Google Scholar]
- 24.Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 Axis in Cancer Progression. Cancers (Basel). 2020;12(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7(5):812–21. [DOI] [PubMed] [Google Scholar]
- 26.Eichmann A, Yuan L, Breant C, Alitalo K, Koskinen PJ. Developmental expression of pim kinases suggests functions also outside of the hematopoietic system. Oncogene. 2000;19(9):1215–24. [DOI] [PubMed] [Google Scholar]
- 27.Yan B, Yau EX, Samanta S, Ong CW, Yong KJ, Ng LK, et al. Clinical and therapeutic relevance of PIM1 kinase in gastric cancer. Gastric Cancer. 2012;15(2):188–97. [DOI] [PubMed] [Google Scholar]
- 28.Weirauch U, Beckmann N, Thomas M, Grunweller A, Huber K, Bracher F, et al. Functional role and therapeutic potential of the pim-1 kinase in colon carcinoma. Neoplasia. 2013;15(7):783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellon M, Lu L, Nicot C. Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood. 2016;127(20):2439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen AM, Grinblat B, Bessler H, Kristt D, Kremer A, Schwartz A, et al. Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma. 2004;45(5):951–5. [DOI] [PubMed] [Google Scholar]
- 31.Li YY, Popivanova BK, Nagai Y, Ishikura H, Fujii C, Mukaida N. Pim-3, a proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad-mediated apoptosis in human pancreatic cancer cell lines. Cancer Res. 2006;66(13):6741–7. [DOI] [PubMed] [Google Scholar]
- 32.Fujii C, Nakamoto Y, Lu P, Tsuneyama K, Popivanova BK, Kaneko S, et al. Aberrant expression of serine/threonine kinase Pim-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int J Cancer. 2005;114(2):209–18. [DOI] [PubMed] [Google Scholar]
- 33.Li YY, Mukaida N. Pathophysiological roles of Pim-3 kinase in pancreatic cancer development and progression. World J Gastroenterol. 2014;20(28):9392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikkers H, Nawijn M, Allen J, Brouwers C, Verhoeven E, Jonkers J, et al. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol Cell Biol. 2004;24(13):6104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Lugt NM, Domen J, Verhoeven E, Linders K, van der Gulden H, Allen J, et al. Proviral tagging in E mu-myc transgenic mice lacking the Pim-1 proto-oncogene leads to compensatory activation of Pim-2. EMBO J. 1995;14(11):2536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu Y, Zhang C, Du E, Wang A, Yang Y, Guo J, et al. Pim-3 is a Critical Risk Factor in Development and Prognosis of Prostate Cancer. Med Sci Monit. 2016;22:4254–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhuang H, Zhao MY, Hei KW, Yang BC, Sun L, Du X, et al. Aberrant expression of pim-3 promotes proliferation and migration of ovarian cancer cells. Asian Pac J Cancer Prev. 2015;16(8):3325–31. [DOI] [PubMed] [Google Scholar]
- 38.Liu B, Wang Z, Li HY, Zhang B, Ping B, Li YY. Pim-3 promotes human pancreatic cancer growth by regulating tumor vasculogenesis. Oncol Rep. 2014;31(6):2625–34. [DOI] [PubMed] [Google Scholar]
- 39.Leroux AE, Biondi RM. Renaissance of Allostery to Disrupt Protein Kinase Interactions. Trends Biochem Sci. 2020;45(1):27–41. [DOI] [PubMed] [Google Scholar]
- 40.Nair JR, Caserta J, Belko K, Howell T, Fetterly G, Baldino C, et al. Novel inhibition of PIM2 kinase has significant anti-tumor efficacy in multiple myeloma. Leukemia. 2017;31(8):1715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Narlik-Grassow M, Blanco-Aparicio C, Cecilia Y, Peregrina S, Garcia-Serelde B, Munoz-Galvan S, et al. The essential role of PIM kinases in sarcoma growth and bone invasion. Carcinogenesis. 2012;33(8):1479–86. [DOI] [PubMed] [Google Scholar]
- 42.Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, et al. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clin Cancer Res. 2014;20(7):1834–45. [DOI] [PubMed] [Google Scholar]
- 43.Sharpe JJ, Cooper TA. Unexpected consequences: exon skipping caused by CRISPR-generated mutations. Genome Biol. 2017;18(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joberty G, Falth-Savitski M, Paulmann M, Bosche M, Doce C, Cheng AT, et al. A Tandem Guide RNA-Based Strategy for Efficient CRISPR Gene Editing of Cell Populations with Low Heterogeneity of Edited Alleles. CRISPR J. 2020;3(2):123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids. 2015;4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015;12(4):673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heck JE, Meyers TJ, Lombardi C, Park AS, Cockburn M, Reynolds P, et al. Case-control study of birth characteristics and the risk of hepatoblastoma. Cancer Epidemiol. 2013;37(4):390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turusov VS, Torii M, Sills RC, Willson GA, Herbert RA, Hailey JR, et al. Hepatoblastomas in mice in the US National Toxicology Program (NTP) studies. Toxicol Pathol. 2002;30(5):580–91. [DOI] [PubMed] [Google Scholar]
- 49.Horton JD, Lee S, Brown SR, Bader J, Meier DE. Survival trends in children with hepatoblastoma. Pediatr Surg Int. 2009;25(5):407–12. [DOI] [PubMed] [Google Scholar]
- 50.Mertens B, Cristina de Araujo Nogueira T, Topalis D, Stranska R, Snoeck R, Andrei G. Investigation of tumor-tumor interactions in a double human cervical carcinoma xenograft model in nude mice. Oncotarget. 2018;9(31):21978–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y, Wang YY, Nakamoto Y, Li YY, Baba T, Kaneko S, et al. Accelerated hepatocellular carcinoma development in mice expressing the Pim-3 transgene selectively in the liver. Oncogene. 2010;29(15):2228–37. [DOI] [PubMed] [Google Scholar]
- 52.Chang JC. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore). 2016;95(1 Suppl 1):S20–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jensen JB, Parmar M. Strengths and limitations of the neurosphere culture system. Mol Neurobiol. 2006;34(3):153–61. [DOI] [PubMed] [Google Scholar]
- 54.Villodre ES, Kipper FC, Pereira MB, Lenz G. Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat Rev. 2016;51:1–9. [DOI] [PubMed] [Google Scholar]
- 55.Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014;135(12):2741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mamun MA, Mannoor K, Cao J, Qadri F, Song X. SOX2 in cancer stemness: tumor malignancy and therapeutic potentials. J Mol Cell Biol. 2020;12(2):85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neradil J, Veselska R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015;106(7):803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bahnassy AA, Fawzy M, El-Wakil M, Zekri AR, Abdel-Sayed A, Sheta M. Aberrant expression of cancer stem cell markers (CD44, CD90, and CD133) contributes to disease progression and reduced survival in hepatoblastoma patients: 4-year survival data. Transl Res. 2015;165(3):396–406. [DOI] [PubMed] [Google Scholar]
- 59.Rikhi RR, Spady KK, Hoffman RI, Bateman MS, Bateman M, Howard LE. Hepatoblastoma: A Need for Cell Lines and Tissue Banks to Develop Targeted Drug Therapies. Front Pediatr. 2016;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costantini S, Di Bernardo G, Cammarota M, Castello G, Colonna G. Gene expression signature of human HepG2 cell line. Gene. 2013;518(2):335–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.